

Abstract
Objective:
In African Americans, we sought to systematically identify coding Alzheimer disease (AD) risk variants at the previously reported AD genome-wide association study (GWAS) loci genes.
Methods:
We identified coding variants within genes at the 20 published AD GWAS loci by whole-exome sequencing of 238 African American participants, validated these in 300 additional participants, and tested their association with AD risk in the combined cohort of 538 and with memory endophenotypes in 319 participants.
Results:
Two ABCA7 missense variants (rs3764647 and rs3752239) demonstrated significant association with AD risk. Variants in MS4A6A, PTK2B, and ZCWPW1 showed significant gene-based association. In addition, coding variants in ZCWPW1 (rs6465770) and NME8 (rs10250905 and rs62001869) showed association with memory endophenotypes.
Conclusions:
Our findings support a role for ABCA7 missense variants in conferring AD risk in African Americans, highlight allelic heterogeneity at this locus, suggest the presence of AD-risk variants in MS4A6A, PTK2B, and ZCWPW1, nominate additional variants that may modulate cognition, and importantly provide a thorough screen of coding variants at AD GWAS loci that can guide future studies in this population.
African American individuals are approximately twice as likely to develop Alzheimer disease (AD) compared to Caucasians1–3; however, genetic studies in African Americans are scarce.4–6 Existing genetic studies provide evidence for genetic heterogeneity between African American and Caucasian participants7–9 (supplemental text at Neurology.org/ng). In the largest African American genome-wide association study (GWAS) conducted in 5,896 participants, besides APOE, only ABCA7 locus achieved genome-wide significance, although the most significant variant was different than that identified in Caucasians.8 A meta-analysis of 74,046 individuals of European ancestry identified or confirmed 20 late-onset Alzheimer disease (LOAD) susceptibility loci,10 none of which have been systematically investigated in African Americans. We performed whole-exome sequencing (WES) in an African American cohort to identify the full spectrum of coding variants in the candidate genes residing at these loci. Our study is a comprehensive coding variant discovery of LOAD candidate risk genes in African American individuals.
METHODS
Participants.
Five hundred fifty unrelated self-reported African American individuals (340 controls and 198 AD cases) were recruited for this study, of which 538 were retained for analysis after quality control (QC). All participants were recruited at Mayo Clinic in Jacksonville, FL, except 1 cognitively normal and 3 AD participants recruited at Wien Center for Alzheimer's Disease and Memory Disorders, Mount Sinai, Miami, FL. WES was performed on 250 DNA samples from this cohort (figure 1). Variants of interest were validated by genotyping or Sanger sequencing (table 1).
Figure 1. Study design.

Whole-exome sequencing (WES) sample quality control (QC) led to exclusion of samples with poor sequence quality, sex ambiguity, or cryptic relatedness. WES single nucleotide polymorphism (SNP) QC led to exclusion of SNPs with a BLAST-like alignment tool (BLAT) score >1, those not resulting in an alteration of the coding sequence, and those outside the 20 late-onset Alzheimer disease (LOAD) genome-wide association study loci candidate genes. Of the remaining 193 variants tested in the final post-QC WES cohort (n = 238), 10 demonstrated association with Alzheimer disease (AD) risk with an uncorrected p < 0.05. After excluding 2 SNPs with strong linkage disequilibrium, the remaining 8 SNPs were confirmed by sequencing or genotyping in an independent validation cohort (n = 300). These 8 SNPs were also evaluated in the combined cohort (n = 538).
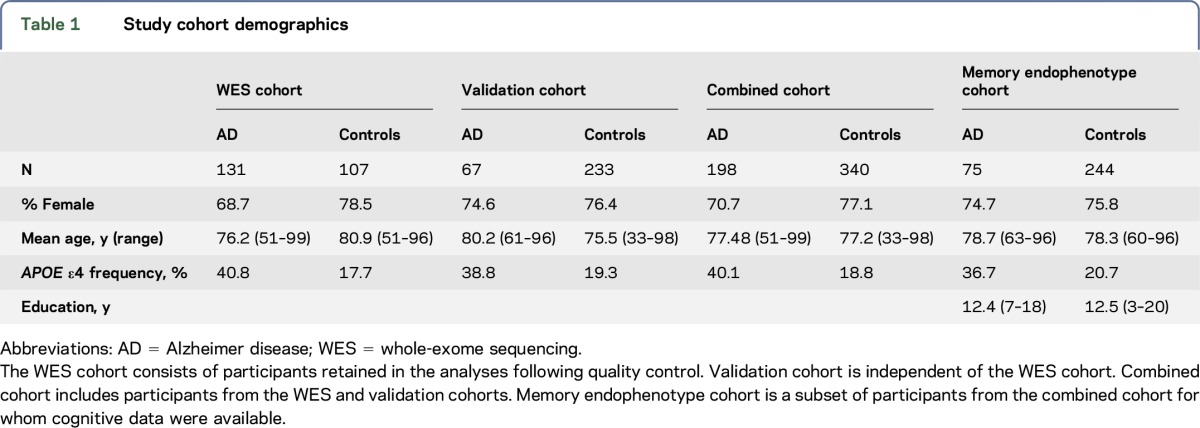
Table 1.
Study cohort demographics
Standard protocol approvals, registrations, and patient consents.
This study was approved by the institutional review boards of all participating institutions, and all participants provided written informed consent for the conducted research.
Whole-exome sequencing.
Exome capture was performed using the Agilent Sure Select V4 + UTR exome capture kit.11 Sequencing was performed on the Illumina HiSeq 2000 platform. The study design, including sample and variant QC, is described in figure 1. Further details of these and population substructure analyses are described in the e-Methods.
Validation.
Variants of interest that achieved nominal significance with AD risk in the WES cohort, either by logistic regression analysis or by Fisher exact test, were genotyped using TaqMan SNP Genotyping Assays or Sanger sequencing. Validation genotyping or sequencing was performed in the combined cohort of 340 African American controls and 198 AD cases, which were then tested for AD-risk association with the coding variants of interest. The ABCA7 intronic variant rs3764650, which shows association with AD risk in a Caucasian LOAD GWAS,12 was also genotyped in our entire African American cohort using a TaqMan assay.
Memory endophenotypes.
Verbal and visual episodic memory associations were tested on a subset of 319 individuals (75 ADs and 244 controls) using the Logical Memory (LM) and the Visual Reproduction (VR) subtests of the Wechsler Memory Scale following standardized instructions.13 The subset of participants included in the memory endophenotype associations were selected merely on the basis of availability of cognitive test results. For each subtest, test performance was assessed using the immediate recall (IR) raw score (LMIR and VRIR), the 30-minute delayed recall (DR) raw score (LMDR and VRDR), and the percent retention (PR) score (LMPR and VRPR). Tests were administered and evaluated by Mayo Clinic neuropsychologists.
Statistical analysis.
The association of the selected variants with AD risk was evaluated by multivariate logistic regression analysis adjusted for age, sex, and APOE ε4 dosage; however, variants with a total minor allele count <10 were analyzed using the Fisher exact test,14 as implemented in PLINK.15 We also performed multivariate logistic regression analysis adjusted for all covariates in the primary analysis plus the first 4 principal components. Haploview was used for linkage disequilibrium (LD) analysis.16 Power analyses were performed in StatsDirect v.2.7.8. Gene-based analysis was conducted using the Sequence Kernel Optimal Unified Association Test17 (e-Methods).
Tests of association between selected variants and the 6 memory endophenotypes (LMIR, LMDR, LMPR, VRIR, VRDR, and VRPR) were performed in R using a multivariate linear regression analysis adjusted for age, sex, APOE ε4 dosage, years of education, and diagnosis.18 Neuropsychological test scores were obtained from the most recent neuropsychological evaluation. Age and diagnosis were defined at the time of the last neuropsychological examination.
RESULTS
WES was performed on 250 African American individuals in our cohort (figure 1). Twelve participants were excluded based on QC; 3 based on poor sequence quality, 5 due to sex ambiguities, and 4 based on identity by descent (IBD) analysis. No population outliers were observed among the remaining participants based on multidimensional scaling analysis15 (figure e-1). The final WES cohort of 131 AD and 107 controls (table 1) has 80% power to detect a variant with control minor allele frequency (MAF) of 0.10 and odds ratio (OR) = 2.96; and with MAF = 0.03 and OR = 5.35 at α = 0.05. The entire cohort composed of 238 post-QC WES participants and an additional 67 AD cases and 233 controls (198 AD and 340 controls) has 80% power to detect a variant with MAF = 0.10 and OR = 2.13; or one with MAF = 0.03 and OR = 3.3 at α = 0.05.
Following QC, 116,376 single nucleotide polymorphisms (SNPs) were retained for analysis. Of these, 193 were in LOAD GWAS candidate genes (table e-1). AD-risk association by multivariate logistic regression analysis identified 7 variants with nominal significance (uncorrected p < 0.05). The Fisher exact test identified 3 additional rare SNPs (table e-2). Secondary analysis of the common variants by multivariate logistic regression analysis that also adjusted for the first 4 principal components did not substantially influence the results (tables e-1 and e-2). Five of these SNPs were in ABCA7, 2 within NME8, and 1 each in CASS4, MS4A6A, and ZCWPW1. We also tested these variants for association with AD risk by comparing their frequencies in the AD participants of our WES cohort to that in the African American population controls from the Exome Sequencing Project.19 Four ABCA7 variants, rs3752232, rs3764647, rs3752239, and rs4147918, showed nominally significant association in this analysis with the same direction of effect as that observed in the WES analysis.
Among the 5 ABCA7 variants, rs3764647 and rs3752232 were in tight LD (r2 = 0.91); as were rs3752239 and rs4147918 (r2 = 0.85) (figure 2). For each pair, the variant with the greater predicted functional impact (rs3752239) or larger OR estimate (rs3764647) was retained for further analysis. Hence, 8 SNPs were validated and subsequently tested for association with AD risk in the combined cohort. Of these, 2 missense variants in ABCA7, rs3764647 and rs3752239 continued to have nominally significant association with AD risk in addition to rs35031530 in CASS4 (table 2), although the latter departed from Hardy-Weinberg equilibrium (p = 0.03) in controls.
Figure 2. Linkage disequilibrium pattern of single nucleotide polymorphisms in ABCA7 in the whole-exome sequencing cohort.

r2 Values are shown on the plot. The Caucasian genome-wide association study (GWAS) single nucleotide polymorphism (SNP) is shown in pink font. The African American GWAS SNP was imputed in our data set (r2 = 0.28) and is shown in light blue font. Given the low imputation quality, the r2 value for this variant may not be accurate, but it is retained in this plot to show its genic location. Variants showing nominal association with Alzheimer disease risk in our data set are represented in navy blue font. aThese 2 ABCA7 variants were in strong linkage disequilibrium (LD) with 2 other ABCA7 variants (b) identified in the whole-exome sequencing and were therefore not included in the validation. bThese 2 ABCA7 variants that were in strong LD with 2 others (a) were retained either because they had a larger odds ratio estimate or greater functional potential.
Table 2.
AD-risk association analyses of SNPs identified in WES of African American participants
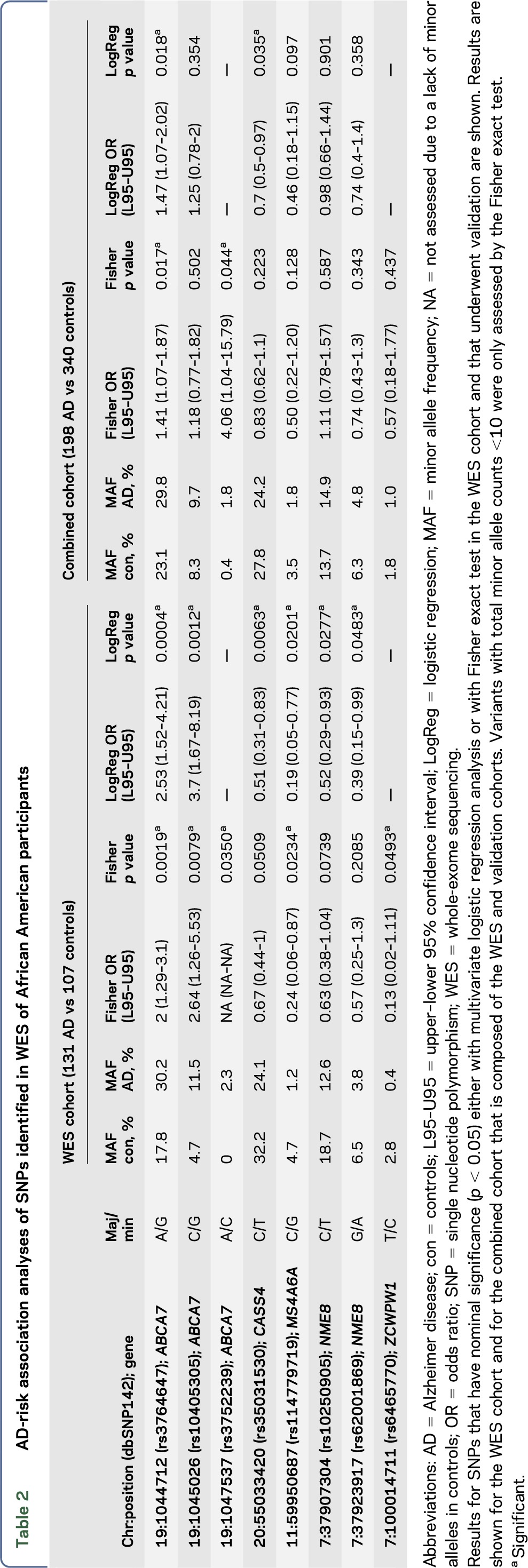
To determine whether the AD-risk association observed with the 2 ABCA7 coding variants accounted for the effect of previously reported ABCA7 GWAS variants, we first evaluated the Caucasian LOAD GWAS ABCA7 risk variant rs376465012 in our entire cohort. This variant was not associated with AD risk in our African American participants (multivariate logistic regression p = 0.13, OR = 1.28). However, rs3764650 is in LD with the ABCA7 coding variant rs3764647 (r2 = 0.77) (figure 2). Conditional analysis adjusting for rs3764650 genotypes abolished the AD-risk association of rs3764647 (conditional p = 0.198, conditional OR = 1.54). In contrast, there is no LD between rs3764650 and the other significant ABCA7 coding variant rs3752239, which remains nominally significant even after accounting for rs3764650 (p = 0.047, OR = 4.65).
Previous AD GWAS in African American participants identified the ABCA7 intronic variant rs115550680 to be significantly associated with AD risk.8 This intronic variant is located at position 1,050,420 of chromosome 19 (GRCh37/hg19 assembly), which is indicated by RepeatMasker to be within a short-interspersed repetitive element of 313 bp.20 Hence, reliable genotyping or sequencing primers could not be designed for rs115550680. Imputation of this position in our WES data using the Michigan Imputation Server (imputationserver.sph.umich.edu/index.html) resulted in low-quality imputation for this locus (r2 = 0.28). When we tested the imputed rs115550680 genotypes for their association with AD risk in our cohort, we observed an OR estimate indicative of increased risk for the minor G allele, although this result did not achieve statistical significance (data not shown).
Gene-based analysis of the 20 LOAD GWAS candidate genes using all variants identified in our WES revealed association for variants in MS4A6A (n = 5 SNPs, p = 0.042) and ZCWPW1 (n = 11 SNPs, p = 0.028) (table e-3). Given that the majority of the missing heritability is thought to reside within low-frequency (5%> MAF >0.5%) and rare (MAF <0.5%) variants,21 we also sought to separately assess these specific classes of variants. Evaluation of the low-frequency variants resulted in a stronger AD-risk association with ZCWPW1 (n = 2 SNPs, p = 0.0099) and with PTK2B variants (n = 2 SNPs, p = 0.042). ABCA7 was the only gene with significant association using rare SNPs (n = 20 SNPs, p = 0.020).
Neuropsychological evaluations were performed in a subset of 319 individuals from our African American cohort (75 AD cases and 244 controls) (table 1). The 8 variants identified by WES that showed nominal AD-risk association were tested for their effects on verbal and visual episodic memory performance (table 3). The 2 NME8 alleles which associate with lower risk of AD in the WES cohort (rs10250905-T; rs62001869-A), but that did not achieve statistically significant association with AD risk in the combined cohort, demonstrated nominally significant (p < 0.05) or suggestive (0.05 < p < 0.1) association with higher scores for VRDR and IR (VRIR) tests. We also observed a trend for better LMIR scores in NME8 rs62001869-A carriers. In addition, ZCWPW1 rs6465770-C, which showed association with decreased risk of AD in the WES cohort but not in the combined cohort, showed nominally significant association with better LMIR and LMDR scores.
Table 3.
Memory endophenotype association analyses of SNPs

DISCUSSION
The goal of this study was to assess genes at previously identified LOAD GWAS loci for coding risk variants in African American participants. This is an AD-risk variant discovery study by WES performed in a sizable cohort of 250 African Americans. A prior WES variant discovery study in African Americans was conducted in 7 patients with AD, followed by genotyping of candidate variants in a larger cohort.7
In our combined African American cohort of 538 individuals, 2 ABCA7 variants, rs3764647 and rs3752239, showed nominal significance for association with AD risk. The common variant, rs3764647, has previously been associated with AD risk in an African American AD GWAS.22 In that study, the ABCA7 variant rs3764650, which demonstrated significant association with AD risk in a LOAD GWAS conducted in Caucasian cohorts,12 showed nominally significant association with AD risk (p = 0.019) with an OR estimate (1.27) similar to that in Caucasians, although the most significant ABCA7 variant was rs3764647 (OR = 1.32, p = 0.0087). Similarly, in our study, we observed only a trend of association with the Caucasian GWAS SNP rs3764650, with an OR estimate (OR = 1.28) almost identical to those in the prior studies12,22; and nominal significance with the common missense variant rs3764647 (OR = 1.47, p = 0.018). In our African American cohort, these 2 variants are in strong LD (r2 = 0.77), such that the association of rs3764647 is abolished when adjusting for rs3764650 genotypes, indicating that in African Americans, the AD-risk association observed with the Caucasian GWAS SNP rs3764650 may at least in part be explained by the coding variant rs3764647.
This finding is distinct from the observations emerging from ABCA7 variant discovery efforts in Caucasian cohorts. Two missense coding SNPs, rs375224612 and rs4147934,23 in modest LD with each other were proposed as functional candidate variants accounting for the ABCA7 locus GWAS signal in Caucasians. Although both of these missense variants were also detected by WES in our cohort, neither had significant AD-risk association in our cohort. More recently, an intronic low-frequency ABCA7 variant, rs78117248, showed association in a Belgian cohort24 even after adjustment for the GWAS variants rs3764650, rs4147929, and rs3752246. Rs78117248 was not assessed in our study, as we only focused on coding variants; however, this variant is monomorphic in African American participants according to dbSNP. These results collectively highlight the allelic heterogeneity of the ABCA7 locus, where different functional risk variants are likely to underlie the GWAS association signal marked by rs3764650, in Caucasian vs African American participants. Our findings support rs3764647 as a functional risk variant underlying this GWAS signal in African American participants. Rs3764647 leads to the amino acid change p.His395Arg in the first extracellular loop of the ABCA7 protein, a region prone to missense mutations. Our study identified AD-risk association with another missense coding SNP, rs3752232 that leads to a p.Thr319Ala change also in the first extracellular loop of ABCA7. While we did not investigate rs3752232 in our larger cohort due to its strong LD with rs3764647 and the slightly better significance of the latter, it remains possible that both of these variants contribute to the risk of AD in African American participants. Although neither rs3764647 nor rs3752232 is predicted to have a deleterious functional consequence by PolyPhen-2 or SIFT,25,26 they may be contributing to a cumulative AD risk via subtle effects on ABCA7 structure or function in the context of one another, additional ABCA7 or other genetic variants. Given our findings, both rs3764647 and rs3752232 merit further assessment by downstream functional assays.
We also found evidence of AD-risk association with a rare missense ABCA7 SNP, rs3752239-C. This variant has an allele frequency of 0.7% in African Americans according to the Exome Variant Server.19 We observed this variant in <1% of the African American controls and ∼2% of the AD cases, leading to an OR estimate of ∼4. AD association with rs3752239 remains significant even after adjusting for the GWAS variant rs3764650, suggesting that this rare variant may confer AD risk in African American participants independently from the common GWAS variant. Furthermore, rs3752239 leads to p.Asn718Thr in the first ABCA7 intracellular domain prior to the first adenosine triphosphate–binding cassette and is predicted to have a possible damaging effect by PolyPhen.25 These results nominate rs3752239 as a rare, putative functional variant that may confer AD risk in African Americans. Rs3752239 is in strong LD with rs4147918, which leads to a p.Gln1686Arg amino acid change in the second ABCA7 transmembrane domain. Although rs4147918 was not assessed in our larger cohort due to this LD and lack of predicted functional effect, it remains possible that this variant may also contribute to AD risk in African Americans, alone or in combination with rs3752239.
Rs3752239 shows association with AD risk in The International Genomics of Alzheimer's Project (IGAP) Stage I10 with p = 0.004, although the minor allele C has a protective estimate in this study of Caucasian participants, which is opposite to our findings. This variant is more common in Caucasians with MAF = 3.7% in Exome Variant Server.19 It is possible that the rs3752239 results in the IGAP population reflect LD with other variants rather than a protective functional effect of this variant in Caucasians. Differential functional effects of rs3752239 in African American vs Caucasian populations are also possible.
The ABCA7 variants that showed significant association in the single-variant analysis in our study had MAFs that fall in the common (MAF >5%) or rare (MAF <0.5%) range. Notably, our gene-based analysis supports the association of multiple rare ABCA7 SNPs with AD risk in African Americans. In addition, our gene-based analysis identified significant associations with SNPs in MS4A6A, PTK2B, and ZCWPW1 that escaped detection by single-variant analysis. For PTK2B, significance was observed only with the set of low-frequency SNPs. ZCWPW1 gene-based analysis shows significance when variants of all frequencies are analyzed, but this result appears to be driven by low-frequency variants in this gene. Hence, there may be additional variants that associate with AD in these genes that escape detection by single-variant analysis approaches.
In this study, we also aimed to assess whether coding variants in the LOAD candidate genes modulate cognitive endophenotypes. We hypothesized that some genetic variants that associate with AD risk in the WES cohort would also influence memory endophenotypes. We observed that the minor alleles of rs6465770 in ZCWPW1, as well as rs10250905 and rs62001869 in NME8 that associate with lower AD risk in the WES cohort also have nominally significant associations with better memory scores. Although these variants did not retain significant AD-risk associations in the larger cohort, the biologically concordant association of their minor alleles with better verbal (rs6465770) or visual (rs10250905 and rs62001869) memory scores is noteworthy. Furthermore, rs6465770 in ZCWPW1 is one of the 2 low-frequency variants that show association in the gene-based analysis. The difference between the memory and AD-risk association results could arise from false-positive or false-negative findings in one of the analyses; or an inherent difference between the overall cohort vs the memory endophenotype subset cohort, although the latter was selected only based on the availability of cognitive results. These findings, which require replication in larger cohorts and longitudinal studies, may imply that these variants may have an influence on the progression of memory decline.
Although informative, this study has limitations. Despite being the largest to date variant discovery effort by WES in African Americans, both our WES and entire cohort have limited power to detect AD-risk associations. None of the variants highlighted in this study would remain significant after corrections for multiple testing for 193 variants in the WES and 8 selected variants in the combined cohort. Likewise, the cohort with cognitive endophenotypes was limited in size, and the cognitive endophenotype associations would not withstand corrections for multiple testing. Given this, the findings from our study require replication in larger independent cohorts. Based on the MAF and OR estimates in our combined cohort, detection of AD-risk association for the rare missense SNP rs3752239 with 80% power at α = 0.05 requires a sample of at least 1,124 AD vs 1,124 controls.
Our study represents a systematic variant discovery conducted by WES in African Americans to identify coding AD-risk variants within genes at the 20 LOAD-risk GWAS loci. In our WES cohort of 238 subjects, of the variants that lead to a coding change, most were in ABCA7, and the only variants that remained nominally significant in the combined cohort of 538 participants were ABCA7 missense variants. Furthermore, the rare ABCA7 SNPs in our study had significant gene-based association. These findings are similar to those observed in Caucasians23,27,28 and highlight the unique tendency of ABCA7 to harbor coding AD-risk variants.
The significant gene-based associations observed for MS4A6A, PTK2B, and ZCWPW1 suggest that there may also be coding AD-risk variants in these genes. Lack of single-variant or gene-based associations with coding variants in the other AD GWAS genes in our study may be multifactorial including insufficient power. It is also possible that risk variants for many of the LOAD genes primarily reside in functional, noncoding regions.29,30 Finally, given that ABCA7 is the only Caucasian AD GWAS locus replicated at genome-wide significance in African Americans,8 lack of identification of coding variants in the other GWAS genes may be due to their lack of influence in this population. This possibility seems unlikely because other Caucasian LOAD GWAS loci, namely CR1, BIN1, EPHA1, and CD33, also have evidence of association in African Americans albeit below genome-wide significance.8
Our findings also highlight the presence of allelic heterogeneity between different ethnic groups and underscore the importance of variant discovery efforts in both Caucasian and non-Caucasian participants. Our study provides support for the enrichment of ABCA7 with coding AD-risk variants, suggests that MS4A6A, PTK2B, and ZCWPW1 may also harbor AD-risk variants, nominates coding variants in ZCWPW1 and NME8 that may be protective by modulating cognition, and importantly reveals a list of coding variants in LOAD candidate genes that can guide future variant discovery and replication efforts in this understudied population.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families for their participation, without whom these studies would not have been possible, and the clinicians, technicians, and administrative staff who helped in the implementation of this study.
GLOSSARY
- AD
Alzheimer disease
- DR
delayed recall
- GWAS
genome-wide association study
- IGAP
The International Genomics of Alzheimer's Project
- IR
immediate recall
- LD
linkage disequilibrium
- LM
Logical Memory
- LOAD
late-onset Alzheimer disease
- MAF
minor allele frequency
- OR
odds ratio
- PR
percent retention
- QC
quality control
- SNP
single nucleotide polymorphism
- VR
Visual Reproduction
- WES
whole-exome sequencing
Footnotes
Supplemental data at Neurology.org/ng
AUTHOR CONTRIBUTIONS
A.N., M.M.C., and N.E.-T. contributed to conception and design of the study. A.N., M.M.C., X.W., J.D.B., T.N., Y.W.A, D.J.S., S.G.Y., M.A., O.P., R.D., M.T.G.C., N.R.G.-R., and N.E.-T. contributed to acquisition and analysis of data. A.N. and N.E.-.T. contributed to drafting the text and preparing the figures.
STUDY FUNDING
This work was supported by the Florida Department of Health, Ed and Ethel Moore Alzheimer's Disease Research Program (5AZ03 to N.E.-T.); Alzheimer's Association (MNIRGD 2013 award to M.M.C.); Mayo Clinic Office of Health Disparities Research (M.M.C.); Mayo Alzheimer's Disease Research Center (P50 AG0016574 to N.E.-T., N.R.G.-R., and S.G.Y.); National Institute on Aging (RF1 AG051504 to N.E.-T. and U01 AG046139 to N.E.-T. and S.G.Y.); National Institute of Neurological Disorders and Stroke (R01 NS080820 to N.E.-T.).
DISCLOSURE
Dr. N'Songo reports no disclosures. Dr. Carrasquillo has received research support from the Alzheimer's Association and Mayo Clinic. Dr. Wang, Mr. Burgess, Ms. Nguyen, Dr. Asmann, and Mr. Serie report no disclosures. Dr. Younkin has received travel funding and a speaker honorarium from Eisai Pharmaceuticals; has received research support from NIH; and receives royalty payments from Mayo for licensing of Tg2576 mouse model of AD. Dr. Allen has received research support from NIH and Mayo Clinic Center for Individualized Medicine. Dr. Pedraza has received research support from NIH. Dr. Duara and Dr. Greig Custo report no disclosures. Dr. Graff-Radford has served on the scientific advisory board of Cytox Consultation; has served on the editorial board of Alzheimer Disease and Therapy; receives publishing royalties from UpToDate; has multicenter treatment study grants from Lilly, TauRx, Biogen, and Axovant; and has received research support from NIA. Dr. Ertekin-Taner has served on the editorial board of the American Journal of Neurodegenerative Disease; holds a patent for Human Monoclonal Antibodies Against Amyloid Beta Protein, and Their Use as Therapeutic Agents Application (pending); has received research support from NIH, CurePSP, Bright Focus, and the Florida Health Ed and Ethel Moore Alzheimer's Disease Research Program Grant; and consulted for Cytox (unpaid). Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Tang MX, Cross P, Andrews H, et al. . Incidence of AD in African-Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology 2001;56:49–56. [DOI] [PubMed] [Google Scholar]
- 2.Green RC, Cupples LA, Go R, et al. . Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA 2002;287:329–336. [DOI] [PubMed] [Google Scholar]
- 3.Plassman BL, Langa KM, Fisher GG, et al. . Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 2007;29:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams MM, Meisel MM, Williams J, Morris JC. An interdisciplinary outreach model of African American recruitment for Alzheimer's disease research. Gerontologist 2011;51(suppl 1):S134–S141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams MM, Scharff DP, Mathews KJ, et al. . Barriers and facilitators of African American participation in Alzheimer's disease biomarker research. Alzheimer Dis Assoc Disord 2010;24(suppl):S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darnell KR, McGuire C, Danner DD. African American participation in Alzheimer's disease research that includes brain donation. Am J Alzheimers Dis Other Demen 2011;26:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Logue MW, Schu M, Vardarajan BN, et al. . Two rare AKAP9 variants are associated with Alzheimer's disease in African Americans. Alzheimers Dement 2014;10:609–618.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reitz C, Jun G, Naj A, et al. . Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E ε4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 2013;309:1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin SC, Carrasquillo MM, Benitez BA, et al. . TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol Neurodegener 2015;10:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. . Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen R, Im H, Snyder M. Whole-exome enrichment with the Agilent SureSelect Human All Exon platform. Cold Spring Harb Protoc 2015;2015:626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollingworth P, Harold D, Sims R, et al. . Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011;43:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wechsler D. WMS-R: Wechsler Memory Scale-Revised. San Antonio, TX: Psychological Corporation; 1987. [Google Scholar]
- 14.Peduzzi P, Concato J, Kemper E, Holford TR, Feinstein AR. A simulation study of the number of events per variable in logistic regression analysis. J Clin Epidemiol 1996;49:1373–1379. [DOI] [PubMed] [Google Scholar]
- 15.Purcell S, Neale B, Todd-Brown K, et al. . PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005;21:263–265. [DOI] [PubMed] [Google Scholar]
- 17.Lee S, Emond MJ, Bamshad MJ, et al. . Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 2012;91:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. ISBN 3-900051-07-0, 2014. [Google Scholar]
- 19.Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP) [online]. Seattle, WA: Exome Variant Server; Available at: evs.gs.washington.edu/EVS/. Accessed in August 2016. [Google Scholar]
- 20.RepeatMasker Open-4.0 [computer program]. 2013–2015. Available at: repeatmasker.org. Accessed in April 2015. [Google Scholar]
- 21.Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet 2014;95:5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Logue MW, Schu M, Vardarajan BN, et al. . A comprehensive genetic association study of Alzheimer disease in African Americans. Arch Neurol 2011;68:1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holton P, Ryten M, Nalls M, et al. . Initial assessment of the pathogenic mechanisms of the recently identified Alzheimer risk Loci. Ann Hum Genet 2013;77:85–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuyvers E, De Roeck A, Van den Bossche T, et al. . Mutations in ABCA7 in a Belgian cohort of Alzheimer's disease patients: a targeted resequencing study. Lancet Neurol 2015;14:814–822. [DOI] [PubMed] [Google Scholar]
- 25.Adzhubei IA, Schmidt S, Peshkin L, et al. . A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 2012;40:W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vardarajan BN, Ghani M, Kahn A, et al. . Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol 2015;78:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinberg S, Stefansson H, Jonsson T, et al. . Loss-of-function variants in ABCA7 confer risk of Alzheimer's disease. Nat Genet 2015;47:445–447. [DOI] [PubMed] [Google Scholar]
- 29.Allen M, Zou F, Chai HS, et al. . Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 2012;79:221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen M, Kachadoorian M, Carrasquillo MM, et al. . Late-onset Alzheimer disease risk variants mark brain regulatory loci. Neurol Genet 2015;1:e15 doi: 10.1212/NXG.0000000000000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.