

Abstract
Chronic low-grade inflammation and cellular stress are important contributors to obesity-linked metabolic dysfunction. Here, we uncover an immune-metabolic role for C1q/TNF-related protein 7 (CTRP7), a secretory protein of the C1q family with previously unknown function. In obese humans, circulating CTRP7 levels were markedly elevated and positively correlated with body mass index, glucose, insulin, insulin resistance index, hemoglobin A1c, and triglyceride levels. Expression of CTRP7 in liver was also significantly upregulated in obese humans and positively correlated with gluconeogenic genes. In mice, Ctrp7 expression was differentially modulated in various tissues by fasting and refeeding and by diet-induced obesity. A genetic loss-of-function mouse model was used to determine the requirement of CTRP7 for metabolic homeostasis. When fed a control low-fat diet, male or female mice lacking CTRP7 were indistinguishable from wild-type littermates. In obese male mice consuming a high-fat diet, however, CTRP7 deficiency attenuated insulin resistance and enhanced glucose tolerance, effects that were independent of body weight, metabolic rate, and physical activity level. Improved glucose metabolism in CTRP7-deficient mice was associated with reduced adipose tissue inflammation, as well as decreased liver fibrosis and cellular oxidative and endoplasmic reticulum stress. These results provide a link between elevated CTRP7 levels and impaired glucose metabolism, frequently associated with obesity. Inhibiting CTRP7 action may confer beneficial metabolic outcomes in the setting of obesity and diabetes.
Keywords: adipokine, obesity, diabetes, insulin resistance, inflammation, oxidative and endoplasmic reticulum stress
circulating levels of glucose and lipids are maintained within a physiological range by the combined action of multiple tissues and organs (35). Tissue cross talk, largely mediated by secreted polypeptide hormones, is critical for integrated control of energy metabolism (34). To understand hormonal control of metabolism, we have previously characterized a highly conserved and novel family of secreted plasma proteins of the C1q family, C1q/TNF-related proteins (CTRP1–15) (33, 41–43, 45–47). Using genetic gain- and loss-of-function mouse models and recombinant protein infusion approaches, we have demonstrated important roles for multiple CTRP family members in regulating peripheral tissue insulin sensitivity, as well as glucose and lipid metabolism and food intake (3, 4, 25–29, 32, 33, 39, 41, 45, 46). In vitro studies also suggest a role for CTRPs in adipocyte differentiation (43) and cell signaling in adipocytes and hepatocytes under basal conditions (40) and in the context of lipid overload (42).
The biological function of CTRP7, however, has remained elusive since its initial identification (47). We have previously shown that Ctrp7 mRNA is increased in adipose tissue of leptin-deficient ob/ob mice at 8 wk (46), an age when ob/ob animals develop hyperglycemia (6). By 12 wk of age, ob/ob mice have substantially reduced hyperglycemia due to a compensatory increase in pancreatic insulin secretion (6). Concomitant with improved blood glucose levels, Ctrp7 expression in ob/ob mice reverts back to levels of lean controls (46). Other studies have also reported upregulated expression of CTRP7 in skeletal muscle of old rats and, conversely, reduced expression in caloric-restricted animals (30). Intriguingly, a recent genome-wide association study found multiple single nucleotide polymorphisms in human CTRP7 that associate with childhood conduct disorder (8), although whether they are causally related remains unknown. Beyond the studies highlighted, however, little is known about CTRP7. Therefore, this study aimed to uncover the physiological role of this conserved secretory protein using a genetic loss-of-function mouse model. The relevance of CTRP7 to human metabolic disorders was further examined using samples obtained from lean controls and obese patients undergoing bariatric surgery.
MATERIALS AND METHODS
Human study design and participants.
A cross-sectional study was conducted from October 2013 to March 2015 at the Johns Hopkins Medical Institutions (Baltimore, MD). Obese patients (BMI >40 kg/m2, or >35 kg/m2 with one or more obesity-associated comorbidities) undergoing bariatric surgery (Roux-en-Y gastric bypass or vertical sleeve gastrectomy) were recruited from the Johns Hopkins Center for Bariatric Surgery. Patients with previous weight loss surgery were excluded. Lean controls (BMI <26 kg/m2) with no history of diabetes or cardiovascular disease were recruited from the Johns Hopkins Hospital. A total of 37 age-, gender-, and ethnicity-matched pairs were included in this study. Participants were verbally briefed about the study and signed written informed consent forms. All human studies were approved by the Johns Hopkins University School of Medicine Institutional Review Board.
Clinical and laboratory measurements.
Body mass index (BMI) was calculated as weight/height2 (kg/m2). Human blood samples were obtained in the morning following an overnight fast. Blood samples were centrifuged at 3,200 g for 7 min, and serum was aliquoted and stored at −80°C for subsequent assays. Fasting glucose, aspartate aminotransferase (AST), alanine aminotransferase (ALT), cholesterol, triglycerides (TG), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and hemoglobin A1c (HbA1c) measurements were performed by the Johns Hopkins Pathology Core Laboratory. Serum insulin levels were measured with a human insulin ELISA (Millipore, Billerica, MA). Insulin resistance was calculated with the homeostatic model assessment of insulin resistance (HOMA-IR) (18). ELISAs were also used to measure levels of human adiponectin (AdipoGen, Incheon, Korea), human CTRP7 (Aviscera Bioscience, Santa Clara, CA; cat. no. SK00396–06 and lot no. 20111325), and leptin (R&D Systems, Minneapolis, MN). Intra-assay coefficients of variation (CVs) were 3.1 ± 0.1 for leptin and 3.4 ± 0.4 for adiponectin. Interassay CVs were 4.3 ± 0.9 for leptin and 4.3 ± 1.2 for adiponectin. For human CTRP7 ELISA, intra-assay precision was 6–8% and interassay precision was 8–12%. The concentrations of CTRP7 in several samples in the lean group were given at 4 ng/ml (the detection limit of the assay) when they were below the sensitivity of the assay.
Human tissue collection and processing.
Obese liver tissues were obtained from the left lateral lobe of the liver and immediately placed into a sterile container containing RNAlater RNA stabilization reagent (Qiagen, Hilden, Germany) until frozen on dry ice and stored at −80°C for future analysis. Normal human liver tissues were obtained through the Liver Tissue Cell Distribution System (Minneapolis, MN), which was funded by National Institutes of Health contract no. HHSN276201200017C. Total RNAs were isolated from human tissues using TRIzol and reverse transcribed using Superscript II RNase H-reverse transcriptase (Invitrogen, Carlsbad, CA). Real-time PCR primers are listed in Table 1. Liver data were normalized to human GAPDH and expressed as relative mRNA levels using the ΔΔCt method.
Table 1.
Primers for quantitative real-time PCR analyses
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| Human CTRP7 | CAAGTTTTGCCATTTGTGCCAG | GGCAAGCCAGGAATGCTGCAG |
| Human GAPDH | TGAAGGTCGGAGTCAACGGATTTGGT | CATGTGGGCCATGAGGTCCACCAC |
| Human PCK1 | AAAACGGCCTGAACCTCTCG | ACACAGCTCAGCGTTATTCTC |
| Human G6PC | ACTGGCTCAACCTCGTCTTTA | CGGAAGTGTTGCTGTAGTAGTCA |
| Ctrp7 | GACGAGTCTTGCCATCTGTGC | TTAGCCTGATTGGCCCGAG |
| Tnf-α | ATGCTGGGACAGTGACCTGG | CCTTGATGGTGGTGCATGAG |
| Mcp-1 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| Il-1β | GCCACCTTTTGACAGTGATGAG | GACAGCCCAGGTCAAAGGTT |
| Il-6 | TTCCATCCAGTTGCCTTCTTG | GAAGGCCGTGGTTGTCACC |
| Cd11c | CTGGATAGCCTTTCTTCTGCTG | GCACACTGTGTCCGAACTCA |
| Ccl3 | TTCTCTGTACCATGACACTCTGC | CGTGGAATCTTCCGGCTGTAG |
| Ccl4 | TTCCTGCTGTTTCTCTTACACCT | CTGTCTGCCTCTTTTGGTCAG |
| Nos2 | GTTCTCAGCCCAACAATACAAGA | GTGGACGGGTCGATGTCAC |
| Mgl2 | GCATGAAGGCAGCTGCTATTGGTT | TAGGCCCATCCAGCTAAGCACATT |
| Ccr2 | ATCCACGGCATACTATCAACATC | CAAGGCTCACCATCATCGTAG |
| Ccr7 | TGTACGAGTCGGTGTGCTTC | GGTAGGTATCCGTCATGGTCTTG |
| F4/80 | CCCCAGTGTCCTTACAGAGTG | GTGCCCAGAGTGGATGTCT |
| Tgf-β1 | CTCCCGTGGCTTCTAGTGC | GCCTTAGTTTGGACAGGATCTG |
| Il-1r1 | GTGCTACTGGGGCTCATTTGT | GGAGTAAGAGGACACTTGCGAAT |
| Cd206 | CTCTGTTCAGCTATTGGACGC | CGGAATTTCTGGGATTCAGCTTC |
| Il-10 | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| Arg1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| Cd68 | TTCTGCTGTGGAAATGCAAG | CAATGATGAGAGGCAGCAAG |
| Retnla | CCAATCCAGCTAACTATCCCTCC | ACCCAGTAGCAGTCATCCCA |
| Sod1 | TGAGGTCCTGCACTGGTAC | CAAGCGGTGAACCAGTTGTG |
| Sod2 | ATCTGTAAGCGACCTTGCTC | GCCTGCACTGAAGTTCAATG |
| Cox-2 | AACCGCATTGCCTCTGAAT | ATTTCTTCCCCCAGCAACCC |
| Nox-4 | CACCAAATGTTGGGCGATTGT | TCTTGTTCTCCTGCTAGGGAC |
| Nqo1 | AGCCAATCAGCGTTCGGTAT | GCCTCCTTCATGGCGTAGTT |
| Col1 | GTGCTCCTGGTATTGCTGGT | AAGGACCATCCCACTGTCTG |
| Col3 | GGGTTTCCCTGGTCCTAAAG | CCTGGTTTCCCATTTTCTCC |
| Col6 | GATGAGGGTGAAGTGGGAGA | CAGCACGAAGAGGATGTCAA |
| Hif-1α | CAAGATCTCGGCGAAGCAA | GGTGAGCCTCATAACAGAAGCTTT |
| Cbf (adipsin) | AGCAGTGG GTGCTCAGTGC | CGTCATCCGTCACTCCATCC |
| Adipoq | ACTGCAACTACCCATAGCCCAT | TGTCGACTGTTCCATGATTCTCC |
| Leptin | GATGGACCAGACTCTGGCAG | AGAGTGAGGCTTCCAGGACG |
| Angptl4 | CCAAGACCATGACCTCCGTG | CGTTGCCGTGGGATAGAGTG |
| Fgf-21 | ACCACACCCTTCCAGCGATT | CGTGGCTACTCCCAGAGCATC |
| Xbp-1 s | CTGAGTCCGAATCAGGTGCAG | GTCCATGGGAAGATGTTCTGG |
| Xbp-1t | TGGCCGGGTCTGCTGAGTCCG | GTCCATGGGAAGATGTTCTGG |
| Chop | CTGGAAGCCTGGTATGAGGAT | CAGGGTCAAGAGTAGTGAAGGT |
| Atf4 | CCTTCGACCAGTCGGGTTTG | CTGTCCCGGAAAAGGCATCC |
| Serpine1 (PAI-1) | TCAGCCCTTGCTTGCCTCAT | GCATAGCCAGCACCGAGGA |
| Pc | CTGAAGTTCCAAACAGTTCGAGG | CGCACGAAACACTCGGATG |
| FoxO1 | CCCAGGCCGGAGTTTAACC | GTTGCTCATAAAGTCGGTGCT |
| FoxA2 | CCCTACGCCAACATGAACTCG | GTTCTGCCGGTAGAAAGGGA |
| G6Pc | CGACTCGCTATCTCCAAGTGA | GTTGAACCAGTCTCCGACCA |
| B2M | TTCTGGTGCTTGTCTCACTGA | CAGTATGTTCGGCTTCCCATTC |
| 36B4 | AGATTCGGGATATGCTGTTGGC | TCGGGTCCTAGACCAGTGTTC |
| β-actin | GGCACCACACCTTCTACAATG | GGGGTGTTGAAGGTCTCAAAC |
Statistical analysis for human data.
Continuous variables were normally distributed and are presented as means ± SE, with ranges. Categorical variables are expressed as proportions (percentage). Student’s t-test was used to compare variables between obese and lean groups. Correlations between CTRP7 and continuous variables [BMI, liver function tests (LFTs), HbA1c, insulin, glucose, HOMA-IR, and cholesterol levels] were analyzed using Pearson’s correlations and linear regression. Linear regression analysis was performed for each independent variable with CTRP7. If variables reached statistical significance in univariate analysis, they were included in multivariate analysis with CTRP7 as a dependent variable. A subset analysis was performed on obese patients with diabetes, hypertension, and hypercholesterolemia. All statistical analyses were performed using STATA statistical software 11.0 (StataCorp LP, College Station, TX). Spearman’s correlation coefficient analysis was used to analyze the associations between hepatic expression of human CTRP7 and glucose-6-phosphatase (G6PC) or phosphoenolpyruvate carboxykinase 1 (PCK1); these analyses were performed using GraphPad Prism software. P < 0.05 was considered statistically significant.
Mouse models.
The Ctrp7 (C1qtnf7)-null mouse strain was created from embryonic stem cell clone 12585C-A2, which was generated by Regeneron Pharmaceuticals (Tarrytown, NY). The KOMP Mouse Biology Program (www.mbp.mousebiology.org) at the University of California, Davis, generated live mice. Ctrp7-knockout (KO) mice were generated and maintained on a C57BL/6N genetic background. Genotyping primers for the Ctrp7 WT allele were TD: forward, 5′-GGCCAATAAGCACCTAGCAATC-3′ and reverse, 5′-TGCAGGTAGATGACTGTGGAC-3′. The size of the amplified WT PCR product was 117 bp. Primers for the KO allele were lacIn: forward 5′-GGTAAACTGGCTCG GATTAGGG-3′ and reverse, 5′-TTGACTGTAGCGGCTGATGTTG-3′. The size of the amplified KO PCR product was 210 bp. To confirm that Ctrp7 mRNA was absent from KO mice, semiquantitative PCR was carried out using Ctrp7-specific primers (Table 1). Semiquantitative PCR primers for control mouse β-actin were forward, 5′- AGTGTGACGTTGACATCCGTA-3′ and reverse, 5′-GCCAGAGCAGTAATCTCCTTCT-3′. A total of 60 ng of reverse-transcribed cDNA templates from adipose and lung tissues were used in 20-μl PCR reactions that consisted of 38 cycles of 95°C denaturation for 15 s, 60°C annealing for 15 s, and 72°C extension for 20 s. PCR products (13 μl) were separated on 2% agarose gels. All KO (−/−) mice and WT (+/+) littermates used in the study were obtained by intercrossing Ctrp7 (+/−) heterozygous mice.
Mice were housed in polycarbonate cages with ad libitum access to water and food in a temperature-controlled room with 12:12-h light-dark cycles. Mice were fed either a high-fat diet (HFD, 60% calories from fat; D12492; Research Diets, New Brunswick, NJ) or a matched control low-fat diet (LFD, 10% calories from fat D12450B; Research Diets). Mice were fed these diets throughout the study, beginning at 5 wk of age. Body weights of WT and Ctrp7-KO mice were measured weekly. At the end of the study, mice were euthanized 2 h after food removal in the morning. Tissues were dissected, weighed, and snap-frozen in liquid nitrogen for RNA and protein extraction or prepared for histology. Additional tissues were stored at −80°C.
For fasting and refeeding studies, C57BL/6J male mice were obtained from Jackson Laboratory (Bar Harbor, ME). Mouse tissues were collected from fasting and refeeding experiments, as previously described (33). For the fasted group, food was removed for 16 h (beginning 10 h into the light cycle), and mice were euthanized 2–3 h into the light cycle. For the refed group, mice fasted for 16 h and were refed with chow pellets for 3 h before being euthanized. Because of the randomness of food intake, an ad libitum-fed group was not included in fasting and refeeding studies. To generate a diet-induced obesity (DIO) model, 4-wk-old C57BL/6J male mice were fed an HFD or control LFD for 12 wk, as previously described (14). All animal experiments were approved by the Animal Care and Use Committee of the Johns Hopkins University School of Medicine.
Mouse body composition analysis.
Body composition of WT and Ctrp7-KO mice was determined using a quantitative NMR instrument (Echo-MRI-100, Echo Medical Systems, Waco, TX) at the Johns Hopkins University School of Medicine mouse phenotyping core facility. Echo-magnetic resonance imaging analyses measured total fat mass, lean mass, and water content.
Indirect calorimetry of mice.
WT and Ctrp7-KO mice fed an LFD or HFD for 26 wk were used for simultaneous assessments of daily body weight change, food intake (corrected for spillage), physical activity, and whole body metabolic profile in an open-flow indirect calorimeter (CLAMS, Columbus Instruments, Columbus, OH). Data were collected for three consecutive days to confirm that mice were acclimated to calorimetry chambers (indicated by stable body weights, food intakes, and diurnal metabolic patterns), and data from the fourth day were analyzed. Rates of oxygen consumption (V̇o2) and carbon dioxide production (V̇co2) in each chamber were measured throughout the studies. Respiratory exchange ratio (RER; V̇co2/V̇o2) was calculated by CLAMS software (version 4.02) to estimate relative oxidation of carbohydrates (RER = 1.0) vs. fats (RER = 0.7), not accounting for protein oxidation. Energy expenditure (EE) was calculated as EE = V̇o2 × [3.815 + (1.232 × RER)]. V̇o2, V̇co2, and EE data were normalized to body mass. Physical activity was measured by infrared beam breaks in the metabolic chamber. Average metabolic values were calculated per subject and averaged across subjects for statistical analysis.
Mouse glucose and insulin tolerance tests.
Glucose and insulin tolerance tests were performed on WT and Ctrp7-KO mice fed either a matched control LFD or HFD for 12 wk. Mice were fasted overnight for 12–14 h, and glucose (1.0 g/kg body wt) was subsequently delivered via oral gavage. Tail vein blood glucose levels were determined at the indicated time points using a glucometer (NovaMax Plus, Billerica, MA). Additional blood samples were drawn before (time 0) and at 15 min after glucose gavage for assessment of insulin levels. Insulin levels were measured using a mouse/rat insulin ELISA kit (Millipore). For insulin tolerance tests, food was removed 2 h before insulin injection. Insulin was then injected ip at 1 U/kg body wt. Blood glucose was measured at the indicated time points. HOMA-IR was calculated on the basis of fasting glucose and insulin concentrations using the following formula: HOMA-IR = [fasting glucose (mM) × fasting insulin (μU/ml)]/22.5 (18). This surrogate index provides a reasonable approximation of the degree of insulin resistance and has been validated against the reference standard glucose clamp for rats (5) and mice (13).
Mouse lipid tolerance test.
Mice were fasted overnight (12–14 h). Blood samples were drawn through the tail vein bleeds at indicated time points. Intralipid (Sigma, St. Louis, MO) (10 μl/g body wt) was delivered orally. Tail vein blood was collected before (time 0) and at 1, 2, 3, and 4 h after lipid gavage. Serum TG levels were quantified using Infinity Triglycerides kit (Thermo Scientific, Waltham, MA).
Extraction and quantification of mouse hepatic lipid contents.
Livers were extracted and 50 mg of liver was homogenized in 500 µl of distilled water. Part of the homogenate (200 µl) was mixed with 1 ml of choloroform:methanol (2:1) and centrifuged at 1,700 rpm for 5 min at 4°C. The chloroform phase was collected and dried in a speed vacuum. Samples were resuspended in tert-butanol:MeOH-Triton-X100 (3:1:1), and triacylglycerol and cholesterol content was quantified using commercially available colorimetric kits (Thermo Scientific).
Mouse histology.
WT and Ctrp7-KO mouse tissues were fixed in 10% formalin at 4°C overnight. Fixed tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) or Masson’s trichrome at the Histology Reference Laboratory at the Johns Hopkins University School of Medicine. Adipocyte cell size distribution in WT and Ctrp7-KO mice was determined using ImageJ software from H&E-stained images of gonadal adipose tissue sections.
Mouse serum and blood chemistry analysis.
Mouse blood samples were collected by tail bleeds or via a submandibular route. Serum samples were separated using Microvette CB 300 (Sarstedt, Newton, NC). Plasma samples were collected using microtainer blood collection tubes (Becton Dickinson, Franklin Lakes, NJ). Blood cell analyses were carried out at the Pathology Phenotyping Core at the Johns Hopkins University School of Medicine. Serum and tissue TG and cholesterol levels were measured using an Infinity kit (Thermo Fisher Scientific, Middletown, VA). Nonesterified free fatty acids were measured using a NEFA-HR kit (Wako Chemicals, Richmond, VA). Insulin, adiponectin, leptin, TNF-α, monocyte chemoattractant protein 1 (MCP-1), and IL-6 were determined using ELISAs and luminex kits from Millipore.
Quantitative real-time PCR analysis.
Liver and adipose tissues were harvested, snap-frozen, and stored at −80°C. RNA was extracted using TRIzol (Invitrogen), and potential DNA contamination was removed with DNase I (New England Biolabs, Ipswich, MA). Random primers and GoScript RT (Promega, Madison, WI) were used to generate cDNA. Quantitative real-time PCR analyses were carried out using iTaq Universal SYBR Green PCR master mix (Applied Biosystems, Life Technologies, Carlsbad, CA) on a CFX Connect system (Bio-Rad Laboratories, Hercules, CA). Data were normalized to β2 microglobulin (B2M), β-actin, or 36B4 [also known as acidic ribosomal phosphoprotein P0 (RPLP0)] to generate a ΔCt value, and then ΔΔCt was obtained by normalizing data to mean ΔCt of the control group (31). All primers used for quantitative PCR analyses are listed in Table 1.
Statistical analysis for mouse data.
Comparisons between data groups were performed using two-tailed Student’s t-tests with 95% confidence intervals or Mann-Whitney U-tests. ANOVA tests were used for analyses of more than two groups. Values were considered significant at P < 0.05. All data are presented as means ± SE.
RESULTS
Patient demographics and baseline clinical characteristics.
Our cohort included 74 patients (12 men and 62 women) with a mean age of 41.3 ± 1.3 yr (range: 23–63 yr). BMI of the lean group (n = 37) was 22.1 ± 0.3 kg/m2 (range: 17.6–26 yr). BMI of the obese group (n = 37) was 46.2 ± 1.2 kg/m2 (range: 35.6–68.6 yr). Both groups included 32 (86%) Caucasians and 5 (14%) Black individuals. Baseline characteristics of all study subjects are summarized in Table 2. Baseline metabolic parameters were within normal ranges for all laboratory tests in the lean group. The obese group showed significantly higher mean BMI, fasting glucose, HbA1c, insulin, HOMA-IR, ALT, TG, and leptin levels when compared with the lean control group, while HDL-C and adiponectin were significantly lower in the obese group compared with the lean group. We found significantly higher circulating CTRP7 levels in the obese group compared with lean controls (238 ± 47 vs. 515 ± 50 ng/ml; P = 0.0001) (Table 2). In the obese group, 11 individuals had a diagnosis of Type 2 diabetes, 24 had hypertension, and 6 had hypercholesterolemia. There was no difference in CTRP7 levels by metabolic diagnosis, but individuals with blood glucose levels >100 mg/dl had significantly elevated CTRP7 levels (P < 0.001) when compared with patients with blood glucose levels <100 mg/dl.
Table 2.
Baseline characteristics of study subjects
| Control (n = 37) [mean (range)] | Obese (n = 37) [mean (range)] | P | |
|---|---|---|---|
| Sex (female) [n (%)] | 31 (84) | 31 (84) | |
| Age, yr | 41.3 (23–63) | 41.6 (23–66) | 0.9 |
| BMI, kg/m2 | 22.1 (17.6–26) | 46.2 (35.6–68.6) | <0.0001 |
| AST, IU/l | 18.7 (12–32) | 17.9 (7–42) | 0.6 |
| ALT, IU/l | 15.8 (10–39) | 23.9 (7–58) | <0.001 |
| Total cholesterol, mg/dl | 178.4 (120–235) | 169.7 (112–239) | 0.18 |
| Triglycerides, mg/dl | 68.7 (34–145) | 150 (40–305) | <0.0001 |
| HDL-C, mg/dl | 69.5 (40–93) | 45.2 (27–79) | <0.0001 |
| LDL-C, mg/dl | 95.1 (34–151) | 95.1 (38–148) | 0.99 |
| Glucose, mg/dl | 77.5 (57–106) | 112.9 (75–259) | <0.0001 |
| Insulin, μU/ml | 2.42 (0.1–6.0) | 7.2 (0.4–17.9) | <0.0001 |
| HOMA-IR | 0.47 (0.02–1.4) | 1.92 (0.1–4.9) | <0.0001 |
| Hemoglobin A1c, % | 5.2 (4.6–5.8) | 6.4 (4.7–10.4) | 0.0001 |
| Adiponectin, ng/ml | 17,381 (5,041–37,197) | 7367 (3304–17304) | <0.0001 |
| CTRP7, ng/ml | 238 (4–1,237) | 515 (17–1079) | <0.0002 |
| Leptin, pg/ml | 11,870 (658–52,585) | 62,133 (12,879–163,543) | <0.0002 |
BMI, body mass index; AST, aspartate aminotransferase; ALT, alanine aminotransferase; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; HOMA-IR, homeostasis model assessment of insulin resistance. P values are calculated by two-tailed t-test.
CTRP7 levels correlate with metabolic parameters.
CTRP7 levels were positively correlated with BMI (P < 0.01), as well as glucose (P < 0.01), insulin (P = 0.03), HOMA-IR (P < 0.01), HbA1c (P < 0.03), TG (P = 0.02), and leptin (P = 0.01) levels and were inversely correlated with HDL (P = 0.04) and adiponectin (P < 0.01) levels. As shown in Table 3, univariate analysis confirmed these results. There was no difference in CTRP7 levels by age, gender, or ethnicity. Multiple regression analyses revealed BMI as a significant predictor of CTRP7 when controlling for age, gender, and ethnicity. Furthermore, metabolic parameters of fasting glucose (β = 3.3; P < 0.01) and HbA1c (β = 76.8; P = 0.03) retained their significance as predictors of CTRP7 in a multivariate model when controlling for age, gender, ethnicity, and BMI.
Table 3.
Univariate and multivariate analyses of relationships between CTRP7 and metabolic parameters
| Univariate Analysis |
Multivariate Analysis |
|||
|---|---|---|---|---|
| β | P | β | P | |
| Sex | 31.7 | 0.76 | 14.6 | 0.8 |
| Age | 1.75 | 0.59 | 2.3 | 0.4 |
| Ethnicity | 111.9 | 0.31 | 79.1 | 0.4 |
| BMI | 9.98 | <0.001 | 9.9 | <0.01 |
| AST | 1.01 | 0.87 | ||
| ALT | 4.25 | 0.26 | ||
| Total cholesterol | −0.06 | 0.97 | ||
| Triglycerides | 1.34 | 0.02 | 0.1 | 0.8 |
| LDL | 0.71 | 0.67 | ||
| HDL | −4.53 | 0.04 | 1.8 | 0.5 |
| Fasting glucose | 4.05 | <0.001 | 3.3 | <0.01 |
| Insulin | 20.1 | 0.03 | 3.7 | 0.7 |
| Hemoglobin A1c | 104.4 | 0.002 | 76.8 | 0.03 |
| HOMA-IR | 96.2 | 0.003 | 51.2 | 0.1 |
| Adiponectin | −0.01 | 0.001 | −0.01 | 0.1 |
| Leptin | 0.001 | 0.06 | ||
β represents unstandardized coefficient; R2 of the multivariate model was 0.20 (P < 0.01). Bolded values indicate significant difference.
Metabolic state modulates Ctrp7 expression in peripheral tissues.
Mouse and human CTRP7 are highly conserved—the full-length protein and globular C1q domain share a striking 96% and 99% amino acid identity, respectively (Fig. 1A). Therefore, we used a mouse model system to investigate the physiological function of CTRP7. We first determined whether acute changes in metabolic state, as in fasting and refeeding, altered expression of Ctrp7 in peripheral tissues. We found that Ctrp7 expression was significantly decreased in liver and skeletal muscle upon refeeding (Fig. 1B). However, refeeding significantly upregulated Ctrp7 transcripts in visceral (epididymal), but not subcutaneous (inguinal), fat depots (Fig. 1B).
Fig. 1.
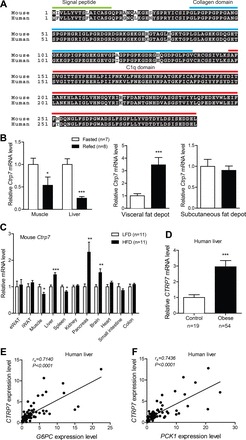
Conservation and metabolic state-dependent modulation of CTRP7 expression. A: sequence alignment of human and mouse CTRP7, with signal peptide, NH2-terminus, collagen domain, and C1q domain indicated. Identical amino acids are shaded in black. B: quantitative real-time PCR analysis of mouse Ctrp7 mRNA expression in skeletal muscle, liver, and visceral (epididymal) and subcutaneous (inguinal) fat depots of fasted (n = 7) and refed (n = 8) mice. C: tissue expression of Ctrp7 mRNA in mice fed a high-fat diet (HFD; n = 11) or a control low-fat diet (LFD; n = 11). D: hepatic expression of human CTRP7 in obese patients (n = 54) and lean controls (n = 19). Expression of human CTRP7 in the liver is positively correlated with glucose-6-phosphatase (G6PC) (E) and phosphoenolpyruvate carboxykinase (PCK1) (F). Data shown are expressed as means ± SE, and statistical analyses represent Mann-Whitney U-tests. *P < 0.05; **P < 0.01; ***P < 0.001.
We next examined expression of Ctrp7 in DIO mice, which represent a model of chronic metabolic stress. Ctrp7 expression in visceral (epididymal) and subcutaneous (inguinal) adipose tissues did not differ in mice fed an HFD for 14 wk compared with mice fed a matched control LFD for the same duration (Fig. 1C). In skeletal muscle and spleen, however, expression of Ctrp7 was significantly reduced in HFD-fed mice relative to LFD-fed mice (Fig. 1C). In contrast, expression of Ctrp7 in liver, pancreas, and brain was significantly upregulated in HFD-fed mice. These data indicate that Ctrp7 expression is modulated by the metabolic state of animals and by different dietary contexts. Given the availability of liver samples, we also assessed expression of human CTRP7 in obese patients and lean controls. Consistent with mouse liver data, we also observed a significant increase in hepatic CTRP7 expression in obese humans (Fig. 1D). Increased hepatic glucose output is a hallmark of diabetes (7). Given that our obese patients are modestly insulin resistant relative to lean controls (Table 2), we also determined the expression levels of hepatic gluconeogenic genes (G6PC and PCK1). Expression of human CTRP7 in liver was positively correlated with the expression levels of both G6PC and PCK1 (Fig. 1, E and F).
Confirmation of Ctrp7-KO mouse model.
We made a null allele of Ctrp7 by deleting 7,262 bp that spans part of exon 2 and most of exon 3 and replacing them with a lacZ and neomycin reporter cassette (Fig. 2A). One set of primers was designed to amplify a region of WT Ctrp7, and another set of primers was designed to amplify a region in lacZ. This enabled us to identify Ctrp7 heterozygous (+/−) and KO (−/−) alleles (Fig. 2B). Because Ctrp7 is abundantly expressed in adipose tissue, we measured the amount of Ctrp7 mRNA in gonadal (epididymal) fat from WT and Ctrp7-KO mice using semiquantitative PCR. As shown in Fig. 2C, no Ctrp7 mRNA was detected in KO mice, thus confirming the loss-of-function mouse model. Ctrp7-KO mice were viable, fertile, born at the expected Mendelian ratio, and developed normally with no gross phenotypes when compared with WT littermates.
Fig. 2.
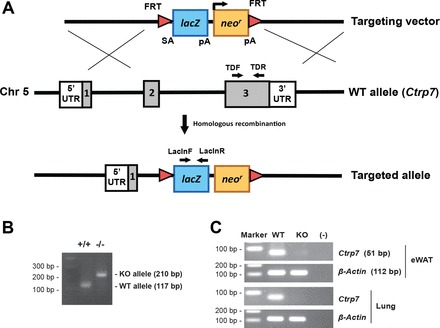
Generation of Ctrp7-KO mice. A: schematic of the gene targeting strategy used to generate Ctrp7-knockout (KO) mice. The majority of Ctrp7 covering exons 2 and 3 was deleted and replaced with a neomycin resistance gene and lacZ reporter cassette. B: PCR genotyping with the indicated primers demonstrated successful production of wild-type (+/+) and homozygous KO (−/−) mice. C: absence of Ctrp7 mRNA in KO mice was confirmed by semiquantitative PCR using Ctrp7-specific primers (Table 1). The same primers were used for real-time PCR.
CTRP7 deficiency has little effect on metabolic parameters.
To evaluate the role of CTRP7 in maintaining energy balance, we fed WT and Ctrp7-KO mice a calorie-dense HFD or matched-control LFD. Given the importance of sex in determining and influencing metabolic outcomes (37), we examined both male and female mice. Body weight of mice was monitored weekly for more than 25 wk. No differences were measured between genotypes of both sexes, regardless of whether animals consumed an LFD or HFD (Fig. 3, A and B). NMR quantification of fat and lean mass also showed no differences between LFD- or HFD-fed WT and Ctrp7-KO mice of both sexes (Fig. 3, C and D). Similarly, percentages of fat and lean mass (normalized to body weight) did not differ between genotypes of both sexes (not shown).
Fig. 3.
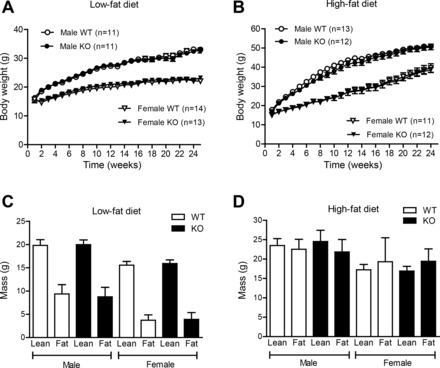
Body weight and body composition of WT and Ctrp7-KO mice. A and B: body weight of male and female wild-type (WT) and Ctrp7 knockout (KO) mice over time. Beginning at 5 wk of age, mice were fed a matched control low-fat diet (LFD; A) or high-fat diet (HFD; B) to induce obesity. NMR analyses of total whole body fat and lean mass in male and female WT and KO mice fed an LFD (C) or HFD (D). Fat and lean mass samples sizes for the LFD group: WT male, n = 15; KO male, n = 10; WT female, n = 14; and KO female, n = 14. Fat and lean mass samples sizes for the HFD group: WT male, n = 12; KO male, n = 12; WT female, n = 11; and KO female, n = 13.
In addition, food intake over a 24-h period did not differ between genotypes of male and female mice (Table 4). Indirect calorimetry analysis of V̇o2, V̇co2, and locomotor activity showed that metabolic rate, EE, RER, and physical activity levels were similar between LFD- and HFD-fed WT and Ctrp7-KO mice of both sexes (Table 4). Examination of tibia length, as well as heart, kidney, liver, visceral (epididymal) fat pad, and subcutaneous (inguinal) fat pad weights also revealed little differences between LFD- and HFD-fed WT and Ctrp7-KO mice of both sexes (Table 5). One exception was that liver weight was significantly less in HFD-fed Ctrp7-KO male mice compared with WT controls.
Table 4.
Indirect calorimetry, activity, and food intake analysis of HFD-fed mice
| Male |
Female |
|||
|---|---|---|---|---|
| WT | KO | WT | KO | |
| n | 6 | 8 | 10 | 13 |
| Food intake, g | 10.6 ± 0.2 | 10.3 ± 0.2 | 2.37 ± 0.13 | 2.56 ± 0.10 |
| V̇o2, ml·kg−1·hour−1 | 2,223 ± 46 | 2,176 ± 53 | 2,691 ± 112 | 2,539 ± 59 |
| V̇co2, ml·kg−1·h−1 | 1,681.3 ± 121 | 1,647 ± 38 | 1,997 ± 81 | 1,927 ± 49 |
| RER (V̇o2/V̇co2) | 0.755 ± 0.01 | 0.75 ± 0.01 | 0.742 ± 0.01 | 0.76 ± 0.01 |
| EE, kcal·kg−1·h−1 | 10.6 ± 0.2 | 10.3 ± 0.25 | 12.73 ± 0.53 | 12.06 ± 0.28 |
| Physical activity (beam breaks) | 26,228 ± 1,291 | 24,607 ± 1,557 | 39,657 ± 3,664 | 34,105 ± 2,948 |
Values are means ± SE; n is number of mice. HFD, high-fat diet; WT, wild-type; KO, knockout; V̇o2, oxygen consumption; V̇co2, CO2 production; RER, respiratory exchange ratio; EE, energy expenditure.
Table 5.
Tibia length and organ weight of WT and Ctrp7-KO mice
| Male |
Female |
|||||||
|---|---|---|---|---|---|---|---|---|
| LFD |
HFD |
LFD |
HFD |
|||||
| WT (n = 6) | KO (n = 6) | WT (n = 11) | KO (n = 11) | WT (n = 12) | KO (n = 12) | WT (n = 11) | KO (n = 9) | |
| Heart, g | 0.16 ± 0.02 | 0.20 ± 0.01 | 0.20 ± 0.01 | 0.22 ± 0.02 | 0.15 ± 0.01 | 0.15 ± 0.01 | 0.16 ± 0.02 | 0.14 ± 0.008 |
| Kidney, g | 0.17 ± 0.01 | 0.18 ± 0.01 | 0.20 ± 0.006 | 0.21 ± 0.02 | 0.12 ± 0.003 | 0.12 ± 0.004 | 0.14 ± 0.005 | 0.14 ± 0.007 |
| Liver, g | 1.58 ± 0.08 | 1.56 ± 0.1 | 3.10 ± 0.1 | 2.57 ± 0.2* | 1.07 ± 0.04 | 1.2 ± 0.08 | 1.30 ± 0.1 | 1.26 ± 0.1 |
| Liver/BW | 0.04 ± 0.001 | 0.04 ± 0.002 | 0.06 ± 0.001 | 0.05 ± 0.002* | 0.01 ± 0.00 | 0.01 ± 0.002 | 0.03 ± 0.002 | 0.03 ± 0.001 |
| eWAT, g | 0.83 ± 0.07 | 0.67 ± 0.07 | 0.77 ± 0.09 | 0.79 ± 0.09 | 0.31 ± 0.04 | 0.38 ± 0.06 | 1.88 ± 0.1 | 1.84 ± 0.1 |
| iWAT, g | 0.65 ± 0.09 | 0.51 ± 0.09 | 1.58 ± 0.2 | 1.28 ± 0.1 | 0.23 ± 0.03 | 0.28 ± 0.04 | 1.36 ± 0.10 | 1.54 ± 0.1 |
| Tibia, mm | 17.8 ± 0.15 | 17.9 ± 0.1 | 19.3 ± 0.2 | 19.5 ± 0.2 | 17.9 ± 0.08 | 17.8 ± 0.1 | 19.2 ± 0.20 | 18.9 ± 0.20 |
Values are means ± SE; n is number of mice. LFD, low-fat diet; HFD, high-fat diet; BW, body weight; eWAT, epididymal white adipose tissue; iWAT, inguinal white adipose tissue.
P < 0.05.
CTRP7 deficiency reduces liver fibrosis and oxidative and endoplasmic reticulum stress.
Despite a 17% reduction in liver weight in HFD-fed Ctrp7-KO male mice, liver histology as well as hepatic TG and cholesterol content (when normalized to protein level) did not differ from WT littermates (Fig. 4, A–C). Serum levels of TG, cholesterol, TG, HDL-C, and LDL-C also did not differ (Fig. 4, D–G). However, the expression of several gluconeogenic genes (Pck1, Pc, G6Pc, and FoxA2) was significantly reduced in livers of HFD-fed Ctrp7-KO male mice relative to WT controls (Fig. 4, H–L). We also performed Masson’s trichrome stain of liver sections to determine the relative degree of fibrosis between HFD-fed WT and Ctrp7-KO male mice. As shown in representative histologic sections, CTRP7 deficiency attenuated the amount of fibrotic collagen (stained blue) in KO livers relative to WT controls (Fig. 5A). Consistent with the liver histology, expression of profibrotic collagen (Col3 and Col6) genes were also markedly reduced in livers of HFD-fed Ctrp7-KO male mice relative to WT controls (Fig. 5, B and C). It is known that HFD-induced obesity promotes hepatic stress. Therefore, we also assessed the levels of multiple genes involved in oxidative and ER stress. Expression of oxidative stress (Sod1, Sod2, Cox2, Nox4, and Nqo1) and ER stress (Xbp1, Chop, and Atf4) genes were significantly reduced in livers of HFD-fed Ctrp7-KO male mice relative to WT controls (Fig. 5, D and E). In the case of oxidative stress, both genes that promote generation (Cox2 and Nox4) and detoxification (Sod1, Sod2, and Nqo1) of reactive oxygen species were significantly downregulated in Ctrp7-KO livers compared with WT controls. Expression of inflammatory genes (Il-1β, Il-6, and Tnf-α,), however, did not differ between the two groups (data not shown).
Fig. 4.
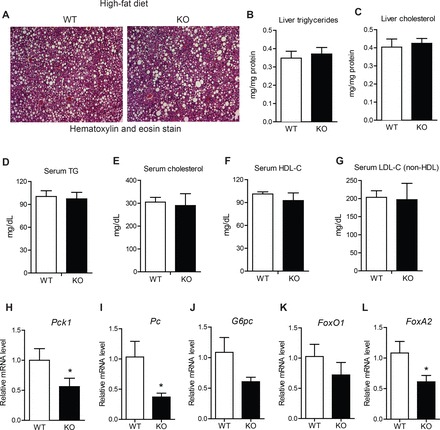
Lipid profiles and reduced hepatic gluconeogenic gene expression in HFD-fed Ctrp7-KO mice. A: representative liver histology (H&E stain; ×100 magnification) of high-fat diet (HFD)-fed wild-type (WT) and knockout (KO) male mice. Liver triglyceride (B) and cholesterol (C) level measurements of HFD-fed WT (n = 10) and KO (n = 11) male mice. Serum triglyceride (D), cholesterol (E), high-density lipoprotein (HDL) cholesterol (F), and low-density lipoprotein (LDL) cholesterol (G) level measurements of HFD-fed WT (n = 5) and KO (n = 8) male mice. H–L: quantitative real-time PCR analysis of expression of gluconeogenic genes (Pck1, Pc, G6Pc, FoxO1, FoxA2) in the liver of WT (n = 11) and KO (n = 11) male mice. Real-time PCR data were normalized to 36B4. *P < 0.05 (Mann-Whitney U-test). Pck1, phosphoenolpyruvate carboxykinase; Pc, pyruvate carboxylase; G6Pc, glucose-6-phosphatase; FoxO1, Forkhead box protein O1; FoxA2, Forkhead box protein A2.
Fig. 5.
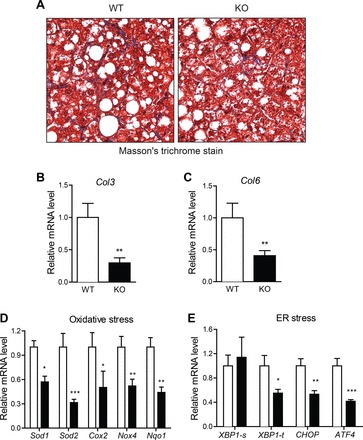
Reduced liver fibrosis and oxidative and ER stress in HFD-fed Ctrp7-KO mice. A: representative liver histology (Masson’s trichrome stain; ×200 magnification) of HFD-fed WT and KO male mice. Collagen-stained blue in histological sections. B–E: quantitative real-time PCR analysis of fibrotic collagen (Col3a1 and Col6a1), oxidative stress (Sod1, Sod2, Cox-2, Nox4, and Nqo1), and endoplasmic reticulum (ER) stress [Xbp1-s (spliced), Xbp1-t (total), Chop, and Atf4] gene expression in liver of WT (n = 11) and KO (n = 11) male mice. Real-time PCR data were normalized to B2M or 36B4. *P < 0.05; **P < 0.01; ***P < 0.005. Col, collagen; Sod, superoxide dismutase; Cox, cyclooxygenase; Nox4, NADPH oxidase 4, Nqo1, NAD(P)H quinone dehydrogenase 1; Xbp, X-Box binding protein 1; Chop, C/EBP homologous protein; Atf4, activating transcription factor 4.
CTRP7 deficiency improves glucose tolerance in the obese state.
To determine the effect of CTRP7 deficiency on glucose homeostasis, we performed oral glucose tolerance tests on LFD- and HFD-fed male and female mice. When fed an LFD, no differences in fasting blood glucose and insulin levels, glucose, and insulin tolerance, and glucose-stimulated insulin secretion were observed between WT and Ctrp7-KO mice of both sexes (Fig. 6, A–D, G–H). However, in mice fed an HFD, loss of CTRP7 significantly reduced fasting blood glucose levels (Fig. 6A) and improved oral glucose tolerance in male, but not female, mice (Fig. 6, E and F). Glucose-stimulated insulin secretion was similar between HFD-fed WT and Ctrp7-KO animals of both sexes (Fig. 6, I and J). The ability of HFD-fed mice to dispose glucose in response to insulin administration (insulin tolerance tests) was not significantly different between WT and Ctrp7-KO mice of both sexes (Fig. 6, K–N). In accordance, insulin-stimulated Akt phosphorylation in adipose tissue, skeletal muscle, and liver was also not different between the two groups (data not shown).
Fig. 6.
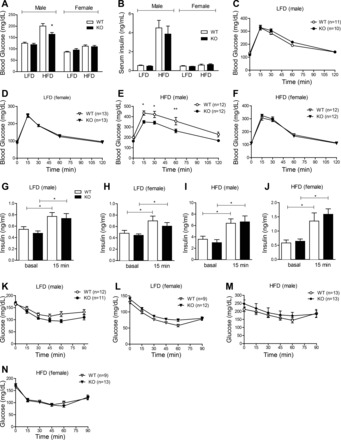
Improved glucose tolerance in Ctrp7-KO mice. Overnight fasting blood glucose (A) and serum insulin (B) levels in male and female WT and KO mice fed a LFD or HFD. Sample sizes for the LFD group: male WT, n = 13; male KO, n = 12; female WT, n = 11; female KO, n = 13. Samples sizes for the HFD group: male WT, n = 10; male KO, n = 10; female WT, n = 10; female KO, n = 10. *P < 0.05 (Student’s t-test). C–F: oral glucose tolerance tests showing blood glucose levels before (at time 0) and after oral glucose load in LFD-fed WT and KO male (C) and female (D) mice, and in HFD-fed WT and KO male (E) and female (F) mice. Two-way ANOVA showed significant difference between glucose levels in HFD-fed WT and KO male mice. Bonferroni post hoc test results are shown in the graphs. *P < 0.05; **P < 0.01. G–J: glucose-stimulated insulin secretion tests showing serum insulin levels before (at time 0) and at 15-min post-glucose gavage in LFD-fed WT and KO male (G) and female (H) mice, and in HFD-fed WT and KO male (I) and female (J) animals. Samples sizes for the LFD group: male WT, n = 10; male KO, n = 10; female WT, n = 10; female KO, n = 10. Samples sizes for the HFD group: male WT, n = 14; male KO, n = 13; female WT, n = 11; female KO, n = 13. K–N: insulin tolerance tests showing blood glucose levels before (at time 0) and after an intraperitoneal injection of insulin in LFD-fed WT and KO male (K) and female (L) mice, and in HFD-fed WT and KO male (M) and females (N) animals. Sample sizes for each group are indicated. *P < 0.05; **P < 0.01 (Student’s t-test).
CTRP7 deficiency does not impact lipid handling.
We also determined whether loss of CTRP7 alters the capacity of Ctrp7-KO mice to handle acute lipid loading in lipid tolerance tests. Serum TG levels were measured over time after oral lipid gavage. Rates of TG clearance following lipid ingestion did not significantly differ between WT and Ctrp7-KO mice of both sexes fed either an LFD or HFD (Fig. 7).
Fig. 7.
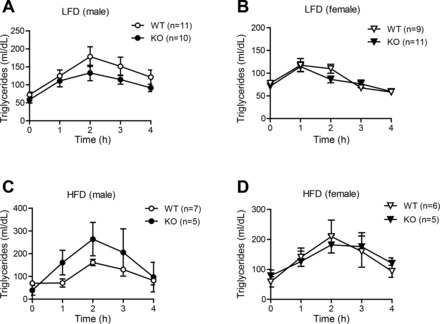
CTRP7 deficiency has little impact on the capacity of KO mice to handle acute lipid challenge. Oral lipid tolerance tests showing serum triglyceride levels before (at time 0) and after oral Intralipid gavage in LFD-fed WT and KO male (A) and female (B) mice, and in HFD-fed WT and KO male (C) and female (D) mice. Samples sizes for the LFD group: male WT, n = 11; male KO, n = 10; female WT, n = 9; female KO, n = 11. Samples sizes for the HFD group: male WT, n = 7; male KO, n = 5; female WT, n = 6; female KO, n = 5.
CTRP7 deficiency reduces inflammation in adipose tissue.
CTRP7 is expressed by adipose tissue and, therefore, could act locally in a paracrine fashion. Because we did not observe any differences in body weight or fat mass, we wanted to determine whether the improvements in glucose tolerance of HFD-fed Ctrp7-KO male mice could be due, in part, to differences in adipose inflammation. Low-grade inflammation within the adipose compartment is a hallmark of obesity in humans (10) and mice (44, 49), and it is mechanistically linked to impaired systemic insulin sensitivity, as well as glucose and lipid metabolism (12). Histological examination of visceral (epididymal) adipose tissue sections revealed greater infiltration of immune cells in Ctrp7-KO mice (Fig. 8A). The distribution of cell size, however, was not different between WT and KO animals (Fig. 8B). When we examined the extent of inflammation in adipose tissue of HFD-fed mice, we found that there was a significantly marked increase in total macrophages in Ctrp7-KO mice, as indicated by 4.5-fold increased expression of the pan-macrophage marker F4/80 (Fig. 8C; P = 0.01).
Fig. 8.
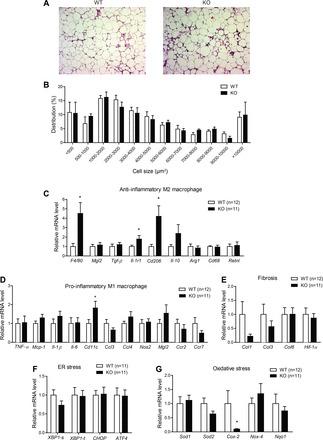
Reduced adipose tissue inflammation in HFD-fed Ctrp7-KO male mice. A: representative adipose histology (H&E stain, ×100 magnification) of WT and KO mice. B: quantification of adipocyte cell size in gonadal fat pad of WT and KO mice. Quantitative real-time PCR analysis of expression of anti-inflammatory M2 macrophage (C) and proinflammatory M1 macrophage (D) marker genes in gonadal adipose tissue of WT and KO male mice. Quantitative real-time PCR analysis of expression of profibrotic (E), ER stress (F), and oxidative stress (G) genes in gonadal adipose tissue of WT and KO male mice are shown. Xbp1-s and Xbp1-t refer to the spliced (s) and total (t) isoforms, respectively. Real-time PCR data were normalized to B2M. *P < 0.05. Mgl2, macrophage galactose N-acetyl-galactosamine specific lectin 2; Tgf-β, transforming growth factor β; Il-1r1, interleukin 1 receptor type 1; Arg1, arginase 1; Rtnla, resistin-like-α; Ccl3, C-C motif chemokine ligand 3; Nos2, nitric oxide synthase 2; Ccr2, C-C motif chemokine receptor 2; Col, collagen; Hif-1α, hypoxia-inducible factor 1α; Xbp-1, X-box binding protein 1; Chop, C/EBP homologous protein; Atf4, activating transcription factor 4; Sod, superoxide dismutase; Nox-4, NADPH oxidase 4; Nqo1, NAD(P)H quinone dehydrogenase 1.
We further assessed whether these macrophages within the fat pad were M1-type, which promote inflammation, or M2-type, which play an anti-inflammatory role. Strikingly, we observed significantly increased expression of M2 macrophage markers Cd206 (4.2-fold) and Il1-r1 (1.8-fold) (Fig. 8C). With the exception of Cd11c, expression of M1 macrophage markers (Ccl3, Ccl4, Nos2, Mgl2, Ccr2, and Ccr7) and major inflammatory genes (Il-1β, Il-6, Tnf-α, and Mcp-1) did not differ between WT and Ctrp7-KO mice (Fig. 8D). Although there was an increase in total as well as M2-type macrophages in the adipose compartment of HFD-fed Ctrp7-KO mice, loss of CTRP7 did not globally affect the immune system; in LFD- and HFD-fed mice of both genotypes and sexes, we did not observe any significant differences in total numbers of circulating red blood cells, white blood cells, lymphocytes, monocytes, neutrophils, eosinophils, and basophils (data not shown).
In addition to inflammation, we also determined the extent of fibrosis, ER stress, and oxidative stress in adipose tissues of HFD-fed WT and Ctrp7-KO mice. The extent of fibrosis and ER stress within visceral (epididymal) fat depots did not differ between genotypes (Fig. 8, E and F). Interestingly, expression of Cox-2, which promotes oxidative stress and inflammation, was reduced five-fold in Ctrp7-KO animals relative to WT controls (Fig. 8G). However, expression of other oxidative stress markers (Sod1, Sod2, Nox4, and Nqo1) did not differ between genotypes.
Impact of CTRP7 deficiency on adipokine expression and circulating levels.
Given reduced proinflammatory and oxidative stress within adipose tissues of HFD-fed Ctrp7-KO male mice, we determined whether these alterations affect the expression and circulating levels of adipokines. At the mRNA level, expression of adipsin (Cfd), adiponectin (Adipoq), leptin (Lp), angiopoietin-like 4 (Angptl4), FGF-21 (Fgf21), plasminogen activator inhibitor-1 (PAI-1; Serpine1), and resistin (Retn) did not differ between HFD-fed WT and Ctrp7-KO mice (Fig. 9A). Consistent with mRNA data, we also did not observe any difference in serum levels of leptin, PAI-1, resistin, or adiponectin (Fig. 9, B–C). However, in contrast to mRNA data, circulating levels of PAI-1 were significantly increased approximately twofold in Ctrp7-KO mice (Fig. 9B). Circulating levels of proinflammatory cytokines (IL-1β, IL-6, TNF-α, and MCP-1) were not different between genotypes (Fig. 9, D–F).
Fig. 9.
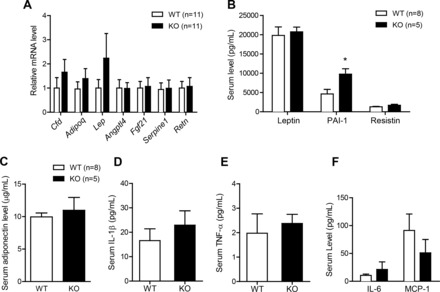
Expression and circulating adipokine levels in HFD-fed WT and Ctrp7-KO male mice. A: quantitative real-time PCR analysis of expression of adipokine genes in gonadal adipose tissue of WT (n = 11) and KO (n = 11) male mice fed a HFD. Real-time PCR data were normalized to B2M. B: serum-level measurements of leptin, PAI-1, and resistin in WT and KO male mice fed an HFD. Serum level measurements of adiponectin (C), IL-1β (D), TNF-α (E), IL-6 and MCP-1 (F) in WT (n = 8) and KO (n = 5) male mice fed an HFD. *P < 0.05. Cfd, complement factor D (also known as adipsin); Adipoq, adiponectin; Lep, leptin; Angptl4, angiopoietin-like 4; Fgf21, fibroblast growth factor 21; Serpine1, serpin family E member 1 (also known as PAI-1); Retn, resistin.
DISCUSSION
CTRP7 is a highly conserved secretory protein found in vertebrates. This is the first study aiming to uncover the biological function of CTRP7 in a physiological context and to provide insights into its relevance to human pathophysiology. Multiple lines of evidence—human studies and functional dissections using KO mouse models—suggest that CTRP7 is a secreted regulator of inflammation and cellular stress, as well as of whole body insulin sensitivity and glucose metabolism. CTRP7 deficiency, in the context of obesity, attenuates insulin resistance and glucose intolerance and dampens adipose tissue inflammation and liver fibrosis, oxidative stress, and ER stress.
In healthy lean volunteers, the average circulating concentration of serum CTRP7 was 238 ± 47 ng/ml. In obese individuals with a BMI >35 kg/m2, circulating CTRP7 levels were markedly elevated (515 ± 50 ng/ml). Furthermore, serum CTRP7 levels were positively correlated with BMI, HOMA-IR, fasting blood glucose and insulin, HbA1c, leptin, and TG levels and inversely correlated with HDL-C and adiponectin levels. Age, gender, and ethnicity did not appear to influence serum CTRP7 levels. Thus, results from our human studies suggest a physiologically relevant link between CTRP7 and metabolism. Furthermore, obesity and insulin resistance increase CTRP7 expression in humans and rodents; in both obese humans and DIO mice, hepatic expression of CTRP7 was significantly elevated relative to lean controls. We also found that the hepatic expression of CTRP7 in obese patients is positively correlated with the expression levels of two key gluconeogenic genes (G6PC and PCK1). Conversely, CTRP7-deficient mice fed an HFD had significantly reduced expression of hepatic gluconeogenic genes. In wild-type DIO mice, we also observed marked increases in Ctrp7 expression in visceral (epididymal), but not subcutaneous (inguinal), fat depots, as well as in the pancreas and brain. The lack of adipose tissue from lean human controls precluded similar analysis in obese subjects. However, elevated circulating levels of CTRP7 in obese humans are likely due to increased expression and secretion of the protein from liver and possibly adipose tissue.
While chronic overnutrition consistently elevated CTRP7 expression and circulating levels in humans and rodents, acute metabolic perturbation (fasting and refeeding) in mice affected Ctrp7 expression in a tissue-specific manner. In visceral fat depots, refeeding following an overnight fast markedly upregulated Ctrp7 mRNA levels. In contrast, refeeding significantly reduced Ctrp7 expression in skeletal muscle and liver. These data suggest that in a normal physiological state, local expression and action of CTRP7 may differ between tissues depending on metabolic contexts, underscoring the potential paracrine and endocrine action of CTRP7 in mediating cell and tissue cross talk. One limitation of our study is the lack of a reliable and specific antibody or ELISA against mouse CTRP7 that can be validated in our KO animals. As such, we could not determine whether tissue and circulating levels of CTRP7 were also correspondingly regulated by different diets or metabolic states in mice.
To establish causal relationships and provide critical genetic evidence that CTRP7, indeed, plays a role in modulating insulin sensitivity and metabolism, we used a loss-of-function mouse model in which Ctrp7 is deleted. When fed a control LFD comparable to standard chow, metabolic phenotypes (body weight and composition, food intake, metabolic rate, glucose, insulin, and lipid tolerance) were indistinguishable between WT and Ctrp7-KO animals. Thus, in a nonobese condition, loss of CTRP7 is dispensable for metabolic control. In contrast, when challenged with an HFD to induce obesity, CTRP7 deficiency significantly reduced insulin resistance, as indicated by lower fasting insulin levels and improved glucose tolerance, independent of body weight, food intake, metabolic rate, energy expenditure, and physical activity levels. The greater rate of glucose disposal in peripheral tissues in response to glucose injection is due to enhanced insulin sensitivity in Ctrp7-KO mice and not from greater insulin secretion, as glucose-stimulated insulin secretion did not differ between genotypes. Insulin normally suppresses expression of hepatic gluconeogenic genes (e.g., Pck1, Pc, and G6Pc) and liver glucose output. Consistently, improved insulin action in Ctrp7-KO mice is also associated with reduced hepatic phosphoenolpyruvate carboxykinase 1 (Pck1), pyruvate carboxylase (Pc), and glucose-6-phosphatase (G6Pc) expression and reduced fasting blood glucose levels. Furthermore, the expression of FoxA2, an important transcriptional regulator of hepatic gluconeogenic gene program (50), was also significantly reduced in Ctrp7-KO liver relative to WT controls. Thus, FoxA2 may link CTRP7 to hepatic gluconeogenic gene expression. The observation that hepatic expression of CTRP7 in obese patients is positively correlated with the expression levels of gluconeogenic genes (G6PC and PCK1) underscores the human relevance of our knockout mouse studies.
Mice fed an HFD develop fatty liver and fibrosis (48), so we determined whether CTRP7 plays a role in these obesity-linked processes that impact systemic metabolism. The degree of steatosis and liver TG and cholesterol content, as well as steady-state serum levels of TG, cholesterol, HDL-C, and LDL-C did not differ between HFD-fed WT and Ctrp7-KO animals. Lipid tolerance tests also indicated comparable capacity of WT and Ctrp7-KO mice to handle and dispose of an acute oral lipid load. Consistent with reduced hepatic fibrosis, as revealed by Masson’s trichrome-stained liver sections, hepatic expression of fibrotic collagen genes (Col3 and Col6) was also markedly reduced in KO mice relative to WT controls. Decreased fibrosis may be associated with reduced oxidative stress of Ctrp7-KO animals, which is thought to promote liver fibrosis (2). However, in contrast to liver, profibrotic collagen gene expression in adipose tissue did not differ between WT and Ctrp7-KO mice. While CTRP7 is expressed by different tissues, the cellular source of CTRP7 remains largely unknown. In the liver, CTRP7 could potentially be synthesized and secreted by hepatocytes, hepatic stellate cells, and/or Kupffer cells. Because loss of CTRP7 attenuates liver fibrosis and fibrotic gene expression and hepatic stellate cells are the major cell type within liver that produces and secretes collagen (9), it is likely that CTRP7 acts on hepatic stellate cells to modulate collagen production.
Chronic high-fat feeding and excess accumulation of fat in the liver are associated with enhanced oxidative (16) and ER stress in hepatocytes (11, 23). In the absence of CTRP7, we observed a substantial reduction in oxidative and ER stress in liver, as indicated by a significant decrease in expression of major oxidative and ER stress genes. Specifically, expression of cyclooxygenase 2 (Cox2) and NADPH oxidase 4 (Nox4), which generate reactive oxygen species (17, 36), was suppressed in Ctrp7-KO mouse livers. Consequently, expression of superoxide dismutases (Sod1 and Sod2) that detoxify reactive oxygen species and NADPH oxidase quinone (Nqo1) that reduces oxygen radical production—some of which are upregulated by oxidative stress (1)—was lower in Ctrp7-KO livers compared with WT controls. This is consistent with an overall reduction in cellular oxidative stress in the livers of CTRP7-deficient animals. A similar reduction in Cox2 expression was also noted in adipose tissue of Ctrp7-KO mice. While the mRNA expression of multiple oxidative stress genes was clearly reduced in the liver of Ctrp7-KO mice compared with WT controls, the level of lipid peroxidation (an index of oxidative stress) in liver was not different between the two groups (data not shown); thus, transcriptional downregulation of genes involved in oxidative stress may correlate with some (e.g., fibrosis) (2), but not all, functional aspects of cellular stress.
To restore homeostasis, ER stress activates an unfolded protein response (UPR) via three parallel pathways regulated by three ER transmembrane proteins—inositol requiring 1 (IRE1), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (38). When these ER transmembrane regulators are activated via phosphorylation or cleavage, they, in turn, activate downstream targets to initiate UPR, including splicing of XBP-1 mRNA by IRE1 and induction of ATF4 and CHOP by PERK (38). In HFD-fed Ctrp7-KO mice, hepatic expression of spliced Xbp-1, Chop, and Atf4 was suppressed relative to WT controls, consistent with an overall decrease in cellular ER stress. It is worth noting that the reduction in ER stress gene expression did not correlate well with the corresponding protein level in liver; for example, although the protein levels of ATF4 and spliced XBP-1 were lower in the Ctrp7-KO liver compared with WT controls (data not shown), the differences fell short of statistical significance.
One of the hallmarks of obesity in both rodents and humans is the presence of chronic low-grade inflammation within the adipose compartment, due largely to the influx of macrophages (10, 12, 44, 49). Two major populations of macrophage are found in the fat depot—proinflammatory M1 and anti-inflammatory M2 macrophages (15, 20, 21). Activated M1 macrophages create an inflammatory milieu within the fat pad by secreting proinflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6), which compromise adipose tissue function (12). Significant improvements in metabolic outcomes, however, can be achieved when macrophages are polarized toward the M2 phenotype (22). Interestingly, in HFD-fed CTRP7-deficient mice, we observed greater recruitment of macrophages to adipose tissue, as indicated by ~4.5-fold higher expression of the pan-macrophage marker gene F4/80 relative to WT controls. Further, it appears that significantly more anti-inflammatory M2 macrophages—with robust expression of Cd206, Il-1r1, and Il-10—are found in the fat pad of Ctrp7-KO mice relative to WT controls. With the exception of Cd11c, expression of other M1 macrophage markers did not differ (Ccl4, Nos2, and Mgl2) or tended to be reduced (Ccl3, Ccr2, and Ccr7) in Ctrp7-KO mice relative to WT controls. The heterogeneity of macrophage subtypes necessitated the need to use multiple markers to define M1 and M2 macrophages within a fat depot. Macrophages that express Cd11c are thought to promote insulin resistance (24), and Cd11c levels are higher in the Ctrp7-KO animals compared with WT control. The significance of this is unclear, since none of the other six M1 markers, nor inflammatory gene (Il-1β, Il-6, Tnf-α, and Mcp-1) expression in adipose tissue were different between genotypes. It may be that the ratio of M1 to M2 is more important in determining the inflammatory status within the fat depot. While there were more adipose tissue macrophages, circulating numbers of lymphoid (T and B cells) and myeloid (monocytes, neutrophils, eosinophils, and basophils) cells in the blood were not different between WT and Ctrp7-KO mice. Overall, our data suggest a better inflammatory profile in the adipose compartment of CTRP7-deficient mice. However, reduction in local inflammation within the fat pad of Ctrp7-KO mice did not alter systemic inflammatory profile, as none of the circulating levels of proinflammatory cytokines (IL-1β, IL-6, TNF-α, and MCP-1) were different between WT and KO animals.
It has become increasingly appreciated that metabolic disease phenotypes and severity can be influenced by the sex of animals (19, 37). For this reason, female WT and Ctrp7-KO mice were also included in our phenotypic analysis. Unlike male mice, loss of CTRP7 is dispensable for metabolic control in female mice fed an LFD or in a DIO state. All whole body metabolic parameters (body weight, adiposity, food intake, metabolic rate, EE, physical activity levels, glucose, insulin, and lipid tolerance) were indistinguishable between WT and Ctrp7-KO female mice. However, sex hormones regulate a variety of physiological processes, so it is not unexpected that the metabolic phenotypes of many KO mouse models, including CTRP7-deficient mice, manifest in male, but not female, mice. Given that this is outside the scope of present studies, future work will uncover whether sex hormones are, indeed, responsible for the differences in phenotypic outcome in response to CTRP7 deficiency.
In summary, we provide the first in vivo characterization of the physiological function of CTRP7. In the context of metabolism, our KO mouse data support a role for CTRP7 in modulating inflammation and cellular stress in the obese state. Our human studies further highlight the relevance of CTRP7 to obesity-linked metabolic dysfunction. Given that CTRP7 is relatively widely expressed across different tissues and is highly conserved among vertebrates, our genetic loss-of-function mouse model will be a valuable tool to uncover additional biological functions of CTRP7 in future studies.
GRANTS
This work was supported in part by a grant from the National Institutes of Health (DK-084171 to G. W. Wong). P. S. Petersen was supported by grants from the Carlsberg Foundation and Danish Council for Independent Research (DFF-4183-00634). R. M. Wolf was supported by grants from the Endocrine Society and the Pediatric Endocrine Society.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
PSP, XL, and RMW contributed equally to the work; PSP, XL, RMW, and GWW contributed to the experimental design; PSP, XL, RMW,SR, HCL, and SYT performed mouse experiments; MAS, THM, and KES performed bariatric surgery and collected human liver and serum samples; PSP, XL, RMW, and GWW analyzed and interpreted the data; and PSP, RMW, and GWW wrote the paper.
ACKNOWLEDGMENTS
We thank Susan Aja for help with indirect calorimetry.
REFERENCES
- 1.Araujo J, Breuer P, Dieringer S, Krauss S, Dorn S, Zimmermann K, Pfeifer A, Klockgether T, Wuellner U, Evert BO. FOXO4-dependent upregulation of superoxide dismutase-2 in response to oxidative stress is impaired in spinocerebellar ataxia type 3. Hum Mol Genet 20: 2928–2941, 2011. doi: 10.1093/hmg/ddr197. [DOI] [PubMed] [Google Scholar]
- 2.Basaranoglu M, Basaranoglu G, Sentürk H. From fatty liver to fibrosis: a tale of “second hit”. World J Gastroenterol 19: 1158–1165, 2013. doi: 10.3748/wjg.v19.i8.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byerly MS, Petersen PS, Ramamurthy S, Seldin MM, Lei X, Provost E, Wei Z, Ronnett GV, Wong GW. C1q/TNF-related protein 4 (CTRP4) is a unique secreted protein with two tandem C1q domains that functions in the hypothalamus to modulate food intake and body weight. J Biol Chem 289: 4055–4069, 2014. doi: 10.1074/jbc.M113.506956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byerly MS, Swanson R, Wei Z, Seldin MM, McCulloh PS, Wong GW. A central role for C1q/TNF-related protein 13 (CTRP13) in modulating food intake and body weight. PLoS One 8: e62862, 2013. doi: 10.1371/journal.pone.0062862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cacho J, Sevillano J, de Castro J, Herrera E, Ramos MP. Validation of simple indexes to assess insulin sensitivity during pregnancy in Wistar and Sprague-Dawley rats. Am J Physiol Endocrinol Metab 295: E1269–E1276, 2008. doi: 10.1152/ajpendo.90207.2008. [DOI] [PubMed] [Google Scholar]
- 6.Coleman DL, Hummel KP. The influence of genetic background on the expression of the obese (Ob) gene in the mouse. Diabetologia 9: 287–293, 1973. doi: 10.1007/BF01221856. [DOI] [PubMed] [Google Scholar]
- 7.DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non-insulin-dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism 38: 387–395, 1989. doi: 10.1016/0026-0495(89)90129-7. [DOI] [PubMed] [Google Scholar]
- 8.Dick DM, Aliev F, Krueger RF, Edwards A, Agrawal A, Lynskey M, Lin P, Schuckit M, Hesselbrock V, Nurnberger J Jr, Almasy L, Porjesz B, Edenberg HJ, Bucholz K, Kramer J, Kuperman S, Bierut L. Genome-wide association study of conduct disorder symptomatology. Mol Psychiatry 16: 800–808, 2011. doi: 10.1038/mp.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med 10: 76–99, 2006. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harman-Boehm I, Blüher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, Shai I, Klöting N, Stumvoll M, Bashan N, Rudich A. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab 92: 2240–2247, 2007. doi: 10.1210/jc.2006-1811. [DOI] [PubMed] [Google Scholar]
- 11.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140: 900–917, 2010. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hotamisligil GS. Inflammation and metabolic disorders. Nature 444: 860–867, 2006. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Muniyappa R, Yan X, Chen H, Yue LQ, Hong EG, Kim JK, Quon MJ. Comparison between surrogate indexes of insulin sensitivity and resistance and hyperinsulinemic euglycemic clamp estimates in mice. Am J Physiol Endocrinol Metab 294: E261–E270, 2008. doi: 10.1152/ajpendo.00676.2007. [DOI] [PubMed] [Google Scholar]
- 14.Lei X, Li Q, Rodriguez S, Tan SY, Seldin MM, McLenithan JC, Jia W, Wong GW. Thromboxane synthase deficiency improves insulin action and attenuates adipose tissue fibrosis. Am J Physiol Endocrinol Metab 308: E792–E804, 2015. doi: 10.1152/ajpendo.00383.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117: 175–184, 2007. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mantena SK, King AL, Andringa KK, Eccleston HB, Bailey SM. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radic Biol Med 44: 1259–1272, 2008. doi: 10.1016/j.freeradbiomed.2007.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marnett LJ, Rowlinson SW, Goodwin DC, Kalgutkar AS, Lanzo CA. Arachidonic acid oxygenation by COX-1 and COX-2. Mechanisms of catalysis and inhibition. J Biol Chem 274: 22903–22906, 1999. doi: 10.1074/jbc.274.33.22903. [DOI] [PubMed] [Google Scholar]
- 18.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28: 412–419, 1985. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 19.Miller VM. Why are sex and gender important to basic physiology and translational and individualized medicine? Am J Physiol Heart Circ Physiol 306: H781–H788, 2014. doi: 10.1152/ajpheart.00994.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11: 723–737, 2011. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol 6: 275–297, 2011. doi: 10.1146/annurev-pathol-011110-130138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447: 1116–1120, 2007. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pagliassotti MJ. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu Rev Nutr 32: 17–33, 2012. doi: 10.1146/annurev-nutr-071811-150644. [DOI] [PubMed] [Google Scholar]
- 24.Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 8: 301–309, 2008. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson JM, Aja S, Wei Z, Wong GW. CTRP1 protein enhances fatty acid oxidation via AMP-activated protein kinase (AMPK) activation and acetyl-CoA carboxylase (ACC) inhibition. J Biol Chem 287: 1576–1587, 2012. doi: 10.1074/jbc.M111.278333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peterson JM, Seldin MM, Tan SY, Wong GW. CTRP2 overexpression improves insulin and lipid tolerance in diet-induced obese mice. PLoS One 9: e88535, 2014. doi: 10.1371/journal.pone.0088535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson JM, Seldin MM, Wei Z, Aja S, Wong GW. CTRP3 attenuates diet-induced hepatic steatosis by regulating triglyceride metabolism. Am J Physiol Gastrointest Liver Physiol 305: G214–G224, 2013. doi: 10.1152/ajpgi.00102.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peterson JM, Wei Z, Seldin MM, Byerly MS, Aja S, Wong GW. CTRP9 transgenic mice are protected from diet-induced obesity and metabolic dysfunction. Am J Physiol Regul Integr Comp Physiol 305: R522–R533, 2013. doi: 10.1152/ajpregu.00110.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterson JM, Wei Z, Wong GW. C1q/TNF-related protein-3 (CTRP3), a novel adipokine that regulates hepatic glucose output. J Biol Chem 285: 39691–39701, 2010. doi: 10.1074/jbc.M110.180695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rohrbach S, Aurich AC, Li L, Niemann B. Age-associated loss in adiponectin-activation by caloric restriction: lack of compensation by enhanced inducibility of adiponectin paralogs CTRP2 and CTRP7. Mol Cell Endocrinol 277: 26–34, 2007. doi: 10.1016/j.mce.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 32.Seldin MM, Lei X, Tan SY, Stanson KP, Wei Z, Wong GW. Skeletal muscle-derived myonectin activates the mammalian target of rapamycin (mTOR) pathway to suppress autophagy in liver. J Biol Chem 289: 36,073–36,082, 2013. doi: 10.1074/jbc.M113.500736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seldin MM, Peterson JM, Byerly MS, Wei Z, Wong GW. Myonectin (CTRP15), a novel myokine that links skeletal muscle to systemic lipid homeostasis. J Biol Chem 287: 11,968–11,980, 2012. doi: 10.1074/jbc.M111.336834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seldin MM, Tan SY, Wong GW. Metabolic function of the CTRP family of hormones. Rev Endocr Metab Disord 15: 111–123, 2014. doi: 10.1007/s11154-013-9255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seldin MM, Wong GW. Regulation of tissue crosstalk by skeletal muscle-derived myonectin and other myokines. Adipocyte 1: 200–202, 2012. doi: 10.4161/adip.20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Fórró L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406: 105–114, 2007. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varlamov O, Bethea CL, Roberts CT Jr. Sex-specific differences in lipid and glucose metabolism. Front Endocrinol (Lausanne) 5: 241, 2015. doi: 10.3389/fendo.2014.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086, 2011. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 39.Wei Z, Lei X, Petersen PS, Aja S, Wong GW. Targeted deletion of C1q/TNF-related protein 9 increases food intake, decreases insulin sensitivity, and promotes hepatic steatosis in mice. Am J Physiol Endocrinol Metab 306: E779–E790, 2014. doi: 10.1152/ajpendo.00593.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei Z, Lei X, Seldin MM, Wong GW. Endopeptidase cleavage generates a functionally distinct isoform of C1q/tumor necrosis factor-related protein-12 (CTRP12) with an altered oligomeric state and signaling specificity. J Biol Chem 287: 35,804–35,814, 2012. doi: 10.1074/jbc.M112.365965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei Z, Peterson JM, Lei X, Cebotaru L, Wolfgang MJ, Baldeviano GC, Wong GW. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J Biol Chem 287: 10,301–10,315, 2012. doi: 10.1074/jbc.M111.303651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei Z, Peterson JM, Wong GW. Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): activation of AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J Biol Chem 286: 15,652–15,665, 2011. doi: 10.1074/jbc.M110.201087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei Z, Seldin MM, Natarajan N, Djemal DC, Peterson JM, Wong GW. C1q/tumor necrosis factor-related protein 11 (CTRP11), a novel adipose stroma-derived regulator of adipogenesis. J Biol Chem 288: 10214–10229, 2013. doi: 10.1074/jbc.M113.458711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Ge G, Spooner E, Hug C, Gimeno R, Lodish HF. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J 23: 241–258, 2009. doi: 10.1096/fj.08-114991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Revett T, Gimeno R, Lodish HF. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-γ agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem J 416: 161–177, 2008. doi: 10.1042/BJ20081240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong GW, Wang J, Hug C, Tsao TS, Lodish HF. A family of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci USA 101: 10302–10307, 2004. doi: 10.1073/pnas.0403760101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wree A, Broderick L, Canbay A, Hoffman HM, Feldstein AE. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol 10: 627–636, 2013. doi: 10.1038/nrgastro.2013.149. [DOI] [PubMed] [Google Scholar]
- 49.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112: 1821–1830, 2003. doi: 10.1172/JCI200319451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Rubins NE, Ahima RS, Greenbaum LE, Kaestner KH. Foxa2 integrates the transcriptional response of the hepatocyte to fasting. Cell Metab 2: 141–148, 2005. doi: 10.1016/j.cmet.2005.07.002. [DOI] [PubMed] [Google Scholar]