

Abstract
Genetic mutations in exons of oncogenes and tumor‐suppressor genes causing qualitative abnormalities result in activation of the oncogenes and inactivation of the tumor‐suppressor genes, thereby causing cancer. In contrast, we have previously demonstrated that decreases in the RB promoter activity by genetic or epigenetic abnormalities can also cause carcinogenesis. In addition, activation and inactivation of a variety of oncogenes and tumor‐suppressor genes finally cause quantitative abnormalities in gene expression. Interestingly, we discovered effective molecular‐targeting agents, such as a novel MEK inhibitor, trametinib, by screening for agents upregulating the expression of cyclin‐dependent kinase inhibitors. In the present review, we focused on the quantitative abnormalities in gene expression with carcinogenesis, and discuss the importance of normalizing the quantitative abnormalities in gene expression with several molecular‐targeting agents.
Keywords: Carcinogenesis, molecular‐targeting therapies, quantitative abnormalities, RB, trametinib
There are a variety of complicated carcinogenic mechanisms. Among them, exon mutations activating oncogenes and inactivating tumor‐suppressor genes resulting in qualitative abnormalities of the product proteins are important. One more essential mechanism is the inactivation of promoter activities of tumor‐suppressor genes by genetic or epigenetic changes resulting in quantitative abnormalities of the product proteins. Interestingly, even qualitative abnormalities of oncogenes or tumor‐suppressor genes finally result in quantitative abnormalities in gene expression as described below.
Silencing of RB gene expression
The RB gene is a representative tumor‐suppressor gene, and mutations and deletions of the exon regions of the gene are observed in not only retinoblastoma, but also many types of malignant tumors. Sakai et al. reported two types of mutations in the promoter region of the RB gene in hereditary retinoblastoma patients (Fig. 1).1 The mutations in the RB promoter region markedly decreased the promoter activity, suggesting that the quantitative abnormality is also important in carcinogenesis. Furthermore, Sakai et al. and another group also found that the promoter region of the RB gene was hypermethylated in retinoblastoma tumors (Fig. 1).2, 3, 4 Subsequently, Ohtani et al. demonstrated that the hypermethylation of the RB promoter region reduced its promoter activity by dissociation of the pivotal transcription factors, activating transcription factor (ATF) and the retinoblastoma binding factor 1 (RBF‐1/E4TF1/GABP) from the core RB promoter region (Fig. 1),5 which was the first demonstration of epigenetic silencing of tumor‐suppressor genes.6, 7 The results indicate that epigenetic abnormalities can cause cancer and that quantitative abnormalities in tumor suppressor genes are essential for carcinogenesis. We therefore hypothesized that agents upregulating the expression of silenced tumor‐suppressor genes may be promising for novel chemotherapeutics.
Figure 1.
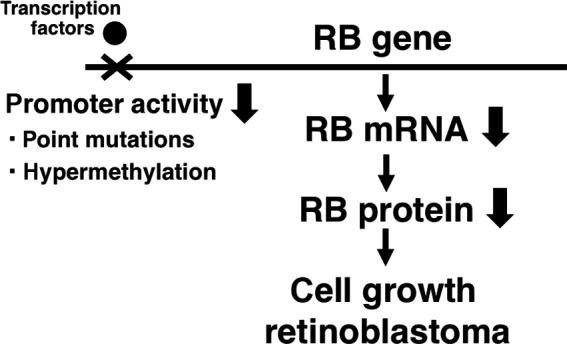
Decreases in the RB promoter activity by genetic or epigenetic abnormalities can cause carcinogenesis.
On the other hand, the p16 gene is also a representative tumor‐suppressor gene and epigenetically silenced by hypermethylation in many types of malignant tumors.8, 9, 10 Indeed, a DNA methyltransferase (DNMT) inhibitor, decitabine, induced the expression of p16 in lung cancer cells.11 At present, decitabine (trade name Dacogen) and another DNMT inhibitor, azacitidine (trade name Vidaza), are used in the treatment of myelodysplastic syndrome. This is consistent with our original hypothesis.
Inactivation of RB protein in many malignancies, which finally increases expression of E2F‐driven genes causing cancer
In addition to inactivation of RB promoter activity, RB protein is also inactivated by phosphorylation. This phosphorylation is caused by CDKs, for example, CDK2, CDK4 and CDK6 with their corresponding cyclins, and CDK inhibitors (CKIs), such as p21, p27, p16, p15, p18 and p19, repress the phosphorylation (Fig. 2).
Figure 2.
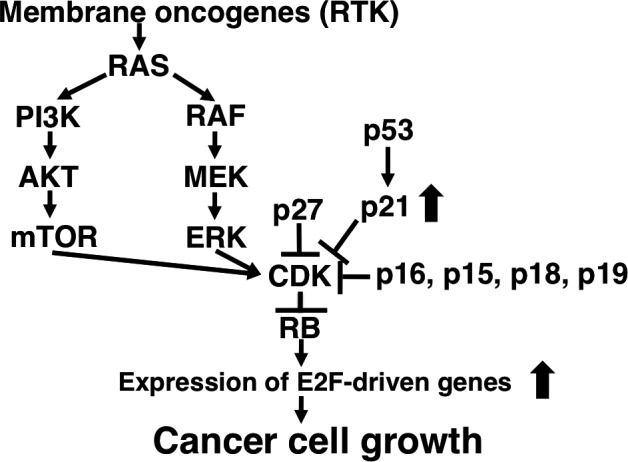
Activated oncogenes and inactivated tumor‐suppressor genes finally activate CDK activity with inactivation of RB function.
As shown in Fig. 2, RB protein is inactivated by activated oncogenes and inactivated tumor‐suppressor genes. For example, RAS genes, such as H‐RAS, K‐RAS and N‐RAS, are representative oncogenes, and the active mutations are observed in a variety types of malignant tumors. As RAS activates both the mitogen‐activated protein kinase (MAPK) pathway, including RAF, MEK and ERK, and the PI3K/AKT/mTOR pathway, mutant RAS constitutively enhances CDK activity through the upregulation of cyclin D1 expression (Fig. 2).12, 13 Oncogenic receptor tyrosine kinases (RTKs), such as epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (Her2), and so on, are transmembrane kinases that act as receptors for extracellular growth factors.14 As RTKs activate RAS function, RTKs also have critical functions in cell proliferation. Indeed, amplification and/or active mutations in RTKs, such as EGFR and Her2, are observed in malignant tumors, resulting in the enhancement of CDK activity with inactivation of RB (Fig. 2).15 In addition, inactivation of the representative tumor‐suppressor genes p53 and p16, the most commonly inactivated tumor‐suppressor genes, also enhance CDK activity with RB inactivation (Fig. 2).16
Taken together, activation of most oncogenes and inactivation of most tumor‐suppressor genes finally activate CDK activity, thereby converting RB protein to the phosphorylated inactivated form.17 Unphosphorylated RB protein is an active form that binds to the transcription factor E2F.18 E2F can transactivate the genes accelerating the cells from G1 phase to S phase at the restriction point (R point),19 such as dihydrofolate reductase, myc, cyclin E, thymidylate synthase and DNA polymerase α, resulting in cellular proliferation (Fig. 2).20 In summary, carcinogenesis is caused by the quantitative abnormalities in gene expression with most malignant tumors.
As CDK activity is regulated by upstream molecules, as mentioned above, we focused on the direct measurement of the CDK activity in clinical samples. As a result, CDK profiling technology, which was named “Cell Cycle Profiling (C2P)” was established in collaboration with Sysmex corporation, Kobe Japan. Using C2P technology, we found that CDK2 activity in more than 70% of gastric cancer and colon cancer tissues was higher than that in adjacent normal tissues.21 This result reflects that various qualitative and/or quantitative abnormalities of oncogenes and/or tumor‐suppressor genes in malignant tumor cells converge on the elevation of CDK activity, resulting in inactivation of RB protein.
Inactivation of tumor‐suppressor gene p53 decreases expression of p53 target genes causing malignancy, and histone deacetylase (HDAC) inhibitors reactivate the expressions of the genes suppressing tumor growth
In addition to RB, p53 is also a representative tumor suppressor gene inactivated in a variety of malignant tumors. As p53 protein acts as a transcription factor and activates the expression of cell cycle‐, DNA repair‐ and apoptosis‐regulating genes as a tumor suppressor, the expression of the target genes is decreased in p53‐mutant malignant tumor cells. Therefore, we tried to screen agents to upregulate the expression of the target genes. First, we found that butyrate induces the expression of a CDK inhibitor, p21, through the Sp1 sites of the promoter in a p53‐independent manner and converts RB protein to the unphosphorylated active form with G1‐cell cycle arrest.22 Subsequently, we demonstrated that HDAC inhibitors also induce the expression of p21 in a similar manner (Fig. 3).23, 24, 25 We also found that an HDAC inhibitor enhances the promoter activities of other p53 target genes, gadd45 and DR5, in p53‐mutated cancer cells in a p53‐independent pathway (Fig. 3).26, 27 Taken together, when expression of p53 target genes is not induced in p53‐mutant cancer cells, HDAC inhibitors can reactivate expression, thereby suppressing cancer. The data above strongly suggest that carcinogenesis can be caused by quantitative abnormalities in gene expression and molecular‐targeting agents, such as HDAC inhibitors, can normalize the quantitative abnormalities.
Figure 3.
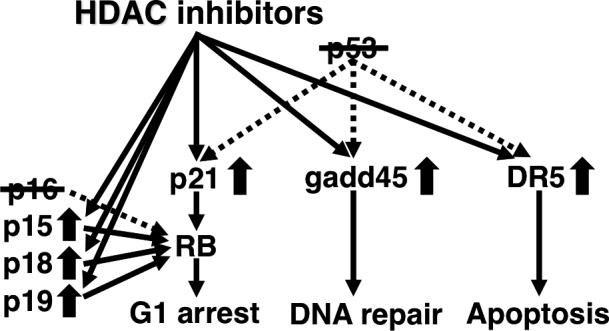
Histone deacetylase (HDAC) inhibitors can normalize the quantitative abnormality in gene expression with p53‐mutated tumors, and can compensate for the inactivated p16 function by increasing the expression of the family genes.
As mentioned previously, the p16 gene is another important tumor‐suppressor gene inactivated in a variety of malignant tumors,8, 9, 10, 28 and p16 converts RB protein to the unphosphorylated active form as a CDK inhibitor. The p16 gene belongs to the INK4 family genes, and we hypothesized that upregulation of other family genes, such as p15, p18, or p19, may compensate for the function of inactivated p16. We then found that HDAC inhibitors can activate the promoter activities of the family genes, such as p15, p18, and p19, converting RB protein to the unphosphorylated active form compensating for inactivated p16 (Fig. 3).29, 30, 31 The results also demonstrate that quantitative normalization by molecular‐targeting agents is useful for treating malignant tumors.
Approved molecular‐targeting agents increase expression of CKIs
As described above, HDAC inhibitors can increase the expression of several CKIs, normalizing the quantitative abnormalities in gene expression in cancer cells (Table 1). Indeed, several HDAC inhibitors, vorinostat (trade name Zolinza), romidepsin (trade name Istodax), panobinostat (trade name Farydak), and belinostat (trade name Beleodaq) have been approved as molecular‐targeting agents for chemotherapy.
Table 1.
Approved molecular‐targeting agents increase expressions of CDK inhibitors (CKIs)
| Target‐molecules | Approved molecular‐targeting agents | Induced CKIs |
|---|---|---|
| HDAC | vorinostat | p21, p15, p18, p19 |
| EGFR kinase | gefitinib | p15 |
| Her2 | trastuzumab | p27 |
| Bcr‐Abl kinase | imatinib | p18 |
| mTOR | everolimus | p27 |
Interestingly, other approved molecular‐targeting agents also increase the expression of CKIs (Table 1). We found that the EGFR kinase inhibitor gefitinib (trade name Iressa) can induce the expression of p15.32 The anti‐Her2 antibody trastuzumab (trade name Herceptin) was shown to upregulate p27 expression.33 The Bcr‐Abl kinase inhibitor imatinib (trade name Gleevec) also upregulates p18 expression.34 Furthermore, an inhibitor of mammalian target of rapamycin (mTOR), everolimus (trade name Afinitor), was reported to induce p27 expression and decreased cyclin D1 expression inhibiting CDK activity.35 These results clearly demonstrate that inhibition of oncogenes by molecular‐targeting agents finally induces the expressions of CKIs, thereby converting RB protein to the unphosphorylated active form.
Screening for novel molecular‐targeting agents regulating expression of CKIs
We therefore hypothesized that screening for agents upregulating expression of CKIs may be useful. As a result of the screening in collaboration with several pharmaceutical companies, we found several potent compounds, including a novel MEK inhibitor trametinib by screening for p15 inducers.36 After submitting the patent, trametinib was in‐licensed by GlaxoSmithKline (GSK) in 2006 and clinically developed. Trametinib was approved as a first‐in‐class MEK inhibitor (trade name Mekinist) in 2013 in the U.S. as a single agent for treatment of BRAF V600E or V600K mutation‐positive unresectable, or metastatic melanoma based on the results of a phase 3 study.37 Drug discovery of the year awarded trametinib from British Pharmacological Society in 2013. Furthermore, combination therapy of trametinib and the BRAF inhibitor dabrafenib was also approved in 2014 in the US based on the results of a phase 1/2 study.38 After trametinib and dabrafenib were in‐licensed by Novartis from GSK in 2015, the combination treatment was approved in Japan in 2016. Against patients with previously treated BRAF‐mutant metastatic non‐small cell lung cancer, 63.2% achieved overall response by the combination therapy with dabrafenib and trametinib in phase 2 trial,39 and the combination therapy for BRAF‐mutant metastatic non‐small cell lung cancer received breakthrough therapy designation from FDA.
In addition, a dual RAF/MEK inhibitor, CH5126766/RO5126766, which is undergoing a clinical phase I trial, was found by screening for p27 inducers,40 and the most potent HDAC inhibitor, OBP‐801/YM753/spiruchostatin A, which is also undergoing a clinical phase I trial, was identified by screening for p21 inducers.41, 42 We are still developing our original screening strategy to find novel molecular‐targeting agents for unmet medical needs in chemotherapy.
Conclusion
In carcinogenesis, both qualitative and quantitative abnormalities are important. However, we focused on the importance of quantitative abnormalities in gene expression because even a variety of qualitative abnormalities finally can also cause quantitative abnormalities in gene expression and molecular‐targeting agents could normalize the quantitative abnormalities. In addition, in our experience, we actually discovered very effective molecular‐targeting agents with our original screening system searching for agents regulating the quantity of several molecules. We therefore emphasize the importance of quantitative abnormalities in gene expression with carcinogenesis and regulating the quantitative abnormalities by effective molecular‐targeting agents.
Disclosure Statement
The authors have no conflict of interest to declare.
Cancer Sci 108 (2017) 570–573
Funding Information
No sources of funding were declared for this study
References
- 1. Sakai T, Ohtani N, McGee TL, Robbins PD, Dryja TP. Oncogenic germ‐line mutations in Sp1 and ATF sites in the human retinoblastoma gene. Nature 1991; 353: 83–6. [DOI] [PubMed] [Google Scholar]
- 2. Sakai T, Toguchida J, Ohtani N, Yandell DW, Rapaport JM, Dryja TP. Allele‐specific hypermethylation of the retinoblastoma tumor‐suppressor gene. Am J Hum Genet 1991; 48: 880–8. [PMC free article] [PubMed] [Google Scholar]
- 3. Greger V, Debus N, Lohmann D, Höpping W, Passarge E, Horsthemke B. Frequency and parental origin of hypermethylated RB1 alleles in retinoblastoma. Hum Genet 1994; 94: 491–6. [DOI] [PubMed] [Google Scholar]
- 4. Ohtani‐Fujita N, Dryja TP, Rapaport JM et al Hypermethylation in the retinoblastoma gene is associated with unilateral, sporadic retinoblastoma. Cancer Genet Cytogenet 1997; 98: 43–9. [DOI] [PubMed] [Google Scholar]
- 5. Ohtani‐Fujita N, Fujita T, Aoike A, Osifchin NE, Robbins PD, Sakai T. CpG methylation inactivates the promoter activity of the human retinoblastoma tumor‐suppressor gene. Oncogene 1993; 8: 1063–7. [PubMed] [Google Scholar]
- 6. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4: 143–53. [DOI] [PubMed] [Google Scholar]
- 7. Hattori N, Ushijima T. Compendium of aberrant DNA methylation and histone modifications in cancer. Biochem Biophys Res Commun 2014; 455: 3–9. [DOI] [PubMed] [Google Scholar]
- 8. Merlo A, Herman JG, Mao L et al 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1995; 1: 686–92. [DOI] [PubMed] [Google Scholar]
- 9. Herman JG, Merlo A, Mao L et al Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995; 55: 4525–30. [PubMed] [Google Scholar]
- 10. Gonzalez‐Zulueta M, Bender CM, Yang AS et al Methylation of the 5′ CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 1995; 55: 4531–5. [PubMed] [Google Scholar]
- 11. Otterson GA, Khleif SN, Chen W, Coxon AB, Kaye FJ. CDKN2 gene silencing in lung cancer by DNA hypermethylation and kinetics of p16INK4 protein induction by 5‐aza 2′deoxycytidine. Oncogene 1995; 11: 1211–6. [PubMed] [Google Scholar]
- 12. Filmus J, Robles AI, Shi W, Wong MJ, Colombo LL, Conti CJ. Induction of cyclin D1 overexpression by activated ras . Oncogene 1994; 9: 3627–33. [PubMed] [Google Scholar]
- 13. Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol 2003; 15: 158–63. [DOI] [PubMed] [Google Scholar]
- 14. Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 2004; 4: 361–70. [DOI] [PubMed] [Google Scholar]
- 15. Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene 2000; 19: 6102–14. [DOI] [PubMed] [Google Scholar]
- 16. Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell 2002; 2: 103–12. [DOI] [PubMed] [Google Scholar]
- 17. Sherr CJ. Cancer cell cycles. Science 1996; 274: 1672–7. [DOI] [PubMed] [Google Scholar]
- 18. Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev 2000; 14: 2393–409. [DOI] [PubMed] [Google Scholar]
- 19. Pardee AB. A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci USA 1974; 71: 1286–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lavia P, Jansen‐Dürr P. E2F target genes and cell‐cycle checkpoint control. BioEssays 1999; 21: 221–30. [DOI] [PubMed] [Google Scholar]
- 21. Ishihara H, Yoshida T, Kawasaki Y et al A new cancer diagnostic system based on a CDK profiling technology. Biochim Biophys Acta 2005; 1741: 226–33. [DOI] [PubMed] [Google Scholar]
- 22. Nakano K, Mizuno T, Sowa Y et al Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53‐negative human colon cancer cell line. J Biol Chem 1997; 272: 22199–206. [DOI] [PubMed] [Google Scholar]
- 23. Sowa Y, Orita T, Minamikawa S et al Histone deacetylase inhibitor activates the WAF1/Cip1 gene promoter through the Sp1 sites. Biochem Biophys Res Commun 1997; 241: 142–50. [DOI] [PubMed] [Google Scholar]
- 24. Sowa Y, Orita T, Minamikawa‐Hiranabe S, Mizuno T, Nomura H, Sakai T. Sp3, but not Sp1, mediates the transcriptional activation of the p21/WAF1/Cip1 gene promoter by histone deacetylase inhibitor. Cancer Res 1999; 59: 4266–70. [PubMed] [Google Scholar]
- 25. Huang L, Sowa Y, Sakai T, Pardee AB. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene 2000; 19: 5712–9. [DOI] [PubMed] [Google Scholar]
- 26. Hirose T, Sowa Y, Takahashi S et al p53‐independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct‐1 and NF‐Y. Oncogene 2003; 22: 7762–73. [DOI] [PubMed] [Google Scholar]
- 27. Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL‐R2 and sensitize apoptosis induced by TRAIL/APO2‐L in human malignant tumor cells. Oncogene 2004; 23: 6261–71. [DOI] [PubMed] [Google Scholar]
- 28. Cairns P, Polascik TJ, Eby Y et al Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nat Genet 1995; 11: 210–2. [DOI] [PubMed] [Google Scholar]
- 29. Hitomi T, Matsuzaki Y, Yokota T, Takaoka Y, Sakai T. p15INK4b in HDAC inhibitor‐induced growth arrest. FEBS Lett 2003; 554: 347–50. [DOI] [PubMed] [Google Scholar]
- 30. Yokota T, Matsuzaki Y, Sakai T. Trichostatin A activates p18INK4c gene: differential activation and cooperation with p19INK4d gene. FEBS Lett 2004; 574: 171–5. [DOI] [PubMed] [Google Scholar]
- 31. Yokota T, Matsuzaki Y, Miyazawa K, Zindy F, Roussel MF, Sakai T. Histone deacetylase inhibitors activate INK4d gene through Sp1 site in its promoter. Oncogene 2004; 23: 5340–9. [DOI] [PubMed] [Google Scholar]
- 32. Koyama M, Matsuzaki Y, Yogosawa S, Hitomi T, Kawanaka M, Sakai T. ZD1839 induces p15INK4b and causes G1 arrest by inhibiting the mitogen‐activated protein kinase/extracellular signal‐regulated kinase pathway. Mol Cancer Ther 2007; 6: 1579–87. [DOI] [PubMed] [Google Scholar]
- 33. Le XF, Pruefer F, Bast RC Jr. HER2‐targeting antibodies modulate the cyclin‐dependent kinase inhibitor p27Kip1 via multiple signaling pathways. Cell Cycle 2005; 4: 87–95. [DOI] [PubMed] [Google Scholar]
- 34. Nishimura N, Furukawa Y, Sutheesophon K et al Suppression of ARG kinase activity by STI571 induces cell cycle arrest through up‐regulation of CDK inhibitor p18/INK4c. Oncogene 2003; 22: 4074–82. [DOI] [PubMed] [Google Scholar]
- 35. Zitzmann K, De Toni EN, Brand S et al The novel mTOR inhibitor RAD001 (everolimus) induces antiproliferative effects in human pancreatic neuroendocrine tumor cells. Neuroendocrinology 2007; 85: 54–60. [DOI] [PubMed] [Google Scholar]
- 36. Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP‐74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo . Int J Oncol 2011; 39: 23–31. [DOI] [PubMed] [Google Scholar]
- 37. Flaherty KT, Robert C, Hersey P et al Improved survival with MEK inhibition in BRAF‐mutated melanoma. N Engl J Med 2012; 367: 107–14. [DOI] [PubMed] [Google Scholar]
- 38. Flaherty KT, Infante JR, Daud A et al Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 2012; 367: 1694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Planchard D, Besse B, Groen HJ et al Dabrafenib plus trametinib in patients with previously treated BRAFV600E‐mutant metastatic non‐small cell lung cancer: an open‐label, multicentre phase 2 trial. Lancet Oncol 2016; 17: 984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ishii N, Harada N, Joseph EW et al Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res 2013; 73: 4050–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shindoh N, Mori M, Terada Y et al YM753, a novel histone deacetylase inhibitor, exhibits antitumor activity with selective, sustained accumulation of acetylated histones in tumors in the WiDr xenograft model. Int J Oncol 2008; 32: 545–55. [PubMed] [Google Scholar]
- 42. Masuoka Y, Nagai A, Shin‐ya K et al Spiruchostatins A and B, novel gene expression enhancing substances produced by Pseudomonas sp. Tetrahedron Lett 2001; 42: 41–4. [Google Scholar]