

Abstract
Epigenetic regulation in hematopoiesis has been a field of rapid expansion. Genome‐wide analyses have revealed, and will continue to identify genetic alterations in epigenetic genes that are present in various types of hematopoietic neoplasms. Development of new mouse models for individual epigenetic modifiers has revealed their novel, sometimes unexpected, functions. In this review, we provide an overview of genetic alterations within epigenetic genes in various types of hematopoietic neoplasms. We then summarize the physiologic roles of these epigenetic modifiers during hematopoiesis, and describe therapeutic approaches targeting the epigenetic modifications. Interestingly, the mutational spectrum of epigenetic genes indicates that myeloid neoplasms are similar to T‐cell neoplasms, whereas B‐cell lymphomas have distinct features. Furthermore, it appears that the epigenetic mutations related to active transcription are more associated with myeloid/T‐cell neoplasms, whereas those that repress transcription are associated with B‐cell lymphomas. These observations may imply that the global low‐level or high‐level transcriptional activity underlies the development of myeloid/T‐cell tumors or B‐cell tumors, respectively.
Keywords: DNA methylation, epigenetics, hematopoiesis, hematopoietic neoplasms, histone modification
Global change in the epigenetic landscape is a hallmark of cancer, and the reversible nature of epigenetic aberrations has led to the emergence of the promising field of epigenetic therapy.1 Hematopoietic neoplasm is one of the major cancer types for which several epigenetic therapies are currently in development. Epigenetic regulation refers to gene regulation achieved by changes in chromatin structure. The basic unit of chromatin, called a nucleosome, consists of an octamer of histone proteins wrapped with DNA. The histone octamer consists of two copies of each of the four core histone proteins: H2A, H2B, H3 and H4. Histone proteins contain a globular C‐terminal domain and an unstructured N‐terminal tail. The N‐terminal histone tails receive a variety of posttranslational modifications, including acetylation, methylation and ubiquitination on specific residues. These histone modifications, together with DNA methylation, regulate gene expression in a highly orchestrated manner without altering the primary DNA sequence. We first summarize the fundamentals of the representative epigenetic modifications (Fig. 1).
Figure 1.
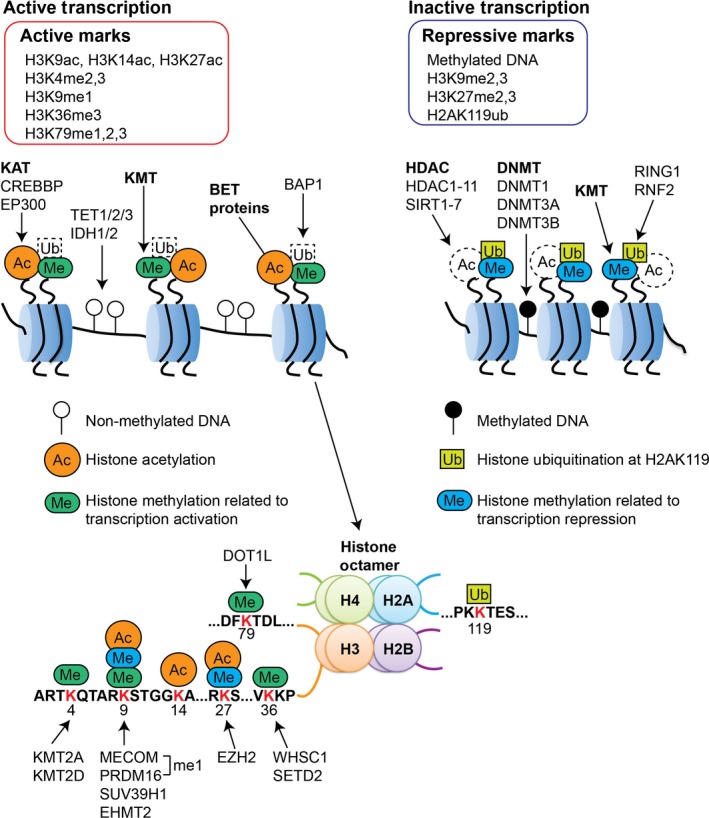
Epigenetic mechanisms to regulate gene expression. DNA is wrapped around histone protein cores composed of an octamer containing two copies of each core histone: H2A, H2B, H3 and H4. Epigenetic patterns are established by a number of mechanisms, including DNA methylation, histone acetylation, histone methylation and histone ubiquitination. DNA methylation is established and maintained by the DNMT enzymes. IDH and TET proteins promote the early steps of active DNA demethylation (see Fig. 2). Histone acetylation is associated with active transcription, and is induced or removed by histone lysine acetyltransferases (KAT) or histone deacetylases (HDAC), respectively. BET proteins bind to acetylated histone to activate transcription. Histone methylation leads to transcriptional activation or repression depending on which residue is modified and the degree of methylation (see the lower part of this figure). Histone ubiquitination at H2AK119 is associated with transcriptional repression.
Fundamentals of Epigenetic Modifications
DNA methylation
DNA methylation, which is associated with heterochromatin formation and transcriptional repression, occurs at the C‐5 carbon of cytosines in DNA to form 5‐methylcytosine (5mC) in mammalian cells.2 There are three major DNA methyltransferases (DNMT) that establish and maintain DNA methylation. DNMT1 plays a key role in maintaining DNA methylation after DNA replication, while DNMT3a and DNMT3b promote genome‐wide de novo DNA methylation. DNA demethylation is a dynamic process involving TET and IDH proteins. The TET enzymes, TET1, TET2 and TET3, catalyze the successive oxidation of 5mC to 5‐hydroxymethylcytosine (5hmC), 5‐formylcytosine (5‐fC) and then 5‐carboxycytosine (5‐caC). These 5mC oxidation products are implicated as intermediates in the conversion of 5mC to unmethylated cytosine, providing a first step in a pathway for active DNA demethylation. TET enzymes need α‐ketoglutarate (α‐KG) for their catalytic function. α‐KG is converted from isocitrate by IDH proteins, IDH1 and IDH2 (Fig. 2).
Figure 2.

Regulation of DNA methylation and demethylation. DNMT enzymes methylate the nucleotide cytosine (5‐methylcytosine, 5‐mC). TET proteins catalyze the oxidation of 5‐mC into 5‐hydroxymethylcytosine (5‐hmC), promoting the demethylation process. IDH proteins promote the conversion from isocitrate to α‐KG, which is required for catalytic function of TET enzymes.
Histone acetylation
Histone lysine acetylation occurs at several different positions in the histone tail (e.g. H3K9, H3K14 and H3K27), and leads to the open structure of chromatin that allows access of transcriptional factors. Consequently, histone acetylation is associated with active transcription. The processes of acetylation and deacetylation are governed by histone lysine acetyltransferases (KAT) and histone deacetylases (HDAC), respectively. KAT include CREBBP (CBP), EP300 (p300), KAT2B (PCAF), KAT5 (Tip60) and KAT6A (MOZ), while HDAC include HDAC1‐11 and SIRT1‐7. Bromodomain and extra‐terminal (BET) proteins, BRD2, BRD3 and BRD4, recognize and bind to the acetylated histone lysine residues to activate transcription.3
Histone methylation
In contrast to histone lysine acetylation, which is generally associated with transcriptional activation, histone lysine methylation leads to transcriptional activation or repression, depending on which residue is modified and the degree of methylation (Fig. 1).4 Methylation of histone H3 lysine 4 (H3K4), H3K36 and H3K79 are associated with transcriptional activation, while dimethylation and trimethylation of H3K9 (H3K9me2, H3K9me3) and H3K27 (H3K27me2, H3K27me3) are associated with transcriptional repression. Monomethylation of H3K9 (H3K9me1) and H3K27 (H3K27me1) are more enriched at active promoters than silent promoters. Histone methylation is regulated by histone lysine methyltransferases (KMT) and lysine demethylases (KDM) targeting specific lysine in histones. H3K4 methylation, which is tightly associated with gene activation, is mediated by SET domain‐containing methyltransferases, such as KMT2A (MLL1) and KMT2D (MLL2). Several enzymes, including SETD2 and WHSC1 (NSD2/MMSET), induce H3K36 methylation. H3K79 is mainly methylated by DOT1L. H3K27 methylation, a well‐known mark of silent chromatin, is induced by EZH1 and EZH2. EZH1/2 is part of the Polycomb Repressive Complex 2 (PRC2) containing SUZ12 and EED. An epigenetic factor ASXL1 was also shown to be associated with PRC2 to increase its function. KMT for H3K9 dimethylations/trimethylations include SUV39H1 and EHMT2 (G9a). In addition, the Prdm family proteins MECOM (EVI1) and PRDM16 were identified as H3K9me1‐specific KMT.5 The methylations at H3K27 and H3K9 together constitute the two main silencing mechanisms in mammalian cells.
Several KDM have been shown to have important roles in hematopoiesis.6 KDM1A (LSD1) removes monomethylated and dimethylated H3K4 and H3K9. KDM2B is a Jumonji (JmjC) domain histone H3K36 di‐demethylase. KDM5A is an α‐ketoglutarate‐dependent JmjC‐containing protein, and acts on dimethylated and trimethylated H3K4. KDM6A (UTX) is another Jmjc protein and removes H3K27me2/me3.
Histone ubiquitination
In mammals, there are two major complexes formed by Polycomb proteins: Polycomb Repressive Complex 1 (PRC1) and PRC2.7 As previously described, PRC2 contains three core subunits (EZH1/2, EED and SUZ12), and catalyzes dimethylation and trimethylation of H3K27. Canonical PRC1 contains four core subunits, BMI1 or MEL18, CBX family proteins, PHC, and an E3 ubiquitin ligase RING1 or RNF2. The PRC2‐mediated histone methylation triggers recruitment of PRC1 in part due to the ability of the CBX subunit to bind to H3K27me3. The RING1 subunit (RING1 or RNF2) then induces mono‐ubiquitination of histone H2A at lysine 119 (H2AK119ub). The H2AK119ub inhibits transcriptional elongation, promotes chromatin compaction, and is associated with transcriptional repression. BAP1, an interacting partner of ASXL1, was identified as a deubiquitinase that removes the ubiquitination on H2AK119.
Genetic Alterations within Epigenetic Modifiers in Hematopoietic Neoplasms
Patient data illuminate the relevance of epigenetic modifications to hematopoietic neoplasms. In this section, we summarize genetic alterations within epigenetic modifiers that are frequently present in hematopoietic neoplasms (Table 1).
Table 1.
Epigenetic modifiers dysregulated in hematopoietic neoplasms
| Genes | Activity | Genetic alteration | Gain‐/Loss‐of‐function | Diseases | Effect of mutations on Transcription | |
|---|---|---|---|---|---|---|
| DNA methylation | DNMT3A | De novo DNA methylation | Mutation (R882 and others), mostly monoallelic | Loss | AML (20%) | Activation |
| MDS (10%) | ||||||
| Mutation (R882 and others), many biallelic mutation | Loss | T‐cell lymphoma/T‐ALL (25%) | ||||
| TET2 | Conversion of 5‐mC to 5‐hmC | Mutation, mostly monoallelic | Loss | AML (20%) | TBD | |
| MDS (20%) | ||||||
| CMML (50%) | ||||||
| B‐cell lymphoma (5%) | ||||||
| T‐cell lymphoma/T‐ALL (25%) | ||||||
| IDH1/IDH2 | A cofactor of TET2 | R132 (IDH1), R140/R172 (IDH2), monoallelic | Gain | AML (10%) | TBD | |
| MDS (3%) | ||||||
| Histone methylation | EZH2 | H3K27 KMT, a member of PRC2 | Mutations, monoallelic or biallelic | Loss | MDS (5%) | Activation |
| T‐ALL (10%) | ||||||
| Mutation (Y641 and others) | Gain | DLBCL (20%) | Repression | |||
| FL (10%) | ||||||
| ASXL1 | Associates with PRC1 and PRC2 | Mutation, monoallelic | TBD | AML (5%) | Activation | |
| CMML (45%) | ||||||
| MDS (20%) | ||||||
| SUZ12 | A member of PRC2 | Mutation | Loss | MDS/T‐ALL (rare) | Activation | |
| EED | A member of PRC2 | Mutation | Loss | MDS/T‐ALL (rare) | Activation | |
| KMT2A (MLL1) | H3K4 KMT | Rearrangements (11q23) | Gain | AML (5%) | Activation | |
| B‐ALL (10%) | ||||||
| PTD | Gain | AML/MDS (5%) | Activation | |||
| KMT2D (MLL2) | H3K4 KMT | Mutation, monoallelic or biallelic | Loss | DLBCL (30%) | Repression | |
| FL (70%) | ||||||
| MECOM | H3K9(me1) KMT | Rearrangements (3q26) | Gain | AML/MDS (rare) | TBD | |
| PRDM16 | H3K9(me1) KMT | Rearrangements [t(1;3)] | Gain | AML/MDS (rare) | TBD | |
| SETD2 | H3K36 KMT | Mutation, monoallelic or biallelic | Loss | AML/B‐ALL (5%) | Repression | |
| WHSC1 | H3K36 KMT | Rearrangements [t(4;14)] | Gain | t(4:14)MM (30%) | TBD | |
| KDM6A (UTX) | H3K27 KDM | Mutation/Deletion | Loss | MM | TBD | |
| T‐ALL (15%) | ||||||
| KDM2B | H3K36 KDM | Mutation/Deletion | Loss | DLBCL (5%) | TBD | |
| Histone acetylation | CREBBP (CBP) | KAT | Rearrangements | Gain | AML (rare) | Activation |
| Mutation/deletion, monoallelic | Loss | DLBCL (15%) | Repression | |||
| FL (40%) | ||||||
| relapsed B‐ALL (20%) | ||||||
| EP300 (p300) | KAT | Rearrangements | Gain | AML (rare) | Activation | |
| Mutation/deletion, monoallelic | Loss | DLBCL (40%) | Repression | |||
| FL (60%) |
ALL, acute lymphoid leukemia; AML, acute myeloid leukemia; CMML, chronic myelomonocytic leukemia; DLBCL, diffuse large B‐cell lymphoma; FL, follicular lymphoma; KDM: histone lysine demethylase; KMT, histone lysine methyltransferase; MDS, myelodysplastic syndrome; MM, multiple myeloma; PTD, partial tandem duplication; TBD, to be determined.
DNMT3A
Mutations in DNMT3A have been frequently found across a range of hematopoietic diseases, including acute myeloid leukemia (AML), myelodysplastic syndromes (MDS) and T‐cell leukemia/lymphoma.8, 9 The type of DNMT3A mutation and the frequency of heterozygous versus homozygous mutations vary among different malignancies. In the myeloid neoplasms, R882 residue is the most frequently mutated (60%), and the mutations are typically heterozygous. The R882 mutations are not so predominant (20%) in T‐cell leukemia/lymphoma. In addition, the frequency of biallelic involvement is very high, indicating that a more complete loss of DNMT3A function is important for the development of T‐cell neoplasms.8
Several lines of evidence indicate that DNMT3A mutations are one of the earliest events during the course of tumor development. DNMT3A mutations have been found at higher variant‐allele frequencies than other accompanying mutations in hematopoietic neoplasms.10 Two independent studies found DNMT3A mutations in human HSC purified from AML patients in the absence of other common leukemia‐associated mutations.11, 12 Recent extensive analyses of genome sequencing data from individuals without hematological diseases clearly showed that hematopoiesis frequently become clonal with age.13, 14, 15 DNMT3A mutations are overwhelmingly the most common mutations in such clonal hematopoiesis. Taken together, DNMT3A mutations appear to give a small advantage to HSC that can lead to their dominance over time, which become foundation of the subsequent development of hematopoietic neoplasms. Interestingly, the clonal hematopoiesis is associated with increases in the risk of not only hematopoietic neoplasms but also cardiovascular diseases.
TET2
Recurrent mutations of TET2 have also been found in a wide range of hematopoietic neoplasms, including AML, MDS, chronic myelomonocytic leukemia (CMML) and T‐cell leukemia/lymphoma.16, 17, 18 In addition, TET2 mutations can be found in premalignant HSC in MDS and AML patients,15 as well as in aged healthy individuals with clonal hematopoiesis.13, 14, 15, 19 TET2 mutations are usually heterozygous and are either missense mutations in the C‐terminal catalytic domain or nonsense⁄frameshift mutations in the N‐terminal region leading to premature truncation before the catalytic core. Thus, disruption of the catalytic activity of TET2 constitutes a critical background for development of hematopoietic neoplasms. In AML, TET2 mutations are mutually exclusive to IDH1/2 mutations (see below) and WT1 mutations, which probably indicates the presence of IDH1/2‐TET2‐WT1 pathway to suppress AML.20, 21
IDH1 and IDH2
Mutations in IDH1 and IDH2 are mostly heterozygous and mutually exclusive.16, 22 In IDH1, almost all the disease‐associated mutations were found at R132, in the enzyme active site. In IDH2, there are two hot spots for mutation: R140 and R172. IDH2‐R140 mutation occurs exclusively in myeloid tumors, whereas the IDH2‐R172 mutation is also found in solid tumors, such as glioma. These mutations are found in up to 10% of cytogenetically normal AML. Metabolite profiling studies revealed that the IDH1/2 mutations gain a function to generate a novel oncometabolite D‐2‐hydroxyglutarate (D2HG) from a‐KG. D2HG inhibits the function of α‐KG‐dependent enzymes such as TET2. Therefore, it is widely believed that IDH mutations induce leukemogenesis mainly by suppressing TET2 function. In line with this notion, mutations in IDH1/2 and TET2 are mutually exclusive in AML, and TET2‐mutated or IDH‐mutated leukemia cells display an overlapping DNA hypermethylation signature that is associated with decreased 5hmC.23 However, there are substantial differences between IDH1/2‐mutated and TET2‐mutated hematopoietic neoplasms. TET2 mutations are more common in MDS/CMML than AML, whereas IDH1/2 mutations are frequently found in AML.16 Unlike TET2 mutations, IDH1/2 mutations are not associated with clonal hematopoiesis in elderly people.13, 14, 15 It should also be noted that TET2 and IDH2 mutations are not exclusive but often coexist in T‐cell lymphomas.16 Thus, IDH1/2 mutations share a common oncogenic function to TET2 mutations, but they also have distinct leukemogenic effects.
EZH2 and other PRC2 genes
EZH2 plays dual roles in the process of tumorigenesis.24 EZH2 mutations at tyrosine residue 641 have been found in B‐cell tumors, including diffuse large B‐cell lymphoma (DLBCL) and follicular lymphoma (FL). The EZH2‐Y641 mutants drive hypermethylation of H3K27, indicating that they are gain‐of‐function mutations. In addition, overexpression of EZH2 has been reported in natural killer/T‐cell (NKT) lymphoma.25 Conversely, loss‐of‐function mutations in EZH2 have been detected in MDS and T‐cell acute lymphoid leukemia (T‐ALL). Mutations in other members of PRC2, EED and SUZ12, were also found in some cases of MDS and T‐ALL. In addition to these mutations in PRC2 genes, several other oncoproteins were shown to inhibit EZH2 function in MDS cells. For example, ASXL1 mutations promote myeloid transformation through loss of PRC2‐mediated gene repression.26, 27 Mutations in a splicing factor SRSF2 induce missplicing of EZH2 to promote MDS development.28 Thus, EZH2 acts as an oncogene in B‐cell and NKT lymphomas, while it acts as a tumor suppressor in MDS and T‐ALL.
ASXL1 and ASXL2
ASXL1 mutations have been frequently found in myeloid neoplasms, including CMML, MDS and AML, but have rarely been found in lymphoid neoplasms.29 Most ASXL1 mutations are heterozygous and exist in exon 12 of the gene, generating C‐terminally truncated mutations. Whether ASXL1 mutations promote myeloid transformation via a gain‐of‐function or loss‐of‐function is under debate. A recent report showed that the mutant ASXL1 proteins are, indeed, expressed in MDS cells, which implies that the mutations are not simple loss‐of‐function mutations.30 ASXL1 mutation is also implicated to clonal hematopoiesis, indicating that it is one of the earliest genetic events during the process of myeloid transformation. Mutations in ASXL2, a homologue of ASXL1, were specifically found in AML with the RUNX1–RUNX1T1 (AML1‐ETO) rearrangement.31
KMT2A (MLL1), KMT2D (MLL2) and KMT2C (MLL3)
Among KMT for H3K4, alterations of KMT2A (MLL1) and KMT2D (MLL2) have been causally implicated in hematopoietic neoplasms. KMT2A gene on chromosome 11q23 is a frequent target of rearrangements. Typically, KMT2A rearrangements result in the loss of the carboxy‐terminal methyltransferase domain and an in‐frame fusion of the amino‐terminal region of KMT2A to 1 of more than 70 known fusion partners.32 Rearrangements of KMT2A are observed in AML and B‐cell acute lymphoid leukemia (B‐ALL). The most common partners for the rearrangements are AF9 in AML and AF4 in B‐ALL. Many fusion partners of KMT2A interact with DOT1L directly or indirectly in complexes that promote transcriptional elongation. DOT1L is a KAT to induce H3K79 methylation. As a result, KMT2A‐fusion proteins gain the ability to induce hypermethylation of H3K79 at the promoter regions of KMT2A target genes. KMT2A gene is also affected by a partial tandem duplication (PTD) of exons near the 5′ end of the KMT2A gene. The KMT2A‐PTD has been found in AML and MDS.
KMT2D (MLL2) has recently received considerable attention because it is one of the most frequently mutated genes in DLBCL and FL.33 KMT2D mutations are predominantly represented by premature stop codons, frameshift insertions or deletions, and splice‐site mutations that are predicted to generate truncated proteins lacking part or all of the C‐terminal domains. Multiple missense mutations have also been found across the KMT2D gene. KMT2D mutations are found either biallelically or monoallelically. The diverse spectrum of KMT2D mutations suggests that they are the loss‐of‐function mutations.
In addition, KMT2C (MLL3) was recently shown to be a haploinsufficient 7q tumor suppressor,34 and a KMT2C gene germ line mutation was found in a pedigree of colorectal cancer and AML.35
MECOM (EVI1) and PRDM16
Rearrangements around MECOM (EVI1) and PRDM16 genes have been detected in a subset of MDS and AML.36, 37 MECOM is located near the chromosome 3q26, and the balanced aberrations involving 3q26 reposition a distal enhancer of GATA2 to ectopically activate MECOM expression.38, 39 PRDM16 is located near the chromosome 1p36, and the reciprocal translocation t(1:3)(q36;q21) induced PRDM16 upregulation. AML with MECOM or PRDM16 rearrangements share many biological features, including micromegakaryocytes, multilineage dysplasia, low myeloperoxidase‐expressing blasts and adverse prognosis.40
WHSC1 (NSD2/MMSET) and SETD2
Genetic alterations of KMT for H3K36 are also found in hematopoietic neoplasms. The translocation t(4; 14)(p16; q32) is one of the most common translocations in multiple myeloma (MM). The t(4; 14) translocation leads to the generation of IgH‐WHSC1 fusion gene to overexpress WHSC1 in MM.41 The epigenetic role of WHSC1 has been a matter of discussion, but a recent report suggested that the principal activity of WHSC1 is promoting H3K36me2.
SETD2 is a KMT that induces H3K36me3, and a recent study detected recurrent mutations in SETD2 in AML and B‐ALL.42 SETD2 mutations are frequently found, but are not restricted, in KMT2A‐rearranged patients. SETD2 mutations were either nonsense or frameshift mutations that truncate the C‐terminus domain. One‐quarter of the SETD2 mutations are biallelic mutations. The mutation spectrum of SETD2 implies that these mutations are loss‐of‐function mutations. Consistent with this notion, a global loss of H3K36me3 was observed in leukemic blasts with SETD2 mutations.
KDM6A (UTX) and KDM2B
Mutations and deletions of several KDM have been found in lymphoid neoplasms. KDM6A (UTX) is a known KDM for H3K27 and is located on chromosome X. Mutations and deletion of KDM6A gene were first identified in multiple cancer cell lines including those of MM.43 A subsequent study found loss‐of‐function mutations within KDM6A gene in male T‐ALL patients. Allelic expression analysis revealed that KDM6A escapes X‐inactivation in female T‐ALL lymphoblasts and normal T cells. Therefore, inactivation of only one single UTX copy in males will contribute to tumor development, while female cells are protected against single copy loss of UTX.44 KDM2B is a H3K36 histone demethylase. Mutations and deletion of KDM2B gene were found in approximately 5% of DLBCL.45
CREBBP (CBP) and EP300 (p300)
The earliest observations linking CREBBP and EP300 to cancer were the identifications of chromosome translocations in AML that disrupt these genes. Those include the t(8;16) and t(8;22) translocations generating KAT6A (MOZ)‐CREBBP and KAT6A‐EP300 fusions. These translocations appear to increase the KAT activity.46 Recently, frequent mutations and/or deletions in CREBBP and EP300 genes were identified in DLBCL and FL.47 These alterations remove or inactivate the KAT coding domain and are usually monoallelic, suggesting that these KAT are haploinsufficient tumor suppressors in B‐cell lymphoma. Taken together, it appears that the enhanced KAT activity promotes myeloid leukemogenesis, while the reduced KAT activity contributes to the development of B‐cell tumors.
Role of Epigenetic Modifiers in Hematopoiesis: Lessons from Mouse Models
The roles of epigenetic modifiers in hematopoiesis have been investigated using a variety of mouse models. Such studies have revealed new and sometimes unexpected roles of individual epigenetic modifiers during hematopoietic development. In this section, we summarize the physiologic roles of the disease‐related epigenetic modifiers in normal and malignant hematopoiesis.
Dnmts (Dnmt1, Dnmt3a, Dnmt3b)
Dnmt1 has been shown to be essential for HSC self‐renewal. In sharp contrast, Dnmt3a deletion in hematopoietic cells led to expansion of adult HSC upon serial transplantation.9 The increased self‐renewal of Dnmt3a‐deficient HSC will explain why loss‐of‐function mutations of DNMT3A are frequently detected in a wide range of hematopoietic neoplasms. Furthermore, a recent study showed that Mx1‐Cre‐mediated Dnmt3a ablation led to the development of myeloproliferative neoplasms (MPN) with myelodysplasia (MDS/MPN), indicating an additional role of Dnmt3a to limit myeloid progenitor expansion in vivo.48 Dnmt3a‐deficient HSC exhibit a global loss of DNA methylation, particularly at the edges of large hypomethylated canyon regions. The hypomethylated regions are enriched for genes associated with HSC self‐renewal, such as Hoxa9, Meis1 and Evi1, which resulted in enhanced expression of these stem cell genes.8 Disruption of Dnmt3b, another de novo DNA methyltransferase, showed only minor effects on HSC. However, combined deletion of Dnmt3a and Dnmt3b resulted in marked expansion of HSC with almost completely blocked hematopoietic differentiation,8 indicating the functional redundancy of these de novo Dnmts in HSC regulation.
Tet enzymes (Tet1, Tet2, Tet3)
Disruption of any of the Tet genes in adult hematopoietic cells leads to increased HSC function as measured by the engrafting potential to recipient mice.18, 49 Loss of Tet2 resulted in a gradual expansion of myeloid‐biased HSC, accompanied with age‐associated extramedullary hematopoiesis. Certain strains of Tet2‐deficient mice developed a CMML‐like disease but with long latency and low penetrance. In contrast to the myeloid‐skewed phenotype of Tet2‐deficient mice, Tet1‐deficiency in hematopoietic cells drove B lymphopoiesis and the late development of a poorly penetrant B‐cell lymphoma. Tet3‐deficiency in hematopoietic cells did not significantly alter the steady‐state hematopoiesis, but augmented the repopulating capacity of HSC. The ablation of Tet1, Tet2 or Tet3 led to a modest decrease in 5hmC levels in bone marrow. Interestingly, mice with combined loss of Tet1 and Tet2 exhibited strikingly decreased incidence and delayed onset of myeloid neoplasms in comparison to Tet2 −/− mice, and instead developed lethal B‐cell neoplasms.50 In contrast, double knockout of Tet2 and Tet3 in mice induced rapid development of an aggressive myeloid leukemia with almost complete loss of 5hmC in the bone marrow.51 Of note, combined loss of Tet2 and Tet3 resulted in the accumulation of DNA damage and impaired DNA repair, indicating the potential role of Tet proteins in the regulation of DNA damage responses. Thus, individual Tet proteins have both distinct and redundant functions in HSC and during hematopoietic differentiation.
Idh1 and Idh2
As described previously, the currently available evidence suggests that IDH1/2 mutations induce D2HG production, and the D2HG‐mediated inhibition of TET enzymes contributes to leukemogenesis. Consistent with this notion, Tet2 knockout and IDH1‐R132H knockin mice showed similar phenotypes, including global 5hmC reduction, altered DNA methylation, impaired hematopoietic differentiation, myeloid skewing and the development of myeloid disorders.16, 22 However, in contrast to the Tet2 knockout mice, IDH1‐R132H knockin mice have reduced numbers of long‐term HSC.52 Mechanistically, it was shown that IDH1–R132H downregulates DNA damage sensor ATM by altering histone methylation, leading to impaired DNA repair and reduced HSC self‐renewal.
For the IDH2 mutations, transgenic mice that express IDH2‐R140Q using a tetracycline‐inducible system were generated.53 The IDH‐R140Q mice showed normal HSC numbers/functions and lineage differentiation, but exhibited increased extramedullary hematopoiesis. Combined expression of IDH2‐R140Q together with HoxA9/Meis1 or FLT3 mutations in hematopoietic cells produced acute leukemia. Importantly, genetic deinduction of IDH2‐R140Q in leukemic cells showed profound growth‐inhibitory effects, indicating the essential role of mutant IDH2 in maintaining leukemogenesis.
Polycomb genes (Bmi1, Rnf2, Ezh2, Eed, Suz12)
Bmi1 represents one of the best‐characterized PRC1 members in the regulation of HSC self‐renewal.24, 54 Bmi1‐deficient mice showed severe postnatal pancytopenia due to progressive depletion of HSC. Conversely, forced expression of Bmi1 in HSC increased self‐renewal of HSC. Bmi1 maintains HSC function by repressing the expression of p16(Ink4a) and p19(Arf) encoded by Cdkn2a gene, and also by regulating mitochondrial function and ROS generation. Bmi1 is also a critical regulator of leukemia stem cells induced by several oncogenes, including MLL‐AF9, HoxA9/Meis1 and E2a‐Pbx1. Rnf2, a core PRC1 member, was shown to restrict the proliferation and differentiation of hematopoietic progenitors by repressing the expression of p16(Ink4a) and Ccnd2.
PRC2 genes also play important roles in the regulation of hematopoiesis.24, 54 Ezh2 overexpression in HSC enhanced their self‐renewal and prevented HSC exhaustion upon serial transplantation. Although Ezh2‐deficient HSC retained almost normal function,55 combined deletion of Ezh1 and Ezh2 abolished the repopulating capacity of HSC. Ezh2 loss in HSC predisposed mice to develop heterogeneous tumors, including MDS, MPN and T‐cell leukemia, after the long latency.56 Disruption of Ezh2 specifically in germinal center (GC) B‐cells using Cγ‐Cre resulted in failure to form GC. Conversely, conditional expression of mutant Ezh2‐Y641N in GC B‐cells induced GC hyperplasia and accelerated lymphomagenesis in cooperation with BCL2.57 These findings clearly indicated the tumor suppressor role of Ezh2 in myeloid and T‐cell tumors, and the oncogenic role of Ezh2 in B‐cell lymphoma. Deletion of Eed resulted in the exhaustion of HSC. Derepressed genes in Eed‐deficient HSC are enriched for H3K27me3 targets, including Cdkn2a, and Cdkn2a deletion in Eed‐knockout mice partially rescued the HSC defect.55 Suz12 is also required for HSC function and lymphopoiesis.58 Thus, both PRC1 and PRC2 members were shown to be involved in the regulation of HSC function and hematopoietic differentiation through the epigenetic control of Polycomb‐target genes, such as Cdkn2a.
Asxl1 and Bap1
Hematopoietic‐specific deletion of Asxl1 results in progressive, multilineage cytopenia and dysplasia, characteristic features of MDS.59, 60 Of note, Asxl1‐deficient HSC exhibited decreased repopulating capacity, which contrasts with the phenotypes of Dnmt3a and Tet2‐deficient HSC that showed enhanced self‐renewal. Thus, despite that ASXL1 mutations are one of the earliest mutations presumably occurring in pre‐leukemic HSC, Asxl1‐deficiency decreased HSC function. Because ASXL1 mutations could have some gain‐of‐function,30, 61 whether conditional expression of mutant Asxl1 also decreases HSC function needs to be investigated. Asxl1 deletion and forced expression of Asxl1 mutations induced global reduction of H3K27me3, upregulation of Hox genes,26, 27, 59 and promote development of MDS and AML in combination with Tet mutations,59 Nras mutations,26, 27 and SETBP1 mutations.62 Bap1 is an Asxl1‐binding partner, and Bap1 deletion in adult mice also caused MDS‐like diseases.63 These findings indicate the important role of Asxl1/Bap1 complex to suppress myeloid transformation.
Kmt2a (Mll1), Kmt2d (Mll2) and Kmt2c (Mll3)
Studies consistently have shown the impaired HSC function in Kmt2a‐deficient mice. Kmt2a regulates multiple HSC genes, including Hoxa9, Meis1, Mecom and Prdm16.64 Several models for KMT2A‐rearranged leukemia have been developed using retroviral transduction and mouse knockin strategies.65 Retrovirus‐mediated transduction of KMT2A‐fusion proteins, such as KMT2A (MLL)‐AF9 and KMT2A‐ENL, can transform not only HSC but also myeloid progenitors to produce AML in vivo. In contrast, KMT2A‐AF9 knockin mice, where oncogene expression is under endogenous regulatory control, efficiently transformed HSC while committed progenitors were transformation‐resistant.66 In addition, some stem cell genes, such as Mecom, were upregulated by KMT2A‐fusions selectively in HSC but not in progenitors.66, 67 These data suggest that cellular origin and oncogene dosage are important for the development of KMT2A‐rearranged AML. Although few strategies have been successful in recapitulating the B‐cell phenotype characteristic of KMT2A‐rearranged childhood leukemia, a recent study showed that human CD34+ cells transduced with KMT2A fused to murine Af4 developed into a pro‐B ALL that recapitulates many features of human disease in immunodeficient mice.68
Disruption of Kmt2d in mouse B cells using Cγ‐Cre showed that loss of Kmt2d led to reduced H3K4 methylation, enhanced germinal center formation, conferred a B cell‐proliferative advantage, and promoted lymphomagenesis in cooperation with Bcl2.69, 70 These findings clearly indicate that Kmt2d acts as a tumor suppressor whose loss facilitates lymphomagenesis by remodeling the epigenetic landscape.
Loss of catalytic function of Kmt2c promoted aberrant myelopoiesis,71 which may contribute to the progression of MDS and AML.
Mecom (Evi1) and Prdm16
Mecom and Prdm16 belong to the Prdm family, and studies using mouse models have shown that loss of either leads to severe defects of HSC activity.72, 73 Interestingly, both Mecom and Prdm16 were shown to be the downstream targets of Kmt2a,64, 67 indicating the critical role of Kmt2a–Mecom/Prdm16 axes in HSC.
Kdms [Kdm1a (Lsd1), Kdm2b, Kdm6a (Utx)]
Physiologic functions of Kdms have become an active area of research in the last few years.6 Conditional deletion of Kdm1a in HSC resulted in severe pancytopenia, impaired granulocytic and erythroid differentiation, accompanied by the increased H3K4 methylation. Kdm2b deletion in HSC resulted in the reduced HSC activity and defective lymphopoiesis. Conversely, ectopic expression of Kdm2b in hematopoietic progenitors favors T lymphocyte commitment.74
Kdm6a (Utx) is located on the X chromosome. Consequently, female Kdm6a knockout mice displayed key features of MDS with chromosomal instability, whereas their male counterparts showed no phenotype.75 The compensatory effect of a homologue Uty in male mice, which is located on the Y chromosome and has no Kdm activity, indicates the demethylase‐independent functions of Kdm6a in HSC and progenitors. In contrast, H3K27me3 demethylation by either Kdm6a or Kdm6b (Jmjd3) is crucial for the terminal steps of T‐cell differentiation.76 The preferential impact of Kdm6a/b on late T‐cell maturation may be linked to the high frequency of KDM6A mutations in T‐ALL.
Kats [Crebbp [Cbp], Ep300 [p300], Kat6a [Moz])
Multiple Kats have been shown to play key roles in hematopoiesis.46 Studies using Crebbp knockout mice showed that loss of Crebbp led to increased apoptosis, differentiation, and quiescence in HSC. Ep300 is more important for hematopoietic differentiation. Kat6a (Moz) has been shown to be a critical regulator in the generation and development of HSC and progenitors. It should be noted that these Kats appear to regulate hematopoiesis partly through acetylation of non‐histone proteins, such as c‐Myb, Gfi1 and Foxp3.46
Therapies Targeting Epigenetic Modifications
Epigenetic aberrations are potentially reversible and can be restored by epigenetic therapies. In this section, we describe drugs targeting aberrant epigenetic modifications in hematopoietic neoplasms that are in various stages of preclinical and clinical development (Fig. 3).
Figure 3.
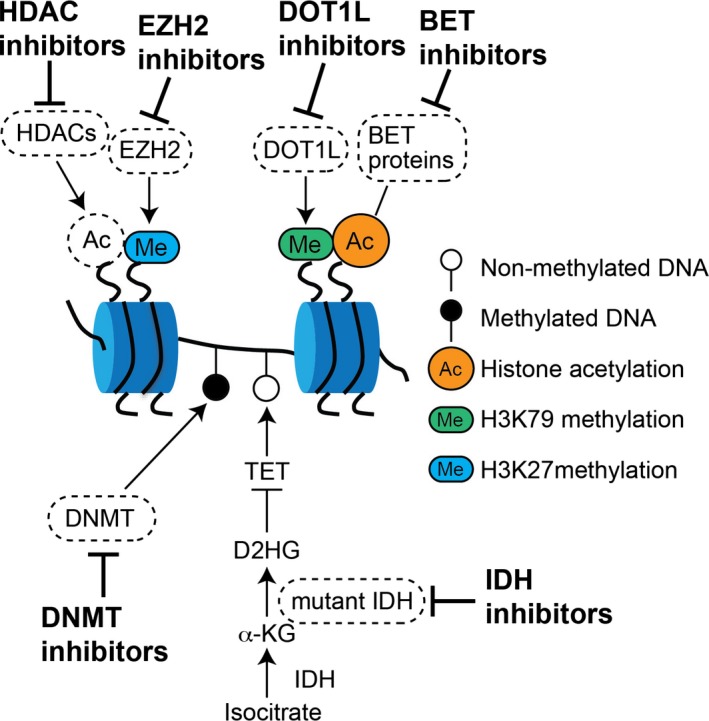
Ongoing and future therapeutic approaches targeting epigenetic modifiers. Epigenetic modifiers can be potential therapeutic targets. Those include DNMT, mutant IDH proteins, histone deacetylases (HDAC), EZH2 and polycomb complexes, the DOT1L enzyme and BET proteins.
DNMT inhibitors
The two hypomethylating agents, azacitidine and decitabine, are currently approved for the treatment of several specific forms of MDS and AML.77 These drugs are considered to inhibit activity of DNMT, to reverse aberrant DNA hypermethylation and to restore expression of previously silenced genes. However, mechanisms of action of these drugs are not fully understood, and there are currently no established biomarkers that predict the response to these drugs. A recent study reported that mutations in DNMT3A and TET2 genes might predict better responsiveness to treatment with the hypomethylating drugs in MDS.78, 79 Given that DNMT3A or TET2 mutations are supposed to be associated with hypomethylation or hypermethylation of DNA, it is not clear why the inhibitors showed efficacy for both types of MDS. Another study reported that somatic mutations did not predict responses to dicitabine in CMML patients, but instead identified differentially methylated regions of DNA that distinguished responders from non‐responders.80 Identification of clinically useful biomarkers to predict response to these drugs will be an important future challenge.
IDH mutant inhibitors
Inhibitors for IDH1 and IDH2 mutants have been developed and are already being tested, either as single agents or in combination, in early clinical trials.77 These drugs, such as AG‐120 for the mutated IDH1 and AG‐221 for the mutated IDH2, are selective inhibitors targeting only mutated IDH proteins but not wild‐type IDH1/2. Therefore, in theory, these inhibitors will be particularly effective on IDH‐mutated leukemia with minimal side effects.
HDAC inhibitors
HDAC inhibitors are the drugs that interfere with the function of histone deacetylase. The most successful clinical application of HDAC inhibitors has been the use of them against T‐cell lymphoma.81 HDAC inhibitors also showed some therapeutic effects on MDS, AML, Hodgkin lymphoma and multiple myeloma.77 However, responses to HDAC inhibitors as a single agent in AML or MDS appear to be modest. Clinical studies using HDAC inhibitors in combination with other agents are ongoing. Again, how or why some types of hematopoietic tumors are sensitive to HDAC inhibitors remains unclear.
BET inhibitors
BET proteins are epigenetic readers that recognize the acetylated lysine residues in histone proteins. BET inhibitors reversibly bind the bromodomain of BET to disrupt protein–protein interaction among BET proteins, acetylated histones and transcription factors. Preclinical studies have shown the therapeutic efficacy of the BET inhibitors in various hematopoietic neoplasms, particularly in AML, multiple myeloma and some lymphomas.82 Future research should include the development of combination therapies with other drugs, and the identification of biomarkers to predict the response.
DOT1L inhibitors
KTM2A fusion proteins were shown to recruit DOT1L to the promoter regions of KTM2A‐target genes. Several small molecules targeting DOT1L have been developed, and showed substantial efficacy against KTM2A‐rearranged leukemia in preclinical models.83 Thus, the DOT1L inhibitors are potential options for treatment of KTM2A‐rearranged leukemia and are currently under clinical investigation. DOT1L inhibitors may also be effective in treating other types of hematopoietic neoplasms, such as AML with DNMT3A mutations.84 Furthermore, it was recently shown that combined pharmacological inhibition of DOT1L and menin‐KTM2A had profound anti‐leukemic activity against NPM1‐mutated AML.85
EZH2 inhibitors
EZH2 have been attractive drug targets for cancer therapy.86 Recent findings of the frequent gain‐of‐function mutations in EZH2 gene indicate that EZH2 represent an ideal therapeutic target in B‐cell lymphoma. Indeed, early success has been achieved using EZH2 inhibitors for treatment of lymphomas bearing EZH2 mutations.87 Given that Polycomb genes establish crosstalk with numerous epigenetic regulators in various types of malignant stem cells, EZH2 inhibitors can potentially be applied to a broader spectrum of hematopoietic neoplasms.
Other epigenetic inhibitors
Inhibition of histone demethylases has also substantial potential to reset the aberrant regulation of gene expression in hematopoietic neoplasms.88 For example, recent studies highlight a potential application of LSD1 inhibition to treat AML.89 Only a few compounds have been developed as KAT (e.g. CREBBP and EP300) inhibitors. Some of them showed the growth‐inhibitory efficacy in solid tumors, but the therapeutic effects of the KAT inhibitors have not been extensively studied in hematopoietic neoplasms. Interestingly, a study showed that pharmacological inhibition of Ep300 was able to abrogate the suppressive functions of Treg cells, and thereby increased tumor immunity.90 Targeting EP300 could, therefore, be a new approach for cancer immunotherapy.
Overview and Concluding Remarks
Despite the tremendous progress that has been achieved, we are still at an early stage in understanding the complex epigenetic regulation in hematopoiesis. This review aims to integrate accumulating knowledge regarding epigenetic (dys)regulation in normal and malignant hematopoiesis. Interestingly, from the epigenetic viewpoint, myeloid neoplasms are very similar to T‐cell neoplasms, whereas B‐cell lymphomas show a distinct pattern of mutation in epigenetic genes. For example, mutations in DNMT3A and TET2 are frequently found in both myeloid and T‐cell tumors, but rarely found in B‐cell tumors. Loss‐of‐function mutations of EZH2 contribute to the development of myeloid and T‐cell tumors, while gain‐of‐function mutations of EZH2 promote B‐cell tumors. The resemblance between myeloid and T‐cell tumors with respect to epigenetic mutations cannot be easily explained by the classic model of binary split differentiation between lymphoid and myeloid lineages. However, several lines of evidence suggest that the separation of the B‐ and T‐cell lymphoid lineages might occur prior to the loss of myeloid potential in the early stages of hematopoiesis.91, 92 In addition, these observations suggest that myeloid and T‐cell neoplasms will be responsive to similar epigenetic therapies, while distinct approaches may be required to treat B‐cell lymphomas.
Of note, the mutational spectrum also indicates that the epigenetic mutations related to active transcription are more associated with myeloid/T‐cell tumors, whereas those that repress transcription are associated with B‐cell tumors. A good example is, again, EZH2. Gain‐of‐function mutations of EZH2 in B‐cell tumors have been shown to enhance H3K27 methylation and repress transcription. Conversely, loss‐of‐function mutations of EZH2 in myeloid and T‐cell tumors have been shown to reduce H3K27 methylation and activate transcription. In addition, rearrangements of CREBBP and EP300 in AML were considered to increase histone acetylation and enhance transcription. In contrast to these rearrangements, loss‐of‐function mutations of CREBBP and EP300, which are supposed to decrease histone acetylation and transcription, are quite prevalent in B‐cell tumors. As summarized in Table1, the list of epigenetic mutations suggests that myeloid and T‐cell tumors tend to be transcriptionally “active,” while B‐cell tumors appear to be transcriptionally “inactive” (Fig. 4). These data may imply that epigenetic mutations found in hematopoietic neoplasms act as general amplifiers or repressors of any given transcriptional state, instead of regulating the expression of specific target genes. This hypothesis could explain why attempts to identify target genes of each epigenetic modifier have been fraught with difficulty and failure.
Figure 4.

Epigenetic characteristics in hematopoietic neoplasms. Genetic alterations in myeloid and T‐cell tumors are generally associated with “active” transcription, while those in B‐cell tumors are more associated with “inactive” transcription. The global low‐level or high‐level transcriptional activity may underlie the development of myeloid/T‐cell tumors or B‐cell tumors, respectively.
Many questions remain to be answered. Biochemically, DNMT3A and TET2 have opposite activity to increase or decrease DNA methylation, respectively. Curiously, however, mutation of either DNMT3A or TET2 leads to similar hematopoietic neoplasms, including AML, MDS and T‐cell tumors. Moreover, mutations of DNMT3A and TET2 can be found in the same malignant clones, indicating the cooperation between these mutations to promote tumor development. Similarly, IDH mutations, which are thought to promote leukemogenesis by inhibiting TET2 function, coexist frequently with DNMT3A mutations in AML and T‐cell tumors. Future research should elucidate the actual role of these epigenetic modifiers in hematopoiesis. Mechanisms of actions of the “epigenetic therapies” warrant further investigation. Earlier studies suggest that the epigenetic drugs reverse aberrant DNA and/or histone modifications, thereby restoring expression of previously silenced tumor suppressor genes. However, accumulating evidence clearly indicates that this concept is not sufficient to fully explain the therapeutic effects of these drugs. Recently, two studies highlighted another mode of action of demethylating agents in solid tumors.93, 94 DNA demethylating agents activated a cellular antiviral program through transcriptional activation of endogenous retroviral sequences. The findings suggested new rational approaches to the use of such agents in immunotherapy, and, indeed, it was shown that inhibition of DNA methylation could sensitize a murine model of melanoma to anti‐CTLA4 immune checkpoint therapy.94 Potential crosstalk between epigenetic therapies and immunotherapies will be an interesting area of future research.
Disclosure Statement
The authors have no conflict of interest to declare.
Cancer Sci 108 (2017) 553–562
Funding Information
Japan Society for the Promotion of Science, (Grant/Award Number: ‘16K15499‘).
References
- 1. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell 2012; 150: 12–27. [DOI] [PubMed] [Google Scholar]
- 2. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet 2013; 14: 204–20. [DOI] [PubMed] [Google Scholar]
- 3. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov 2014; 13: 337–56. [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev 2001; 15: 2343–60. [DOI] [PubMed] [Google Scholar]
- 5. Pinheiro I, Margueron R, Shukeir N et al Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 2012; 150: 948–60. [DOI] [PubMed] [Google Scholar]
- 6. Andricovich J, Kai Y, Tzatsos A. Lysine‐specific histone demethylases in normal and malignant hematopoiesis. Exp Hematol 2016; 44: 778–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackledge NP, Rose NR, Klose RJ. Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nat Rev Mol Cell Biol 2015; 16: 643–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer 2015; 15: 152–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Celik H, Kramer A, Challen GA. DNA methylation in normal and malignant hematopoiesis. Int J Hematol 2016; 103: 617–26. [DOI] [PubMed] [Google Scholar]
- 10. Welch JS, Ley TJ, Link DC et al The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150: 264–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shlush LI, Zandi S, Mitchell A et al Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506: 328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Corces‐Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A 2014; 111: 2548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie M, Lu C, Wang J et al Age‐related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20: 1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Genovese G, Kahler AK, Handsaker RE et al Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371: 2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jaiswal S, Fontanillas P, Flannick J et al Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371: 2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inoue S, Lemonnier F, Mak TW. Roles of IDH1/2 and TET2 mutations in myeloid disorders. Int J Hematol 2016; 103: 627–33. [DOI] [PubMed] [Google Scholar]
- 17. Nakajima H, Kunimoto H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci 2014; 105: 1093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016; 30: 733–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Busque L, Patel JP, Figueroa ME et al Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012; 44: 1179–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y, Xiao M, Chen X et al WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell 2015; 57: 662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rampal R, Alkalin A, Madzo J et al DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep 2014; 9: 1841–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 2013; 3: 730–41. [DOI] [PubMed] [Google Scholar]
- 23. Figueroa ME, Abdel‐Wahab O, Lu C et al Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18: 553–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takamatsu‐Ichihara E, Kitabayashi I. The roles of Polycomb group proteins in hematopoietic stem cells and hematological malignancies. Int J Hematol 2016; 103: 634–42. [DOI] [PubMed] [Google Scholar]
- 25. Yan J, Ng SB, Tay JL et al EZH2 overexpression in natural killer/T‐cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood 2013; 121: 4512–20. [DOI] [PubMed] [Google Scholar]
- 26. Abdel‐Wahab O, Adli M, LaFave LM et al ASXL1 mutations promote myeloid transformation through loss of PRC2‐mediated gene repression. Cancer Cell 2012; 22: 180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inoue D, Kitaura J, Togami K et al Myelodysplastic syndromes are induced by histone methylation‐altering ASXL1 mutations. J Clin Invest 2013; 123: 4627–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim E, Ilagan JO, Liang Y et al SRSF2 Mutations contribute to myelodysplasia by mutant‐specific effects on exon recognition. Cancer Cell 2015; 27: 617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gelsi‐Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol 2012; 5: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Inoue D, Matsumoto M, Nagase R et al Truncation mutants of ASXL1 observed in myeloid malignancies are expressed at detectable protein levels. Exp Hematol 2016; 44: 172–6.e1. [DOI] [PubMed] [Google Scholar]
- 31. Micol JB, Duployez N, Boissel N et al Frequent ASXL2 mutations in acute myeloid leukemia patients with t(8;21)/RUNX1–RUNX1T1 chromosomal translocations. Blood 2014; 124: 1445–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Neff T, Armstrong SA. Recent progress toward epigenetic therapies: the example of mixed lineage leukemia. Blood 2013; 121: 4847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morin RD, Mendez‐Lago M, Mungall AJ et al Frequent mutation of histone‐modifying genes in non‐Hodgkin lymphoma. Nature 2011; 476: 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen C, Liu Y, Rappaport AR et al MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014; 25: 652–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li WD, Li QR, Xu SN et al Exome sequencing identifies an MLL3 gene germ line mutation in a pedigree of colorectal cancer and acute myeloid leukemia. Blood 2013; 121: 1478–9. [DOI] [PubMed] [Google Scholar]
- 36. Goyama S, Kurokawa M. Evi‐1 as a critical regulator of leukemic cells. Int J Hematol 2010; 91: 753–7. [DOI] [PubMed] [Google Scholar]
- 37. Morishita K. Leukemogenesis of the EVI1/MEL1 gene family. Int J Hematol 2007; 85: 279–86. [DOI] [PubMed] [Google Scholar]
- 38. Groschel S, Sanders MA, Hoogenboezem R et al A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014; 157: 369–81. [DOI] [PubMed] [Google Scholar]
- 39. Yamazaki H, Suzuki M, Otsuki A et al A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 2014; 25: 415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matsuo H, Goyama S, Kamikubo Y, Adachi S. The subtype‐specific features of EVI1 and PRDM16 in acute myeloid leukemia. Haematologica 2015; 100: e116–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie Z, Chng WJ. MMSET: role and therapeutic opportunities in multiple myeloma. Biomed Res Int 2014; 2014: 636514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu X, He F, Zeng H et al Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet 2014; 46: 287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Haaften G, Dalgliesh GL, Davies H et al Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet 2009; 41: 521–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van der Meulen J, Sanghvi V, Mavrakis K et al The H3K27me3 demethylase UTX is a gender‐specific tumor suppressor in T‐cell acute lymphoblastic leukemia. Blood 2015; 125: 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pasqualucci L, Trifonov V, Fabbri G et al Analysis of the coding genome of diffuse large B‐cell lymphoma. Nat Genet 2011; 43: 830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun XJ, Man N, Tan Y, Nimer SD, Wang L. The role of histone acetyltransferases in normal and malignant hematopoiesis. Front Oncol 2015; 5: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pasqualucci L, Dominguez‐Sola D, Chiarenza A et al Inactivating mutations of acetyltransferase genes in B‐cell lymphoma. Nature 2011; 471: 189–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guryanova OA, Lieu YK, Garrett‐Bakelman FE et al Dnmt3a regulates myeloproliferation and liver‐specific expansion of hematopoietic stem and progenitor cells. Leukemia 2016; 30: 1133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. TET proteins and 5‐methylcytosine oxidation in hematological cancers. Immunol Rev 2015; 263: 6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhao Z, Chen L, Dawlaty MM et al Combined loss of Tet1 and Tet2 promotes B cell, but not myeloid malignancies, in mice. Cell Rep 2015; 13: 1692–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. An J, Gonzalez‐Avalos E, Chawla A et al Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun 2015; 6: 10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Inoue S, Li WY, Tseng A et al Mutant IDH1 downregulates ATM and alters DNA repair and sensitivity to DNA damage independent of TET2. Cancer Cell 2016; 30: 337–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kats LM, Reschke M, Taulli R et al Proto‐oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 2014; 14: 329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sashida G, Iwama A. Epigenetic regulation of hematopoiesis. Int J Hematol 2012; 96: 405–12. [DOI] [PubMed] [Google Scholar]
- 55. Xie H, Xu J, Hsu JH et al Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental‐stage‐specific manner. Cell Stem Cell 2014; 14: 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mochizuki‐Kashio M, Aoyama K, Sashida G et al Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1‐dependent manner. Blood 2015; 126: 1172–83. [DOI] [PubMed] [Google Scholar]
- 57. Beguelin W, Popovic R, Teater M et al EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013; 23: 677–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee SC, Miller S, Hyland C et al Polycomb repressive complex 2 component Suz12 is required for hematopoietic stem cell function and lymphopoiesis. Blood 2015; 126: 167–75. [DOI] [PubMed] [Google Scholar]
- 59. Abdel‐Wahab O, Gao J, Adli M et al Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med 2013; 210: 2641–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang J, Li Z, He Y et al Loss of Asxl1 leads to myelodysplastic syndrome‐like disease in mice. Blood 2014; 123: 541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Balasubramani A, Larjo A, Bassein JA et al Cancer‐associated ASXL1 mutations may act as gain‐of‐function mutations of the ASXL1‐BAP1 complex. Nat Commun 2015; 6: 7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Inoue D, Kitaura J, Matsui H et al SETBP1 mutations drive leukemic transformation in ASXL1‐mutated MDS. Leukemia 2015; 29: 847–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dey A, Seshasayee D, Noubade R et al Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 2012; 337: 1541–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Artinger EL, Mishra BP, Zaffuto KM et al An MLL‐dependent network sustains hematopoiesis. Proc Natl Acad Sci U S A 2013; 110: 12000–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yokoyama A. Molecular mechanisms of MLL‐associated leukemia. Int J Hematol 2015; 101: 352–61. [DOI] [PubMed] [Google Scholar]
- 66. Chen W, Kumar AR, Hudson WA et al Malignant transformation initiated by Mll‐AF9: gene dosage and critical target cells. Cancer Cell 2008; 13: 432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Arai S, Yoshimi A, Shimabe M et al Evi‐1 is a transcriptional target of mixed‐lineage leukemia oncoproteins in hematopoietic stem cells. Blood 2011; 117: 6304–14. [DOI] [PubMed] [Google Scholar]
- 68. Lin S, Luo RT, Ptasinska A et al Instructive role of MLL‐fusion proteins revealed by a model of t(4;11) pro‐b acute lymphoblastic leukemia. Cancer Cell 2016; 30: 737–49. [DOI] [PubMed] [Google Scholar]
- 69. Zhang J, Dominguez‐Sola D, Hussein S et al Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 2015; 21: 1190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ortega‐Molina A, Boss IW, Canela A et al The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med 2015; 21: 1199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Arcipowski KM, Bulic M, Gurbuxani S, Licht JD. Loss of Mll3 catalytic function promotes aberrant myelopoiesis. PLoS ONE 2016; 11: e0162515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Goyama S, Yamamoto G, Shimabe M et al Evi‐1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell 2008; 3: 207–20. [DOI] [PubMed] [Google Scholar]
- 73. Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol 2010; 12: 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Andricovich J, Kai Y, Peng W, Foudi A, Tzatsos A. Histone demethylase KDM2B regulates lineage commitment in normal and malignant hematopoiesis. J Clin Invest 2016; 126: 905–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Thieme S, Gyarfas T, Richter C et al The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood 2013; 121: 2462–73. [DOI] [PubMed] [Google Scholar]
- 76. Manna S, Kim JK, Bauge C et al Histone H3 lysine 27 demethylases Jmjd3 and Utx are required for T‐cell differentiation. Nat Commun 2015; 6: 8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016; 127: 42–52. [DOI] [PubMed] [Google Scholar]
- 78. Traina F, Visconte V, Elson P et al Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014; 28: 78–87. [DOI] [PubMed] [Google Scholar]
- 79. Itzykson R, Kosmider O, Cluzeau T et al Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011; 25: 1147–52. [DOI] [PubMed] [Google Scholar]
- 80. Meldi K, Qin T, Buchi F et al Specific molecular signatures predict decitabine response in chronic myelomonocytic leukemia. J Clin Invest 2015; 125: 1857–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rangwala S, Zhang C, Duvic M. HDAC inhibitors for the treatment of cutaneous T‐cell lymphomas. Future Med Chem 2012; 4: 471–86. [DOI] [PubMed] [Google Scholar]
- 82. Chaidos A, Caputo V, Karadimitris A. Inhibition of bromodomain and extra‐terminal proteins (BET) as a potential therapeutic approach in haematological malignancies: emerging preclinical and clinical evidence. Ther Adv Hematol 2015; 6: 128–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Daigle SR, Olhava EJ, Therkelsen CA et al Selective killing of mixed lineage leukemia cells by a potent small‐molecule DOT1L inhibitor. Cancer Cell 2011; 20: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rau RE, Rodriguez BA, Luo M et al DOT1L as a therapeutic target for the treatment of DNMT3A‐mutant acute myeloid leukemia. Blood 2016; 128: 971–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kuhn MW, Song E, Feng Z et al Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov 2016; 6: 1166–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med 2016; 22: 128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. McCabe MT, Ott HM, Ganji G et al EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature 2012; 492: 108–12. [DOI] [PubMed] [Google Scholar]
- 88. Thinnes CC, England KS, Kawamura A, Chowdhury R, Schofield CJ, Hopkinson RJ. Targeting histone lysine demethylases – Progress, challenges, and the future. Biochim Biophys Acta 2014; 1839: 1416–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mould DP, McGonagle AE, Wiseman DH, Williams EL, Jordan AM. Reversible inhibitors of LSD1 as therapeutic agents in acute myeloid leukemia: clinical significance and progress to date. Med Res Rev 2015; 35: 586–618. [DOI] [PubMed] [Google Scholar]
- 90. Liu Y, Wang L, Predina J et al Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat Med 2013; 19: 1173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bell JJ, Bhandoola A. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature 2008; 452: 764–7. [DOI] [PubMed] [Google Scholar]
- 92. Wada H, Masuda K, Satoh R et al Adult T‐cell progenitors retain myeloid potential. Nature 2008; 452: 768–72. [DOI] [PubMed] [Google Scholar]
- 93. Roulois D, Loo Yau H, Singhania R et al DNA‐demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 2015; 162: 961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chiappinelli KB, Strissel PL, Desrichard A et al Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 2015; 162: 974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]