

Abstract
Deregulation of the canonical Wnt signaling pathway plays an important role in human tumorigenesis through the accumulation of β‐catenin and subsequent transactivation of TCF7L2. Although some of the consequences associated with the accumulated β‐catenin have been clarified, the comprehensive effect of activated β‐catenin/TCF7L2 transcriptional complex on tumorigenesis remains to be elucidated. To understand the precise molecular mechanisms underlying colorectal cancer, we searched for genes regulated by the complex in colorectal tumors. We performed expression profile analysis of HCT116 and SW480 colon cancer cells treated with β‐catenin siRNAs, and ChIP‐sequencing using anti‐TCF7L2 antibody. Combination of these data with public microarray data of LS174 cells with a dominant‐negative form of TCF7L2 identified a total of 11 candidate genes. In this paper, we focused on FERM domain‐containing protein 5 (FRMD5), and confirmed that it is regulated by both β‐catenin and TCF7L2. An additional reporter assay disclosed that a region in intron1 transcriptionally regulated the expression of FRMD5. ChIP assay also corroborated that TCF7L2 associates with this region. These data suggested that FRMD5 is a novel direct target of the β‐catenin/TCF7L2 complex.
Keywords: Colorectal cancer, FRMD5, TCF7L2, Wnt signaling, β‐catenin
Colorectal carcinogenesis involves deregulation of several pathways including Wnt, RAS‐MAPK, PI3K‐AKT, TP53, and mismatch‐repair pathways.1 The impairment of Wnt signaling is one of the earliest steps because APC mutations are frequently detected in adenomas of the colon as well as carcinomas, suggesting that this pathway plays a critical role in the tumorigenesis. Wnt/β‐catenin or canonical Wnt signaling is a highly conserved signal‐transduction pathway, controlling embryonic development and adult stem cell renewal through the regulation of downstream genes.2, 3 It has been shown that intestinal epithelium cells have a high turnover rate4 and that Wnt/β‐catenin signaling contributes to the tissue homeostasis through the control of their proliferation.5 The activity of Wnt signaling is strictly regulated at many levels. In normal condition, cytoplasmic level of β‐catenin, a key component of the canonical Wnt signaling, is down‐regulated through its phosphorylation by a destruction complex including APC, GSK‐3β, and AXIN1/2, and subsequent ubiquitin‐dependent degradation.6 Upon the binding of Wnt ligands with Frizzled receptors, Dishevelled (Dvl/Dsh) induces suppression of GSK‐3β, which results in its stabilization and activation of T‐cell factor/lymphoid enhancer factor (TCF/LEF) through the interaction with β‐catenin.7 In colorectal tumors, mutations in APC or the β‐catenin gene itself lead to abnormal accumulation of β‐catenin. Abundantly expressed in colon epithelial cells, TCF7L2, one of the TCF/LEF family members, forms a complex with β‐catenin and subsequently activates transcription of its downstream genes. Although previous studies have identified a number of Wnt target genes such as MYC and CCND1,8, 9 the entire downstream players have not been clarified.
To understand the comprehensive role of canonical Wnt signaling in the development and progression of colorectal cancer (CRC), we attempted to identify novel target genes of Wnt/β‐catenin signaling. In this study, we have integrated data of genome‐wide TCF7L2 chromatin occupancy into the expression profile data that are regulated by both β‐catenin and TCF7L2. These analyses revealed that FERM domain‐containing protein 5 (FRMD5) is a novel target of the β‐catenin/TCF7L2 complex.
Materials and Methods
Cell culture
Human CRC cell lines, HCT116, HCT‐15, SW480, DLD‐1, LoVo, Caco‐2, LS174T, and HT‐29 were purchased from the American Type Culture Collection (Manassas, VA, USA). All cells were grown in appropriate media (McCoy's 5a Medium Modified for HCT116 and HT‐29; RPMI‐1640 for HCT‐15 and DLD‐1; Leibovitz's L‐15 for SW480, F‐12K for LoVo; EMEM for Caco‐2 and LS174T) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA), and antibiotic/antimycotic solution (Sigma, St. Louis, MO, USA).
Identification of genes regulated by the β‐catenin/TCF7L2 complex
Human CTNNB1‐specific siRNA (ON‐TARGETplus SMARTpool siRNA, L‐003482‐00) and control siRNA (ON‐TARGETplus Non‐targeting Pool #D‐001810‐10) were purchased (GE Dharmacon, Lafayette, CO, USA). HCT116 and SW480 cells that show transactivated β‐catenin/TCF7L2 complex due to either mutation in β‐catenin or APC, respectively, were treated with the pooled β‐catenin siRNA or control siRNA using Lipofectamine RNAiMAX (Thermo Fisher Scientific). RNA was extracted from the cells using RNeasy Plus mini Kit (Qiagen, Venlo, Netherlands), and subsequently expression profiles were analyzed by SurePrint G3 Unrestricted Gene Expression 8 × 60K microarray (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's protocol. In addition, microarray data of LS174T cells treated with a dominant negative form of TCF7L2 were obtained from Gene Expression Omnibus (GSE46465, https://www.ncbi.nlm.nih.gov/geo/). For validation, real‐time PCR was performed using qPCR Kapa SYBR Fast ABI Prism Kit (Kapa Biosystems, Wilmington, MA, USA) on StepOnePlus (Thermo Fisher Scientific). cDNA was synthesized using Transcriptor First Strand cDNA Synthesis Kit (Roche diagnostics, Basel, Switzerland), with RNA extracted from both HCT116 and SW480 cells treated with β‐catenin siRNA (siβ‐catenin #9 sense: 5′‐GAUCCUAGCUAUCGUUCUU‐3′; siβ‐catenin #10 sense: 5′‐UAAUGAGGACCUAUACUUA‐3′) or control siRNA. Primer sets used for the PCR are shown in Table S1.
Chromatin immunoprecipitation with high throughput sequencing (ChIP‐seq)
HCT116 cells were cross‐linked with 1% formaldehyde for 10 min at room temperature and quenched with 0.4 M glycine. Chromatin extracts were sheared by micrococcal nuclease digestion, and subsequently protein‐DNA complexes were immunoprecipitated with 10 μg of anti‐TCF7L2 antibody (#05‐511, EMD; Millipore, Billerica, MA, USA). Normal mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used as a negative control. The precipitated protein‐DNA complexes were purified by the conventional DNA extraction method, and the DNAs were subjected to next generation sequencing or quantitative PCR analysis. The next generation sequencing was carried out by Ion Proton System according to the manufacturer's protocol (Thermo Fisher Scientific), and the data were analyzed using Hotelling Observer, a shape‐based peak caller in CLC Genomics Workbench version 6.0 (Qiagen). In the calling process, peaks were primarily filtered by reads >49, normalized difference <0.33, and FDR<0.1%. Peaks located in the loci or flanking regions (5′ and 3′) of genes were further selected for the screening. ChIP‐qPCR was performed using a set of primers (forward primer, 5′‐CGCCAGATGCTCAACTAGAA‐3′ and reverse primer; 5′‐CAATCTCTTGCTGCCACAAA‐3′). Primer sets of intron1 and an intergenic region of RNF43 were used as a positive and a negative control, respectively.10
Gene expression analysis
RNA was extracted from HCT116 cells treated with two different FRMD5 siRNAs (siFRMD5 #2 sense: 5′‐CUUACAUCCUUCAAGCGGA‐3′, siFRMD5 #3 sense: 5′‐GACAGAUAGCAAUGAGCGA‐3′) or control siRNA (ON‐TARGETplus Non‐targeting Pool #D‐001810‐10, GE Dharmacon), and subsequently used for the gene expression profile analysis by microarray with SurePrint G3 Unrestricted Gene Expression 8 × 60K microarray (Agilent Technologies). For validation, real‐time PCR was performed with a set of primers shown in Table S1. To elucidate the role of FRMD5 as a downstream target of canonical Wnt signaling in colon cancer, we selected genes whose expression levels were commonly altered more than two‐fold by the FRMD5 siRNA and the β‐catenin siRNA in HCT116 cells. Additionally, gene set enrichment analysis was performed using MSigDB (Molecular signatures database, http://www.broadinstitute.org/gsea/msigdb/index.jsp) with gene sets derived from Canonical pathways, BioCarta, KEGG, and Reactome.
Reporter plasmids and luciferase assay
Reporter plasmids containing a putative promoter region of FRMD5 were constructed by cloning its 5′‐flanking region (−1255 to +112) into the XhoI and BglII restriction enzyme sites of pGL4.14 (Promega, Madison, WI, USA). Reporter plasmids containing a putative enhancer region of FRMD5 were constructed by cloning a region within FRMD5 intron1 (hg19‐chr15:44,449,571‐44,450,548) into KpnI and XhoI restriction enzyme sites of pGL4.23 (Promega). The putative promoter and enhancer regions were amplified by PCR using genomic DNA from healthy volunteers as a template with a set of primers (forward: 5′‐CCGCTCGAGCAGCATTAATGTTCTATGTT‐3′, and reverse: 5′‐GGAAGATCTCCAGGCACCTGCACCAT‐3′), and another set (forward: 5′‐CGGGGATCCGACAGGGCTTAAGGTCACAAC‐3′, and reverse: 5′‐CCGCTCGACTGGACCTTCAGACTGCTCTT‐3′), respectively. Mutant reporter plasmids were prepared by the substitution from CTTTCA to CGCTCA in the putative TCF7L2 binding sites using the reporter plasmid as a template with a set of primers (forward: 5′‐TGGATTTTCCTTTTCGCTCATCTCCTGAATTG‐3′, and reverse: 5′‐CAATTCAGGAGATGAGCGAAAAGGAAAATCCA‐3′) and Pfu DNA polymerase (Agilent Technologies). CRC cells seeded on 12‐well plates were transfected with the reporter plasmids together with pRL‐null (Promega) using FuGENE 6 reagent (Promega). The cells were harvested 48 h after transfection, and reporter activities were measured by dual luciferase system (TOYO B‐Net, Tokyo, Japan). For the knockdown of β‐catenin, HCT116 cells were transfected with 10 nM of CTNNB1 siRNA (siβ‐catenin #9 or siβ‐catenin #10) at 6 h after seeding, and incubated for an additional 48 h.
Western blotting
Total protein was extracted from cultured cells using radioimmunoprecipitation assay buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Nonidet P‐40, 0.1% SDS) supplemented with a Protease Inhibitor Cocktail Set III (Calbiochem, San Diego, CA, USA). Protein concentration was determined by BCA Protein Assay Kit (Thermo Fisher Scientific). Protein (30–50 μg/lane) was separated by 10% SDS‐PAGE and transferred to nylon transfer membranes. Primary antibodies used for western blotting were anti‐FRMD5 (HPA011746, Sigma), anti‐E‐Cadherin (ab1416, Abcam, Cambridge, UK) and anti‐β‐actin (a5441, Sigma) antibodies. Horseradish peroxidase‐conjugated goat anti‐mouse or anti‐rabbit IgG (GE Healthcare, Buckinghamshire, UK) served as the secondary antibody for the ECL Detection System (GE Healthcare).
Cell cycle analysis
HCT116, DLD‐1, LS174T and HCT‐15 cells treated with/without siFRMD5 #2 and siFRMD5 #3 were harvested by trypsinization, fixed with 70% ethanol, and stored at −20°C until use. The cells were rehydrated with phosphate‐buffered saline (PBS), treated with ribonuclease A (2 mg/mL) at 37°C for 30 min, and incubated with propidium iodide (20 μg/mL) in PBS at room temperature for 30 min. After filtration with nylon mesh, the cells were applied for FACS analysis (FACSCalibur, Becton Dickinson, Franklin Lakes, NJ, USA).
Expression of FRMD5 in non‐cancerous epithelial cell
Frozen sections (10 μm in thickness) of non‐cancerous colon tissues were fixed in ice‐cold 70% ethanol for 1 min, washed with water for 30 s, and subsequently stained with hematoxylin and eosin. Epithelial cells in the mucosa were collected from crypt bases and crypt tips by laser microdissection using LMD 7000 (Leica Microsystems, Bensheim, Germany). Total RNA was extracted from the collected cells using miRNeasy mini kit (Qiagen) according to the manufacturer's instructions. Concentration of RNA was assessed by Qubit2 fluorometer (Invitrogen, CA, USA).
Transwell migration assay
HCT116 cells were transfected with siFRMD5 or control siRNA, and maintained in McCoy's 5a Medium Modified medium with 10% FBS for an additional 24 h. Then the cells were starved in medium without FBS for 24 h. Transwell migration assay was performed using Transwell chambers (Corning, Corning, NY, USA) with 8.0 μm pore size. Approximately 1.5 × 105 cells were seeded on the upper surface of the Transwell membranes, incubated in McCoy's 5a Medium Modified medium without FBS for 24 h at 37°C. The lower surface of the Transwell membrane was immersed to the medium with 10% FBS. The Transwell membrane was then fixed with methanol for 2 min and stained by hematoxylin solution for 10 min. Four microscopic fields were randomly chosen for the analysis.
Statistical analysis
Statistical analysis was carried out by R (https://www.r-project.org/). The unpaired Student's t‐test and Dunnett's test were used to determine the significance of experimental data.
Results
Identification of FRMD5 as a novel target gene of the β‐catenin/TCF7L2 complex
In order to find novel target genes of the β‐catenin/TCF7L2 complex, expression profile analysis was performed using HCT116 and SW480 cells that have increased transcriptional activity of β‐catenin/TCF7L2 complex due to either mutation in β‐catenin or APC, respectively. RNA was extracted from the cells treated with pooled β‐catenin or control siRNA. We selected genes whose expression levels were decreased in the cells treated with β‐catenin siRNA to less than two‐thirds of the cells with control siRNA, and identified a total of 2671 and 2886 genes in HCT116 and SW480 cells, respectively. Additional analysis of public microarray data discovered a total of 1118 genes whose expression levels were decreased to less than two‐thirds by a dominant‐negative form of TCF7L2 (dnTCF7L2) in LS174T cells. These data consequently identified a total of 134 genes that were commonly down‐regulated by the suppression of β‐catenin or TCF7L2 (Fig. 1a).
Figure 1.
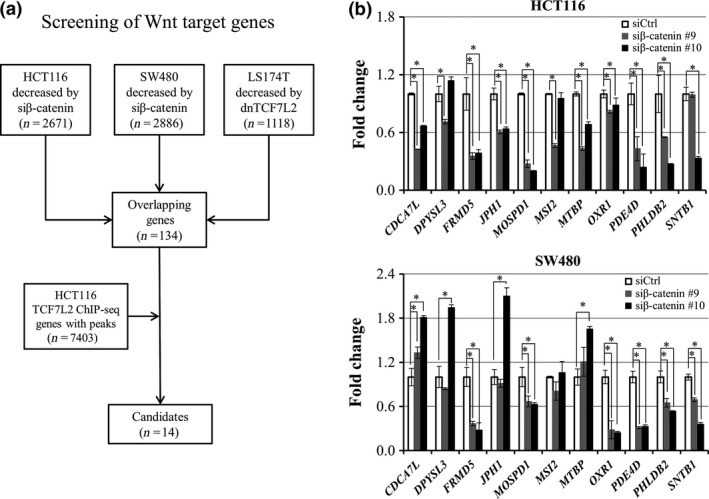
Identification of novel direct target genes of β‐catenin/TCF7L2. (a) Strategy for the identification of candidate target genes of β‐catenin/TCF7L2 complex. (b) Relative expression of the 11 candidate genes in HCT116 (left) and SW480 (right) cells treated with β‐catenin siRNA (gray and black boxes) compared to the cells treated with control siRNA (open boxes). Quantitative PCR was performed in triplicate (mean ± SD) and result was normalized by GAPDH. An asterisk indicates statistical significance (Dunnett's test, P < 0.05) between β‐catenin siRNA and control siRNA.
To identify genes that are directly regulated by the β‐catenin/TCF7L2 complex, we carried out ChIP‐seq of TCF7L2 in HCT116 cells (Fig. 1a). As a result, we identified a total of 7403 peaks that have significantly increased read‐counts compared with the background in the genome. Among the 134 candidate genes, 14 genes harbored at least one peak of the 7403 within a region between 10 kb upstream and downstream of the gene (Table 1). It is of note that the 14 genes included AXIN2, RNF43, and MYC, three well‐known direct targets of the complex.
Table 1.
Candidates of Wnt target gene
| Gene symbol | Fold change in microarray data | ||
|---|---|---|---|
| HCT116 with β‐catenin siRNA | SW480 with β‐catenin siRNA | LS174T with dnTCF7L2 | |
| AXIN2 a | −19.3 | −4.8 | −5.0 |
| CDCA7L | −1.9 | −1.6 | −2.0 |
| DPYSL3 | −1.6 | −1.5 | −2.1 |
| FRMD5 | −2.7 | −1.8 | −2.0 |
| JPH1 | −2.5 | −2.3 | −1.7 |
| MOSPD1 | −2.0 | −1.9 | −1.7 |
| MSI2 | −5.3 | −1.9 | −1.8 |
| MTBP | −2.2 | −1.7 | −1.7 |
| MYC a | −1.5 | −4.1 | −2.3 |
| OXR1 | −1.8 | −8.6 | −1.6 |
| PDE4D | −1.6 | −3.7 | −1.6 |
| PHLDB2 | −2.1 | −2.1 | −2.7 |
| RNF43 a | −5.4 | −1.8 | −2.7 |
| SNTB1 | −2.4 | −2.2 | −1.8 |
Reported target gene of β‐catenin/TCF7L2 complex.
We next focused on the remaining 11 genes and evaluated their expression using RNA extracted from HCT116 and SW480 cells treated with two β‐catenin siRNAs (Fig. 1b). Quantitative PCR analysis revealed that expression of PDE4D, PHLDB2, MOSPD1, and FRMD5 was significantly decreased with the two siRNAs in both cell lines. Expression of JPH1, CDCA7L, and MTBP was decreased with the two siRNAs in HCT116 but not in SW480. We first focused on FRMD5 because its function has not been studied in colon cancer cells. Consistent with the qPCR data, decreased FRMD5 protein was confirmed by western blot analysis in HCT116 cells (Fig. 2a). Another western blot analysis disclosed abundant expression of FRMD5 in HCT116, HCT‐15, DLD‐1, Caco‐2, HT‐29, and LS174T cells, and limited expression in SW480 and LoVo cells (Fig. 2b).
Figure 2.
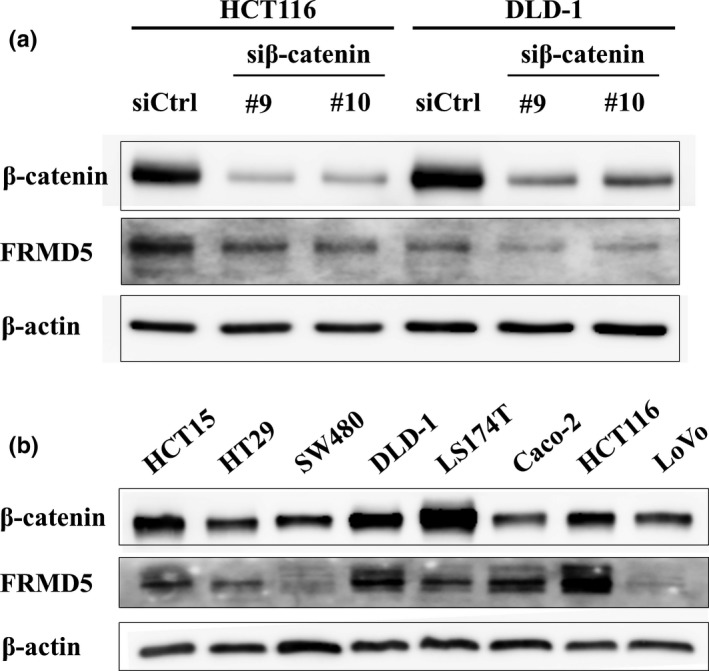
Expression of FRMD5 in CRC cell lines. (a) Reduced expression of FRMD5 protein by the knockdown of β‐catenin in HCT116 and DLD‐1 cells. (b) FRMD5 expression in eight CRC cell lines. Expression of β‐actin served as an internal control.
Expression of FRMD5 in colon cancer tissues
To clarify the involvement of FRMD5 in colorectal carcinogenesis, we searched for FRMD5 expression in ONCOMINE database (www.oncomine.org) (Fig. S2). Four of eight microarray datasets (TCGA Colorectal, Sabates‐Bellver Colon, Gaedcke Colorectal and Hong Colorectal datasets) showed significant increase of FRMD5 expression (≥2.0‐fold) in CRC tissues compared with normal colonic tissues (P < 0.01). In addition, three of the eight datasets (Skrzypczak Colorectal, Skrzypczak Colorectal 2 and Kaiser Colon datasets) displayed a slight increase of FRMD5 expression (<1.5‐fold) in CRC tissues. Although one dataset (Graudens Colon) revealed decreased expression of FRMD5 in CRC tissue, seven of the eight datasets showed elevated FRMD5 expression in CRC supporting our findings that the expression of FRMD5 is induced through the activation of the β‐catenin/TCF7L2 complex in CRC cells. In addition, we further investigated the relationship between the expression of FRMD5 and that of known Wnt target genes, AXIN2 and RNF43 in three microarray datasets (TCGA Colorectal, Sabates‐Bellver Colon, and Gaedcke Colorectal datasets). As a result, positive association between FRMD5 and AXIN2 was observed in Sabates‐Bellver Colon (r = 0.57) and Gaedcke Colorectal datasets (r = 0.56) (Fig. S3). In addition, similar positive association was found between FRMD5 and RNF43 in Sabates‐Bellver Colon (r = 0.55) and Gaedcke Colorectal datasets (r = 0.53). These data are completely consistent with the notion that FRMD5 is a downstream gene of Wnt signaling pathway. However, the positive correlation was not detected between FRMD5 and AXIN2 (r = 0.03) or FRMD5 and RNF43 (r = −0.02) in TCGA Colorectal dataset.
Expression of FRMD5 in non‐cancerous epithelial cells in the colon
Activation of Wnt signaling has been observed in the intestinal stem cells that express LGR5. The LGR5‐positive stem cells are mainly localized in the crypt bases of intestinal mucosa,11 and diminish the expression of LGR5 during their differentiation and movement along crypt‐villus axis.12 Hence, we investigated the expression of FRMD5 in crypt bases and tips of colonic mucosa. Consistently, we found approximately 20‐fold higher levels of LGR5 expression in crypt base cells compared with differentiated cells in the villi by quantitative PCR analysis. In addition, we observed 5.8‐fold higher expression of FRMD5 in crypt bases than tips, suggesting that FRMD5 is also augmented by the activation of Wnt signaling in normal colon mucosa (Fig. S1).
Identification of a TCF7L2‐interacting region in FRMD5
Since our ChIP‐seq data identified a TCF7L2‐binding peak located in a region (hg19; chr15:44,449,680‐44,450,487) within FRMD5 intron1 (Fig. 3a), we additionally searched public ChIP‐seq data in Encyclopedia of DNA Elements (ENCODE: www.encodeproject.org). Consistent with our results, data of HCT‐116 cells (wgENCODEEH000629) also demonstrated a TCF7L2‐interacting peak (hg19; chr15:44,449,944‐44,450,042) in intron1. Additional ChIP‐qPCR analysis corroborated that the DNA immunoprecipitated with anti‐TCF7L2 antibody contained 89.4‐fold higher concentration of the intronic region compared to that with control IgG. RNF43 intron1 which was used as a positive control showed 296.4‐fold higher concentration of DNA compared with control IgG (Fig. 3b).
Figure 3.
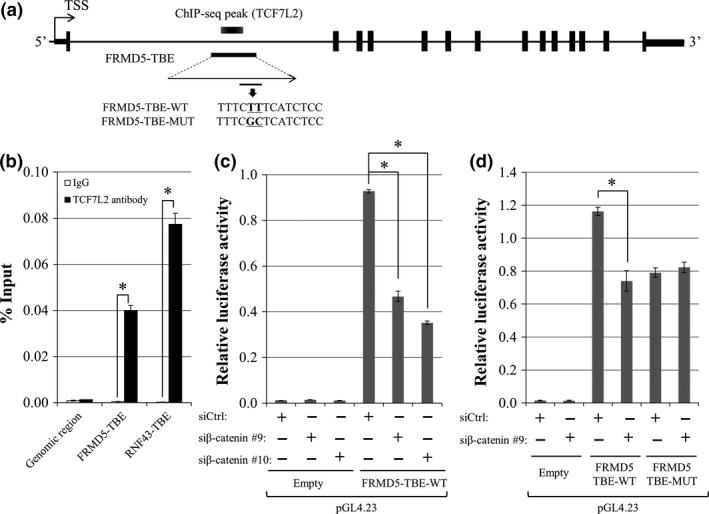
Identification of an FRMD5‐interacting region and its transcriptional activity. (a) Genomic structure of FRMD5 and the candidate FRMD5‐interacting region in intron1. (b) Association of the candidate region with TCF7L2 was analyzed by ChIP‐qPCR with anti‐TCF7L2 antibody. A TCF7L2‐binding region in RNF43 was used as a positive control. Quantitative PCR was performed in triplicate (mean ± SD). (c) Transcriptional activity of the candidate region. Reporter assay was performed using reporter plasmids containing the region with/without β‐catenin siRNA in HCT116 cells. (d) Luciferase activities of the wild type and mutant reporter plasmids. Luciferase activities were measured in triplicate (mean ± SD). An asterisk indicates statistical significance (Student's t‐test or Dunnett's test, P < 0.05).
Enhancer activity of the TCF7L2‐interacting region
The ENCODE data additionally depicted a peak in the 5′‐flanking region (hg19; chr15:44,487,635‐44,487,877) of FRMD5 which our ChIP‐seq data failed to identify. To analyze the transcriptional activity of the 5′‐flanking region and the TCF7L2‐interacting region in the intron1, we prepared two reporter plasmids, one containing a region between −1255 and +112 (pGL4.14‐FRMD5‐promoter), and the other containing the interacting region (pGL4.23‐FRMD5‐TBE‐WT). The results of dual luciferase assay in HCT116 cells showed that the activity of pGL4.14‐FRMD5‐promoter was 12.9‐fold higher than that of the control plasmid (pGL4.14‐Empty). However, the reporter activity was unchanged by the knockdown of β‐catenin (Fig. S4), suggesting that the transcriptional activity of the 5′‐flanking region is not associated with the β‐catenin/TCF7L2 complex. On the other hand, the reporter activity of pGL4.23‐FRMD5‐TBE‐WT was 77.5‐fold higher compared with the control plasmid (pGL4.23‐Empty), suggesting an enhancer activity of the region. Importantly, the activity of pGL4.23‐FRMD5‐TBE‐WT was reduced to 50.3% and 37.9% by β‐catenin siRNA #9 and #10, respectively (Fig. 3c). These data tempted us to speculate that the enhancer activity is regulated by the β‐catenin/TCF7L2 complex.
We therefore searched further for TCF7L2‐binding elements (TBEs) in this region using a motif search for transcription factor‐binding motifs, JASPAR (http://jaspar.genereg.net/). Motifs of putative TBEs with JASPAR score≥10 were picked as candidates. As a result, we identified one putative TCF7L2 binding element (TBE), 5′‐TTTCTTTCATCTCC‐3′, with a JASPAR score of 14.5 in the region (Fig. 3a). We additionally prepared a mutant reporter plasmid (pGL4.23‐FRMD5‐TBE‐MUT) by substituting TTTCTTTCATCTCC to TTTCGCTCATCTCC in pGL4.23‐FRMD5‐TBE‐WT, and reporter assay was performed. As expected, the reporter activity of pGL4.23‐FRMD5‐TBE‐MUT was significantly repressed to 63.6% compared with the activity of pGL4.23‐FRMD5‐TBE‐WT. In addition, the activity of the mutant was unchanged by the treatment with β‐catenin siRNA (Fig. 3d).
Role of FRMD5 as a downstream target of Wnt signaling
To uncover the role of FRMD5 in CRC, we carried out expression profile analysis of HCT116 cells treated with/without FRMD5 siRNA. As expected, expression of FRMD5 was decreased to 10.3‐fold and 12.4‐fold by the treatment with siFRMD5 #2 and siFRMD5 #3, respectively, compared with control siRNA (Fig. 4a). We selected genes whose expression levels were commonly decreased or increased more than two‐fold by the FRMD5 siRNAs. As a result, we identified a total of 189 commonly downregulated genes and 226 commonly upregulated genes by the two FRMD5 siRNAs. Among the genes, we further selected genes whose expression was consistently altered by β‐catenin siRNA in HCT116 cells to elucidate the function of FRMD5 as a downstream target of canonical Wnt signaling. Consequently, we found a total of 36 and 53 genes that were either downregulated or upregulated, respectively, by FRMD5 siRNA as well as β‐catenin siRNA (Tables S2 and S3). Additional qPCR analysis validated altered expression in four (MYB, PIGC, RPRML, and DTL) of the five top downregulated genes and three (GRM8, CAMP, and KRT80) of the five top upregulated genes (Fig. 4b). The 36 and 53 genes were applied for gene set enrichment analysis, which identified ten enriched gene sets such as DNA replication, cell cycle, and extracellular matrix (ECM) (Table 2). The gene set associated with DNA replication and cell cycle included CDC45, CDT1, POLA2, MCM10, and SMC1A, which are all in the 36 downregulated genes. Meanwhile the gene set associated with ECM included WNT7A, WNT7B, ANGPTL4, LEFTY1, TCHH, EGLN3, PLAU and PLXDC1, which are all in the 53 up‐regulated genes. These results indicated that FRMD5 may play an important role in the regulation of DNA replication and cell cycle, and/or the interaction with extracellular matrix through these genes.
Figure 4.
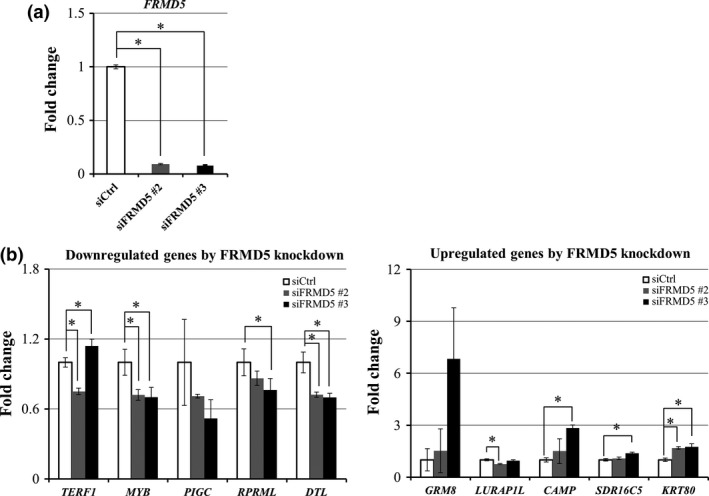
Genes with altered expression by the knockdown of FRMD5. (a) Decreased expression of FRMD5 in response to FRMD5 siRNA #2 and #3. (b) Top five downregulated genes (left) and top five upregulated genes (right) by FRMD5 siRNA in HCT116. Quantitative PCR was performed in triplicate using RNA from the cells treated with FRMD5 siRNA #2 (gray) or #3 (black), or control siRNA (white). Relative expression levels of the ten genes are shown in the histogram (mean ± SD) and result was normalized by GAPDH. An asterisk indicates statistical significance (Dunnett's test, P < 0.05) between FRMD5 siRNA and control siRNA.
Table 2.
Gene sets enrichment analysis of genes regulated by FRMD5
| Gene Set | Description | Genes in Overlap | P value | FDR q value |
|---|---|---|---|---|
| Reactome activation of the pre‐replication complex [31] | Genes involved in Activation of the pre‐replicative complex | 4 | 2.1 e‐7 | 2.79 e‐4 |
| NABA_MATRISOME_ASSOCIATED [753] |
Ensemble of genes encoding ECM‐associated proteins including ECM‐affilaited proteins, ECM regulators and secreted factors |
9 | 4.35 e‐6 | 2.77 e‐3 |
| NABA_MATRISOME [1028] | Ensemble of genes encoding extracellular matrix and extracellular matrix‐associated proteins | 10 | 7.54 e‐6 | 2.77 e‐3 |
| REACTOME_MITOTIC_M_M_G1_PHASES [172] | Genes involved in Mitotic M‐M/G1 phases | 5 | 1.03 e‐5 | 2.77 e‐3 |
| REACTOME_M_G1_TRANSITION [81] | Genes involved in M/G1 Transition | 4 | 1.04 e‐5 | 2.77 e‐3 |
| REACTOME_DNA_REPLICATION [192] | Genes involved in DNA Replication | 5 | 1.75 e‐5 | 3.89 e‐3 |
| NABA_SECRETED_FACTORS [344] | Genes encoding secreted soluble factors | 6 | 2.37 e‐5 | 4.5 e‐3 |
| REACTOME_E2F_MEDIATED_REGULATION _OF_DNA_REPLICATION [35] | Genes involved in E2F mediated regulation of DNA replication | 3 | 2.74 e‐5 | 4.55 e‐3 |
| REACTOME_G1_S_TRANSITION [112] | Genes involved in G1/S Transition | 4 | 3.75 e‐5 | 5.53 e‐3 |
| REACTOME_CELL_CYCLE [421] | Genes involved in Cell Cycle | 6 | 7.26 e‐5 | 9.65 e‐3 |
[]: the total number of genes in gene sets.
FRMD5 regulates cell cycle in a cell‐context dependent manner
Because the expression profile showed that FRMD5 is associated with cell cycle regulation and DNA replication, we carried out cell cycle profile analysis, focusing on S phase change in HCT116, DLD‐1, HCT‐15, and LS174T cells (Fig. 5). The result showed that compared with HCT116 cells treated with siControl, the cells treated with siFRMD5 had decreased S phase (33.3% and 36.1% by siFRMD5 #2, and #3, respectively, compared with 47.9% by siControl). Meanwhile, DLD‐1 showed marginal decrease of S phase by FRMD5 #2, and #3 (30.3% and 27.4%, respectively, compared with 32.7% by siControl). In LS174T and HCT‐15 cells, knockdown of FRMD5 did not induce significant changes in S phase, indicating that FRMD5 may regulate cell cycle in a cell‐context dependent manner.
Figure 5.
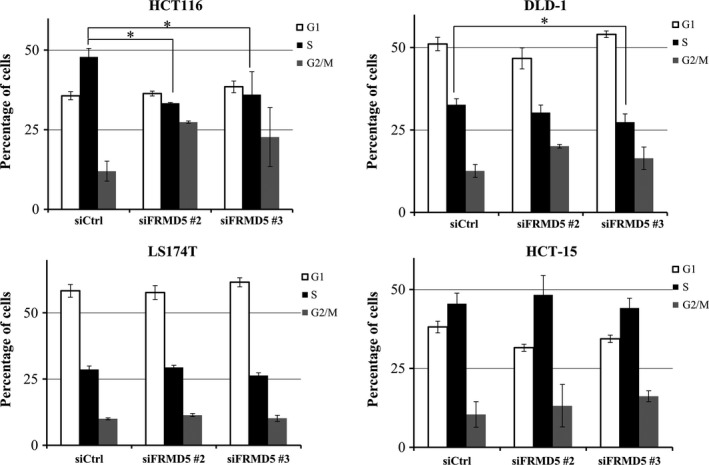
FRMD5 regulates cell cycle in a cell context‐dependent manner. HCT116, DLD‐1, LS174T, and HCT‐15 cells were treated with FRMD5 siRNA #2, #3 or control siRNA. G1 (white), S (black) and G2 (grey) phases are measured by FACS. Percentage (%) of each phase was showed in the histogram (mean ± SD). An asterisk indicates statistical significance (Dunnett's test, P < 0.05) between FRMD5 siRNA and control siRNA.
Discussion
FRMD5, a member of the FERM (4.1/ezrin/radixin/moesin) protein family, encodes a 570 amino acids‐protein containing an N‐terminal FERM domain. It was first identified as a gene down‐regulated by mutant p53R273H protein.13 In this study, we showed that FRMD5 is a novel direct target gene of β‐catenin/TCF7L2 in CRC cells. The data of ONCOMINE indicated that tumor tissue had higher expression level of FRMD5 than that of normal tissue. Although two of three datasets (Sabates‐Bellver Colon and Gaedcke Colorectal datasets) showed a positive association of FRMD5 expression with that of AXIN2 and RNF43, a dataset (TCGA Colorectal dataset) did not reveal any relationship in the expression between FRMD5 and AXIN2 or between FRMD5 and RNF43. Sabates‐Bellver Colon and Gaedcke Colorectal datasets were comprised of paired tumor and normal tissues, but TCGA data were composed of unpaired tumor and normal tissues and the number of normal tissues is limited. Therefore variation of unpaired normal samples might cause the negative correlation in TCGA dataset. We also detected abundant expression of FRMD5 in four of six CRC cell lines carrying a mutation in APC or CTNNB1. Consistent with the previous report, limited expression of FRMD5 was observed in SW480 cells that carries a mutated p53 (p53R273H) allele.14 These data indicates that several factors including β‐catenin/TCF7L2 and mutant p53R273H will play a role in FRMD5 expression.
In intron1 of FRMD5, we identified a region that was responsible for the expression of FRMD5. An additional search of histone modifications in the ENCODE database indicated that this region encompasses high levels of H3K4 mono‐methylation and H3K27 acetylation and low levels of H3K4 tri‐methylation, and that the same region was overlapped by DNaseI hypersensitivity peak clusters (Fig. S5). Reportedly, high levels of H3K4 mono‐methylation with low levels H3K4 tri‐methylation indicate enhancer regions15, 16 and augmented acetylation of H3K27 is a marker for active enhancer rather than poised enhancer.17 In addition, DNaseI hypersensitivity sites is linked to the regions with open chromatin, and thus may serve as binding regions for transcription factors.18 These data are consistent with the notion that this region interacts with β‐catenin/TCF7L2 complex, and functions as an enhancer.
The biological function of FRMD5 remains to be fully elucidated. It was reported that FRMD5 stabilizes E‐cadherin through the interaction with p120‐catenin in lung H1299 cancer cells,19 and that FRMD5 represses epithelial‐mesenchymal transition (EMT) and cell motility in H1299 cancer cells through interacting with integrin β5.20 However, knockdown of FRMD5 did not repress expression of E‐cadherin in HCT116 cells (Fig. S6). Moreover, knockdown of FRMD5 did not show consistent change of cell migration in HCT116 cells (Fig. S7). Treatment with siFRMD5 #2 increased migration of HCT116 cells but that with siFRMD5 #3 decreased migration (Fig. S7). Therefore, it is difficult to conclude that FRMD5 is involved in EMT or cell migration in colon cancer cells. Further investigation is needed to clarify the role of FRMD5 in carcinogenesis.
In our study, expression profile analysis detected enriched gene sets of DNA replication, cell cycle, and extracellular matrix in HCT116 cells treated with siFRMD5. Additional qPCR analysis validated that CDT1 and POLA2 were downregulated by the knockdown of FRMD5 and that LEFTY1 was upregulated by the knockdown (Fig. S8). Notably, CDT1 encodes chromatin licensing factor 1 that plays an essential role in the initiation of DNA replication21 together with origin recognition complex‐associated (ORCA).22 Minichromosome maintenance complex component 10 (MCM10) and DNA‐polymerase alpha 2 (POLA2) are components of DNA polymerase α‐primase complex,23 which plays a crucial role in eukaryotic DNA replication. Previous studies showed that MCM10 is recruited to the replication sites24 and regulates the stability and association of DNA polymerase‐alpha.25 POLA2 enhances the protein level of p180, a catalytic subunit of DNA polymerase‐alpha 1 (POLA1),26 and increases DNA polymerase activity. Therefore, enhanced expression of CDT1, MCM10, and POLA2 may facilitate DNA replication in the proliferating cancer cells. Regarding the gene set of ECM, left‐right determination factor 1 (LEFTY1) encodes a secreted ligand of TGF‐β family member, and is involved in the determination of leftness during development. It is reported that expression of LEFTY1 is reduced in metastasized colorectal tumors in the liver compared with their primary tumors.27 Therefore, elevated expression of FRMD5 may additionally play a role in the progression and/or metastasis of colon cancer cells through the reduced expression of LEFTY1.
We also found that knockdown of FRMD5 decreased the expression of MYB, a well‐known oncogene. It was reported that Myb blocks differentiation of colon epithelial cells28 and regulates the renewal of intestinal stem cells,29 and that Myb is often overexpressed in colon cancer with poor prognosis.30 Consistently, we found that colorectal cancer patients with a high expression level of FRMD5 showed poor prognosis compared to patients with a low expression level (PrognoScan, www.abren.net/PrognoScan/, dataset GSE17536 and GSE17537)31 (Fig. S9). Interestingly, a recent study revealed that intestinal‐specific activation of Myb in pro‐carcinogen‐induced mouse model accelerated the tumorigenesis in colon by expanding the intestinal stem cell pool.32 Therefore, elevated expression of FRMD5 may render malignant properties to colorectal cancer cells through the induction of MYB and subsequent induction of stemness in the tumor cells.
In summary, our results demonstrated that FRMD5 was a novel target of the Wnt signaling pathway, and a region in intron1 was responsible for the regulation of FRMD5 transcription. Expression profile analysis and subsequent gene enrichment analysis revealed that FRMD5 may be involved in the regulation of DNA replication, cell cycle, and ECM in CRC. Further studies are essential to clarify the role played by FRMD5 in colorectal tumorigenesis.
Disclosure Statement
We have no financial relationships to disclose.
Supporting information
Table S1. Primers for real‐time qPCR.
Table S2. Downregualted genes by the knockdown of FRMD5.
Table S3. Upregualted genes by the knockdown of FRMD5.
Fig. S1. Expression of LGR5 and FRMD5 in non‐cancerous mucosa.
Fig. S2. Expression of FRMD5 in colon cancer tissues.
Fig. S3. Association of the expression of FRMD5 with AXIN2 and RNF43.
Fig. S4. Transcriptional activity of the 5′‐flanking region of FRMD5.
Fig. S5. Histone modifications around the TCF7L2‐interactiong region.
Fig. S6. Expression of E‐Cadherin after FRMD5 knockdown.
Fig. S7. Cell migration assay after FRMD5 knockdown.
Fig. S8. Validation of candidate genes in GO analysis.
Fig. S9. Prognosis analysis of FRMD5 in CRC patients.
Acknowledgment
We thank Seira Hatakeyama, Yuko Yamaguchi, and Rika Koubo (The University of Tokyo) for their technical assistance. This work was supported in part by the Grant‐in‐Aid (#16H01569) from the Japan Society for the Promotion of Science..
Cancer Sci 108 (2017) 612–619
Funding Information
This work was supported in part by the Grant‐in‐Aid (#16H01569) from the Japan Society for the Promotion of Science.
References
- 1. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang J, Sinha T, Wynshaw‐Boris A. Wnt signaling in mammalian development: lessons from mouse genetics. Cold Spring Harb Perspect Biol. 2012; 4: a007963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ring A, Kim Y‐M, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev 2014; 10: 512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev 2005; 19: 877–90. [DOI] [PubMed] [Google Scholar]
- 5. Fevr T, Robine S, Louvard D, Huelsken J. Wnt/beta‐catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol Cell Biol 2007; 27: 7551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu C, Li Y, Semenov M et al Control of β‐catenin phosphorylation/degradation by a dual‐kinase mechanism. Cell 2002; 108: 837–47. [DOI] [PubMed] [Google Scholar]
- 7. Chien AJ, Conrad WH, Moon RT. A Wnt survival guide: from flies to human disease. J Invest Dermatol. 2009; 129: 1614–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang S, Li Y, Wu Y, Shi K, Bing L, Hao J. Wnt/β‐catenin signaling pathway upregulates c‐Myc expression to promote cell proliferation of P19 teratocarcinoma cells. Anat Rec (Hoboken). 2012; 295: 2104–13. [DOI] [PubMed] [Google Scholar]
- 9. Tetsu O, McCormick F. Beta‐catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999; 398: 422–6. [DOI] [PubMed] [Google Scholar]
- 10. Takahashi N, Yamaguchi K, Ikenoue T, Fujii T, Furukawa Y. Identification of two Wnt‐responsive elements in the intron of RING finger protein 43 (RNF43) gene. PLoS One 2014; 9(1): e86582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. BioEssays 2002; 24(1): 91–8. [DOI] [PubMed] [Google Scholar]
- 12. Gregorieff A, Pinto D, Begthel H, Destrée O, Kielman M, Clevers H. Expression pattern of Wnt signaling components in the adult intestine. Gastroenterology 2005; 129(2): 626–38. [DOI] [PubMed] [Google Scholar]
- 13. Brázdová M, Quante T, Tögel L et al Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res 2009; 37: 1486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rochette PJ, Bastien N, Lavoie J, Guérin SL, Drouin R. SW480, a p53 double‐mutant cell line retains proficiency for some p53 functions. J Mol Biol 2005; 352(1): 44–57. [DOI] [PubMed] [Google Scholar]
- 15. Heintzman ND, Stuart RK, Hon G et al Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007; 39(3): 311–18. [DOI] [PubMed] [Google Scholar]
- 16. Barski A, Cuddapah S, Cui K et al High‐resolution profiling of histone methylations in the human genome. Cell 2007; 129: 823–37. [DOI] [PubMed] [Google Scholar]
- 17. Creyghton MP, Cheng AW, Welstead GG et al Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 2010; 107: 21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem 1988; 57(1): 159–97. [DOI] [PubMed] [Google Scholar]
- 19. Wang T, Pei X, Zhan J, Hu J, Yu Y, Zhang H. FERM‐containing protein FRMD5 is a p120‐catenin interacting protein that regulates tumor progression. FEBS Lett 2012; 586: 3044–50. [DOI] [PubMed] [Google Scholar]
- 20. Hu J, Niu M, Li X et al FERM domain‐containing protein FRMD5 regulates cell motility via binding to integrin β5 subunit and ROCK1. FEBS Lett 2014; 588: 4348–56. [DOI] [PubMed] [Google Scholar]
- 21. Zhong W, Feng H, Santiago FE, Kipreos ET. CUL‐4 ubiquitin ligase maintains genome stability by restraining DNA‐replication licensing. Nature 2003; 423: 885–9. [DOI] [PubMed] [Google Scholar]
- 22. Shen Z, Chakraborty A, Jain A et al Dynamic association of ORCA with prereplicative complex components regulates DNA replication initiation. Mol Cell Biol 2012; 32: 3107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu W, Ukomadu C, Jha S et al Mcm10 and And‐1/CTF4 recruit DNA polymerase to chromatin for initiation of DNA replication. Genes Dev 2007; 21: 2288–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Izumi M, Yatagai F, Hanaoka F. Localization of human Mcm10 is spatially and temporally regulated during the S phase. J Biol Chem 2004; 279: 32569–77. [DOI] [PubMed] [Google Scholar]
- 25. Ricke RM, Bielinsky A‐K. Mcm10 regulates the stability and chromatin association of DNA polymerase‐α. Mol Cell 2004; 16(2): 173–85. [DOI] [PubMed] [Google Scholar]
- 26. Mizuno T, Ito N, Yokoi M et al The second‐largest subunit of the mouse DNA polymerase alpha‐primase complex facilitates both production and nuclear translocation of the catalytic subunit of DNA polymerase alpha. Mol Cell Biol 1998; 18: 3552–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naba A, Clauser KR, Whittaker CA et al Extracellular matrix signatures of human primary metastatic colon cancers and their metastases to liver. BMC Cancer 2014; 14(1): 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramsay RG, Ciznadija D, Sicurella C et al Colon epithelial cell differentiation is inhibited by constitutive c‐Myb expression or mutant APC plus activated RAS. DNA Cell Biol 2005; 24(1): 21–9. [DOI] [PubMed] [Google Scholar]
- 29. Cheasley D, Pereira L, Lightowler S, Vincan E, Malaterre J, Ramsay RG. Myb Controls Intestinal Stem Cell Genes and Self‐Renewal. Stem Cells 2011; 29: 2042–50. [DOI] [PubMed] [Google Scholar]
- 30. Ramsay RG, Barton AL, Gonda TJ. Targeting c‐Myb expression in human disease. Expert Opin Ther Targets. 2003; 7(2): 235–48. [DOI] [PubMed] [Google Scholar]
- 31. Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta‐analysis of the prognostic value of genes. BMC Med Genomics 2009; 2(1): 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Malaterre J, Pereira L, Putoczki T et al Intestinal‐specific activatable Myb initiates colon tumorigenesis in mice. Oncogene 2016; 35: 2475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers for real‐time qPCR.
Table S2. Downregualted genes by the knockdown of FRMD5.
Table S3. Upregualted genes by the knockdown of FRMD5.
Fig. S1. Expression of LGR5 and FRMD5 in non‐cancerous mucosa.
Fig. S2. Expression of FRMD5 in colon cancer tissues.
Fig. S3. Association of the expression of FRMD5 with AXIN2 and RNF43.
Fig. S4. Transcriptional activity of the 5′‐flanking region of FRMD5.
Fig. S5. Histone modifications around the TCF7L2‐interactiong region.
Fig. S6. Expression of E‐Cadherin after FRMD5 knockdown.
Fig. S7. Cell migration assay after FRMD5 knockdown.
Fig. S8. Validation of candidate genes in GO analysis.
Fig. S9. Prognosis analysis of FRMD5 in CRC patients.