

Abstract
Gilbert’s syndrome in humans is derived from a polymorphism (TA repeat) in the hepatic UGT1A1 gene that results in decreased conjugation and increased levels of unconjugated bilirubin. Recently, we have shown that bilirubin binds directly to the fat-burning nuclear peroxisome proliferator-activated receptor-α (PPARα). Additionally, we have shown that serine 73 phosphorylation [Ser(P)73] of PPARα decreases activity by reducing its protein levels and transcriptional activity. The aim of this study was to determine whether humanized mice with the Gilbert’s polymorphism (HuUGT*28) have increased PPARα activation and reduced hepatic fat accumulation. To determine whether humanized mice with Gilbert’s mutation (HuUGT*28) have reduced hepatic lipids, we placed them and C57BL/6J control mice on a high-fat (60%) diet for 36 wk. Body weights, fat and lean mass, and fasting blood glucose and insulin levels were measured every 6 wk throughout the investigation. At the end of the study, hepatic lipid content was measured and PPARα regulated genes as well as immunostaining of Ser(P)73 PPARα from liver sections. The HuUGT*28 mice had increased serum bilirubin, lean body mass, decreased fat mass, and hepatic lipid content as well as lower serum glucose and insulin levels. Also, the HuUGT*28 mice had reduced Ser(P)73 PPARα immunostaining in livers and increased PPARα transcriptional activity compared with controls. A chronic but mild endogenous increase in unconjugated hyperbiliubinemia protects against hepatic steatosis through a reduction in Ser(P)73 PPARα, causing an increase in PPARα transcriptional activity.
Keywords: Gilbert’s syndrome, bilirubin, peroxisome proliferator-activated receptor-α, fatty liver, nonalcoholic fatty liver disease
obesity is a worldwide epidemic that may be caused by a combination of several factors, including genetics, overeating, and a sedentary lifestyle. Obesity is also a significant contributing factor in the development of hepatic steatosis and insulin resistance, which make the liver vulnerable to increased oxidative stress and inflammation that may lead to nonalcoholic fatty liver disease (NAFLD) (15, 31, 39, 53, 55). Several population studies have demonstrated a negative correlation between plasma bilirubin levels and the development of metabolic and cardiovascular diseases (9–11, 21, 43, 45, 54, 55). Plasma bilirubin levels are partially regulated through the conjugation of bilirubin in the liver by the enzyme uridine diphosphate glucuronosyltransferase-1A (UGT1A1) (8, 13, 52). TA polymorphisms in UGT1A1 give rise to Gilbert’s syndrome, which is characterized by moderately elevated plasma bilirubin levels affecting ~5–10% of the population. Gilbert’s syndrome is commonly due to the UGT1A1*28 allele that generates A(TA)7 TAA (UGT1A1*28 allele) instead of the normal A(TA)6TAA sequence (UGT1A1*1 allele). This TA polymorphisms in the promoter region of the gene leads to decreased UGT1A1 transcription, resulting in reduced enzymatic activity and moderate hyperbilirubinemia (22, 50). It has recently been reported that plasma bilirubin levels are negatively correlated with the development of hepatic steatosis, which suggests that moderate hyperbilirubinemia may be protective against liver injury (23, 46).
Recently, we have shown that bilirubin can bind directly to the nuclear receptor transcription factor PPARα (51), which causes the burning of fat by increasing genes for β-oxidation. PPARα is important for whole body fatty acid homeostasis and is protective against hepatic steatosis and NAFLD (42). Furthermore, we showed that a liver-specific knockout of biliverdin reductase A (BVRA), the enzyme that reduces biliverdin to bilirubin (44), caused hepatic steatosis in mice (26). In the BVRA liver-specific knockout, we showed that PPARα protein levels are significantly reduced in the liver due to the hyperphosphorylation of serine 73 [Ser(P)73], which increased ubiquitination and decreased transcriptional activity (26). In a human study with Gilbert’s syndrome, it was recently shown that they had higher levels of serine 12 (Ser12) phosphorylation of PPARα in peripheral blood mononucleated cells (PBMCs) (41). The role of how S er12 affects PPARα activity is unknown, but Barger et al. (3) showed that serines in the NH2 terminus of PPARα are important for transcriptional activity.
In these studies, we wanted to test a humanized mouse model of Gilbert’s syndrome that had a transgenic expression of the human UGT1A1*28 allele (HuUGT*28) in mice whose endogenous Ugt1 gene had been inactivated (6, 20). The HuUGT*28 mice exhibit early neonatal hyperbilirubinemia, which tapers off after weaning to moderate hyperbilirubinemia in adulthood. We found that the UGT1A1*28 mice had elevated bilirubin and were protected against diet-induced hepatic steatosis as well as subsequent insulin resistance. Furthermore, we found that the UGT1A1*28 mice had significantly less Ser(P)73, which resulted in higher PPARα protein levels and transcriptional activity.
METHODS
Animals.
The experimental procedures and protocols of this study conformed to the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center in accordance with the NIH Guide for the Care and Use of Laboratory Animals. All mice had free access to food and water ad libitum. Animals were housed in a temperature-controlled environment with a 12-h dark-light cycle. Gilbert’s mice UGT1A1*28 (TgUGTA1*28)Ugt−/− were obtained from Dr. Robert H. Tukey at the University of California San Diego as described originally (6, 20). These mice were maintained by homozygous breeding on a C57BL/6J genetic background. Age-matched C57BL/6J (stock no. 000664) were purchased from The Jackson Laboratory (Bar Harbor, ME). Studies were performed on 4-wk-old male mice housed initially under standard conditions with full access to standard mouse chow and water. After this, mice were switched with full access to a 60% high-fat diet (diet no. D12492; Research Diets, New Brunswick, NJ) for 36 wk and allowed access to water. This diet contains 60% of its total kilocalories from fat and 20% from carbohydrates derived from mainly from maltodextrin 10 (12%) and sucrose (6.8%).
Body composition.
Body composition changes were assessed at 6-wk intervals throughout the study using magnetic resonance imaging (EchoMRI-900TM; Echo Medical System, Houston, TX). MRI measurements were performed in conscious mice placed in a thin-walled plastic cylinder, with a cylindrical plastic insert added to limit movement of the mice. Mice were briefly submitted to a low-intensity electromagnetic field where fat mass, lean mass, free water, and total water were measured.
Oxygen consumption, respiratory exchange rate, and motor activity.
At 32 wk after the start of the experimental protocol, mice were placed individually in an acrylic cage (16 × 24 × 17 cm) equipped with a metabolic monitoring system (AccuScan System; Harvard Apparatus, Holliston, MA) for measurements of oxygen consumption (V̇o2) and motor activity as described previously (14, 29). V̇o2 was determined daily (for 2 min every 10-min interval) and expressed as the 24-h average normalized to lean body mass as determined by EchoMRI. Motor activity was determined using infrared light beams mounted in the cages in x-, y-, and z-axes.
Food consumption.
Food consumption was measured during week 18 of the experimental protocol in mice housed individually. The total amount of food was weighed daily in the morning and averaged for each mouse to obtain a 24-h food consumption measurement. Daily 24-h food consumption measurements were then averaged over 1 wk.
Hepatic lipid measurement.
Liver triglyceride was measured biochemically from 100 mg of liver tissue, as described previously (26, 51). Samples from individual mice were run in duplicate and averaged, and the averages of individual mice were then used to obtain group averages. Hepatic lipids were also measured by Oil Red O staining, as described previously (26, 51). Measurements were obtained from three individual animals per group. Data are presented as the average ± SE of the percent Oil Red O staining for each group.
Fasting glucose and insulin.
Following an 8-h fast, a blood sample was obtained via orbital sinus under isoflurane anesthesia. Blood glucose was measured using an Accu-Chek Advantage glucometer (Roche, Mannheim, Germany). Fasting plasma insulin concentrations were determined by ELISA (Linco Insulin ELISA kit), as described previously (14, 29).
Glucose tolerance test.
For glucose tolerance tests (GTTs), mice were subjected to fasting overnight (∼16 h), and d-glucose (1 g/kg of body weight) was injected intraperitoneally. Blood glucose was monitored at 0, 15, 30, and 45 min after glucose injection. Blood glucose was measured using a glucometer, as described above. GTT was performed on mice during week 28 of the high-fat diet.
Measurement of total and direct bilirubin.
Total and direct bilirubin were measured separately from 50 μl of plasma using commercially available kits (Total Bilirubin and Direct Bilirubin; Wako Diagnostics, Mountain View, CA). Direct bilirubin was measured by the reaction of bilirubin glucuronate with sulphodiazonium salt to form colored derivative azobilirubin. The color intensity of formed azobilirubin measured at 540–550 nm is proportional to direct bilirubin concentration in the sample. Total bilirubin was measured, utilizing vanadate as an oxidizing agent to oxidize all of the bilirubin in the sample to biliverdin. The reaction monitors the bilirubin absorbance before and after the vanadate oxidation. All reactions were performed in duplicate, with standards supplied by the manufacturer and the data presented as mg/dl.
Measurement of aminotransferase, albumin, and cholesterol.
We used the VetScanVS2 Chemistry Analyzer, an in vitro veterinary diagnostic system (Abaxis, Union City, CA), with the VetScan Mammalian Liver Profile reagent rotor (www.abaxis.com) to determine alanine aminotransferase (ALT) activity, albumin (ALB), and total cholesterol (CHOL) in heparinized plasma. The reagent rotor sample chamber was loaded with 120 μl of plasma and the rotor inserted. Sample values were compared with standardized mouse ranges, with internal quality control (QC) monitors on the analyzer, rotor, and sample before and during analysis to ensure reliable, consistent results. Each of the individual rotors used for analysis passed all QC parameters as indicated when the test was complete.
Quantitative real-time PCR analysis.
Total RNA was harvested from WT and HuUGT*28 livers using a Qiagen Tissue Lyser LT (Qiagen) and then extraction by a 5-Prime PerfectPure RNA Tissue Kit (Fisher Scientific). Total RNA was read on a NanoDrop 2000 spectrophotometer (ThermoFisher Scientific, Wilmington, DE), and cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR amplification of the cDNA was performed by quantitative real-time PCR using TrueAmp SYBR Green qPCR SuperMix (Alkali Scientific). The thermocycling protocol consisted of 5 min at 95°C, 40 cycles of 15 s, at 95°C, and 30 s at 60°C and finished with a melting curve ranging from 60 to 95°C to allow distinction of specific products. Normalization was performed in separate reactions with primers to GAPDH mRNA (Table 1).
Table 1.
Gene names and primer sequences
| Name | Gene | Forward | Reverse |
|---|---|---|---|
| FAS | Fasn | GCTGCTGTTGGAAGTCAGC | AGTGTTCGTTCCTCGGAGTG |
| SREBP-1 | Srebf1 | CACCAGCATAGGCGAAGGA | AGTGTGCGGCCTGTGGAT |
| ACC | Acaca | GAATCTCCTGGTGACAATGCTTATT | GGTCTTGCTGAGTTGGGTTAGCT |
| PPARγ2 | Pparg | AAACTCTGGGAGATTCTCCTGTTG | GAAGTGCTCATAGGCAGTGCA |
| FGF21 | Fgf21 | CCTCTAGGTTTCTTTGCCAACAG | AAGCTGCAGGCCTCAGGAT |
| Cyp4A10 cytochrome P450 | Cyp4a10 | GACAAGGACCTACGTGCTGAGG | CTCATAGCAAATTGTTTCCCA |
| Cyp4A12 cytochrome P450 | Cyp4a12 | GAGTCCTATGAAAGAGTGCC | CTGGAAGCCCAGCAGAAGGTG |
| CYP4A14 cytochrome P450 | Cyp4a14 | CCCACAGGGACATGCAGATTAG | CACACAGAGCTCGGAAGACC |
FAS (Fasn), fatty acid synthase; SREBP-1 (Srebf1), sterol regulatory element-binding protein 1; ACC (Acaca), acetyl-coenzyme A carboxylase-α; PPARγ2 (Pparg), peroxisome proliferator-activated receptor-γ2; FGF21 (Fgf21), fibroblast grotwh factor 21; Cyp4A10 (Cyp4a10), cytochrome 450 4A10; Cyp4A12 (Cyp4a12), cytochrome 450 4A12; CYP4A14 (Cyp4a14), cytochrome 450 4A14.
Gel electrophoresis and Western blotting.
Western blotting was done as described previously (26, 51). Membranes were incubated overnight at 4°C with the following antibodies: PPARα (sc-9000; Santa Cruz Biotechnology, Santa Cruz, CA) or β-actin (ab6276; Abcam, Cambridge, MA). After three washes in TBS + 0.1% Tween-20, the membrane was incubated with an infrared anti-rabbit (IRDye 800, green) or anti-mouse (IRDye 680, red) secondary antibody labeled with IRDye infrared dye (LI-COR Biosciences) (1:10,000 dilution in TBS) for 2 h at 4°C. Immunoreactivity was visualized and quantified by infrared scanning in the Odyssey system (LI-COR Biosciences).
Hepatic immunostaining.
Immunofluorescence was performed on formalin-fixed, paraffin-embedded liver sections cut at 5-μm thickness. Sections were deparaffinized, hydrated, washed in PBS, and blocked in 5% normal donkey serum (NDS) for 1 h at 4°C. Sections were incubated with primary antibody in 5% NDS overnight at 4°C and washed in PBS. Primary antibodies include a mouse monoclonal PPARα antibody (cat. no. ab2779 1:400; Abcam) and a rabbit phospho-S73-PPARα antibody that was made as described previously (26). Antibody labeling was visualized using the Alex Fluor 488 and 594 secondary antibodies (Invitrogen, Carlsbad CA) incubated for 2 h at a room temperature at 1:1,000 dilution in 5% NDS. Following a final wash in PBS and DAPI counterstaining, samples were coverslipped with Fluoromount-G mounting media (Southern Biotech, Birmingham, AL). Samples were imaged using a Leica TCS SP5 Laser Scanning Confocal Microscope (Leica Microsystems, Buffalo Grove, IL). Images were acquired in the XYZ plane in 1-µm steps with a HCX PL APO CS 0.70 Dry ×20 objective in sequential scan mode to eliminate any spectral overlap in the individual fluorophores. Selected images are a two-dimensional projection of the image stack.
Statistical analysis.
Data were analyzed with Prism 6 (GraphPad Software, San Diego, CA) using analysis of variance combined with Tukey’s posttest to compare pairs of group means or unpaired t-tests. Results are expressed as means ± SE. Additionally, one-way ANOVA with a least significant difference post hoc test was used to compare mean values between multiple groups, and a two-tailed, two-way ANOVA was utilized in multiple comparisons, followed by the Bonferroni post hoc analysis to identify interactions. P ≤ 0.05 or smaller was considered statistically significant.
RESULTS
HuUGT*28 mice exhibit alterations in body composition, fat mass, and plasma bilirubin levels.
Weekly body weights were obtained over the entire 36-wk experimental protocol. HuUGT*28 mice exhibited significant differences in body weight early on (weeks 3–7) and late (weeks 29–33) in response to a high-fat diet but did not exhibit a significant difference in body weight over the majority of the time on a high-fat diet (Fig. 1A). HuUGT*28 mice exhibited a 100% increase in plasma total bilirubin levels and unconjugated bilirubin levels (Fig. 1B). Interestingly, conjugated bilirubin levels were not different between HuUGT*28 mice and control C57 mice. (Fig. 1B). The increase in total and unconjugated bilirubin levels was associated with decreased levels of epididymal, visceral, and total fat (Fig. 1C) and alterations in percent fat (Fig. 1D) and lean (Fig. 1E) mass in HuUGT*28 as compared with control C57 mice. Differences in body composition at early time points (6 and 12 wk) were also present between the groups (data not shown). These alterations in body composition were not due to any difference in food intake, activity; oxygen consumption, or heat production between HuUGT*28 and control mice (Fig. 2).
Fig. 1.

Body weight, body fat, and serum bilirubin levels in HuUGT*28 and control C57 mice fed a high-fat diet. A: weekly body weight over the study. B: plasma total, conjugated, and unconjugated bilirubin levels in 30-wk-old mice. C: fat mass as measured directly at the end of the experimental protocol. D: body fat as determined by noninvasive echoMRI. E: lean mass as determined by noninvasive echoMRI. *P < 0.05 vs. control C57 mice (means ± SE; n = 4–5/group).
Fig. 2.
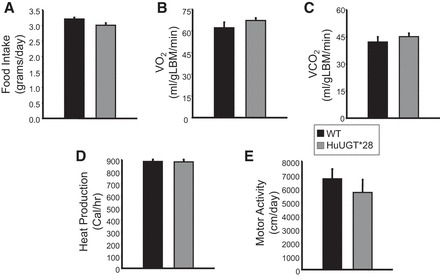
Food intake and indices of metabolism in HuUGT*28 and control C57 mice fed a high-fat diet. A: food intake. B: oxygen consumption (V̇o2) normalized to lean body mass. C: CO2 production (V̇co2) normalized to lean body mass. D: heat production. E: motor activity.
HuUGT*28 mice are protected against high-fat diet-induced hepatic steatosis.
Liver histology as determined by hematoxylin and eosin staining revealed decreased hepatic fat accumulation in the HuUGT*28 mice due to the reduced amount of white areas that were observed in the images (Fig. 3A). To determine whether this was directly related to liver function, we measured total cholesterol levels and alanine aminotransferase (ALT) activity, both of which were significantly decreased in HuUGT*28 compared with control C57 mice (Fig. 3B), indicating no hepatic dysfunction in the HuUGT*28 mice. The albumin (ALB) levels were not significantly different between the mouse groups (Fig. 3C). Liver function is reduced with the chronic accumulation of fatty acids and triglycerides (TGs). To determine the level of fat in the liver, we performed Oil Red O staining of liver sections that were obtained from each group at the end of the experimental protocol. The Oil Red O staining showed that HuUGT*28 mice had significantly (P < 0.05) less lipid accumulation in the liver (Fig. 3D). As a confirmation, we also measured the hepatic TG levels, and the HuUGT*28 mice had significantly (P < 0.05) fewer TGs accumulated in the liver (Fig. 3D). The progression of hepatic lipid accumulation is linked to de novo production of fatty acids by the Fasn gene that produces fatty acid synthase (FAS), the Acaca gene for acetyl-CoA carboxylase (ACC), the Srebf1 gene for sterol regulatory element-binding protein 1 (SREBP-1), and the Pparg gene for the nuclear receptor PPARγ (26, 27, 39, 57). The HuUGT*28 mice exhibited reduced expression of these genetic markers that regulate fatty acid production and accumulation in the liver (Fig. 3E). ACC drives catalyzation of the initial steps of lipid synthesis by carboxylation of acetyl-coenzyme A (acetyl-CoA) to malonyl-CoA. Also, a reduction in SREBP-1 mRNA levels, which is a transcription factor that enhances genes related to lipid and cholesterol production, may explain why the cholesterol levels were reduced in the HuUGT*28 mice. The overall production of long-chain fatty acids in the liver is maintained by FAS, and levels are directly related to the TG pool. Corresponding with lipid accumulation, FAS was significantly (P < 0.05) lower in the liver of HuUGT*28 mice compared with the C57 mice (Fig. 3E). PPARγ, which is a known player in the development of hepatic steatosis, was also reduced in the HuUGT*28 mice. Overall, these results indicate that the HuUGT*28 polymorphism may protect from liver dysfunction by a reduction in chronic lipid accumulation.
Fig. 3.

HuUGT*28 mice are protected against high-fat diet-induced hepatic steatosis. A: hematoxylin and eosin staining of liver sections (scale bar, 50 μm). B and C: biochemical measurements of hepatic total cholesterol and alanine aminotransferase (ALT; B) as well as albumin (ALB; C). D: Oil Red O staining of liver sections and densitometry (scale bar, 20 μm) and hepatic triglyceride levels. E: real-time PCR measurement of hepatic fatty acid synthase (FAS), sterol regulatory element-binding protein-1 (SREBP1), acetyl-CoA carboxylase (ACC), and peroxisome proliferator-activated receptor-γ2 (PPARγ2). *P < 0.05 vs. control C57 mice; (means ± SE; n = 4–5/group).
HuUGT*28 mice exhibit increased hepatic PPARα and increased expression of its target genes.
The levels of PPARα protein in the liver were significantly increased in HuUGT*28 compared with control C57 mice (Fig. 4A). Next, we compared the levels of PPARα phosphorylation of serine 73 [Ser(P)73] by immunostaining of liver sections with our newly developed Ser(P)73 PPARα antibody (described in Ref. 26). In agreement with our Western blot data, HuUGT*28 mice exhibited markedly increased levels of total PPARα and, more importantly, significantly decreased levels of phosphorylation of Ser(P)73 PPARα (Fig. 4B). PPARα is a transcription factor that regulates hundreds of genes, and in the liver, major targets include genes in the fat-burning peroxisomal and microsomal oxidation pathway, which include the Fgf21 gene for the fibroblast growth factor 21 (FGF21) and the cytochrome P450 4A (Cyp4A genes) enzymes (2, 47, 48). Recently, we have shown that increasing Ser(P)73 of PPARα increases ubiquitination and reduces protein levels and transcriptional activity (26). To determine whether PPARα levels were associated with increased expression of these known target genes, the expression levels of FGF21 and the Cyp4A isoforms were measured in the livers of HuUGT*28 and control C57 mice. Indeed, expression of FGF21, Cyp4A10, Cyp4A12, and Cyp4A14 were all significantly (P < 0.05) increased in HuUGT*28 compared with control C57 mice (Fig. 4C). Together, these results show that increasing plasma bilirubin levels reduces Ser(P)73 of PPARα, increasing protein levels, and transcriptional activity.
Fig. 4.
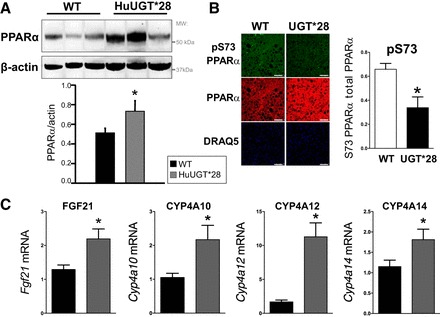
PPARα levels and target gene expression are increased in HuUGT*28 mice. A: Western blot and densitometry of hepatic protein levels of PPARα. B: immunostaining of total and serine 73 phosphorylation PPARα (pS73) in the liver. C: real-time PCR measurement of hepatic expression of fibroblast growth factor 21 (FGF21), cytochrome 450 4A10 (CYP4A10), cytochrome 450 4A12 (CYP4A12), and cytochrome 450 4A14 (CYP4A14). *P < 0.05 vs. control C57 mice (means ± SE; n = 4–5/group). WT, wild type; DRAQ5, deep red anthraquinone 5.
HuUGT*28 mice are resistant to the metabolic effects of a high-fat diet.
Fasting blood glucose and insulin levels were obtained from each of the groups of mice at 6-wk intervals starting at 18 wk on the high-fat diet. Blood glucose levels in HuUGT*28 mice were significantly lower than the corresponding levels in control C57 mice at all time points analyzed (Fig. 5A). In fact, fasting blood glucose levels in the HuUGT*28 mice remained close to normal levels (~100 mg/dl) through the entire length of the study (Fig. 5A). Differences in fasting blood glucose levels between the groups were also evident at earlier time points in the study (data not shown). In a similar fashion, fasting insulin levels were also significantly lower in the HuUGT*28 mice compared with the control C57 mice at all time points analyzed (Fig. 5B). Finally, HuUGT*28 mice exhibited significantly enhanced glucose tolerance compared with control C57 mice on a high-fat diet (Fig. 5C).
Fig. 5.

HuUGT*28 mice are protected against high-fat diet-induced hyperglycemia, hyperinsulinemia, and insulin resistance. A: fasting blood glucose measured at 6-wk intervals starting from week 18 on high-fat diet. B: fasting insulin measured at 6-wk intervals starting from week 18 on high-fat diet. C: intraperitoneal glucose tolerance test (GTT) measured in week 28 of the high-fat diet [area under the curve (AUC)]. *P < 0.05 vs. control C57 mice (means ± SE; n = 4–5/group).
DISCUSSION
Obesity is an emerging worldwide epidemic, with rates of obesity increasing by more than 25% in adults and more than 45% in children in the last 25 yr. Despite these alarming global increases in obesity rates, the development of novel treatments for obesity is lacking. Several population studies have demonstrated a negative correlation between plasma bilirubin levels and components of the metabolic syndrome in both obese and overweight patient populations (12, 30, 56, 58). Gilbert’s syndrome is a genetic condition resulting in moderate hyperbilirubinemia. Although individuals with Gilbert’s polymorphisms have previously been reported to be protected from cardiovascular disease, the effect of this mutation on the development of obesity and high-fat diet-induced hepatic insulin resistance and steatosis is not known (36). Recent studies have shown that bilirubin treatments in mice reduced insulin and glucose levels (37, 60). Here, we show that mice expressing the human Gilbert’s HuUGT*28 polymorphism are protected from high-fat diet-induced hepatic steatosis and insulin resistance due to an elevation of bilirubin and PPARα activity. The HuUGT*28 mice exhibit alterations in body composition showing less body fat and leaner mass compared with control mice on a high-fat diet despite no significant changes in food intake, oxygen consumption, or activity. Whereas the levels of total bilirubin and unconjugated bilirubin were clearly elevated in the HuUGT*28 mice compared with the control C57 mice, the plasma levels of bilirubin in the C57 mice utilized in this study are on the high side of normal that has been reported previously for this strain (37). The bilirubin levels in the present study need to be interpreted with consideration until additional measurements of plasma bilirubin by more sensitive methods such as HPLC/LCMS are reported in these strains to ensure accuracy in values. Regardless, the HuUGT*28 polymorphism clearly demonstrates a novel mouse model for investigating moderately elevated bilirubin levels and the relation to obesity and hepatic lipid accumulation.
NAFLD is a complication of obesity and one of the leading causes of liver injury (40). Hepatic lipid accumulation, although benign, can set the stage for the development of a more serious condition like nonalcoholic steatohepatitis (NASH) if left untreated or in response to a second “hit” to the liver, such as increased oxidative stress or inflammation (1, 7). Hepatic lipid accumulation was significantly attenuated in the moderately hyperbilirubinemic HuUGT*28 mice compared with the control C57 mice. Fatty acid production in the liver is maintained by FAS, which generates de novo synthesis of lipids that can act as intracellular signaling molecules that are capable of regulating genes involved in fat storage, such as ACC and SREBP-1. ACC is the rate-liming enzyme that catalyzes lipids for synthesis involving the carboxylation of acetyl-CoA to malonyl-CoA, which is one of the initial steps in the storage of fat and intrahepatic production of cholesterol. The suppression of the fatty acid synthesis genes in the HuUGT*28 mice is most likely the positive action of bilirubin on PPARα and suppression of PPARγ, which caused a decrease in hepatic steatosis. Importantly, the HuUGT*28 mice had reduced Ser(P)73 of PPARα, which increases protein levels and transcriptional activity (26). Our finding of reduced hepatic steatosis in the moderately hyperbilirubinemic HuUGT*28 mice is in agreement with similar studies in human patient populations that have demonstrated a negative relationship between plasma bilirubin levels and the incidence of NAFLD in both men and women (33) as well as higher levels of S12 phosphorylation of PPARα in patients with known Gilbert’s syndrome (41), which increases transcriptional activity (3). The HuUGT*28 mice exhibited significant decreases in liver triglyceride levels compared with control C57 mice in response to a chronic high-fat diet. Furthermore, the HuUGT*28 mice exhibited significantly lower plasma cholesterol levels after 30 wk on a high-fat diet. The decrease in plasma cholesterol in the HuUGT*28 mice is similar to the effect observed in individuals with Gilbert’s polymorphism (4, 5, 56). The effect of lower plasma cholesterol in the HuUGT*28 mice may also contribute to the increased insulin sensitivity, which was also observed in dietary-induced obese mice with the treatment of bilirubin (37).
Bilirubin is a well-recognized antioxidant as well as an anti-inflammatory molecule. More importantly, we have recently shown a novel action of bilirubin as a nuclear hormone receptor agonist that activates PPARα by binding directly to the ligand-binding domain of the protein (51). PPARα enhances genes in the liver for the burning of fat through the peroxisomal and microsomal oxidation pathways that include FGF21 and the Cyp4A cytochrome P450 enzymes (2, 47, 48). PPARα levels were increased in the liver of HuUGT*28 mice along with the Cyp4A enzymes and FGF21, which suggest that the PPARα transcriptional activity was significantly increased. The ability of increased levels of FGF21 to reverse hepatic steatosis has been reported in several studies (17, 27, 34). Although we did observe increased levels of hepatic FGF21 in HuUGT*28 mice, these mice did not display an increase in metabolism or change in body weight typically associated with activation of FGF21. Furthermore, Mölzer et al. (41) showed that FGF21 levels were not significantly changed in humans with Gilbert’s [they reported that FGF21 levels for Gilbert’s was 0.35 (n = 52) and 0.10 μg/ml for control (n = 51), P = 0.086)]. Also, Molzer et al. (41) demonstrated that serine 112 of PPARγ was hyperphosphorylated in humans with Gilbert’s syndrome, which inhibits transcriptional activity and reduces adipogenesis (28). Bilirubin most likely reduces lipid accumulation by PPARα activation and a reduction in PPARγ transcriptional activity. This is supported by the treatment of wild-type C57 mice with bilirubin for 1 wk and reduction in fat mass and increased lean mass (51). Similarly, the HuUGT*28 mice displayed a significant decrease in fat mass and an increase in lean mass. Also in support of this concept is that bilirubin treatment in PPARα-knockout mice had no effect on fat or lean mass (51).
Humans that have the TA repeat polymorphism for Gilbert’s syndrome do have reduced body weights and increased phosphorylation of AMPK (p-AMPK), which is an energy and metabolism biomarker (41). We have shown that elevating bilirubin levels via the heme oxygenase pathway increased p-AMPK as well as PPARα expression and transcriptional activity, which reduced body weight and hepatic steatosis (27). We also showed that BVRA-knockout mice on a high-fat diet have the manifestation of hepatic steatosis and a reduction of PPARα in liver (25, 26). Liu et al. (37) did find that bilirubin treatment in diet-induced obese (DIO) mice reduced body weight and liver fat. Furthermore, they found that PPARγ expression was reduced in the liver of DIO mice, as has been reported previously, but increased with bilirubin treatments in the DIO mice. However, the bilirubin levels were significantly higher in the DIO mice but were not increased with bilirubin treatment. PPARγ has been shown to induce lipid accumulation in hepatocyte cell lines (49) and livers of mice (59). Under steatotic conditions within the liver, PPARγ is upregulated (18) and PPARα suppressed (24, 27, 32, 38, 42). Importantly, hepatic PPARγ expression was reduced in the HuUGT*28 mice, which we have shown previously with bilirubin treatments and a reduction in lipid accumulation in 3T3-L1 adipocytes (51). Long-term bilirubin exposure in Gilbert’s polymorphism may cause a reduction in hepatic lipid accumulation by suppression of PPARγ and activation of PPARα and p-AMPK.
HuUGT*28 mice exhibited a remarkable ability to preserve hepatic insulin sensitivity in response to high-fat diet feeding. Fasting blood glucose levels of the HuUGT*28 mice remained in the normal range at all time points measured, and fasting insulin levels remained constant throughout the entire study. There are several potential mechanisms by which moderate hyperbilirubinemia observed in the HuUGT*28 mice could mediate this phenotype. Studies in type 1 diabetic NOD mice treated with an inducer of heme oxygenase-1 (HO-1) (one of whose end products is bilirubin), as well as in severely hyperbilirubinemic Gunn rats, have shown that bilirubin can protect islet cells of the pancreas through an antioxidant-dependent mechanism (19, 35). It is possible that similar antioxidant properties of bilirubin play a role in the maintenance of hepatic insulin sensitivity in response to chronic high-fat diet feeding. Bilirubin has also been demonstrated to improve insulin sensitivity in obese leptin receptor-deficient db/db mice through attenuation of endoplasmatic reticulum (ER) stress (16). Finally, bilirubin is a ligand for nuclear hormone receptor PPARα and may improve insulin sensitivity through activation of hepatic PPARα activity, which was lost in PPARα-knockout mice (51). The Gilbert’s polymorphism in the UGT1A1 enzyme causes an increase in plasma bilirubin levels, which in turn activates PPARα to reduce lipid accumulation and lower cholesterol levels. Targeting of UGT1A1 may prove to be successful therapy for the obese and in hepatic steatosis.
PERSPECTIVES
The results of several human population studies have clearly established a relationship between plasma bilirubin levels and the protection from cardiovascular disease, diabetes, diabetic nephropathy, and hepatic steatosis. Patients with Gilbert’s syndrome have also been reported to be protected from the development of cardiovascular disease as well. Although the protective aspects of moderate hyperbilirubinemia have been established in population studies and studies from individuals with Gilbert’s polymorphisms, the mechanisms by which increased plasma levels of bilirubin can preserve liver insulin sensitivity and protect against hepatic steatosis are unclear. Recent evidence has demonstrated that bilirubin is a positive modulator of PPARα and suppressor of PPARγ activity, which contributes to the alteration in body composition in the HuUGT*28 mice as well as in mice with the loss of hepatic BVRA (25, 26). However, the specific genes regulated by bilirubin interaction with PPARα are not known and will need to be addressed in future studies (25). The HuUGT*28 mouse model is an ideal candidate to elucidate the relative contributions of each of these pathways to the anti-diabetic and anti-steatotic actions of moderate hyperbilirubinemia. Methods of increasing plasma bilirubin (e.g., inhibition of UGT1A1) or exogenous delivery of bilirubin may serve as a potential treatment for hepatic steatosis and NAFLD before this condition progresses to a more severe state of NASH.
GRANTS
This work was supported by the University of Toledo deArce-Memorial Endowment Fund (T. D. Hinds). Research reported in this publication was also supported, in whole or in part, by the National Institutes of Health (L32-MD-009154 to T. D. Hinds), the National Heart, Lung, and Blood Institute (K01-HL-125445 to T. D. Hinds and PO1-HL-051971 and HL-088421 to (D. E. Stec), and the National Institute of General Medical Sciences (P20-GM-104357 to D. E. Stec). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.D.H., P.A.H., and D.E.S. conceived and designed research; T.D.H., P.A.H., M.W.H., A.N.-K., and D.E.S. performed experiments; T.D.H., P.A.H., M.W.H., A.N.-K., and D.E.S. analyzed data; T.D.H., P.A.H., A.N.-K., and D.E.S. interpreted results of experiments; T.D.H. and D.E.S. prepared figures; T.D.H. and D.E.S. drafted manuscript; T.D.H., P.A.H., M.W.H., A.N.-K., and D.E.S. edited and revised manuscript; T.D.H., P.A.H., M.W.H., A.N.-K., and D.E.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We gratefully acknowledge the Analytical and Assay Core Laboratory in the Department of Physiology and Biophysics at the University of Mississippi Medical Center.
REFERENCES
- 1.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 10: 330–344, 2013. doi: 10.1038/nrgastro.2013.41. [DOI] [PubMed] [Google Scholar]
- 2.Barclay TB, Peters JM, Sewer MB, Ferrari L, Gonzalez FJ, Morgan ET. Modulation of cytochrome P-450 gene expression in endotoxemic mice is tissue specific and peroxisome proliferator-activated receptor-alpha dependent. J Pharmacol Exp Ther 290: 1250–1257, 1999. [PubMed] [Google Scholar]
- 3.Barger PM, Browning AC, Garner AN, Kelly DP. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: a potential role in the cardiac metabolic stress response. J Biol Chem 276: 44495–44501, 2001. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 4.Boon AC, Hawkins CL, Bisht K, Coombes JS, Bakrania B, Wagner KH, Bulmer AC. Reduced circulating oxidized LDL is associated with hypocholesterolemia and enhanced thiol status in Gilbert syndrome. Free Radic Biol Med 52: 2120–2127, 2012. doi: 10.1016/j.freeradbiomed.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bulmer AC, Verkade HJ, Wagner KH. Bilirubin and beyond: a review of lipid status in Gilbert’s syndrome and its relevance to cardiovascular disease protection. Prog Lipid Res 52: 193–205, 2013. doi: 10.1016/j.plipres.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Cai H, Nguyen N, Peterkin V, Yang YS, Hotz K, La Placa DB, Chen S, Tukey RH, Stevens JC. A humanized UGT1 mouse model expressing the UGT1A1*28 allele for assessing drug clearance by UGT1A1-dependent glucuronidation. Drug Metab Dispos 38: 879–886, 2010. doi: 10.1124/dmd.109.030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caldwell S. NASH (Nonalcoholic steatohepatitis): A case of multiorganelle failure. Free Radic Biol Med 75, Suppl 1: S6, 2014. doi: 10.1016/j.freeradbiomed.2014.10.839. [DOI] [PubMed] [Google Scholar]
- 8.Chen G, Ramos E, Adeyemo A, Shriner D, Zhou J, Doumatey AP, Huang H, Erdos MR, Gerry NP, Herbert A, Bentley AR, Xu H, Charles BA, Christman MF, Rotimi CN. UGT1A1 is a major locus influencing bilirubin levels in African Americans. Eur J Hum Genet 20: 463–468, 2012. doi: 10.1038/ejhg.2011.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheriyath P, Gorrepati VS, Peters I, Nookala V, Murphy ME, Srouji N, Fischman D. High Total Bilirubin as a Protective Factor for Diabetes Mellitus: An Analysis of NHANES Data From 1999 – 2006. J Clin Med Res 2: 201–206, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chin HJ, Cho HJ, Lee TW, Na KY, Oh KH, Joo KW, Yoon HJ, Kim YS, Ahn C, Han JS, Kim S, Jeon ES, Jin DC, Kim YL, Park SH, Kim CD, Song YR, Kim SG, Kim YG, Lee JE, Oh YK, Lim CS, Lee SK, Chae DW, Cho WY, Kim HK, Jo SK; Progressive REnal disease and Medical Informatics and gEnomics Research (PREMIER) members . The mildly elevated serum bilirubin level is negatively associated with the incidence of end stage renal disease in patients with IgA nephropathy. J Korean Med Sci 24, Suppl 1: S22–S29, 2009. doi: 10.3346/jkms.2009.24.S1.S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin HJ, Song YR, Kim HS, Park M, Yoon HJ, Na KY, Kim Y, Chae DW, Kim S. The bilirubin level is negatively correlated with the incidence of hypertension in normotensive Korean population. J Korean Med Sci 24, Suppl 1: S50–S56, 2009. doi: 10.3346/jkms.2009.24.S1.S50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi SH, Yun KE, Choi HJ. Relationships between serum total bilirubin levels and metabolic syndrome in Korean adults. Nutr Metab Cardiovasc Dis 23: 31–37, 2013. doi: 10.1016/j.numecd.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Clarke DJ, Moghrabi N, Monaghan G, Cassidy A, Boxer M, Hume R, Burchell B. Genetic defects of the UDP-glucuronosyltransferase-1 (UGT1) gene that cause familial non-haemolytic unconjugated hyperbilirubinaemias. Clin Chim Acta 266: 63–74, 1997. doi: 10.1016/S0009-8981(97)00167-8. [DOI] [PubMed] [Google Scholar]
- 14.Csongradi E, Docarmo JM, Dubinion JH, Vera T, Stec DE. Chronic HO-1 induction with cobalt protoporphyrin (CoPP) treatment increases oxygen consumption, activity, heat production and lowers body weight in obese melanocortin-4 receptor-deficient mice. Int J Obes 36: 244–253, 2012. doi: 10.1038/ijo.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology 114: 842–845, 1998. doi: 10.1016/S0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 16.Dong H, Huang H, Yun X, Kim DS, Yue Y, Wu H, Sutter A, Chavin KD, Otterbein LE, Adams DB, Kim YB, Wang H. Bilirubin increases insulin sensitivity in leptin-receptor deficient and diet-induced obese mice through suppression of ER stress and chronic inflammation. Endocrinology 155: 818–828, 2014. doi: 10.1210/en.2013-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, Badman MK, Martinez-Chantar ML, Maratos-Flier E. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology 139: 456–463, 2010. doi: 10.1053/j.gastro.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Escalona-Nandez I, Guerrero-Escalera D, Estanes-Hernández A, Ortíz-Ortega V, Tovar AR, Pérez-Monter C. The activation of peroxisome proliferator-activated receptor γ is regulated by Krüppel-like transcription factors 6 & 9 under steatotic conditions. Biochem Biophys Res Commun 458: 751–756, 2015. doi: 10.1016/j.bbrc.2015.01.145. [DOI] [PubMed] [Google Scholar]
- 19.Fu YY, Kang KJ, Ahn JM, Kim HR, Na KY, Chae DW, Kim S, Chin HJ. Hyperbilirubinemia reduces the streptozotocin-induced pancreatic damage through attenuating the oxidative stress in the Gunn rat. Tohoku J Exp Med 222: 265–273, 2010. doi: 10.1620/tjem.222.265. [DOI] [PubMed] [Google Scholar]
- 20.Fujiwara R, Nguyen N, Chen S, Tukey RH. Developmental hyperbilirubinemia and CNS toxicity in mice humanized with the UDP glucuronosyltransferase 1 (UGT1) locus. Proc Natl Acad Sci USA 107: 5024–5029, 2010. doi: 10.1073/pnas.0913290107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghem C, Sarmento-Leite RE, de Quadros AS, Rossetto S, Gottschall CA. Serum bilirubin concentration in patients with an established coronary artery disease. Int Heart J 51: 86–91, 2010. doi: 10.1536/ihj.51.86. [DOI] [PubMed] [Google Scholar]
- 22.Gil J, Sąsiadek MM. Gilbert syndrome: the UGT1A1*28 promoter polymorphism as a biomarker of multifactorial diseases and drug metabolism. Biomarkers Med 6: 223–230, 2012. doi: 10.2217/bmm.12.4. [DOI] [PubMed] [Google Scholar]
- 23.Giral P, Ratziu V, Couvert P, Carrié A, Kontush A, Girerd X, Chapman MJ. Plasma bilirubin and gamma-glutamyltransferase activity are inversely related in dyslipidemic patients with metabolic syndrome: relevance to oxidative stress. Atherosclerosis 210: 607–613, 2010. doi: 10.1016/j.atherosclerosis.2009.12.026. [DOI] [PubMed] [Google Scholar]
- 24.Goto T, Lee JY, Teraminami A, Kim YI, Hirai S, Uemura T, Inoue H, Takahashi N, Kawada T. Activation of peroxisome proliferator-activated receptor-alpha stimulates both differentiation and fatty acid oxidation in adipocytes. J Lipid Res 52: 873–884, 2011. doi: 10.1194/jlr.M011320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinds TD Jr, Adeosun SO, Alamodi AA, Stec DE. Does bilirubin prevent hepatic steatosis through activation of the PPARα nuclear receptor? Med Hypotheses 95: 54–57, 2016. doi: 10.1016/j.mehy.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinds TD Jr, Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, AlAmodi AA, Hankins MW, Vanden Heuvel JP, Stec DE. Biliverdin Reductase A Attenuates Hepatic Steatosis by Inhibition of Glycogen Synthase Kinase (GSK) 3β Phosphorylation of Serine 73 of Peroxisome Proliferator-activated Receptor (PPAR) α. J Biol Chem 291: 25179–25191, 2016. doi: 10.1074/jbc.M116.731703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinds TD Jr, Sodhi K, Meadows C, Fedorova L, Puri N, Kim DH, Peterson SJ, Shapiro J, Abraham NG, Kappas A. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity (Silver Spring) 22: 705–712, 2014. doi: 10.1002/oby.20559. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Hinds TD Jr, Stechschulte LA, Cash HA, Whisler D, Banerjee A, Yong W, Khuder SS, Kaw MK, Shou W, Najjar SM, Sanchez ER. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-γ (PPARγ). J Biol Chem 286: 42911–42922, 2011. doi: 10.1074/jbc.M111.311662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hosick PA, AlAmodi AA, Storm MV, Gousset MU, Pruett BE, Gray W III, Stout J, Stec DE. Chronic carbon monoxide treatment attenuates development of obesity and remodels adipocytes in mice fed a high-fat diet. Int J Obes 38: 132–139, 2014. doi: 10.1038/ijo.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenko-Pražnikar Z, Petelin A, Jurdana M, Žiberna L. Serum bilirubin levels are lower in overweight asymptomatic middle-aged adults: an early indicator of metabolic syndrome? Metabolism 62: 976–985, 2013. doi: 10.1016/j.metabol.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 31.John K, Marino JS, Sanchez ER, Hinds TD Jr. The glucocorticoid receptor: cause of or cure for obesity? Am J Physiol Endocrinol Metab 310: E249–E257, 2016. doi: 10.1152/ajpendo.00478.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komatsu M, Kimura T, Yazaki M, Tanaka N, Yang Y, Nakajima T, Horiuchi A, Fang ZZ, Joshita S, Matsumoto A, Umemura T, Tanaka E, Gonzalez FJ, Ikeda S, Aoyama T. Steatogenesis in adult-onset type II citrullinemia is associated with down-regulation of PPARα. Biochim Biophys Acta 1852: 473–481, 2015. doi: 10.1016/j.bbadis.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwak MS, Kim D, Chung GE, Kang SJ, Park MJ, Kim YJ, Yoon JH, Lee HS. Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol 18: 383–390, 2012. doi: 10.3350/cmh.2012.18.4.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li L, Chao QG, Ping LZ, Xue C, Xia ZY, Qian D, Shi-ang H. The prevalence of FOXP3+ regulatory T-cells in peripheral blood of patients with NSCLC. Cancer Biother Radiopharm 24: 357–367, 2009. doi: 10.1089/cbr.2008.0612. [DOI] [PubMed] [Google Scholar]
- 35.Li M, Peterson S, Husney D, Inaba M, Guo K, Kappas A, Ikehara S, Abraham NG. Long-lasting expression of HO-1 delays progression of type I diabetes in NOD mice. Cell Cycle 6: 567–571, 2007. doi: 10.4161/cc.6.5.3917. [DOI] [PubMed] [Google Scholar]
- 36.Lin JP, O’Donnell CJ, Schwaiger JP, Cupples LA, Lingenhel A, Hunt SC, Yang S, Kronenberg F. Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham Heart Study. Circulation 114: 1476–1481, 2006. doi: 10.1161/CIRCULATIONAHA.106.633206. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Dong H, Zhang Y, Cao M, Song L, Pan Q, Bulmer A, Adams DB, Dong X, Wang H. Bilirubin Increases Insulin Sensitivity by Regulating Cholesterol Metabolism, Adipokines and PPARγ Levels. Sci Rep 5: 9886, 2015. doi: 10.1038/srep09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu Y, Liu X, Jiao Y, Xiong X, Wang E, Wang X, Zhang Z, Zhang H, Pan L, Guan Y, Cai D, Ning G, Li X. Periostin promotes liver steatosis and hypertriglyceridemia through downregulation of PPARα. J Clin Invest 124: 3501–3513, 2014. doi: 10.1172/JCI74438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marino JS, Stechschulte LA, Stec DE, Nestor-Kalinoski A, Coleman S, Hinds TD Jr. Glucocorticoid Receptor β Induces Hepatic Steatosis by Augmenting Inflammation and Inhibition of the Peroxisome Proliferator-activated Receptor (PPAR) α. J Biol Chem 291: 25776–25788, 2016. doi: 10.1074/jbc.M116.752311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masuoka HC, Chalasani N. Nonalcoholic fatty liver disease: an emerging threat to obese and diabetic individuals. Ann N Y Acad Sci 1281: 106–122, 2013. doi: 10.1111/nyas.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mölzer C, Wallner M, Kern C, Tosevska A, Schwarz U, Zadnikar R, Doberer D, Marculescu R, Wagner KH. Features of an altered AMPK metabolic pathway in Gilbert’s Syndrome, and its role in metabolic health. Sci Rep 6: 30051, 2016. doi: 10.1038/srep30051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montagner A, Polizzi A, Fouché E, Ducheix S, Lippi Y, Lasserre F, Barquissau V, Régnier M, Lukowicz C, Benhamed F, Iroz A, Bertrand-Michel J, Al Saati T, Cano P, Mselli-Lakhal L, Mithieux G, Rajas F, Lagarrigue S, Pineau T, Loiseau N, Postic C, Langin D, Wahli W, Guillou H. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 65: 1202–1214, 2016. doi: 10.1136/gutjnl-2015-310798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Novotný L, Vítek L. Inverse relationship between serum bilirubin and atherosclerosis in men: a meta-analysis of published studies. Exp Biol Med (Maywood) 228: 568–571, 2003. doi: 10.1177/15353702-0322805-29. [DOI] [PubMed] [Google Scholar]
- 44.O’Brien L, Hosick PA, John K, Stec DE, Hinds TD Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol Metab 26: 212–220, 2015. doi: 10.1016/j.tem.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perlstein TS, Pande RL, Creager MA, Weuve J, Beckman JA. Serum total bilirubin level, prevalent stroke, and stroke outcomes: NHANES 1999-2004. Am J Med 121: 781–788.e1, 2008. doi: 10.1016/j.amjmed.2008.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puri K, Nobili V, Melville K, Corte CD, Sartorelli MR, Lopez R, Feldstein AE, Alkhouri N. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 57: 114–118, 2013. doi: 10.1097/MPG.0b013e318291fefe. [DOI] [PubMed] [Google Scholar]
- 47.Rakhshandehroo M, Knoch B, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res 2010: pii: 612089, 2010. doi: 10.1155/2010/612089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rakhshandehroo M, Sanderson LM, Matilainen M, Stienstra R, Carlberg C, de Groot PJ, Müller M, Kersten S. Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res 2007: 26839, 2007. doi: 10.1155/2007/26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. PPARγ2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am J Physiol Endocrinol Metab 288: E1195–E1205, 2005. doi: 10.1152/ajpendo.00513.2004. [DOI] [PubMed] [Google Scholar]
- 50.Schwertner HA, Vítek L. Gilbert syndrome, UGT1A1*28 allele, and cardiovascular disease risk: possible protective effects and therapeutic applications of bilirubin. Atherosclerosis 198: 1–11, 2008. doi: 10.1016/j.atherosclerosis.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 51.Stec DE, John K, Trabbic CJ, Luniwal A, Hankins MW, Baum J, Hinds TD Jr. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PLoS One 11: e0153427, 2016. doi: 10.1371/journal.pone.0153427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sundararaghavan VL, Sindhwani P, Hinds TD Jr. Glucuronidation and UGT isozymes in bladder: new targets for the treatment of uroepithelial carcinomas? Oncotarget, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 52: 1836–1846, 2010. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 54.Vítek L. Impact of serum bilirubin on human diseases. Pediatrics 115: 1411–1412, 2005. doi: 10.1542/peds.2004-1796. [DOI] [PubMed] [Google Scholar]
- 55.Wagner KH, Wallner M, Mölzer C, Gazzin S, Bulmer AC, Tiribelli C, Vitek L. Looking to the horizon: the role of bilirubin in the development and prevention of age-related chronic diseases. Clin Sci (Lond) 129: 1–25, 2015. doi: 10.1042/CS20140566. [DOI] [PubMed] [Google Scholar]
- 56.Wallner M, Marculescu R, Doberer D, Wolzt M, Wagner O, Vitek L, Bulmer AC, Wagner KH. Protection from age-related increase in lipid biomarkers and inflammation contributes to cardiovascular protection in Gilbert’s syndrome. Clin Sci (Lond) 125: 257–264, 2013. doi: 10.1042/CS20120661. [DOI] [PubMed] [Google Scholar]
- 57.Warrier M, Hinds TD Jr, Ledford KJ, Cash HA, Patel PR, Bowman TA, Stechschulte LA, Yong W, Shou W, Najjar SM, Sanchez ER. Susceptibility to diet-induced hepatic steatosis and glucocorticoid resistance in FK506-binding protein 52-deficient mice. Endocrinology 151: 3225–3236, 2010. doi: 10.1210/en.2009-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu Y, Li M, Xu M, Bi Y, Li X, Chen Y, Ning G, Wang W. Low serum total bilirubin concentrations are associated with increased prevalence of metabolic syndrome in Chinese. J Diabetes 3: 217–224, 2011. doi: 10.1111/j.1753-0407.2011.00138.x. [DOI] [PubMed] [Google Scholar]
- 59.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor γ1 (PPARγ1) overexpression. J Biol Chem 278: 498–505, 2003. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 60.Zelenka J, Dvořák A, Alán L, Zadinová M, Haluzík M, Vítek L. Hyperbilirubinemia Protects against Aging-Associated Inflammation and Metabolic Deterioration. Oxid Med Cell Longev 2016: 6190609, 2016. doi: 10.1155/2016/6190609. [DOI] [PMC free article] [PubMed] [Google Scholar]