

Abstract
SMARCB1/INI1 is one of the core subunit proteins of the ATP‐dependent SWI/SNF chromatin remodeling complex, and is identified as a potent and bona fide tumor suppressor. Interactions have been demonstrated between SMARCB1/INI1 and key proteins in various pathways related to tumor proliferation and progression: the p16‐RB pathway, WNT signaling pathway, sonic hedgehog signaling pathway and Polycomb pathway. Initially, no detectable SMARCB1/INI1 protein expression was found in malignant rhabdoid tumor cells, whereas all other kinds of tumor cells and non‐tumorous tissue showed SMARCB1/INI1 protein expression. Therefore, immunohistochemical testing for the SMARCB1/INI1 antibody has been considered useful in confirming the histologic diagnosis of malignant rhabdoid tumors. However, recently, aberrant expression of SMARCB1/INI1 has been found in various tumors such as epithelioid sarcomas, schwannomatosis, synovial sarcomas, and so on. In addition, it has been reported that aberrant expression can be classified into three patterns: complete loss, mosaic expression and reduced expression. Although the various pathways related to mechanisms of tumorigenesis and tumor proliferation are complexly intertwined, the clarification of these mechanisms may contribute to therapeutic strategies in SMARCB1/INI1‐deficient tumors. In terms of pathological classifications, SMARCB1/INI1‐deficient tumors may be re‐classified by genetic backgrounds.
Keywords: Atypical teratoid/rhabdoid tumor, chromatin remodeling, Malignant rhabdoid tumor, SMARCB1/INI1, SWI/SNF
SMARCB1 (SWI/SNF‐related matrix‐associated actin‐dependent regulator of chromatin subfamily B member 1), which is also named INI1 (integrase interactor 1), is one of the core subunit proteins in the SWI/SNF (SWItch/Sucrose Non‐Fermentable) ATP‐dependent chromatin remodeling complex encoded at chromosomal position 22q11.2.1 SMARCB1 was first identified as a gene essential for glucose‐repressible genes in Saccharomyces cerevisiae.2 Characterization of the SMARCB1/INI1 gene revealed that it encodes glutamine‐ and proline‐rich domains characteristic of activation domains.3 As for the protein, a yeast 2‐hybrid screen designed to identify host proteins that could interact with HIV‐1 integrase first, identified the human homologue of SNF5, which was then named SMARCB1,4 and a human polypeptide corresponding in sequence to yeast SNF5 was isolated using a yeast 2‐hybrid screen in WI38 human fibroblasts.5
SMARCB1/INI1 is ubiquitously expressed in the nuclei of all normal cells.6 Disruption of SMARCB1/INI1 expression in mice results in early embryonic lethality: SMARCB1/INI1‐null embryos die between 3.5 and 5.5 days post‐coitum.7 SMARCB1/INI1 heterozygous‐deficient mice and those with conditional ablation of SMARCB1/INI1 develop aggressive cancer including rhabdoid‐like tumors and T‐cell lymphomas at a median onset of only 11 weeks.8, 9, 10
This embryonic lethality is rapid compared with other tumor suppressors. For example, p53 inactivation leads to cancer at 20 weeks, p19Arf loss at 38 weeks, and p16Ink4a loss at 60 weeks. Thus, the rapid onset and complete penetrance of cancer following inactivation of SMARCB1/INI1 establishes this gene as a potent and bona fide tumor suppressor.11, 12
Function of SMARCB1/INI1
Role of SMARCB1/INI1 in the p16‐RB pathway
The p16 tumor suppressor protein functions as an inhibitor of CDK4 and CDK6, the D‐type cyclin‐dependent kinases that initiate phosphorylation of the retinoblastoma tumor suppressor protein (Rb) and activate the E2F transcription factor.13 Rb represses gene transcription, required for the transition from G0/G1 to S phase, by directly binding to the transactivation domain of E2F.14 Thus, p16 has the capacity to arrest cells in the G1‐phase of the cell cycle.13
Reintroduction of SMARCB1/INI1 into malignant rhabdoid tumor cell lines having SMARCB1/INI1 deficiency induced the accumulation of cells in G0/G1, and, in some cases, cell senescence or apoptosis.15, 16 These findings resulted from G0/G1 cell cycle arrest associated with transcriptional repression of Cyclin D1, induction of P16, and hypophosphorylation of RB.16, 17 This repression of Cyclin D1 transcription was associated with direct recruitment of HDAC activity to the Cyclin D1 promoter.16, 17, 18
Previous studies have suggested that SMARCB1/INI1 suppresses tumor progression by signaling through the p16INK4a and retinoblastoma tumor suppressors to negatively regulate cell cycle progression from G0/G1 to the S‐phase.19 It was recently reported that SMARCB1/INI1 signals via the p16INK4a‐Rb‐E2F pathway regulate chromosomal stability, suggesting a new function in tumor suppression for this chromatin‐remodeling protein.20
Role of SMARCB1/INI1 in the canonical WNT pathway
Traditionally, WNT signaling pathways have been characterized by two large categories: the canonical WNT (or β‐catenin‐dependent) and non‐canonical WNT (or β‐catenin‐independent) pathways. Biologically, the canonical WNT signaling pathway usually plays important roles regulating cell fate, proliferation and survival, and its aberrant activation is found in several types of human cancer, whereas non‐canonical WNT signaling is more associated with differentiation, cell polarity and migration.21
SMARCB1/INI1 deficiency in the developing limb mesenchyme leads to aberrant activation of the canonical WNT pathway and to phenotypic defects consistent with WNT/β‐catenin overexpression.22 In SMARCB1/INI1‐deficient tumors, WNT targets are elevated compared with those in the normal cerebellum, and aberrant activation of β‐catenin target genes occurs independently of canonical WNT pathway activation.22 Thus, SMARCB1/INI1 deficiency causes aberrant activation of the WNT signaling pathway and results in phenotypic defects consistent with WNT/β‐catenin overexpression.22
Role of SMARCB1/INI1 in the sonic hedgehog signaling pathway
The sonic hedgehog (Shh) signaling pathway is a major regulator of cell differentiation, cell proliferation, and tissue polarity.23 Tumorigenesis, tumor progression and therapeutic response have all been shown to be impacted by the Shh signaling pathway.23 Hedgehog signal transduction is initiated by the binding of Hh proteins to the Patched 1 protein (Ptch1). Ptch1 inhibits the activity of a smoothened (SMO) protein that activates factors downstream of the Hh signaling pathway when those ligands are not bound to Ptch1.24, 25 SMO stimulates a signaling cascade that results in the activation of the glioma‐associated oncogene homolog (GLI) family of zinc finger transcription factors (GLI1, GLI2, and GLI3), when those ligands are bound to Ptch1.24, 25
SMARCB1/INI1 was found to localize to the upstream regions of the transcription start sites of GLI1 and Ptch1. Sh‐RNA‐mediated knockdown of SMARCB1/INI1 leads to upregulation of the GLI1 and Ptch1 expressions, and to activation of the Shh signaling pathway.26 Conversely, re‐expression of SMARCB1/INI1 in malignant rhabdoid tumor cell lines represses GLI1 expression.26 Clinical cases of primary SMARCB1/INI1‐deficient tumors (malignant rhabdoid tumor and atypical teratoid/rhabdoid tumor) showed enrichment of gene expression associated with Shh signaling pathway activation and GLI1 overexpression signatures which often possess activating mutations in the Shh signaling pathway.26 Therefore, SMARCB1/INI1 is identified as one of the top regulators of GLI1, and is a key mediator of Shh signaling pathway.26
Role of SMRCB1/INI1 in the Polycomb pathway
Activations of Polycomb proteins contribute to epigenetically based gene silencing during the developmental processes of proliferation, and it has been suggested that these proteins may serve important roles during oncogenic transformation.27 Polycomb proteins form two distinct multiprotein repressive complexes, PRC1 and PRC2. EZH2, which is the functional enzymatic component of PRC2, is highly expressed in various cancers, and is often correlated with tumor progression and poor prognosis, although the mechanisms underlying the upregulation of EZH2 are poorly understood.27 EZH2 plays an important role as the catalytic subunit in PRC2 and mediates gene silencing by catalyzing the trimethylation of histone 3 lysine 27 (H3K27me3) at the promoter regions of target genes.27, 28
SMARCB1/INI1‐deficient tumor samples also express higher levels of EZH2.27 EZH2 transcription is directly repressed by SMARCB1/INI1 in mouse embryonic fibroblasts.27 SMARCB1/INI1 deficiency leads to broad repression of lineage‐specific Polycomb‐regulated genes, and this repression is dependent on the presence of EZH2.27 SMARCB1/INI1 deficiency causes elevated levels of H3K27me3 at lineage‐specific Polycomb targets.27 Thus, SMARCB1/INI1 deficiency mechanistically leads to elevated expression and recruitment of EZH2 to Polycomb targets, the trimethylation of histone 3 lysine 27, and the ultimate repression of Polycomb genes in SMARCB1‐deficient fibroblasts and cancers.27 In SMARCB1/INI1‐deficient malignant rhabdoid tumors, inhibition of EZH2 functions as a SMARCB1/INI1 surrogate and derepresses neural differentiation genes, cell cycle inhibitors, and tumor suppressors while reducing GLI1, Patch1, MYC and EZH2.29
Other targets of SMARCB1/INI1
It has been reported that the other targets of SMARCB1/INI1 are c‐MYC and Aurora A. C‐MYC, which is a regulator gene that codes for a transcription factor, plays a role in cell cycle progression, apoptosis and cell transformation. C‐MYC is known to be significantly upregulated in SMARCB1/INI1‐deficient malignant rhabdoid tumors.30 Recruitment of the SWI/SNF complex, mediated by the interaction of INI1 with c‐MYC, facilitates the transcription of a discrete subset of c‐MYC target genes, especially those involved in apoptosis, which might explain the tumor‐suppressor activity of SMARCB1/INI1.31, 32
Aurora A, which is a member of a family of mitotic serine/threonine kinases, is implicated with important processes during mitosis and meiosis, the proper functionings of which are critical for healthy cell proliferation. Aurora A is a direct downstream target of SMARCB1/INI1‐mediated repression in malignant rhabdoid tumor cells, and the loss of SMARCB1/INI1, which is required for their survival, leads to aberrant overexpression of Aurora A in these tumors.33
SMARCB1/INI1‐deficient tumors
In 1990, monosomy 22 as the only cytogenetic abnormality was found in three cases of atypical teratoid/rhabdoid tumors.34 In 1998, positional cloning and sequence analysis of malignant rhabdoid tumors eventually identified mutations, deletions and other somatic alterations in the SMARCB1/INI1 gene.35 After that, aberrant expression of the SMARCB1/INI1 protein has been reported to occur in various tumors.36, 37, 38, 39 At present, three patterns of aberrant SMARCB1/INI1 expression‐ complete loss, mosaic expression and reduced expression‐ have been identified (Table 1).
Table 1.
SMARCB1/INI1‐deficient tumors
| Complete loss group |
| Malignant rhabdoid tumor (atypical teratoid/rhabdoid tumor) |
| Epithelioid sarcoma |
| Renal medullary carcinoma |
| Epithelioid malignant peripheral nerve sheath tumor |
| Myoepithelial tumor |
| Extraskeletal myxoid chondrosarcoma |
| Pediatric chordoma |
| Pancreas undifferentiated rhabdoid carcinoma |
| Sinonasal basaloid carcinoma |
| Rhabdoid carcinoma of the gastrointestinal tract |
| Mosaic expression group |
| Schwannomatosis |
| Gastrointestinal stromal tumor |
| Ossifying fibromyxoid tumor |
| Reduced expression group |
| Synovial sarcoma |
Complete loss groups
Malignant rhabdoid tumor
Malignant rhabdoid tumors are classified as tumors of uncertain differentiation. Most such tumors present at birth or develop in infancy, and occur in the central nervous system, kidney and soft tissue.40 Almost all malignant rhabdoid tumors show complete loss of SMARCB1/INI1 expression (Fig. 1a,b).41, 42, 43, 44 In a small minority of cases, although SMARCB1/INI1 expression is preserved, SMARCA4/BRG1 is completely lost.45 SMARCB1/INI1 is inactivated homozygously in the majority cases of this kind of tumor by deletions and/or mutations.41, 46 However, about 20% cases with loss of SMARCB1/INI1 protein expression also showed no alteration at either the DNA or RNA level, and the mechanism of the inactivation of the SMARCB1/INI1 gene product was not clarified.41, 46
Figure 1.
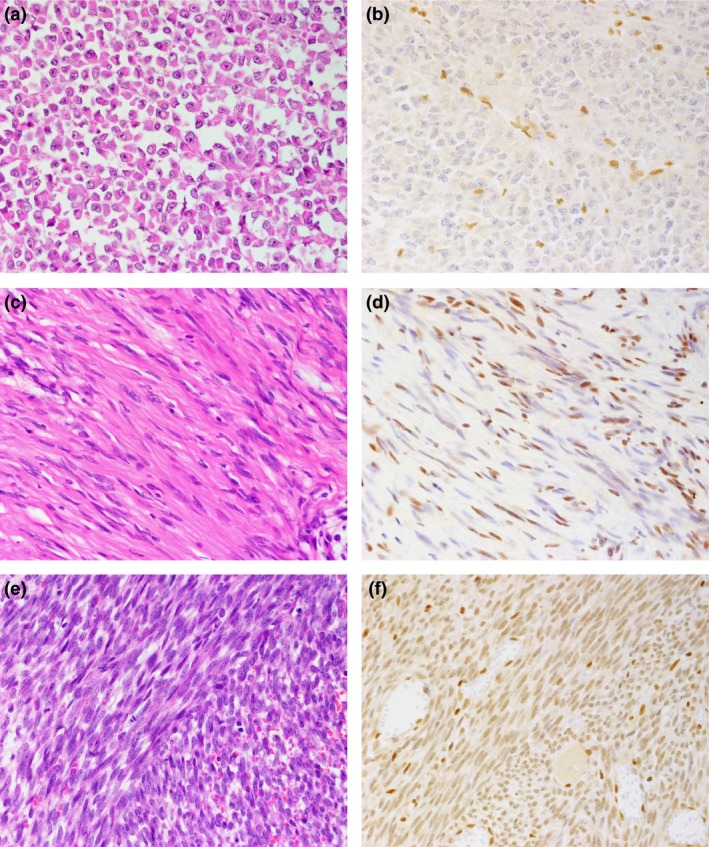
Hematoxylin–eosin histologic (a, c, e) and SMARCB1/INI1 (b, d, f) immunohistochemical findings. (a, b) Malignant rhabdoid tumor (2‐year‐old male; kidney). No nuclear expression of SMARCB1/INI1 protein is observed in tumor cells, whereas infiltrating lymphocytes or vascular endothelial cells disclose immunoreactivity (b). (c, d) Schwannomatosis (48‐year‐old woman; cauda equina). SMARCB1/INI1 protein expression is focally reduced with a mixture of nuclear‐positive and nuclear‐negative tumor cells, showing mosaic pattern (d). (e, f) Synovial sarcoma (22‐year‐old woman; abdominal wall). The tumor cells showed reduced expression of SMARCB1/INI1 protein compared with the positive control, which included infiltrating lymphocytes and entrapped normal tissue (f).
Epithelioid sarcoma
Epithelioid sarcoma is a rare soft tissue tumor displaying an uncertain line of differentiation. Two clinicopathologic subtypes are recognized: the conventional‐type, characterized by its proclivity for distal extremities and a pseudo‐granulomatous growth pattern; and proximal‐type, which arises mainly in the proximal extremities or truncal regions, and consists of nests and sheets of large epithelioid cells.47 Complete loss of SMARCB1/INI1 expression is found in 76–100% cases of proximal‐type and 81–93% cases of conventional‐type epithelioid sarcoma.48, 49, 50, 51, 52 The ratio of gene alteration at either the DNA or RNA level causing SMARCB1/INI1 protein inactivation varies widely between 0% and 58% in conventional‐type or between 19% and 100% in proximal‐type cases.48, 52, 53, 54 In addition, it is suggested that microRNAs such as miR193a‐5p, miR‐206, miR‐381 and miR‐671‐5p may have the potential to inhibit SMARCB1 mRNA in epithelioid sarcoma.55, 56, 57
Pancreatic undifferentiated rhabdoid carcinoma
Undifferentiated carcinoma of the pancreas that are predominantly (>50%) composed of rhabdoid cells is very rare with only approximately 60 cases reported previously in the medical literature.58 Agaimy et al.58 reported that 4 of the 14 this carcinoma shows the complete loss of SMARCB1/INI1 immunoexpression, and these all four cases are monomorphic anaplastic histology, whereas the remaining 10 cases are pleomorphic giant cell histology. In SMARCB1/INI1 deficient cases, three of the four cases lacked KRAS alterations (mutations and/or amplifications).58
SMARCB1/INI1‐deficient carcinoma of the sinonasal or gastrointestinal tract
SMARCB1/INI1‐deficiency has been also found in extremely small numbers of carcinoma of the sinonasal or gastrointestinal tract.59, 60, 61 Agaimy et al.59, 60, 61 reported three cases of sinonasal SMARCB1/INI1‐deficient basal cell carcinoma and five cases of SMARCB1/INI1‐negative rhabdoid carcinoma of the gastrointestinal tract.
Age/sex of these sinonasal basal cell carcinomas were as follows: 35 years old (years)/female; 52 years/male; 28 years/female.59 Histological features showed a few scattered rhabdoid cells, basaloid “blue” appearance, papilloma‐like exophytic component, extensive pagetoid surface growth with prominent denuding features, and replacement of underlying mucous glands mimicking an inverted papilloma.59 High‐risk human papilloma virus infection was negative in all cases.59
Age/sex/primary site of these rhabdoid carcinomas of gastrointestinal tract were as follows: 32 years old (years)/male/large intestine; 54 years/male/esophago‐gastric junction; 58 years/male/antrum; 79 years/male/cecum; 66 years/male/stomach body.61 Histological features showed rhabdoid cells and anaplastic large ones.61 Spindle‐shaped sarcomatoid cells or medullary structures were also found in each of one case.61
Other tumors
Complete loss of SMARCB1/INI1 protein expression has been reported to occur in all renal medullary carcinomas, about half of epithelioid malignant peripheral nerve sheath tumors, some myoepithelial tumor and some extraskeletal myxoid chondrosarcomas.6, 62, 63, 64, 65 Recently, pediatric chordoma with SMARCB1/INI1‐deficiency cases have also been reported.66 These tumors are known to have rhabdoid cells, which are characterized by the existence of a large eosinophilic inclusion within the cytoplasm, eccentric nuclei and prominent nucleoli.
Mosaic groups
Schwannomatosis
Schwannoma is benign peripheral nerve sheath neoplasms composed exclusively of Schwann cells.67 Schwannomatosis, which is a familial or sporadic syndrome, is classified into two major categories according to the absence of vestibular schwannomas and neurofibromatosis type 2 (NF2) pathology.68 Most cases of familial schwannomatosis (14/15; 93%) and NF2‐associated schwannomas (10/12; 83%), and some cases of sporadic schwannomatosis (10/18; 55%) show mosaic patterns of SMARCB1/INI1 protein expression (Fig. 1c,d).69 Genetically, most schwannomatosis patients show missense or splice‐site mutations of SMARCB1/INI1 genes at germline.70, 71 These mutations cause the replacement of an important amino‐acid residue or the in‐frame deletion or insertion of amino‐acid residues, resulting in the synthesis of a SMARCB1/INI1 protein with altered activity.70
Gastrointestinal stromal tumor
Gastrointestinal stromal tumor (GIST), which is the specific KIT‐positive mesenchymal tumor of the gastrointestinal tract, demonstrates a gain‐of‐function mutation of the KIT gene or the PDGFRA gene.72, 73 About half of GIST cases (17/27; 63%) show mosaic patterns of SMARCB1/INI1 protein expression.74 Genetically, among the 27 informative cases, 19 (70%) showed LOH of at least one of the microsatellite markers on 22q11.23 including the SMARCB1/INI1 gene.74 In another study, four of the seven metastatic GIST cases harbored a heterozygous deletion of part or the entire arm of chromosome 22, on which SMARCB1 is located.75
Ossifying fibromyxoid tumor
Ossifying fibromyxoid tumors, characterized by a lobular proliferation of small bland round cells with a peripheral shell of woven bone, are classified as tumors of uncertain differentiation.76, 77 Immunohistochemically, the mosaic pattern of SMARCB1/INI1 was noted in 14 of 19 (74%) cases.78 Genetically, although epigenetic events such as posttranslational modifications or small deletions or mutations are not detectable by FISH, five of seven cases showed an aberrant signal in the SMARCB1/INI1 gene by FISH. Five cases showed a hemizygous deletion of both SMARCB1/INI1 and PANX2 (the control probe) in >50% of cells, and three of those five had a second population of cells showing two signals for SMARCB1/INI1 and one signal for the control, suggesting loss of one copy of the 22q telomeric region.78 No cases with homozygous deletion of the SMARCB1/INI1 gene were found.78
Reduced group
Synovial sarcoma
Synovial sarcoma, which is classified as a tumor of uncertain differentiation, has three major histological subtypes: the monophasic type, biphasic type and poorly differentiated type.79 Genetically, a fusion of the SS18 gene to an SSX family member as the result of a chromosomal translocation, t(X;18), is recognized.79, 80, 81 Kohashi et al.81 first identified reduced SMARCB1/INI1 immuno‐expression in the majority of cases of synovial sarcoma (Fig. 1e,f). Kadoch et al.82 clarified the mechanism of reduced expression of the SMARCB1 protein: SS18‐SSX integrates into SWI/SNF complex, and wild‐type SS18 and SMARCB1 are displaced from the complex; then SMARCB1 is proteosomally degraded.
Conclusion
SMARCB1/INI1 plays an important role in various interwoven factors in several pathways (Fig. 2), and different cancers show different aberrant expression patterns of its protein. Although the several pathways related to mechanisms of tumorigenesis and tumor proliferation are intertwined in complex ways, the clarification of these mechanisms may contribute to therapeutic strategies in SMARCB1/INI1‐deficient tumors. In terms of pathological classification, SMARCB1/INI1‐deficient tumors may be re‐classified by their genetic backgrounds.
Figure 2.
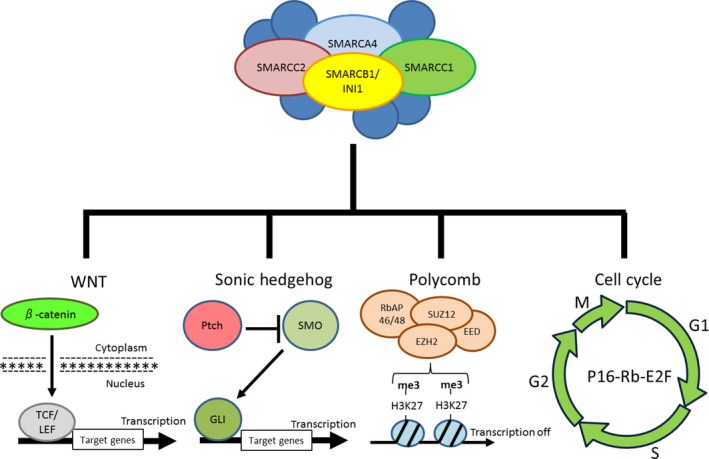
Target genes and pathways implicated in the tumor suppressor activity of SMARCB1/INI1.
Disclosure Statement
The authors have no conflict of interest to declare.
Acknowledgments
This study was supported by JSPS KAKENHI Grant Numbers 25293088 and 26460435. The English used in this article was revised by KN International (http://www.kninter.com/).
Cancer Sci 108 (2017) 547–552
Funding Information
Japan Society for the Promotion of Science (25293088; 26460435).
References
- 1. Roberts CW, Biegel JA. The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther 2009; 8: 412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abrams E, Neigeborn L, Carlson M. Molecular analysis of SNF2 and SNF5, genes required for expression of glucose‐repressible genes in Saccharomyces cerevisiae . Mol Cell Biol 1986; 6: 3643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Laurent BC, Treitel MA, Carlson M. The SNF5 protein of Saccharomyces cerevisiae is a glutamine‐ and proline‐rich transcriptional activator that affects expression of a broad spectrum of genes. Mol Cell Biol 1990; 10: 5616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalpana GV, Marmon S, Wang W et al Binding and stimulation of HIV‐1 integrase by a human homolog of yeast transcription factor SNF5. Science 1994; 266: 2002–6. [DOI] [PubMed] [Google Scholar]
- 5. Muchardt C, Sardet C, Bourachot B et al A human protein with homology to Saccharomyces cerevisiae SNF5 interacts with the potential helicase hbrm. Nucleic Acids Res 1995; 23: 1127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hollmann TJ, Hornick JL. INI1‐deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol 2011; 35: e47–63. [DOI] [PubMed] [Google Scholar]
- 7. Guidi CJ, Sands AT, Zambrowicz BP et al Disruption of Ini1 leads to peri‐implantation lethality and tumorigenesis in mice. Mol Cell Biol 2001; 21: 3598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roberts CW, Orkin SH. The SWI/SNF complex–chromatin and cancer. Nat Rev Cancer 2004; 4: 133–42. [DOI] [PubMed] [Google Scholar]
- 9. Roberts CW, Galusha SA, McMenamin ME et al Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci USA 2000; 97: 13796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts CW, Leroux MM, Fleming MD et al Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2002; 2: 415–25. [DOI] [PubMed] [Google Scholar]
- 11. Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene 2009; 28: 1653–68. [DOI] [PubMed] [Google Scholar]
- 12. Kim KH, Roberts CW. Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet 2014; 207: 365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ohtani N, Yamakoshi K, Takahashi A et al The p16INK4a‐RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest 2004; 51: 146–53. [DOI] [PubMed] [Google Scholar]
- 14. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene 2006; 25: 5220–7. [DOI] [PubMed] [Google Scholar]
- 15. Ae K, Kobayashi N, Sakuma R et al Chromatin remodeling factor encoded by ini1 induces G1 arrest and apoptosis in ini1‐deficient cells. Oncogene 2002; 21: 3112–20. [DOI] [PubMed] [Google Scholar]
- 16. Versteege I, Medjkane S, Rouillard D et al A key role of the hSNF5/INI1 tumour suppressor in the control of the G1‐S transition of the cell cycle. Oncogene 2002; 21: 6403–12. [DOI] [PubMed] [Google Scholar]
- 17. Betz BL, Strobeck MW, Reisman DN et al Re‐expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 2002; 21: 5193–203. [DOI] [PubMed] [Google Scholar]
- 18. Zhang ZK, Davies KP, Allen J et al Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol 2002; 22: 5975–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imbalzano AN, Jones SN. Snf5 tumor suppressor couples chromatin remodeling, checkpoint control, and chromosomal stability. Cancer Cell 2005; 7: 294–5. [DOI] [PubMed] [Google Scholar]
- 20. Vries RG, Bezrookove V, Zuijderduijn LM et al Cancer‐associated mutations in chromatin remodeler hSNF5 promote chromosomal instability by compromising the mitotic checkpoint. Genes Dev 2005; 19: 665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang K, Wang X, Zhang H et al The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest 2016; 96: 116–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mora‐Blanco EL, Mishina Y, Tillman EJ et al Activation of β‐catenin/TCF targets following loss of the tumor suppressor SNF5. Oncogene 2014; 33: 933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rimkus TK, Carpenter RL, Qasem S et al Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel) 2016; 8: pii: E22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol 2005; 6: 306–17. [DOI] [PubMed] [Google Scholar]
- 25. Paces‐Fessy M, Boucher D, Petit E et al The negative regulator of Gli, suppressor of fused (Sufu), interacts with SAP18, Galectin3 and other nuclear proteins. Biochem J 2004; 378: 353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jagani Z, Mora‐Blanco EL, Sansam CG et al Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog‐Gli pathway. Nat Med 2010; 16: 1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilson BG, Wang X, Shen X et al Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010; 18: 316–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pasini D, Di Croce L. Emerging roles for Polycomb proteins in cancer. Curr Opin Genet Dev 2016; 36: 50–8. [DOI] [PubMed] [Google Scholar]
- 29. Knutson SK, Warholic NM, Wigle TJ et al Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA 2013; 110: 7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gadd S, Sredni ST, Huang CC et al Rhabdoid tumor: gene expression clues to pathogenesis and potential therapeutic targets. Lab Invest 2010; 90: 724–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stojanova A, Penn LZ. The role of INI1/hSNF5 in gene regulation and cancer. Biochem Cell Biol 2009; 87: 163–77. [DOI] [PubMed] [Google Scholar]
- 32. Cheng SW, Davies KP, Yung E et al c‐MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet 1999; 22: 102–5. [DOI] [PubMed] [Google Scholar]
- 33. Lee S, Cimica V, Ramachandra N et al Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res 2011; 71: 3225–35. [DOI] [PubMed] [Google Scholar]
- 34. Biegel JA, Rorke LB, Packer RJ et al Monosomy 22 in rhabdoid or atypical tumors of the brain. J Neurosurg 1990; 73: 710–4. [DOI] [PubMed] [Google Scholar]
- 35. Versteege I, Sévenet N, Lange J et al Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998; 394: 203–6. [DOI] [PubMed] [Google Scholar]
- 36. Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 2011; 11: 481–92. [DOI] [PubMed] [Google Scholar]
- 37. Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet 2014; 166C: 350–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yaniv M. Chromatin remodeling: from transcription to cancer. Cancer Genet 2014; 207: 352–7. [DOI] [PubMed] [Google Scholar]
- 39. Margol AS, Judkins AR. Pathology and diagnosis of SMARCB1‐deficient tumors. Cancer Genet 2014; 207: 358–64. [DOI] [PubMed] [Google Scholar]
- 40. Oda Y, Biegel JA. Extrarenal rhabdoid tumour In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press, 2013; 228–9. [Google Scholar]
- 41. Kohashi K, Oda Y, Yamamoto H et al Highly aggressive behavior of malignant rhabdoid tumor: a special reference to SMARCB1/INI1 gene alterations using molecular genetic analysis including quantitative real‐time PCR. J Cancer Res Clin Oncol 2007; 133: 817–24. [DOI] [PubMed] [Google Scholar]
- 42. Kohashi K, Tanaka Y, Kishimoto H et al Reclassification of rhabdoid tumor and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression: three subtypes of rhabdoid tumor according to their histological features. Mod Pathol 2016; 29: 1232–42. [DOI] [PubMed] [Google Scholar]
- 43. Hoot AC, Russo P, Judkins AR et al Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra‐renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol 2004; 28: 1485–91. [DOI] [PubMed] [Google Scholar]
- 44. Sigauke E, Rakheja D, Maddox DL et al Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: an immunohistochemical study with implications for diagnosis. Mod Pathol 2006; 19: 717–25. [DOI] [PubMed] [Google Scholar]
- 45. Hasselblatt M, Gesk S, Oyen F et al Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 2011; 35: 933–5. [DOI] [PubMed] [Google Scholar]
- 46. Biegel JA, Tan L, Zhang F et al Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 2002; 8: 3461–7. [PubMed] [Google Scholar]
- 47. Oda Y, Dal Cin P, Laskin WB. Epithelioid sarcoma In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press, 2013; 216–8. [Google Scholar]
- 48. Modena P, Lualdi E, Facchinetti F et al SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 2005; 65: 4012–9. [DOI] [PubMed] [Google Scholar]
- 49. Chbani L, Guillou L, Terrier P et al Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol 2009; 131: 222–7. [DOI] [PubMed] [Google Scholar]
- 50. Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal‐type epithelioid sarcoma. Am J Surg Pathol 2009; 33: 542–50. [DOI] [PubMed] [Google Scholar]
- 51. Flucke U, Slootweg PJ, Mentzel T et al Re: Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor: direct evidence of mutational inactivation of SMARCB1/INI1 in epithelioid sarcoma. Hum Pathol 2009; 40: 1361–2. [DOI] [PubMed] [Google Scholar]
- 52. Kohashi K, Izumi T, Oda Y et al Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol 2009; 40: 349–55. [DOI] [PubMed] [Google Scholar]
- 53. Sullivan LM, Folpe AL, Pawel BR et al Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol 2013; 26: 385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Papp G, Changchien YC, Péterfia B et al SMARCB1 protein and mRNA loss is not caused by promoter and histone hypermethylation in epithelioid sarcoma. Mod Pathol 2013; 26: 393–403. [DOI] [PubMed] [Google Scholar]
- 55. Kohashi K, Yamamoto H, Kumagai R et al Differential microRNA expression profiles between malignant rhabdoid tumor and epithelioid sarcoma: miR193a‐5p is suggested to downregulate SMARCB1 mRNA expression. Mod Pathol 2014; 27: 832–9. [DOI] [PubMed] [Google Scholar]
- 56. Jamshidi F, Bashashati A, Shumansky K et al The genomic landscape of epithelioid sarcoma cell lines and tumours. J Pathol 2016; 238: 63–73. [DOI] [PubMed] [Google Scholar]
- 57. Papp G, Krausz T, Stricker TP et al SMARCB1 expression in epithelioid sarcoma is regulated by miR‐206, miR‐381, and miR‐671‐5p on Both mRNA and protein levels. Genes Chromosom Cancer 2014; 53: 168–76. [DOI] [PubMed] [Google Scholar]
- 58. Agaimy A, Haller F, Frohnauer J et al Pancreatic undifferentiated rhabdoid carcinoma: KRAS alterations and SMARCB1 expression status define two subtypes. Mod Pathol 2015; 28: 248–60. [DOI] [PubMed] [Google Scholar]
- 59. Agaimy A, Koch M, Lell M et al SMARCB1(INI1)‐deficient sinonasal basaloid carcinoma: a novel member of the expanding family of SMARCB1‐deficient neoplasms. Am J Surg Pathol 2014; 38: 1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Agaimy A, Rau TT, Hartmann A et al SMARCB1 (INI1)‐negative rhabdoid carcinomas of the gastrointestinal tract: clinicopathologic and molecular study of a highly aggressive variant with literature review. Am J Surg Pathol 2014; 38: 910–20. [DOI] [PubMed] [Google Scholar]
- 61. Agaimy A, Daum O, Märkl B et al SWI/SNF complex‐deficient undifferentiated/rhabdoid carcinomas of the gastrointestinal tract: a series of 13 cases highlighting mutually exclusive loss of SMARCA4 and SMARCA2 and frequent co‐inactivation of SMARCB1 and SMARCA2. Am J Surg Pathol 2016; 40: 544–53. [DOI] [PubMed] [Google Scholar]
- 62. Cheng JX, Tretiakova M, Gong C et al Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol 2008; 21: 647–52. [DOI] [PubMed] [Google Scholar]
- 63. Kohashi K, Oda Y, Yamamoto H et al SMARCB1/INI1 protein expression in round cell soft tissue sarcomas associated with chromosomal translocations involving EWS: a special reference to SMARCB1/INI1 negative variant extraskeletal myxoid chondrosarcoma. Am J Surg Pathol 2008; 32: 1168–74. [DOI] [PubMed] [Google Scholar]
- 64. Gleason BC, Fletcher CD. Myoepithelial carcinoma of soft tissue in children: an aggressive neoplasm analyzed in a series of 29 cases. Am J Surg Pathol 2007; 31: 1813–24. [DOI] [PubMed] [Google Scholar]
- 65. Jo VY, Fletcher CD. Epithelioid malignant peripheral nerve sheath tumor: clinicopathologic analysis of 63 cases. Am J Surg Pathol 2015; 39: 673–82. [DOI] [PubMed] [Google Scholar]
- 66. Antonelli M, Raso A, Mascelli S et al SMARCB1/INI1 involvement in pediatric chordoma: a mutational and immunohistochemical analysis. Am J Surg Pathol 2017; 41: 56–61. [DOI] [PubMed] [Google Scholar]
- 67. Antonescu CR, Perry A, Woodruff JM. Schwannoma (including variants) In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press, 2013; 170–2. [Google Scholar]
- 68. Carter JM, O'Hara C, Dundas G et al Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma‐like” schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol 2012; 36: 154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Patil S, Perry A, Maccollin M et al Immunohistochemical analysis supports a role for INI1/SMARCB1 in hereditary forms of schwannomas, but not in solitary, sporadic schwannomas. Brain Pathol 2008; 18: 517–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hulsebos TJ, Kenter S, Siebers‐Renelt U et al SMARCB1 involvement in the development of leiomyoma in a patient with schwannomatosis. Am J Surg Pathol 2014; 38: 421–5. [DOI] [PubMed] [Google Scholar]
- 71. Boyd C, Smith MJ, Kluwe L et al Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin Genet 2008; 74: 358–66. [DOI] [PubMed] [Google Scholar]
- 72. Miettinen MM, Corless CL, Debiec‐Rychter M et al Gastrointestinal stromal tumours In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press, 2013; 164–7. [Google Scholar]
- 73. Miettinen M, Lasota J, Sobin LH. Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long‐term follow‐up and review of the literature. Am J Surg Pathol 2005; 29: 1373–81. [DOI] [PubMed] [Google Scholar]
- 74. Yamamoto H, Kohashi K, Tsuneyoshi M et al Heterozygosity loss at 22q and lack of INI1 gene mutation in gastrointestinal stromal tumor. Pathobiology 2011; 78: 132–9. [DOI] [PubMed] [Google Scholar]
- 75. Saponara M, Urbini M, Astolfi A et al Molecular characterization of metastatic exon 11 mutant gastrointestinal stromal tumors (GIST) beyond KIT/PDGFRα genotype evaluated by next generation sequencing (NGS). Oncotarget 2015; 6: 42243–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Miettinen MM, Kawashima H, Weiss SW. Ossifying fibromyxoid tumour In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press, 2013; 206–7. [Google Scholar]
- 77. Endo M, Kohashi K, Yamamoto H et al Ossifying fibromyxoid tumor presenting EP400‐PHF1 fusion gene. Hum Pathol 2013; 44: 2603–8. [DOI] [PubMed] [Google Scholar]
- 78. Graham RP, Dry S, Li X et al Ossifying fibromyxoid tumor of soft parts: a clinicopathologic, proteomic, and genomic study. Am J Surg Pathol 2011; 35: 1615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Suurmeijer AJH, de Bruijn D, van Geurts Kessel A et al Synovial sarcoma In: Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, eds. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press, 2013; 213–5. [Google Scholar]
- 80. Ito J, Asano N, Kawai A et al The diagnostic utility of reduced immunohistochemical expression of SMARCB1 in synovial sarcomas: a validation study. Hum Pathol 2016; 47: 32–7. [DOI] [PubMed] [Google Scholar]
- 81. Kohashi K, Oda Y, Yamamoto H et al Reduced expression of SMARCB1/INI1 protein in synovial sarcoma. Mod Pathol 2010; 23: 981–90. [DOI] [PubMed] [Google Scholar]
- 82. Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18‐SSX oncogenic fusion in synovial sarcoma. Cell 2013; 153: 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]