

Abstract
We previously showed that the CD82/signal transducer and activator of transcription/interleukin‐10 (IL‐10) axis is activated in CD34+/CD38− AML cells that favor the bone marrow microenvironment. The present study explored the novel biological function of IL‐10 in regulation of expression of adhesion molecules in AML cells and found that exposing AML cells to IL‐10 induced expression of E‐cadherin, but not other adhesion molecules, including VLA4, CD29, and LFA1. Downregulation of E‐cadherin with an siRNA suppressed the adhesion of leukemia cells to bone marrow‐derived mesenchymal stem cells and enhanced the anti‐leukemia effect of cytarabine. A microRNA (miRNA) database search identified an miR‐9 as a candidate miRNA binding onto the 3′‐UTR of E‐cadherin and regulating its expression. Notably, treatment of leukemia cells with IL‐10 decreased miR‐9 expression through hypermethylation of the miR‐9 CpG islands. In addition, downregulation of DNA methyltransferase 3A by siRNAs decreased E‐cadherin expression in parallel with an increase in levels of miR‐9 in leukemia cells. Notably, short hairpin RNA‐mediated IL‐10 downregulation impaired engraftment of human AML cells and enhanced the anti‐leukemia effect of cytarabine in conjunction with miR‐9 upregulation and E‐cadherin downregulation in a human AML xenograft model. Taken together, the IL‐10/E‐cadherin axis may be a promising therapeutic target for treating AML.
Keywords: AML, DNMT3A, E‐cadherin, IL‐10, miR‐9
Interleukin‐10 is a pleiotropic immunoregulatory cytokine that inhibits cytokine secretion and the effector function of T cells, monocytes, and macrophages.1 Interleukin‐10 exerts its actions through a heterodimeric membrane receptor composed of a binding chain (IL‐10R1) and a transducing chain (IL‐10R2). Interaction between IL‐10R1 and IL‐10R2 activates a series of intracellular signaling molecules including the STATs.2, 3, 4
Human BM contains a higher frequency of CD4+CD25+FoxP3+ regulatory T cells, which produce IL‐10, than other secondary lymphoid organs,5 suggesting that the amount of IL‐10 in the BM may be greater than that in other organs. The treatment of allogeneic HSPCs with an anti‐IL‐10R antibody led to a ~90% reduction in the number of surviving allogeneic HSPCs compared with control IgG antibody treatment.6 Interleukin‐10 significantly increased the colony‐forming ability of HSPCs in association with an increase in levels of anti‐apoptotic Bcl‐2 proteins.7 These observations suggest the important role of IL‐10 in survival of HSPCs.
We previously showed that CD82 inhibits the activity of MMP9 and enhances the adhesion of AML cells to the BM microenvironment.8 We also found that CD82 stimulates production of IL‐10 through STAT5 signal transduction pathways in AML cells.9
CD82 acts as a tumor suppressor and inhibits tumor metastasis. Forced expression of CD82 inhibits cytokine‐stimulated phosphorylation of β‐catenin and stabilizes the E‐cadherin/β‐catenin complex, resulting in strengthened hemophilic intercellular cell adhesion in non‐small‐cell lung cancer cells.10 In addition, positive correlation between CD82 and E‐cadherin expression was noted in non‐small‐cell lung cancer.11 Thus, it is reasonable to assume that CD82 may regulate the expression of E‐cadherin in AML cells.
Mature miRNAs are endogenous, single‐stranded, non‐protein‐coding small RNAs measuring 19–25 nt in length, which suppress the expression of target proteins by pairing with the 3′‐UTR of target mRNAs.12, 13, 14 In carcinogenesis, miRNAs are classified as either oncogenic or tumor suppressor miRs.15, 16 Recently, tumor suppressor miRNAs have been shown to be silenced by aberrant DNA hypermethylation in cancers.17, 18, 19 In humans, there are three independent miR‐9 genes (miR‐9‐1 on chromosome 1, miR‐9‐2 on chromosome 5, and miR‐9‐3 on chromosome 15), with identical mature miR‐9 sequences. In different cancers, miR‐9 can function either as an oncomir or a tumor suppressor miRNA, depending on the type of cancer.20, 21 For instance, overexpression of miR‐9 can enhance metastasis or invasion in metastatic brain tumors or glioblastoma, indicating its oncogenic role.22, 23, 24 MicroRNA‐9 interacts with the 3′‐UTR of E‐cadherin and downregulates its expression, which induces β‐catenin nuclear translocation and subsequently upregulation of c‐Myc and CD44.25 MicroRNA‐9 overexpression can induce epithelial–mesenchymal transition (EMT) and promote tumor metastasis through E‐cadherin downregulation in esophageal squamous cell carcinoma.25 Abnormal miR‐9‐1 and miR‐9‐3 methylation and their downregulation are frequently reported in many cancers.20, 26, 27
The present study examines the relationship between the CD82/STAT5/IL‐10 axis and E‐cadherin expression in AML cells. We also explore the biological function of IL‐10/E‐cadherin in AML cells.
Materials and Methods
Cell sample collection
Each study participant provided informed written consent, and the study was approved by the Kochi University ethics committee (Nankoku, Japan). Leukemia cells were isolated from patients with AML (n = 15; Table 1) who were classified, according to the WHO classification system, as having AML without maturation (cases 14 and 15), AML with maturation (cases 3, 5, and 8–12), acute myelomonocytic leukemia (cases 6 and 7), acute monocytic leukemia (cases 1, 2, and 13), or therapy‐related AML (case 4). Normal BM MNCs were isolated from healthy volunteers (n = 5). The WHO classification system was categorized by use of cytogenetic or molecular genetic abnormalities, and that these genetic changes form clinicopathologic–genetic entities (Table 1).28, 29
Table 1.
Characteristics of 15 patients with AML
| Patient no. | Sex | WHO leukemia classification | Cytogenetics |
|---|---|---|---|
| 1 | M | Acute monocytic leukemia | 45, X, −Y, ?t(3;11;10)(q29;q23;p12) [18]/46, XY [2] |
| 2 | M | Acute monocytic leukemia | 46, XY, add(8)(p11.2), ?t(10;11)(p12;q23) [8]/46, XY [12] |
| 3 | M | AML with maturation | 44, XY, del(5)(q?), −17, −19, i(21)(q10), −22, −22, +mar1, +mar2 [2]/45, idem, add(11)(q23), add(12)(p11.2), +16, +19, add(20)(p13), +21, −i(21), −mar2 [16] |
| 4 | M | Therapy‐related AML | 46, XY [20] |
| 5 | F | AML with maturation | 46, XX [20] |
| 6 | F | Acute myelomonocytic leukemia | 46, XX [20] |
| 7 | F | Acute myelomonocytic leukemia | 46, XX, t(6;21)(q23;q22) [3]/46, XX, add(18)(p11.2) [1]/46, XX, t(5;12)(q15;q24.1) [1]/46, XX [5]/46, XY[2] |
| 8 | F | Acute myelomonocytic leukemia | 46, XX, t(10;17)(p15;q23) [1]/46, XX, del(2)(q?), add(3)(p13), add(4)(q21), −8, add(9)(p13), add(12)(p11.2), +mar [1]/46, XY[14] |
| 9 | F | AML with maturation | 46, XX, del(11)(q?) [4]/46, XX, del(9)(q?) [2]/46, XX[14] |
| 10 | M | AML with maturation | 47, XY, −5, add(7)(p11.2), +8, −18, −21, +mar1, +mar2, +mar3, +mar4 [2]/44, idem, add(6)(p11), −8, add(13)(q14), −17, −mar3, −mar4, +mar5 [7] |
| 11 | M | AML with maturation | 46, XY [20] |
| 12 | F | AML with maturation | 46, XX[5] |
| 13 | M | Acute monocytic leukemia | 45, X, −Y, ins(1;?)(q21;?),?t(3;11;10)(q29;q23;p12),der(5)t(5;12)(q13;p11.2),t(5;12) [20] |
| 14 | M | AML without maturation | 46, XY [20] |
| 15 | F | AML without maturation | 49, XX, +5, +17, +18 [2]/56, idem, +X, +5, +8, +9, +12, +13 [13]/46, XX, del(6)(q?) [4] /46, XX[1] |
Pt, patient; F, female; M, male; WHO, World Health Organization (leukemia classification).
Cell culture
The acute monocytic leukemia cell line MOLM13, carrying an ITD of the juxtamembrane domain of FLT3 (FLT3/ITD), was kindly provided by Dr. Yoshinobu Matsuo (Fujisaki Cell Center, Okayama, Japan).30 The leukemia cell lines THP‐1 and MV4‐11 (FLT3‐ITD+) were obtained from ATCC (Manassas, VA, USA). UE6E7T‐3 human BM‐derived MSCs were obtained from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan).
Pharmacological inhibition
Interleukin‐10 was purchased from Miltenyi Biotec (Bergisch Gladbach, Germany).
Western blot analysis
Western blot analysis was carried out as described previously.31 Protein concentrations were quantified using a Bio‐Rad assay (Bio‐Rad Laboratories, CA, USA). Proteins were resolved in a 10% SDS polyacrylamide gel and transferred to an Immobilon PVDF membrane (Amersham, Arlington Heights, IL, USA). Primary anti‐IL‐10 (Abcam, Cambridge, UK), anti‐E‐cadherin (BioLegend, San Diego, CA, USA), and anti‐GAPDH (Abcam) antibodies were used to sequentially probe the membrane.
RNA isolation and real‐time RT‐PCR
Total RNA was extracted from leukemia cells and reverse transcribed according to the manufacturer's instructions (PrimeScript RT reagent kit; Takara, Shiga, Japan). Real‐time RT‐PCR was carried out using Power SYBR Green PCR Master Mix (Applied Biosystems, Warrington, UK), a StepOnePlus Real‐Time PCR System, and the following thermocycling conditions: 95°C for 10 min and 40 cycles at 95°C for 15 s, 60°C for 1 min. Expression of the 18S gene was used for normalization purposes. The sequences of the primers used are listed in Table 2.
Table 2.
Polymerase chain reaction primers
| Gene | Direction | Primer |
|---|---|---|
| IL10 | Forward | 5′‐AGAACAGCTGCACCCACTTC‐3′ |
| Reverse | 5′‐GCATCACCTCCTCCAGGTAA‐3′ | |
| CDH1 | Forward | 5′‐TGCCCAGAAAATGAAAAAGG‐3′ |
| Reverse | 5′‐GTGTATGTGGCAATGCGTTC‐3′ | |
| DNMT3A | Forward | 5′‐CACACAGAAGCATATCCAGGAGTG‐3′ |
| Reverse | 5′‐AGTGGACTGGGAAACCAAATACCC‐3′ | |
| 18S | Forward | 5′‐AAACGGCTACCACATCCAAG‐3′ |
| Reverse | 5′‐CCTCCAATGGATCCTCGTTA‐3′ |
MicroRNA target prediction
TargetScan (http://www.targetscan.org/) and miRBase (http://www.mirbase.org/) were used to identify putative miRNAs regulating expression of E‐cadherin.
MicroRNA expression
Expression of miRNA was analyzed using a Mir‐X miRNA quantitative RT‐PCR SYBR Kit (Catalog #638314; Clontech Laboratories, Mountain View, CA, USA), following the manufacturer's instructions. MicroRNA levels were normalized to U6 mRNA expression, and relative miR quantities were determined by the ΔΔC t method. The sequence of the 5′ primers used to detect miR‐9‐5p was as follows: 5′‐TCTTTGGTTATCTAGCTGTATGA‐3′. The 3′ primer is the mRQ 3′ Primer supplied with the kit.
Methylation analysis
DNA (300 ng) was isolated from leukemia cells and treated with bisulfite using an MethylEasy Xceed Rapid DNA Bisulphite Modification Kit (Takara), according to the supplier's protocol. The sequences of the methylation‐specific primers for miR9‐3 followed the previous study:32 forward, 5′‐GGTGTTAGGACGTACGGAAC‐3′; and reverse, 5′‐TACCCGAATCCTAAAACGC‐3′. The sequences of primers specific for unmethylated miR9‐3 DNA were: forward, 5′‐GGTGTTAGGATGTATGGAAT‐3′; and reverse, 5′‐TACCCAAATCCTAAAACAC‐3′. Amplification was carried out using the EpiScope MSP Kit (Takara) in a MyCycler thermal cycler (Bio‐Rad Laboratories). The cycle conditions were as follows: 95°C initial activation for 30 s followed by denaturation for 45 cycles of 98°C for 5 s, annealing at 55°C for 30 s, and a final extension at 72°C for 1 min.
Overexpression of miR‐9 precursor
A precursor miR‐9 mimic (Sigma, Deisenhofen, Germany) was transfected into MOLM13, MV4‐11, and THP‐1 cells using the INTERFERin transfection reagent (Polyplus‐transfection, New York, NY, USA), according to the manufacturers’ instructions. A non‐targeting oligonucleotide mimic was used as a negative control.
Small interfering RNAs and transfections
A control siRNA and two siRNAs against DNMT3A were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Sigma, respectively. THP‐1 cells were transfected with either the control or DNMT3A siRNAs (300 nM) using an Amaxa Electroporator Nucleofector II (Wako Pure Chemical Industries, Osaka, Japan) and the Nucleofector Kit V (program V‐001), as previously described.31
Fluorescent microscopy
THP‐1 cells were transfected with a GFP vector (0.5 μg) using an Amaxa Electroporator Nucleofector II (Wako Pure Chemical Industries) and the Nucleofector Kit V (program V‐001), as previously described.31 Subsequently, GFP‐positive THP‐1 cells were transfected with either control or E‐cadherin siRNAs (300 nM) using the same procedure. After 24 h, the cells were cocultured with UE6E7T‐3 BM‐MSCs and treated with AraC and/or IL‐10. After 48 h, floating cells were removed and the remaining adherent cells were washed twice with PBS. The GFP‐positive cells were analyzed by fluorescent microscopy (BZ‐9000; KEYENCE, Osaka, Japan). Cell counts were undertaken using BZ‐II Dynamic Cell Count software (KEYENCE).
Bone marrow transplantation
NOD.Cg‐Rag1 tm1Mom Il2rg tm1Wjl/SzJ mice (stock number 007799) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA)8 and bred in a pathogen‐free environment in accordance with the guidelines of the Kochi University School of Medicine; 6‐week‐old female and male animals were used for the experiments. Human MOLM13 cells (5 × 106/mouse) transfected with a control shRNA or IL‐10‐specific shRNA were injected into the tail vein. At 1 week post‐AML cell transplantation, the mice were treated with AraC (20 mg/kg; 3 days/week for 2 weeks); control mice received PBS (100 μL/mouse, 3 days/week for 2 weeks). Cell engraftment was analyzed by flow cytometry after staining PBMCs with a phycoerythrin‐conjugated anti‐human CD82 mAb (BioLegend). On days 18 and 28 post‐transplantation, mice were killed and the BM was removed. Bone marrow cells were flushed from the femurs using 25‐gauge needles (Becton Dickinson Biosciences, New Jersey, USA) and collected to extract mRNA. Human cell engraftment was analyzed by flow cytometry after staining BM cells with a phycoerythrin‐conjugated anti‐human CD82 mAb (BioLegend). Plasma was separated by centrifugation, and mRNA was isolated from plasma using the NucleoSpin miRNA Plasma Kit (Takara). The levels of hsa‐miR‐9 in plasma were assessed using a Mir‐X miRNA qRT‐PCR SYBR Kit (Clontech Laboratories), following the manufacturer's instructions.
Statistical analysis
Student's t‐test was used when comparing results between two groups. All statistical analyses were carried out using PRISM statistical analysis software (GraphPad Software, San Diego, CA, USA) and the results were considered significant in cases where the P‐value was <0.05, or highly significant when the P‐value was <0.01.
Results
Effect of IL‐10 on E‐cadherin expression in leukemia cells
The exposure of leukemia cells (MOLM13, MV4‐11, and THP‐1 cells) to IL‐10 (100 ng/mL, 48 h) increased E‐cadherin expression at both the mRNA and protein levels (Figs 1a,S1). E‐cadherin expression was also upregulated in UE6E7T‐3 BM‐MSCs following treatment with IL‐10 (Fig. 1b). The levels of both IL‐10 and E‐cadherin in BM‐MNCs isolated from AML patients (n = 15) were higher than those in BM‐MNCs isolated from healthy volunteers (n = 4) at both the mRNA and protein levels (Fig. 1c). Western blot analyses also found that BM‐MNCs isolated from AML patients (n = 6) and AML cell lines (MOLM13, MV4‐11, and THP‐1) express detectable the levels of IL‐10 and E‐cadherin proteins. However, these proteins were not detectable in BM‐MNCs isolated from healthy volunteers (Fig. 1d).
Figure 1.
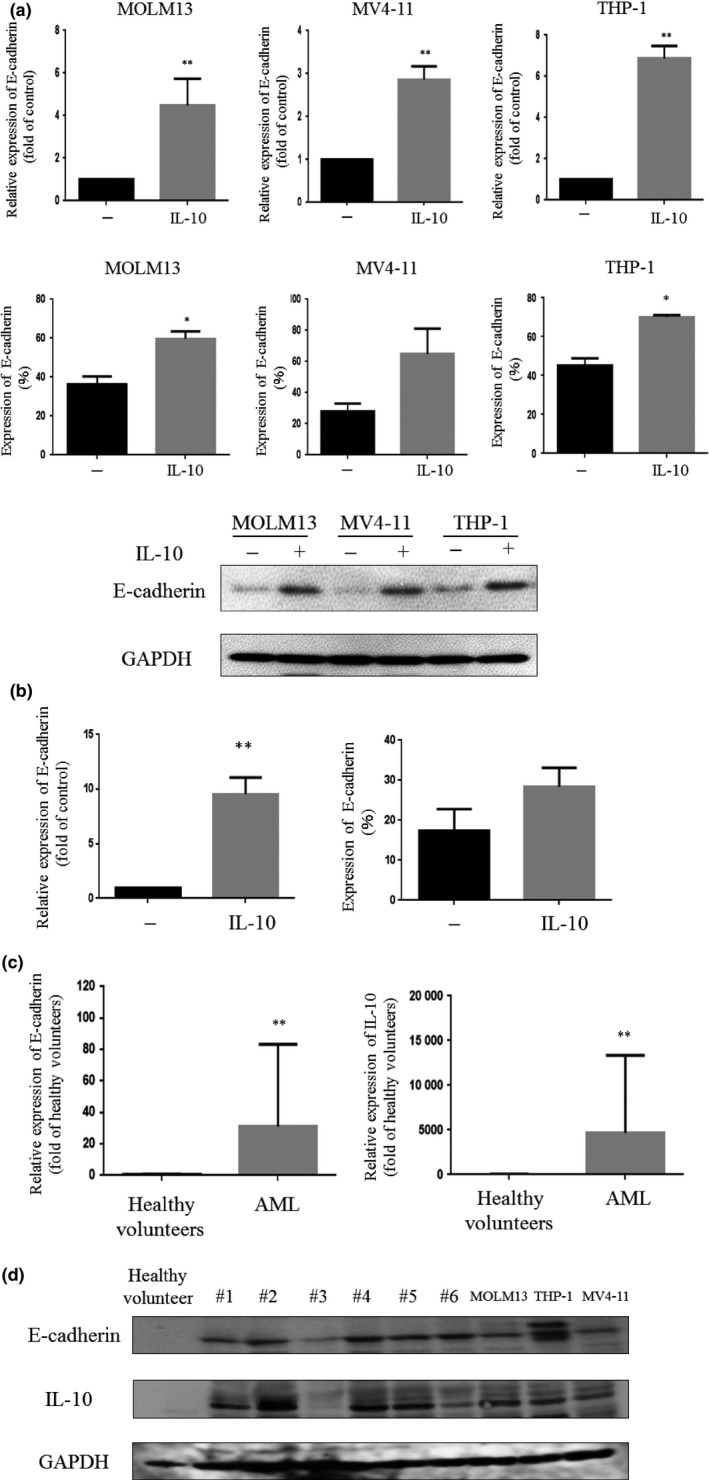
Effect of interleukin‐10 (IL‐10) treatment on E‐cadherin expression in AML cells. (a) MOLM13, MV4‐11, and THP‐1 cells were treated with IL‐10 (100 ng/mL) for 48 h and analyzed for E‐cadherin expression by real‐time RT‐PCR, flow cytometry, or Western blot. (b) UE6E7T‐3 human bone marrow (BM)‐derived mesenchymal stem cells were treated with IL‐10 (100 ng/mL) for 48 h and analyzed for E‐cadherin expression by real‐time RT‐PCR. (c) E‐cadherin and IL‐10 mRNA levels in AML cells isolated from patients (n = 15) and BM mononuclear cells isolated from healthy volunteers (n = 4). (d) AML cells isolated from patients (#1–#6, n = 6), BM mononuclear cells isolated from a healthy volunteer (n = 1), and MOLM13, THP‐1, and MV4‐11 cells were harvested and analyzed for IL‐10, E‐cadherin, and GAPDH expression by Western blot. *P < 0.05; **P < 0.01. −, untreated cells.
Effect of IL‐10 on miR‐9 expression in leukemia cells
To verify the mechanisms by which IL‐10 increases levels of E‐cadherin in AML cells, we focused on miRNA. We identified homology between miR‐9 and the 3′‐UTR of the human E‐cadherin gene by searching mRNA databases (Fig. 2a). These observations suggested that miR‐9 might bind to 3′‐UTR of E‐cadherin and suppress its expression. We first examined whether IL‐10 modulates expression of miR‐9 in AML cells. Exposure of AML cells to IL‐10 downregulated miR‐9 expression by nearly half in these cells (Fig. 2b). We next examined whether miR‐9 regulates expression of E‐cadherin. As expected, forced expression of miR‐9 in AML cells significantly downregulated levels of E‐cadherin at both mRNA and protein levels (Fig. 2c–e). These observations suggested that IL‐10 decreases levels of miR‐9, leading to upregulation of E‐cadherin. Notably, the levels of miR‐9 in BM‐MNCs isolated from AML patients (n = 15) were significantly lower than those in BM cells isolated from healthy volunteers (n = 4, Fig. 2f).
Figure 2.
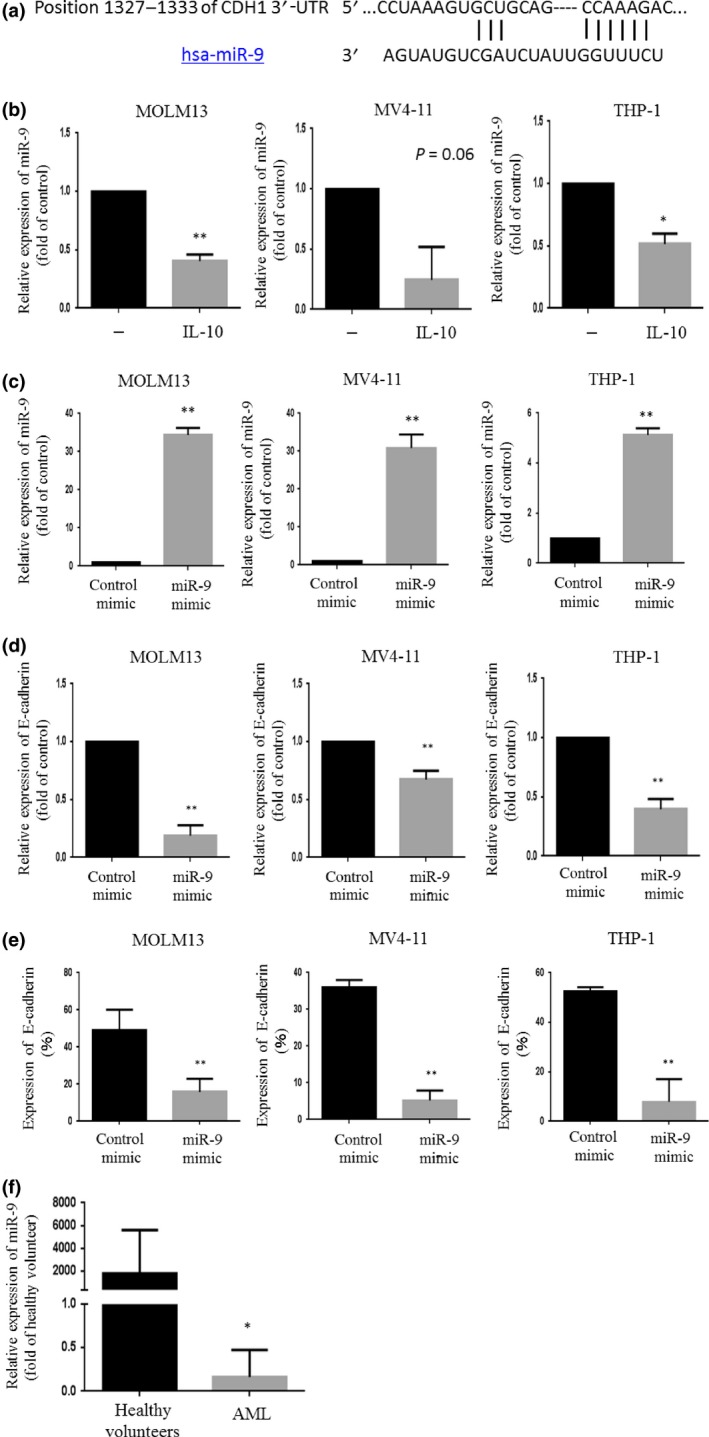
(a) Sequence of microRNA‐9 (miR‐9) and its potential binding site in the E‐cadherin 3′‐UTR. (b) Expression of miR‐9 in leukemia cells treated with interleukin‐10 (IL‐10) was analyzed using a Mir‐X miRNA qRT‐PCR SYBR Kit. The results shown represent the mean ± SD of three experiments carried out in triplicate. (c) Expression of miR‐9 in leukemia cells transfected with either a control mimic or an miR‐9 mimic was analyzed using a Mir‐X miRNA qRT‐PCR SYBR Kit to measure the levels of the indicated gene. The results shown represent the mean ± SD of three experiments carried out in triplicate. (d, e) E‐cadherin expression in leukemia cells transfected with either a control or miR‐9 mimic was analyzed by real time RT‐PCR and flow cytometry. (f) miR‐9 levels in AML cells isolated from patients (n = 15) and bone marrow mononuclear cells isolated from healthy volunteers (n = 4). *P < 0.05; **P < 0.01.
Effect of IL‐10 on the methylation status of miR‐9 CpG islands in leukemia cells
We next examined whether the expression of miR‐9 was regulated by epigenetic DNA methylation changes in the miR‐9 gene. To test this possibility, we examined the methylation status of miR‐9 CpG islands in leukemia cells by methylation‐specific PCR. Interestingly, exposing leukemia cells to IL‐10 increased the levels of DNTM3A in both mRNA and protein (Fig. 3a,b). Concomitantly, the hypermethylation of miR‐9‐3 CpG islands was noted in these leukemia cells (Fig. 3c). These observations suggested that IL‐10 downregulates miR‐9 expression through DNMT3A activity, resulting in E‐cadherin induction. As expected, the downregulation of DNMT3A by siRNAs decreased E‐cadherin expression in association with an increase in miR‐9 expression in leukemia cells (Fig. 3d–g).
Figure 3.
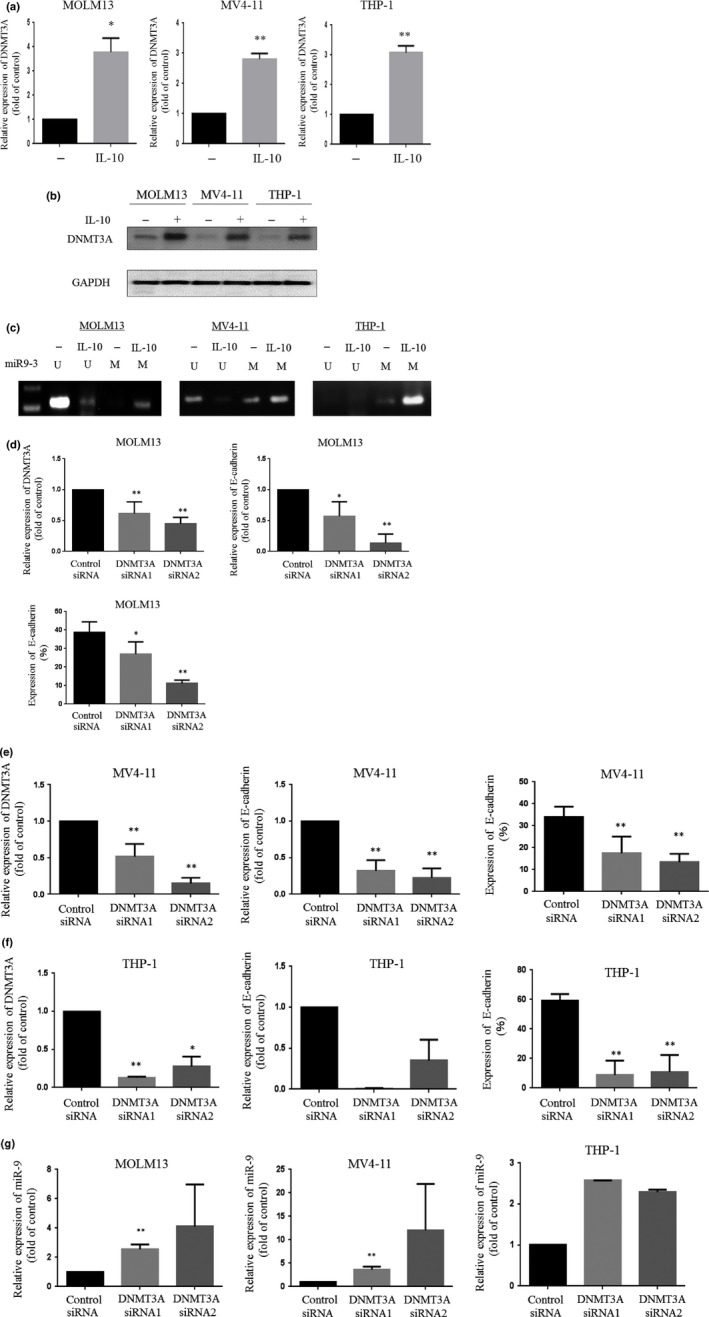
Effect of interleukin‐10 (IL‐10) on DNA methyltransferase 3A (DNMT3A) expression in leukemia cells. (a) DNMT3A expression in leukemia cells treated with IL‐10 was analyzed by real‐time RT‐PCR. (b) Western blot analysis. Leukemia cells treated with IL‐10 were harvested, and subjected to Western blot analysis to monitor the levels of the indicated proteins. (c) Methylation‐specific PCR. Leukemia cells were treated with IL‐10, and DNA was extracted from the cells. DNA with methylated CpG was processed using a MethylEasy Xceed Rapid DNA Bisulphite Modification Kit. The recovered DNA was amplified by PCR on miR‐9‐3 CpG islands. The experiments were repeated three times. M, methylation of the gene promoter; U, unmethylated gene promoter. (d–f) DNMT3A expression in MOLM13 (d), MV4‐11 (e), and THP‐1 (f) cells transfected with either control siRNA or DNMT3A siRNAs was analyzed by real‐time RT‐PCR. E‐cadherin expression in leukemia cells transfected with either control or DNMT3A siRNAs was analyzed by real‐time RT‐PCR and flow cytometry. (g) Expression of miR‐9 in leukemia cells transfected with either control or DNMT3A siRNAs was analyzed using an Mir‐X miRNA qRT‐PCR SYBR Kit to measure the levels of the indicated gene. The results shown represent the mean ± SD of three experiments carried out in triplicate. *P < 0.05; **P < 0.01.
Role of E‐cadherin in adhesion of leukemia cells
We next examined the biological function of E‐cadherin in leukemia cells. The GFP‐expressing THP‐1 cells were transfected with either control or E‐cadherin siRNAs (Fig. 4a), which were then incubated with AraC and/or IL‐10 in plates coated with UE6E7T‐3 BM‐MSCs. After 48 h, the number of GFP‐positive adherent cells was analyzed by fluorescent microscopy. The results showed that treatment with IL‐10 increased the number of adherent cells in control siRNA transfectants. The exposure of these cells to AraC decreased the number of adherent cells; however, the number of adherent cells increased in the presence of IL‐10 (Fig. 4b,c). In contrast, IL‐10 was not able to enhance the adhesion of THP‐1 cells to UE6E7T‐3 BM‐MSCs when E‐cadherin was depleted by siRNA. Nor did IL‐10 counteract the ability of AraC to detach leukemia cells from MCS cells in E‐cadherin‐depleted THP‐1 cells (Fig. 4c). Importantly, the number of adherent E‐cadherin siRNA‐transfected THP‐1 cells was less than that of control siRNA transfectants (Fig. 4c).
Figure 4.
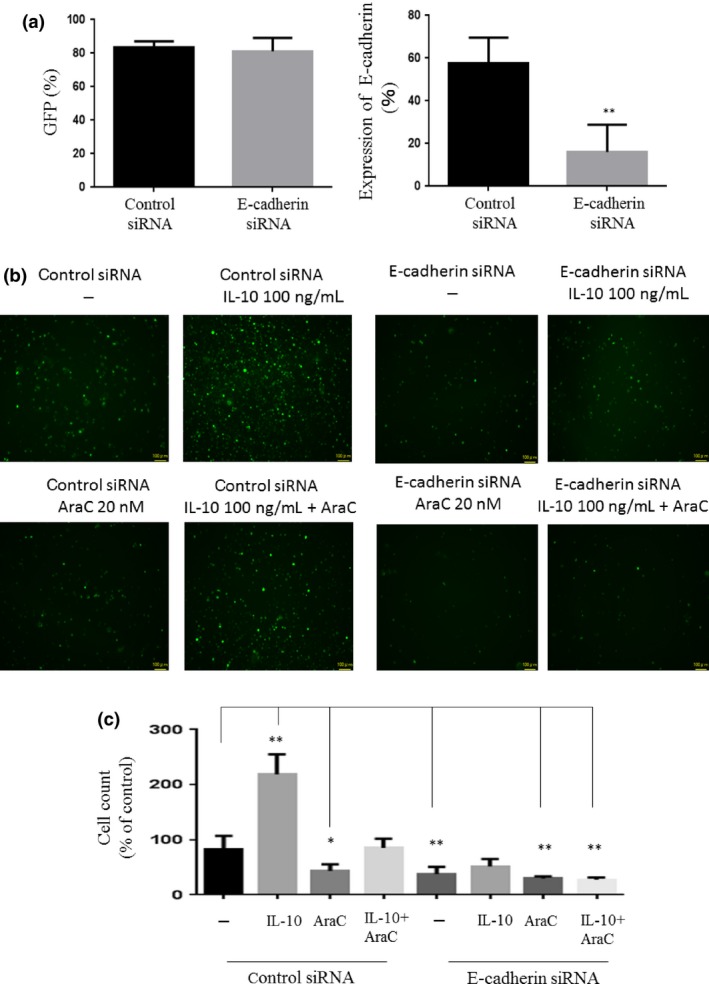
Function of E‐cadherin in leukemia cells. (a) GFP‐positive THP‐1 cells were transfected with control or E‐cadherin‐specific siRNA and analyzed for GFP and E‐cadherin expression by flow cytometry. **P < 0.01. (b) GFP‐positive THP‐1 cells were cocultured with UE6E7T‐3 human bone marrow‐derived mesenchymal stem cells and then treated with cytarabine (AraC) and/or interleukin‐10 (IL‐10). After 48 h, floating cells were removed and adherent cells were washed twice. GFP‐positive cells were analyzed by fluorescence microscopy. (c) Cell counts were undertaken using BZ‐II Dynamic Cell Count software.
Effect of IL‐10 on leukemia cell engraftment in vivo
Interleukin‐10 downregulation by an shRNA decreased the levels of E‐cadherin in MOLM13 cells to approximately 25% (Fig. S2). We next explored the effect of IL‐10/E‐cadherin on AML cell growth in severely immunocompromised mice. MOLM13 cells (5 × 106) transduced with control shRNA or an IL‐10‐specific shRNA were transplanted i.v. into NOD.Cg‐Rag1 tm1Mom Il2rg tm1Wjl/SzJ mice. Treatment of these mice with AraC (20 mg/kg; days 1, 3, 5, 8, 10, and 12; Fig. 5a) was initiated on day 8 of transplantation. Kaplan–Meier survival curves indicated that the mice transplanted with IL‐10‐depleted MOLM13 cells (hereafter referred to as IL‐10 shRNA mice) survived longer than the mice transplanted with control shRNA transduced MOLM13 cells (hereafter referred to as control shRNA mice, Fig. 5a). The treatment with AraC only slightly prolonged the survival of control shRNA mice (Fig. 5a). Of note, treatment of IL‐10 shRNA mice with AraC significantly prolonged their survival compared with untreated IL‐10 shRNA mice (Fig. 5a, P < 0.01). We also examined the MOLM13 leukemic cell burden in the transplanted mice. On day 18 of transplantation, engraftment of MOLM13 cells was severely impaired in IL‐10 shRNA mice compared with control shRNA mice, as judged by quantification of human CD82 expression by FACS (PBMCs: control shRNA, 33%; IL‐10 shRNA, 15%) (BM: control shRNA, 71%; IL‐10 shRNA, 29%) (Fig. 5b). Moreover, the treatment of either control shRNA or IL‐10 shRNA mice with AraC decreased leukemia cell numbers in both peripheral circulation and BM (PBMCs: control shRNA, 2%; IL‐10 shRNA, 3%) (BM: control shRNA, 14%; IL‐10 shRNA, 6%) (Fig. 5b). All of control shRNA and IL‐10 shRNA mice died because of leukemia by day 28 of transplantation. Fewer MOLM13 leukemia cells were noted in IL‐10 shRNA mice treated with AraC than control shRNA mice treated with AraC in both peripheral circulation and BM (PBMCs: control shRNA, 17%; IL‐10 shRNA, 7%) (BM: control shRNA, 46%; IL‐10 shRNA, 11%) (Fig. 5c). Simultaneously, we measured the levels of IL‐10, DNMT3A, and E‐cadherin in MOLM13 cells isolated from BM of control shRNA and IL‐10 shRNA mice on day 18 of transplantation to examine the effect of depletion of IL‐10 by an shRNA on expression of these genes in vivo. Real‐time RT‐PCR found that IL‐10 was effectively downregulated in MOLM13 cells removed from IL‐10 shRNA mice in parallel with a decrease in levels of DNMT3A and E‐cadherin (Fig. 5d). Real‐time RT‐PCR using murine plasma found that expression of miR‐9 was significantly elevated in plasma isolated from IL‐10 shRNA mice compared with that in plasma removed from control shRNA mice (Fig. 5e).
Figure 5.
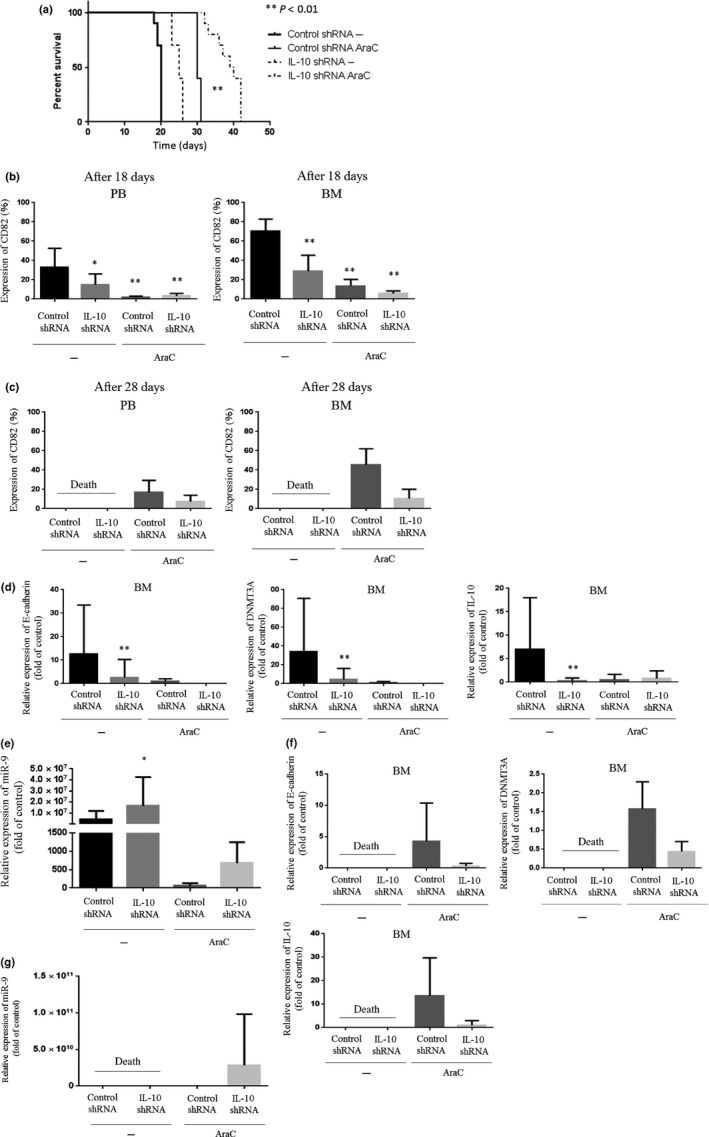
Effect of interleukin‐10 (IL‐10) on leukemia cell engraftment in immunodeficient mice and mouse survival. MOLM13 cells transduced with control shRNA or IL‐10‐specific shRNA were transplanted into NOD.Cg‐Rag1 tm1Mom Il2rg tm1Wjl/SzJ mice (control shRNA mice or IL‐10 shRNA mice) by tail vein injections (n = 10 mice/group). The treatment was initiated after 1 week of transplantation. The mice received either cytarabine (AraC) (n = 10; 20 mg/kg, 3 days/week for 2 weeks) or PBS (n = 10; 100 μL, 3 days/week for 2 weeks). (a) Kaplan–Meier analysis of mouse survival (n = 10). At day 18 (b) and 28 (c) of transplantation, peripheral blood (PB) and bone marrow (BM) cells were collected and incubated with anti‐CD82 antibody. Positive cells were analyzed by flow cytometry (n = 10). BM cells were collected on day 18 (d) and 28 (f) post‐transplantation and analyzed for IL‐10, DNA methyltransferase 3A (DNMT3A), and E‐cadherin mRNA expression by real‐time RT‐PCR (n = 10). Plasma was collected on day 18 (e) and 28 (g) post‐transplantation and analyzed for microRNA‐9 (miR‐9) expression by real‐time RT‐PCR (n = 10). *P < 0.05; **P < 0.01.
Discussion
The present study showed that AML cells aberrantly express immunosuppressive cytokine IL‐10 and an adhesion molecule E‐cadherin. Real‐time RT‐PCR analyses revealed that AML cells also express receptors for IL‐10 (data not shown). Because of the paucity of the sample number (n = 15), we were not able to identify the specific leukemia subtype that highly expresses these molecules. Also, close correlation between IL‐10 and E‐cadherin expression in AML cells was not noted (data not shown); nevertheless, exposing AML cells to IL‐10 significantly increased levels of E‐cadherin (Fig. 1a), suggesting the critical roles of IL‐10 in regulation of E‐cadherin expression. We previously showed that IL‐10 activates STAT5 that binds to the promoter region of the IL‐10 gene, resulting in IL‐10 production in AML cells.9 Interleukin‐10 produced by AML cells may support their survival by an autocrine mechanism. We also showed that STAT5A bound to the promoter region of DNMT3A gene and modulated the levels of DNMT3A in AML cells.33 As we found that IL‐10 elevated the expression of DNMT3A in AML cells, we strongly postulated that IL‐10‐induced hypermethylation and downregulation of miR‐9 in AML was most likely due to DNMT3A activity (Figs 2b,3c). Downregulation of DNMT3A by siRNAs increased expression of miR‐9 in parallel with a decrease in levels of E‐cadherin (Fig. 3g). In addition, forced expression of miR‐9 in AML cells decreased expression of E‐cadherin, whose 3′‐UTR contains binding sites for miR‐9. Collectively, IL‐10 increases levels of E‐cadherin through DNMT3A‐mediated downregulation of miR‐9 (Fig. 6). Regulatory T cells infiltrated in BM produce IL‐10, which may also mediate intracellular signal pathways leading to upregulation of E‐cadherin in AML cells in a paracrine manner.
Figure 6.
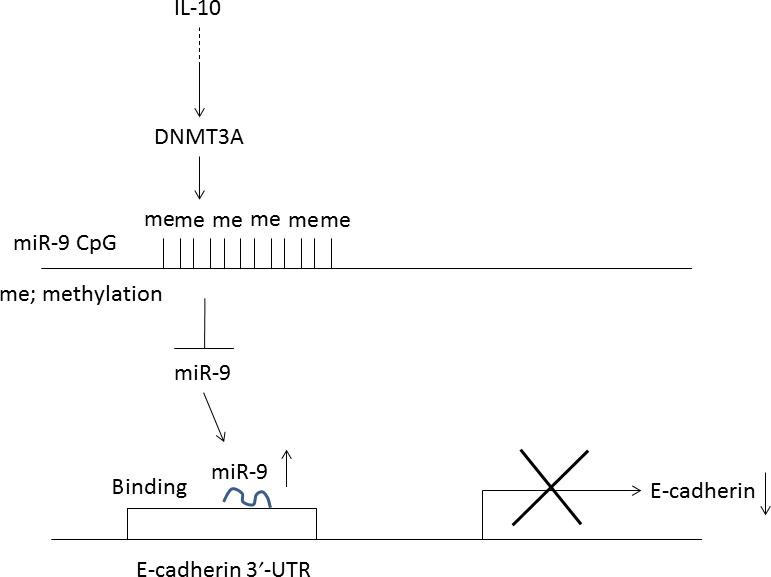
Interleukin‐10 ( IL‐10) regulates E‐cadherin expression through microRNA‐9 (miR‐9). DNMT3A, DNA methyltransferase 3A; me, methylation.
Proteomic analyses exploring therapeutic molecules aberrantly expressed in the chemotherapy‐resistant CD34+/CD38− immature AML cells identified CD82.8 CD34+/CD38− leukemia cells are enriched with leukemia stem cells, characterized by self‐renewal as well as differentiation capacity.34, 35 Leukemia stem cells are able to survive even after cytotoxic chemotherapy by interacting with the BM microenvironment comprised of vascular endothelium, osteoblasts, and stromal cells.36 E‐cadherin may play a role in the adhesion of leukemia stem cells to the BM microenvironment, the so‐called niche. The present study found that downregulation of E‐cadherin by knockdown IL‐10 by an shRNA potentiated the cytotoxic effects of AraC and prolonged the survival of MOLM13‐bearing mice (Fig. 5). We have recently proposed another therapeutic strategy targeting the CD82/STAT5/IL10 axis: the use of an antibody against CD82 mobilized the AML cells into peripheral circulation and potentiated the cytotoxic effects of AraC and significantly prolonged the survival of human AML bearing mice.37 Blockade of CD82 by an antibody might downregulate the expression of E‐cadherin through the IL‐10/DNMT3A pathway. Another candidate involved in leukemia cell mobilization by an anti‐CD82 antibody is MMP9. Downregulation of CD82 by shRNAs activated MMP9 and stimulated migration of leukemia cells in vitro.8 However, at the present time, precise molecular mechanisms by which CD82 regulates MMP activity remain unknown.
Previous findings showed that ectopic overexpression of miR‐9 inhibited the proliferative, migratory, and invasive capacities of nasopharyngeal carcinoma cells in vitro and in vivo. Overexpression of miR‐9 reduced the levels of CXCR4 by binding to the 3′‐UTR of CXCR4 in nasopharyngeal carcinoma cells.38 Interaction between HSCs and stromal cells is regulated by CXCR4/SDF‐1. The CXCR4 antagonist AMD3100 mobilizes leukemia cells into the peripheral circulation, which were then sensitized to the in vivo effects of cytotoxic chemotherapy.39 Thus, miR‐9 could induce the migration of leukemia cells through downregulation of CXCR4.
CD82 inhibits p38 MAPK and increases expression of EZH2, the polycomb group member in AML cells. The elevated expression of EZH2 was associated with histone H3 lysine 27 trimethylation in the promoter region of tumor suppressor genes such as PTEN and their downregulation.40 Activation of EZH2 may also cause downregulation of miR‐9 in AML cells.
The frequency of aberrant DNA methylation among AML patients was 13–69% for E‐cadherin.41, 42, 43 This pattern was markedly different between each patient. As the treatment of IL‐10 increased the levels of DNMT3A in leukemia cells, upregulation of DNMT3A could induce the methylation of the E‐cadherin promoter region, resulting in suppression of E‐cadherin expression. Contrary to our expectation, downregulation of DNMT3A decreased the levels of E‐cadherin in leukemia cells (Fig. 3d–f). These observations suggested that E‐cadherin expression might be repressed by various means including transcription factors, miRNAs, and epigenetic regulation.
Taken together, our results showed that IL‐10 positively regulated the expression of E‐cadherin by downregulation of miR‐9 in leukemia cells. Interleukin‐10 may be a promising molecular target for eradicating AML cells.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- AraC
cytarabine
- BM
bone marrow
- CXCR4
C‐X‐C chemokine receptor type 4
- DNMT3A
DNA methyltransferase 3A
- EZH2
enhancer of zeste homolog 2
- HSPC
hematopoietic stem and progenitor cell
- IL
interleukin
- ITD
internal tandem duplication
- miRNA
microRNA
- MNC
mononuclear cell
- MSC
mesenchymal stem cell
- STAT
signal transducer and activator of transcription
Supporting information
Fig. S1. Fluorescence‐activated cell sorting of leukemia cells.
Fig. S2. Interleukin‐10 (IL‐10) and E‐cadherin mRNA expression by real‐time RT‐PCR. **P < 0.01.
Acknowledgments
This work was supported in part by the Takeda Science Foundation (to C.N.) and the Japan Society for the Promotion of Science (KAKENHI Grant No. 14446690 to C.N.).
Cancer Sci 108 (2017) 685–695
Funding Information
Takeda Science Foundation; Japan Society for the Promotion of Science (14446690).
References
- 1. Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin‐10 and related cytokines and receptors. Annu Rev Immunol 2004; 22: 929–79. [DOI] [PubMed] [Google Scholar]
- 2. Liu Y, Wei SH, Ho AS, de Waal Malefyt R, Moore KW. Expression cloning and characterization of a human IL‐10 receptor. J Immunol 1994; 152: 1821–9. [PubMed] [Google Scholar]
- 3. Finbloom DS, Winestock KD. IL‐10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 alpha and STAT3 complexes in human T cells and monocytes. J Immunol 1995; 155: 1079–90. [PubMed] [Google Scholar]
- 4. Kotenko SV, Krause CD, Izotova LS, Pollack BP, Wu W, Pestka S. Identification and functional characterization of a second chain of the interleukin‐10 receptor complex. EMBO J 1997; 16: 5894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zou L, Barnett B, Safah H et al Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res 2004; 64: 8451–5. [DOI] [PubMed] [Google Scholar]
- 6. Fujisaki J, Wu J, Carlson AL et al In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem‐cell niche. Nature 2011; 474: 216–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weber‐Nordt RM, Henschler R, Schott E et al Interleukin‐10 increases Bcl‐2 expression and survival in primary human CD34 + hematopoietic progenitor cells. Blood 1996; 88: 2549–58. [PubMed] [Google Scholar]
- 8. Nishioka C, Ikezoe T, Yang J et al CD82 regulates STAT5/IL‐10 and supports survival of acute myelogenous leukemia cells. Int J Cancer 2014; 134: 55–64. [DOI] [PubMed] [Google Scholar]
- 9. Nishioka C, Ikezoe T, Furihata M et al CD34⁺/CD38− acute myelogenous leukemia cells aberrantly express CD82 which regulates adhesion and survival of leukemia stem cells. Int J Cancer 2013; 132: 2006–19. [DOI] [PubMed] [Google Scholar]
- 10. Abe M, Sugiura T, Takahashi M, Ishii K, Shimoda M, Shirasuna K. A novel function of CD82/KAI‐1 on E‐cadherin‐mediated homophilic cellular adhesion of cancer cells. Cancer Lett 2008; 266: 163–70. [DOI] [PubMed] [Google Scholar]
- 11. Shiwu WU, Lan Y, Wenqing S, Lei Z, Yisheng T. Expression and clinical significance of CD82/KAI1 and E‐cadherin in non‐small cell lung cancer. Arch Iran Med 2012; 15: 707–12. [PubMed] [Google Scholar]
- 12. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6: 857–66. [DOI] [PubMed] [Google Scholar]
- 13. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116: 281–97. [DOI] [PubMed] [Google Scholar]
- 15. Chen CZ. MicroRNAs as oncogenes and tumor suppressors. N Engl J Med 2005; 353: 1768–71. [DOI] [PubMed] [Google Scholar]
- 16. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol 2007; 302: 1–12. [DOI] [PubMed] [Google Scholar]
- 17. Wong KY, Huang X, Chim CS. DNA methylation of microRNA genes in multiple myeloma. Carcinogenesis 2012; 33: 1629–38. [DOI] [PubMed] [Google Scholar]
- 18. Yim RLH, Kwong YL, Wong KY, Chim CS. DNA methylation of tumor suppressive miRNAs in non‐Hodgkin's lymphomas. Front Genet 2012; 3: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Esteller M. Epigenetics in cancer. N Engl J Med 2008; 358: 1148–59. [DOI] [PubMed] [Google Scholar]
- 20. Tsai KW, Liao YL, Wu CW et al Aberrant hypermethylation of miR‐9 genes in gastric cancer. Epigenetics 2011; 6: 1189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang H, Qi M, Li S et al microRNA‐9 targets matrix metalloproteinase 14 to inhibit invasion, metastasis, and angiogenesis of neuroblastoma cells. Mol Cancer Ther 2012; 11: 1454–66. [DOI] [PubMed] [Google Scholar]
- 22. Ma L, Young J, Prabhala H et al miR‐9, a MYC/MYCN‐activated micro‐ RNA, regulates E‐cadherin and cancer metastasis. Nat Cell Biol 2010; 12: 247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nass D, Rosenwald S, Meiri E et al MiR‐92b and miR‐9/9* are specifically expressed in brain primary tumors and can be used to differentiate primary from metastatic brain tumors. Brain Pathol 2009; 19: 375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schraivogel D, Weinmann L, Beier D et al CAMTA1 is a novel tumour suppressor regulated by miR‐9/9* in glioblastoma stem cells. EMBO J 2011; 30: 4309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song Y, Li J, Zhu Y et al MicroRNA‐9 promotes tumor metastasis via repressing E‐cadherin in esophageal squamous cell carcinoma. Oncotarget 2014; 5: 11669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hildebrandt MA, Gu J, Lin J et al Hsa‐miR‐9 methylation status is associated with cancer development and metastatic recurrence in patients with clear cell renal cell carcinoma. Oncogene 2010; 29: 5724–8. [DOI] [PubMed] [Google Scholar]
- 27. Lehmann U, Hasemeier B, Christgen M et al Epigenetic inactivation of microRNA gene hsa‐mir‐9‐1 in human breast cancer. J Pathol 2008; 214: 17–24. [DOI] [PubMed] [Google Scholar]
- 28. Döhner H, Estey EH, Amadori S et al Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010; 115: 453–74. [DOI] [PubMed] [Google Scholar]
- 29. Vardiman JW, Thiele J, Arber DA et al The 2008 revision of the WHO Classification of Myeloid Neoplasms and Acute Leukemia: rationale and important changes. Blood 2009; 114: 937–51. [DOI] [PubMed] [Google Scholar]
- 30. Matsuo Y, MacLeod RA, Uphoff CC et al Two acute monocytic leukemia (AML‐M5a) cell lines (MOLM‐13 and MOLM‐14) with interclonal phenotypic heterogeneity showing MLL‐AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia 1997; 11: 1469–77. [DOI] [PubMed] [Google Scholar]
- 31. Nishioka C, Ikezoe T, Yang J, Nobumoto A, Tsuda M, Yokoyama A. Downregulation of miR‐217 correlates with resistance of Ph(+) leukemia cells to ABL tyrosine kinase inhibitors. Cancer Sci 2014; 105: 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lujambio A, Calin GA, Villanueva A et al A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 2008; 105: 13556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takeuchi A, Nishioka C, Ikezoe T, Yang J, Yokoyama A. STAT5A regulates DNMT3A in CD34(+)/CD38(−) AML cells. Leuk Res 2015; 39: 897–905. [DOI] [PubMed] [Google Scholar]
- 34. Clarke MF, Dick JE, Dirks PB et al Cancer stem cells‐perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66: 9339–44. [DOI] [PubMed] [Google Scholar]
- 35. Ishikawa F, Yoshida S, Saito Y et al Chemotherapy‐resistant human AML stem cells home to and engraft within the bone‐marrow endosteal region. Nat Biotechnol 2007; 25: 1315–21. [DOI] [PubMed] [Google Scholar]
- 36. Tabe Y, Konopleva M. Role of microenvironment in resistance to therapy in AML. Curr Hematol Malig Rep 2015; 10: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nishioka C, Ikezoe T, Yokoyama A. Blockade of CD82 by a monoclonal antibody potentiates anti‐leukemia effects of AraC in vivo. Cancer Med 2015; 4: 1426–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu J, Luo H, Liu X et al miR‐9 targets CXCR4 and functions as a potential tumor suppressor in nasopharyngeal carcinoma. Carcinogenesis 2014; 35: 554–63. [DOI] [PubMed] [Google Scholar]
- 39. Nervi B, Ramirez P, Rettig MP et al Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood 2009; 113: 6206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nishioka C, Ikezoe T, Yang J, Yokoyama A. Tetraspanin family member, CD82, regulates expression of EZH2 via inactivation of p38 MAPK signaling in leukemia cells. PLoS ONE 2015; 10: e0125017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Melki JR, Vincent PC, Clark SJ. Concurrent DNA hypermethylation of multiple genes in acute myeloid leukemia. Cancer Res 1999; 59: 3730–40. [PubMed] [Google Scholar]
- 42. Galm O, Wilop S, Lüders C et al Clinical implications of aberrant DNA methylation patterns in acute myelogenous leukemia. Ann Hematol 2005; 84: 39–46. [DOI] [PubMed] [Google Scholar]
- 43. Ekmekci CG, Gutiérrez MI, Siraj AK, Ozbek U, Bhatia K. Aberrant methylation of multiple tumor suppressor genes in acute myeloid leukemia. Am J Hematol 2004; 77: 233–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Fluorescence‐activated cell sorting of leukemia cells.
Fig. S2. Interleukin‐10 (IL‐10) and E‐cadherin mRNA expression by real‐time RT‐PCR. **P < 0.01.