

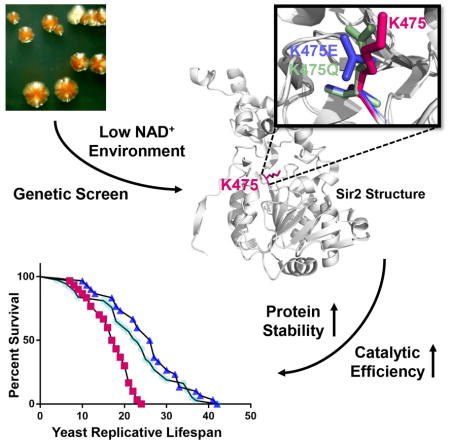
Summary
Sirtuin enzymes depend on NAD+ to catalyze protein deacetylation. Therefore, the lowering of NAD+ during aging leads to decreased sirtuin activity and may speed up aging processes in laboratory animals and humans. In this study, we used a genetic screen to identify two mutations in the catalytic domain of yeast Sir2 that allow the enzyme to function in an NAD+-depleted environment. These mutant enzymes give rise to a significant increase of yeast replicative lifespan and increase deacetylation by the Sir2 ortholog, SIRT1, in mammalian cells. Our data suggest that these mutations increase the stability of the conserved catalytic sirtuin domain, thereby increasing the catalytic efficiency of the mutant enzymes. Our approach to identifying sirtuin mutants that permit function in NAD+-limited environments may inform the design of small molecules that can maintain sirtuin activity in aging organisms.
eTOC
Ondracek et al. report the identification of two adaptive mutations in yeast Sir2 that allow the enzyme to function in an NAD+-depleted environment by enhancing protein stability and increasing enzyme catalysis, which increases yeast replicative lifespan and deacetylation of in vivo substrates by the Sir2 ortholog, SIRT1, in mammalian cells.
Introduction
Originally discovered for its role in yeast genomic silencing, Sir2 is a nicotinamide adenine dinucleotide (NAD+) dependent deacetylase that mediates gene silencing and aging (Imai et al. 2000). Sir2 related proteins, or sirtuins, especially the mammalian Sir2 isoform SIRT1, have been shown to modulate numerous diseases in mouse models, including metabolic disorders, neurodegeneration, cardiovascular disease, cancers, and aging (Imai et al. 2014; Guarente 2013).
Acetylation of specific lysine (Lys) residues in the amino-terminus of histone proteins is recognized as a fundamental epigenetic mark that affects transcriptional regulation (Rothbart & Strahl 2014). Histone acetylation is generally associated with transcriptional activation, as Lys residues in histone proteins neutralize negative charges along the DNA backbone. Deacetylation therefore results in chromatin condensation, which is associated with transcriptional repression and gene silencing. Post-translational modifications, including deacetylation, also recruit regulatory molecules to chromatin and determine the transcriptional status of genes (Allis & Jenuwein 2016). Importantly, sirtuins also deacetylate many non-histone proteins, such as transcription factors and metabolic enzymes, and can thus exert a profound effect on cellular physiology.
Because sirtuins are dependent on NAD+, it is possible that cellular processes modulated by sirtuins, including aging, are influenced by changes in cellular NAD+ levels. We, and others (Smith et al. 2000; Lin & Guarente 2003), have shown that mutations in or loss of NPT1 in NAD+ biosynthetic pathways reduce NAD+ concentrations resulting in a loss of silencing at telomeres and rDNA, as well as significantly decreasing lifespan. Importantly, NAD+ levels decline in humans and other organisms as they age, (Massudi et al. 2012; Braidy et al. 2011) reducing the activity of SIRT1 and likely all seven mammalian sirtuins. Since loss of sirtuins has been implicated in accelerated aging (Ghosh & Zhou 2015) and age-related diseases (Bonkowski & Sinclair 2016), we hypothesized that if sirtuins could function in the NAD+-depleted environment of aging, the health benefits of these enzymes could be restored. Our approach was to use mutagenesis and a genetic screen to identify mutations that allow yeast Sir2 function to persist in a low NAD+ setting. We identified two mutations in SIR2 that extend replicative lifespan (RLS) and increase enzymatic activity in low NAD+. The mutations appear to cause conformational changes that improve the catalytic efficiency of Sir2, and this effect is conserved when the mutations are installed in the conserved, analogous residue in human SIRT1. Our experimentally-derived data is augmented by predictive protein modeling which supports our hypothesis that these mutations increase rigidity of the catalytic core and stabilize the mutant enzymes. Our findings may thus identify a structural target for a class of SIRT1 activating compounds that allow activity to persist at low NAD+ levels.
Results
Strains used to select Sir2 mutants
The main pathways involved in NAD+ biosynthesis in yeast and other organisms are de novo synthesis of NAD+ from tryptophan (requiring Qpt1) and the salvage pathway generating NAD+ from nicotinamide (requiring Pnc1) and nicotinic acid (requiring Npt1) (Bedalov et al. 2003; Lin & Guarente 2003; Revollo et al. 2004). In our initial attempts to produce yeast with low enough levels of NAD+ to inhibit Sir2 activity, we knocked-out either QPT1 or NPT1. Only knockout of NPT1 reduced NAD+ levels enough to affect activity, consistent with earlier studies. This strain also contained deletions of the SIR2 and ADE2 genomic loci and had an integrated copy of ADE2 at either the ribosomal DNA (rDNA) or telomeres (both locations work comparably). When transformed with a single copy of an ARS-CEN plasmid expressing SIR2, red colonies form due to silencing of ADE2, while vector control colonies not expressing Sir2 are white.
NAD+ levels were decreased in npt1Δ yeast ~3–4 fold, resulting in a significant decrease in lifespan (Fig. 1A, B). We next assessed Sir2 activity by silencing of the integrated ADE2 marker at the telomeres and rDNA. Wildtype (WT) yeast expressing Sir2 resulted in normal silencing, indicated phenotypically by red colonies (Fig. 1C1, D1), while yeast lacking SIR2 (sir2Δ) were unable to silence ADE2 at either telomeres or rDNA, resulting in white colonies (Fig. 1C2, D2). Yeast lacking NPT1 (npt1Δ) resulted in white colonies, as NAD+ levels were too low for Sir2 to function (Fig. 1C3, D3), and as expected, double knockout yeast lacking SIR2 and NPT1 (sir2Δ npt1Δ) resulted in white colonies (Fig. 1C4, D4). We were able to rescue the single mutants with plasmids expressing SIR2 or NPT1(Fig. 1C5, 6; D5, 6), thus resulting in red colonies. Double knockout strains (sir2Δ npt1Δ) were not rescued with plasmids expressing either SIR2 or NPT1, resulting in white colonies (Fig. 1C7, 8; D7, 8). These results indicate that a threshold level of NAD+ is required for Sir2 to function in yeast. We used our double knockout strain (sir2Δ npt1Δ) to select for sirtuin mutants capable of functioning in NAD+-depleted cells.
Figure 1. Lifespan and sirtuin activity are affected by NAD+ loss and specific sirtuin mutants restore activity in npt1Δ mutants.

Fig. 1A: NAD+ levels are significantly decreased in the npt1Δ yeast. Experiments were performed in duplicate. Differences between means are statistically significant (p<0.0001).
Fig. 1B: Survival of rDNA::ADE2 and tel::ADE2 yeast strains in NAD+-sufficient and limited backgrounds (npt1Δ). Median NAD+-deficient lifespan for rDNA::ADE2=19 generations; sufficient NAD+ lifespan=26; median NAD+-deficient lifespan for tel::ADE2=19; sufficient NAD+ lifespan=30.5 (25–30 mother cells per strain). Significance between NAD+-sufficient and insufficient yeast strains (p<0.0001) determined using Log-rank (Mantel-Cox) test.
Figs. 1C, D: SIR2 activity assessed by chromatin silencing of integrated ADE2 marker in WT, SIR2, NPT1, and sir2Δ npt1Δ deficient yeast (C: tel::ADE2, D: rDNA::ADE2). 1. WT yeast 2. sir2Δ, SIR2 deficient yeast 3. npt1Δ, NPT1 deficient yeast 4. sir2Δ npt1Δ, SIR2 NPT1 deficient. Rescue deletion strains with plasmids carrying SIR2 or NPT1. 5. sir2 + pRS316-SIR2. 6. npt1+ pRS316-NPT1. 7. sir2Δ npt1Δ + pRS316-NPT1. 8. sir2Δ npt1Δ + pRS316-SIR2. Note the sir2Δ npt1Δ mutant is not rescued to red color by pRS316-SIR2.
Figs. 1E, F: sir2Δ npt1Δ deficient yeast (E: tel::ADE2, F: rDNA::ADE2) transformed with WT SIR2 and mutant K475E (1E) or K475Q (1F) SIR2 plasmids. WT SIR2 transformed strains (1, 2) remain white, indicating loss of silencing and Sir2 activity. K475E or K475Q SIR2 transformed strains (3, 4) result in red colonies, indicating silencing of ADE2 and active Sir2 in NAD+-deficient yeast.
Fig. 1G: Multiple protein sequence alignment of sequences within the Rossman fold of sirtuin proteins using COBALT. Lys residues at conserved site (K475 in S. cerevisiae Sir2) are indicated in red; C. elegans (Sir-2.1), M. musculus (SIRT1), and H. Sapiens (SIRT1).
Figs. 1H, I: Protein blots of integrated Sir2 yeast. Western blot of Sir2 proteins isolated from sir2Δ npt1Δ deletion strain, rDNA::ADE2 (H Top). Western blot of Sir2 proteins isolated from sir2Δ deletion strain, rDNA::ADE2 (I Bottom). α-Tubulin loading control. Survival of Sir2 WT and K475E, K475Q mutants (H): sir2Δ npt1Δ background (NAD+-insufficient tel::ADE2, rDNA::ADE2); (I): sir2Δ background (NAD+-sufficient tel::ADE2, rDNA::ADE2). Figures represent data from a single, representative experiment.
Fig. 1J: Median survival of NAD+-insufficient (npt1Δ) and NAD+-sufficient yeast with integrated K475, K475E, or K475Q SIR2. Median lifespan determined using Graphpad Prism and survival curves determined to be significantly different between WT and mutant sirtuin enzymes using the Log-rank (Mantel-Cox) test.
Identification of Sir2 mutants that remain active in an NAD+-deficient environment
To test whether sirtuin activity could evolve to persist in low NAD+ concentrations, we screened for mutations in the SIR2 gene that potentiate function of the sirtuin in the npt1Δ strain. The SIR2 gene was mutagenized along its entire conserved domain (residues 157–546) by error-prone PCR. Conditions were experimentally derived to generate ~1–2 mutations/gene and to reduce mutational bias, as monitored by DNA sequencing of individual clones after mutagenesis. When the mutagenized SIR2 plasmid was introduced into sir2Δ npt1Δ double mutant strains with ADE2 at the rDNA or telomeres, red colonies appeared rarely; of ~30,000 colonies screened six mutants were identified. The isolated plasmids retransformed the sir2Δ npt1Δ mutants to produce red colonies. The six SIR2 genes were sequenced and every mutant contained a change of Lys (K) 475 to either glutamic acid (Glu, E) or glutamine (Gln, Q). In four mutants, additional single-point mutations were present, but in two cases, the K475E and K475Q represented single amino acid (AA) changes, indicating that they conferred the phenotype. In Fig. 1E, F, we show the two yeast tester strains with ADE2 at rDNA and telomeres lacking sir2Δ npt1Δ now transformed with WT, K475E (Fig. 1E), and K475Q (Fig. 1F) SIR2 plasmids. WT SIR2 transformed colonies were white (Fig. 1E, F), indicating loss of silencing and Sir2 activity. K475E and K475Q SIR2 transformed colonies were red (Fig. 1E, F), indicating silencing of ADE2 and active Sir2 in NAD+-deficient cells.
Importantly, K475 lies in the Rossmann fold of the NAD+ binding domain of sirtuins, and the known sirtuin structures indicate that this Lys is in proximity to the NAD+ and substrate binding clefts (Min et al. 2001). This residue is conserved in Sir2 orthologs among many species including yeast, worm, mice, and humans (Fig. 1G). However, comparing sequences of the yeast Sir2 and seven human sirtuin paralogs (Fig. S1) revealed that although K475 is conserved in SIRT1 (K444) and SIRT7 (K272), the other five sirtuins contain either Glu (SIRT3 E323), Gln (SIRT2 Q228; SIRT4 Q264, SIRT6 Q218), or valine (SIRT5 V253). Notably SIRT5 is a desuccinylase and not a deacetylase (Park et al. 2013). That six of the seven mammalian sirtuins contain K, E, or Q at this site was intriguing as these AAs possess different physiochemical properties (+, −, polar uncharged), raising the possibility that this position might play a direct role in catalysis or may alter interactions with surrounding residues to influence catalytic differences among sirtuin paralogs.
Mutant Sir2 extends lifespan in both NAD+-insufficient and NAD+-sufficient yeast
We next determined the RLS of yeast with the K475E or K475Q mutations integrated using pRS306 plasmids. We verified protein expression and determined that levels of WT and mutant proteins were similar in both NAD+-deficient (Fig. 1H Top) and NAD+-sufficient (Fig. 1I Bottom) conditions. In both NAD+-deficient (Fig. 1H) and NAD+-sufficient (Fig. 1I) strains the K475 mutations significantly extended life span compared to WT SIR2. In both figures I show representative life span curves; one from each reporter strain used in this study, but combining at least three independent experiments with 20–50 yeast cells each. Both median and maximal lifespan significantly increased (p<0.05) with the mutant enzymes (Fig. 1J; Supplemental).
Kinetic analyses of yeast mutant enzymes
To explain how these mutations confer both enhanced survival and increased silencing activity, we characterized the sirtuin mutants biochemically. We expressed ~90% pure His-tagged fusion WT and mutant Sir2 proteins (concentration verified by Coomassie staining on an SDS-PAGE protein gel (Fig. 2A)). The deacetylation rate (concentration of deacetylated product produced over time (Fig. S2)) was determined for WT and mutant Sir2 proteins at saturating peptide (200 μM) or NAD+ (1 mM) concentrations against increasing concentrations of NAD+ (0, 0.01, 0.02, 0.05, 0.1, 0.25, 0.5, 1.0 mM) or peptide (0, 0.05, 0.1, 0.25, 0.5, 1.0 mM). Steady state parameters (KM, kcat) and catalytic efficiency (kcat/KM) were obtained using nonlinear regression of the Michaelis-Menten equation (Fig. 2B, C). Values obtained from two independent replicates are indicated by red circles (WT K475), teal (K475Q), and blue (K475E). Experimentally derived kinetic analyses are described in Fig. 2G and in Supplementary. Overall, the turnover of the mutant enzymes was improved (increased kcat) and KM values for peptide (K475E) and NAD+ (K475Q) were lowered, which may also contribute to increased activity in vivo. While our initial hypothesis was that the K475 mutant enzymes would function by lowering KM values for NAD+, our most consistent finding is an overall increase in catalytic efficiency, which may be driving the persistence of their activities in low NAD+ environments.
Figure 2. Kinetic parameters for deacetylation by WT and mutant yeast and human enzymes.

Fig. 2A: Coomassie stained protein gel of purified Sir2 proteins for kinetic analyses. Blot shows >90% pure proteins and densitometric analysis guided use of matched protein concentrations for kinetic analyses.
Figs. 2B, C: Enzymatic activity of WT and mutant sirtuins measured by deacetylation of 11-mer peptide H3K14ac in saturating NAD+ (or peptide) at varying NAD+ concentrations (B) or varying peptide concentrations (C) at multiple time points and fit to the Michaelis–Menton equation.
Fig. 2D: Protein gel of purified human proteins for assay based kinetic analyses, as for panel A.
Figs. 2E, F: Enzymatic activity of WT and mutant sirtuins measured by deacetylation of FDL® substrate in saturating peptide with varying concentrations of NAD+ (E) or saturating NAD+ and varying peptide (F). Deacetylation reactions were fit to the Michaelis–Menton equation.
Fig. 2G: Yeast Sir2 and Human SIRT1 deacetylation kinetics. Top: kcat and KM values were determined for Sir2 K475 and mutants Sir2 K475E or K475Q. Bottom: kcat and KM values were determined for SIRT1 K444 and mutants SIR2 K444E using the FDL® SIRT1 Assay. Calculated parameters determined from nonlinear regression fits to the Michaelis-Menton eq v=[(kcat/Km)[S]]/(1+[S]/Km))(n≥2, ±SD of the mean).
Kinetic analyses of human mutant enzymes
To determine whether these effects were limited only to yeast Sir2 or extend to other species, we expressed WT SIRT1 K444 and the K444E (K444 corresponds to K475 in Sir2) mutant as His-tagged fusion proteins. We focused exclusively on the K444E mutant in SIRT1 protein as this mutation conferred the greatest effect in Sir2 protein. We followed similar extraction and purification methods as with the yeast proteins to yield ~90% pure proteins (Fig. 2D).
The steady state parameters (KM and kcat) and catalytic efficiency (kcat/KM) were determined using the Fluorde-lys® (FDL®) SIRT1 Assay, which measures deacetylation of an acetyl-Lys substrate (FDL®) comprising AAs 379–382 of human p53 (RHKK(Ac)). We determined the velocities of the WT and mutant enzyme catalyzed reactions with increasing concentrations of FDL® substrate (0, 0.01, 0.02, 0.05, 0.1, 0.25, 0.5, and 1.0 mM) and saturating concentrations of NAD+ (3.0 mM), as well as with increasing concentrations of NAD+ (0, 0.01, 0.05, 0.1, 0.25, 0.5, 1.0, 2.5, and 5.0 mM) and saturating concentrations of FDL® substrate (1.0 mM). Multiple independent reactions were performed. Representative sets of kinetic data for the SIRT1 K444 and SIRT1 K444E catalyzed reactions are shown in Figures 2E, F. Significant differences in the Vmax between WT and mutant enzymes were apparent and the experimentally derived kinetic analyses are described (Fig. 2G Bottom; Supplemental). Our results indicate that K444E SIRT1 is catalytically more active than WT (higher kcat) and has a significantly lower KM for both substrates. Thus, it appears that enhanced catalytic efficiency is the conserved effect of the K to E mutation in yeast Sir2 and mammalian SIRT1. Additional kinetic analyses were performed using the SIRTainty assay to corroborate the FDL® assay. Representative data from K444 and K444E catalyzed reactions performed with the SIRTainty Assay are shown in Fig. S3. Results show increased catalytic efficiency by the K444E enzyme, corroborating results from the FDL® SIRT1 Assay.
We next examined the effect of the mutations on the actions of three compounds, the activator resveratrol and inhibitors suramin and nicotinamide. The NAD+ binding pocket of sirtuins is divided into three regions (Min et al. 2001): the adenine ribose of NAD+ binds the A site; nicotinamide ribose binds B; and the reaction product nicotinamide binds C (Avalos et al. 2005). Suramin binds within the B and C pockets, inhibiting sirtuin reactions by blocking NAD+ access to the catalytic domain. Using the FDL® SIRT1 Assay, we inhibited the sirtuin reaction using a range of suramin (0–1000 μM). Results showed IC50 values of 2.99 μM for K444 and 68.02 μM for K444E (Fig. S4A), indicating that mutant K444E is much more resistant to inhibition by suramin. We next determined the effects of nicotinamide on WT and mutant SIRT1 and found IC50 values of 43.05 μM for K444 and 34.27 μM for K444E (Fig. S4B), indicating at best a slight increase of inhibition of mutant SIRT1 by nicotinamide. We also assessed the effects of resveratrol on the mammalian K444E mutant. The activation peak was shifted from 1 mM to 10 mM in the mutant. Further, the K444E mutant could not be activated by resveratrol to the degree of WT SIRT1 (Fig. S4C). In a recent paper the structure of SIRT1 in complex with resveratrol and a 7-amino-4-methylcoumarin-containing peptide showed binding of a resveratrol trimer in three different conformations, one of which directly interacts with K444 (Cao et al. 2015). We hypothesize that the K444E mutation disrupts this interaction thus explaining the decreased binding and activation by resveratrol.
Mutant SIRT1 enzymes result in decreased acetylation of in vivo SIRT1 substrates
After determining the catalytic differences of both yeast and human mutant proteins in vitro, we wanted to know whether these mutations extended their effects to organisms beside yeast. To determine how mutant human SIRT1 would affect downstream targets in an in vivo cell culture model we evaluated differences in sirtuin activity by measuring the steady state acetylation status of in vivo substrates. We thus performed transient transfections of human epithelial kidney cells (293T) with SIRT1 K444, K444E, and K444Q plasmids. Transfecting 1 μg of each plasmid resulted in increased SIRT1 protein expression relative to endogenous protein expression (Fig. 3A). A pan acetyl-Lys blot of total protein revealed that cells transiently transfected with K444E or K444Q appeared to have less acetylation than K444 of multiple cellular proteins, most notably those in the 10–20 kDa range, where histone proteins run on 4–15% SDS-PAGE gels. It is known that in vivo recruitment of SIRT1 to target promoters leads to gene silencing through histone deacetylation (Vaquero et al. 2007), particularly at H4K16 and H3K9. We specifically probed for levels of H4K16ac and found decreased acetylation by the K444E mutant (Fig. 3B), indicating that specific human substrates can be differentially deacetylated by mutant sirtuins.
Figure 3. SIRT1 expression and activity in vivo.

Fig. 3A, B. Cell lysates prepared from 293T cells transfected with SIRT1 K444, K444E, K444Q plasmids. Protein blots probed for hSIRT1, pan acetyl Lys, α-Tubulin (A), and total histone 4 (H4) and H4K16Ac (B). Quantification of SIRT1 protein expression performed using ImageJ, relative to tubulin and normalized to 1. Quantification of H4K16ac relative to total H4 performed using ImageJ and normalized to 1.
Fig 3C: Cell lysates prepared from SIRT1-null MEFs stably transfected with K444, K444E, K444Q SIRT1 lentiviral expression vectors (two independent viral infections). Protein blots probed for SIRT1, H4K16ac, H4 and α-Tubulin. Quantification performed using ImageJ and normalized: SIRT1 levels relative to α-Tubulin; H4K16ac levels relative to H4. Mutant protein expression levels are relative to WT expression that is normalized to 1. SDs indicate differences between stable lines.
Fig. 3D: Scatter dot plots of RT-qPCR data of SIRT1 mRNA expression in WT MEFs, SIRT1-null MEFs, and stable MEF cells expressing SIRT1 WT, K444E, or K444Q. SIRT1 expression is relative to GAPDH and Actin mRNA and normalized to WT MEF levels of SIRT1. Performed one-way ANOVA statistical test used with Dunnett’s multiple comparisons test to detect differences in cell types relative to SIRT1 WT condition. 95% confidence intervals are indicated. Statistically significant differences (p<0.05) are indicated in the Supp. Table.
We next used SIRT1-null mouse embryonic fibroblasts (MEFs) to produce stable cell lines expressing K444, K444E, or K444Q. Fig. 3C shows representative blots of two independent sets of cell lines, illustrating a significant reduction in the acetylation of H4K16ac in cells expressing SIRT1 K444E and to a slightly lesser extent, K444Q. We observed higher levels of the mutant SIRT1 proteins in these stable lines. To determine whether this was due to differences in gene expression, we performed real time RT-qPCR and found all three stable cell lines expressed similar levels of SIRT1 RNA (Fig 3D; Supp. Table). This finding suggested that the mutant proteins might be more stable than WT, as we address below, and indicates that the increased deacetylation of H4K16 by SIRT1 mutants may be due to higher catalytic activity plus increased protein levels.
Predictive protein modeling suggests enhanced structural stability of mutant sirtuin proteins
To gain mechanistic insight into how the mutations might result in more catalytically active enzymes, and whether the stability of the protein could be altered due to the mutations, we used predictive protein modeling to examine how these single AA changes may affect enzyme stability and function. The conserved domain sequence (residues 231–555) of Sir2 was submitted to the ENCoM web server (Frappier et al. 2015; Supplemental) and ΔΔG values were calculated for WT and compared to K475 mutant enzymes, with negative ΔΔG values indicating increased thermodynamic structural rigidity. Strikingly, both K475E and K475Q mutations increased rigidity across the entire 231–555 domain in a rank order that follows their increased kcat values (Fig. 4A). Next, we used ENCoM to calculate how each K475 mutation affected the rigidity at each residue of the 231–555 domain compared to WT using the area under the curve (AUC) of receiver operator curves (ROC). AUCs were distributed from [0.00–0.5] (blue to red), where an AUC=0 corresponds to cases where the mutants had the most pronounced rigidifying effect (blue) and AUC=0.5 represents cases where the mutants have an average effect (red) (Fig. 4B). This analysis reveals regions of increased rigidity by both mutants compared to WT encompassing two main regions, A and B (B includes K475 itself). These regions are mapped onto the Sir2 structure indicating the average effect of both K475E and K475Q mutations and reveal a close proximity of K475 in region B and R275 in region A (Fig. 4C: region A=teal, region B=blue). The structure indeed predicts that K475 and R275 exhibit a repulsive charge interaction (Fig. 4D). Moreover, we hypothesize that in the K475 mutants attractive charge-charge (K475E) or hydrogen bonding (K475Q) can now occur, which may increase the catalytic cleft stability (Fig. 4D). Notably, R275 is at the acetyl-Lys binding cleft and is highly conserved in SIR2 orthologs.
Figure 4. Effect of mutations on sirtuin rigidity and predictive modeling of Sir2 structure.

Fig. 4A: Graph of calculated ΔΔG (difference in Gibbs free energy between WT and mutant) for WT and mutant Sir2 protein residues 231–555 versus experimentally derived kcat values (Fig. 2). A strong correlation between ΔΔG and kcat is evident.
Fig. 4B: Heatmap of ΔΔG values for each residue of mutant versus WT. Blue residues indicate negative ΔΔG and increased rigidity in mutant, white residues indicate no differences, red residues indicate decreased rigidity. Two distinct regions, A and B of residues positively affected by the K475 mutation are indicated. Average trace indicates the calculated average ΔΔG values as a result of K475E, K475Q mutations combined.
Fig. 4C: Structure of Sir2 with regions A (blue) and B (teal). Note proximity of K475 and R275, within regions B and A respectively.
Fig. 4D: Schematic of Sir2 catalytic domain with putative chemical interactions between residues Ser473, Arg275 and Lys475 and Glu475.
Fig. 4E Top: AUC heatmap represents the residue-by-residue rigidity rank of the K475E, K475Q mutants versus all other possible AA substitutions (6156) in the 231–555 domain (blue=more rigid, white=less rigid).
Fig. 4E Bottom: Conservation heatmap represents the conservation among sirtuin sequences analyzed using CONSURF (blue=high conservation; red=no conservation).
Fig. 4F: AUC (Fig. 4E) projected onto Sir2.
Fig. 4G: Conservation map (Fig. 4E) projected onto Sir2. Note the large overlap between Figs. 4F and 4G, notably within A, B regions.
Since K475 and R275 are highly conserved residues and it is well established that conserved residues are often crucial for protein function, we examined the conservation level of 150 sirtuin and sirtuin-related sequences using CONSURF (Ashkenazy et al. 2016). Residues forming the NAD+ and protein substrate binding pocket were identified by contact greater than 10 Å using a Voronoi surface algorithm between ligand and residues within the crystal structure (McConkey et al. 2002). Blue residues indicate high conservation and red no conservation among species (Fig. 4E). Note that the most conserved regions in the AUC plot include regions A and B of Fig. 4B.
Next we compared the stabilization effects of the K475 mutations with all 6156 possible AA substitutions across the 231–555 domain (324 residues multiplied by 19 AAs; minus STOP residues). We calculated a ΔΔG score for each of the 6156 mutations versus WT, and ranked their ΔΔG effects at each residue compared to the K475 mutations again using AUCs of the ROC (Fig. 4E). Blue indicates that the K475 mutants had the most pronounced rigidifying effect compared to the 6156 changes, and white indicates that the mutants have an average effect compared to the other 6156 mutants. The strongest rigidifying effects of the K475 mutations compared to the other 6156 possible changes again are in regions A and B. These same regions are projected onto the structure (Fig. 4F) distributed from more rigid (blue) to less rigid (white) and correspond well with a projection of the most highly conserved residues onto the structure (Fig. 4G). This structural analysis gives strong theoretical evidence that the K475 mutations increase the rigidity of the catalytic domain, possibly by creating charge or ionic interactions with R275, and that these changes in local dynamic properties may contribute to increased stability and increased catalytic efficiency in mutant enzymes. This might occur, for example, by reducing the time the protein is in a breathing (inactive) state.
Mutant human SIRT1 shows increased protein stability in vivo and in vitro
The findings of ENCoM analysis predict that the mutant SIRT1 proteins display increased stability in vivo. We therefore assayed whether the higher levels of SIRT1 mutants were a result of increased protein stability. Various studies have measured SIRT1 half-life at between 4 and 12 hr (Gao et al. 2011; Ford et al. 2008) after using cycloheximide (CHX) to inhibit protein synthesis (Stocklein & Piepersberg 1980). SIRT1-null MEF lines expressing K444, K444E, and K444Q SIRT1 were treated with DMSO (solvent control) (Fig. 5A) or CHX (Fig. 5B) and protein lysates were collected at 0, 1, 2, 4, 8, 12 hr post-treatment. Our results show that WT SIRT1 protein starts declining at 4 hr, with a significant loss of protein expression at 8 and 12 hr post-treatment (Fig. 5B). In contrast, both mutant SIRT1 proteins only start to decline at 8 hr; remaining >3 fold higher at 8 and 12 hr than WT protein (Fig. 5C, D). We also performed thermal shift assays of purified WT and mutant Sir2 proteins to determine the melting temperature (Tm) and thus stability of the enzymes. We determined that the Tm increased in a rank order with highest average Tm of 48.4°C for K475 Sir2, 50.9°C for K475Q, and 52.8°C for K475E (Fig. S5A, B). Thus, both mutant proteins demonstrate evidence of increased thermostability in vitro and are also significantly more stable in cells than WT protein.
Figure 5. In vivo effect of human SIRT1 mutations on protein expression and stability.

Fig. 5A, B: SIRT1-null MEFs stably transfected with K444, K444E, K444Q SIRT1 lentiviral expression vectors. Cells treated with CHX (representative of three independent experiments shown) or DMSO (one experiment) for increasing time lengths. Loss of SIRT1 protein expression evaluated via Western blotting and densitometry.
Fig. 5C: Optical density of SIRT1 protein quantified using ImageJ, normalized to β-actin, and protein expression levels at t=0 (relative to 1) were plotted. Data represent the mean ± SD of three independent experiments. Lines drawn using linear regression of data sets using GraphPad Prism.
Fig. 5D: Data at 8, 12 hr post CHX treatment indicating significant (p<0.001) differences between mutant and WT proteins (2way ANOVA, Dunnet’s multiple comparisons test) (K444 vs. K444E p=0.0004, K444 vs. K444Q p=0.0002).
Discussion
In this study we used a genetic screen to produce sirtuins capable of functioning in an NAD+-depleted environment. Sirtuins share a highly conserved catalytic domain consisting of distinct sites that bind NAD+ and protein substrates, and are sensitive to cellular NAD+ levels. As NAD+ levels decrease with age, NAD+-dependent sirtuins decrease in activity. We thus genetically engineered yeast to have a 3–4 fold reduction in NAD+ levels, which was sufficient to abolish the red colony color due to Sir2-mediated silencing of an ADE2 gene. After mutagenesis across the conserved domain of SIR2, we screened for red transformants and identified two adaptive mutations in residue K475, which restored Sir2 activity in an NAD+-limited environment and extended yeast RLS. The in vitro kinetics analyses indicated modestly decreased KM for NAD+ by the K475Q mutant and for peptide by the K475E mutant. However, the most consistent effect observed in the mutant sirtuins was a 2–3 fold increase in kcat, indicating a significant increase in catalytic efficiency.
Importantly, K475 is conserved in all Sir2 orthologs. Replacing K444 (analogous to Sir2 K475) of human SIRT1 with E also increased the kcat of the human enzyme, while lowering the KM for both substrates. Moreover, the K444E and K444Q mutants gave rise to a higher degree of deacetylation in human and murine cells than WT. In particular, histone H4K16, a known substrate for Sir2 orthologs, was more deacetylated in cells expressing the mutants. These findings indicate a striking functional conservation in this catalytically-placed Sir2 Lys, and show that the increase in catalytic efficiency conferred by this mutation extends to mammalian SIRT1.
To gain mechanistic insight into how our mutations result in more catalytically efficient enzymes, we looked into the structural changes that might occur using predictive protein modeling. The dynamic regulation of biochemical processes in the cell involves entropic effects and molecular forces between interacting groups, such as between sirtuins and their substrates (Leckband & Israelachvili 2001). The analysis of our mutants predicts a decreased ΔΔG, suggesting increased rigidity of the catalytic core. This analysis also suggests that the Lys mutations may exert their effects by introducing a favorable interaction with a conserved Arg (R275 in Sir2). Increased catalytic site rigidity can enhance stability and catalysis of some enzymes (Xie et al. 2014). Consistent with the predicted increase in stability, we demonstrated increased melting temperatures of mutant Sir2 enzymes in vitro and increased half-lives of mutant human sirtuins in cultured mammalian cells compared to WT protein. There are examples of improved enzymes produced using methods similar to ours, such as LovD, a natural enzyme that was transformed using a genetic screen (Jiménez-Osés et al. 2014) into a variant 1000-fold more efficient at synthesizing the drug, simvastatin, than WT LovD. This variant achieved enhanced catalysis as measured by a significant increase of kcat, not through significant changes of binding affinity (KM). The mutations altered the conformational dynamics of the enzyme’s catalytic domain, leading to enhanced rigidity and higher thermostability. Notably, whereas our mutant sirtuins contain only one mutated residue and increased catalytic efficiency by 2–3 fold, mutant LovD alters 29 residues. It is plausible that sirtuin enzymes can be optimized further by including changes predicted by our modeling.
While our nicotinamide inhibition does not show significant differences between WT or mutant enzymes, we do demonstrate differential effects of mutant sirtuins compared to WT in response to resveratrol and suramin. For suramin, a large increase in the Ki was observed, which may be due to the predicted higher rigidity of the catalytic core. For example, if breathing of this structure is required for suramin to bind, a lower affinity might be expected for the mutants. Of course, a more rigid catalytic domain could also inhibit the binding of certain Sir2 substrates, which require breathing of the catalytic core. In this regard, it will be of interest to examine the effects of SIRT1 K444E and K444Q mutations on many cellular substrates by a proteomics approach. For resveratrol there appeared to be both an increase in Km and a decrease of maximal activation. Both may be due to the fact that structural studies show that resveratrol contacts K444 (Cao et al., 2015).
In yeast, integrated copies of the mutant SIR2 genes gave rise to increased silencing compared to WT. In this case, the expression levels of all proteins were indistinguishable, so we attribute the hypermorphic phenotype of the mutants to their altered catalytic properties. In mammalian cells, however, stable lines showed slightly higher levels of mutant proteins compared to WT. Thus we cannot be certain whether the increase in deacetylation observed in mammalian cells expressing SIRT1 mutants is driven by higher catalytic activity, higher protein levels, or both. The higher protein levels are consistent with the mutations conferring a higher intrinsic stability. Differences in the protein turnover machinery or negative feedback regulation (Michel et al. 2005) are possible explanations for why higher mutant protein levels were not observed in yeast.
The conserved effect of the mutations suggests a possible strategy for increasing SIRT1 activity separate from the use of conventional SIRT1 activating compounds. These compounds, including polyphenols like resveratrol and additional compounds under development at GSK, work by lowering the KM of the enzyme for the protein substrate. We imagine that an assay similar to ours could screen for small molecules that activate yeast Sir2 in low NAD+ cells to reveal lead compounds that function like our Lys mutations and increase the catalytic efficiency of the enzyme. Such molecules may provide another translational path for activating SIRT1. Moreover, they may also activate SIRT7, which also has a Lys at the analogous position as K444 in SIRT1, and thereby exert a unique effect on cells.
In summary, we discovered that mutations in the previously uninvestigated residue, K475, to E or Q result in catalytically improved enzymes. This Lys is conserved in all Sir2 orthologs, and these mutations exert similar effects on the human enzyme. Computational and experimental methods suggest that the mutations increase the stability of two regions in the catalytic core of the enzyme. We hypothesize that by increasing the rigidity and thus the stability of the catalytic domain, the K475E and K475Q enzymes breathe less and are locked into an active state, allowing faster deacetylation. Since the mutant Sir2 orthologs might function in the NAD+-depleted setting of aging, our findings may potentiate a strategy for treating diseases of aging.
Experimental Procedures (Details in Supplemental)
NAD+ Quantification
NAD+ quantification was performed using the NAD/NADH Assay (Promega) following the manufacturer’s protocol and yeast specific methods. NAD+ levels are indicated in relative light units (RLU), relative to total protein expression.
Yeast Replicative Lifespan Analysis
Micromanipulation and RLS analysis of yeast were performed with 20–50 mother cells/experiment and carried out at least three times.
Sir2 Deacetylation Assay
Deacetylation of a synthetic 11-mer peptide corresponding to Lys 14 of histone H3 plus surrounding residues by WT and mutant Sir2. Reaction products were separated by HPLC on a Kinetex XB-C18 column and product conversion was determined by comparing the ratio of deacetylated to acetylated peptide substrate.
SIRT1 Activity: Fluor-de-lys® Assay
The steady state parameters (Km and kcat) and catalytic efficiency (kcat/Km) of recombinant human SIRT1 were determined using the FDL® SIRT1 Fluorometric Assay. All rate measurements were obtained from three or more replicates and performed independently at least two times.
RNA Extraction and Quantitative Real-Time PCR
Total RNA was extracted from cells by the RNeasy Mini Kit. cDNA was synthesized using the RETROscript kit. All PCR reactions were performed in triplicate with SYBR Green in a Light Cycler 480. Relative RNA expression levels were estimated using the ΔΔCT method with normalization to GAPDH and β-actin.
Inhibition of de Novo Protein Synthesis
MEFs were treated with 150 μg/ml cycloheximide or DMSO. Total protein samples were harvested and protein concentration verified using BCA. Total protein was loaded on precast protein gels (Biorad) and analyzed via Western blotting.
Statistical Methods
Graphs were made and statistical analyses performed using GraphPad Prism. Statistical analysis of yeast lifespan was determined using the log-rank (Mantel-Cox) test. Kinetic analyses were performed using the Michaelis-Menton model. Statistical analysis for RT-qPCR was determined using 2-way ANOVA, Dunnett’s multiple comparisons test. Inhibition studies were analyzed using nonlinear exponential decay. Analyses of paired values of data were performed with paired two-tailed t-tests.
Supplementary Material
Highlights.
Adaptive mutations in yeast Sir2 permit function with low NAD+
Structural rigidity of catalytic domain and overall protein stability is enhanced
Catalytic efficiency is increased in mutant enzymes
Mutants increase yeast replicative lifespan and deacetylation of mammalian substrates
Acknowledgments
This work was supported by grants from NIH and the Glenn Foundation for Medical Research to LG and by National Institute of General Medical Sciences Grants to CW. Thanks to Jonathan Renn and David Colino for critical input, and Luke Brezovec and Virginia A. Adams for lab assistance. Thanks to Su-Ju Lin and Matt Kaeberlein for sharing yeast strains and Stephen Bell for providing plasmids. LG consults for GSK, Segterra, Sebelius and is a founder of Elysium Health.
Footnotes
Author Contributions
CRO developed project ideas, performed experiments, and wrote the manuscript; VF assisted with predictive protein modeling; AER assisted with Sir2 kinetic analyses; CW provided laboratory, equipment, and consultation; LG developed project ideas.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nature Rev Genet. 2016;17(8):487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44(W1):W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos JL, et al. Mechanism of Sirtuin Inhibition by Nicotinamide: Altering the NAD+ Cosubstrate Specificity of a Sir2 Enzyme. Mol Cell. 2005;17(6):855–868. doi: 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Bedalov A, et al. NAD+-Dependent Deacetylase Hst1p Controls Biosynthesis and Cellular NAD+ Levels in S. cerevisiae. Mol Cell Biol. 2003;23(19):7044–7054. doi: 10.1128/MCB.23.19.7044-7054.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol. 2016;17(11):679–690. doi: 10.1038/nrm.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, et al. Age Related Changes in NAD+ Metabolism Oxidative Stress and Sirt1 Activity in Wistar Rats A. Xu, ed. PLoS ONE. 2011;6(4):e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cao D, et al. Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 2015;29(12):1316–1325. doi: 10.1101/gad.265462.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford J, et al. JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle. 2008;7(19):3091–3097. doi: 10.4161/cc.7.19.6799. [DOI] [PubMed] [Google Scholar]
- Frappier V, Najmanovich R. Vibrational entropy differences between mesophile and thermophile proteins and their use in protein engineering. Prot Sci. 2015;24(4):474–483. doi: 10.1002/pro.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, et al. Sirtuin 1 (SIRT1) Protein Degradation in Response to Persistent c-Jun N-terminal Kinase 1 (JNK1) Activation Contributes to Hepatic Steatosis in Obesity. J Biol Chem. 2011;286(25):22227–22234. doi: 10.1074/jbc.M111.228874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Zhou Z. SIRTain regulators of premature senescence and accelerated aging. Protein Cell. 2015;6(5):322–33. doi: 10.1007/s13238-015-0149-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27(19):2072–2085. doi: 10.1101/gad.227439.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, et al. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24(8):464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403(6771):795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Jiménez-Osés G, et al. The role of distant mutations and allosteric regulation on LovD active site dynamics. Nat Chem Biol. 2014;10(6):431–6. doi: 10.1038/nchembio.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leckband D, Israelachvili J. Intermolecular forces in biology. Q Rev Biophys. 2001;34(2):105. doi: 10.1017/s0033583501003687. [DOI] [PubMed] [Google Scholar]
- Lin S-J, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol. 2003;15(2):241–246. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- Massudi H, et al. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PloS One. 2012;7(7):e42357. doi: 10.1371/journal.pone.0042357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey BJ, Sobolev V, Edelman M. Quantification of protein surfaces, volumes and atom-atom contacts using a constrained Voronoi procedure. Bioinformatics. 2002;18(10):1365–1373. doi: 10.1093/bioinformatics/18.10.1365. [DOI] [PubMed] [Google Scholar]
- Michel AH, et al. Spontaneous rDNA copy number variation modulates Sir2 levels and epigenetic gene silencing. Genes Dev. 2005;19(10):1199–210. doi: 10.1101/gad.340205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, et al. Crystal Structure of a SIR2 Homolog–NAD Complex. Cell. 2001;105(2):269–279. doi: 10.1016/s0092-8674(01)00317-8. [DOI] [PubMed] [Google Scholar]
- Park J, et al. SIRT5-Mediated Lys Desuccinylation Impacts Diverse Metabolic Pathways. Mol Cell. 2013;50(6):919–930. doi: 10.1016/j.molcel.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279(49):50754–63. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839(8):627–43. doi: 10.1016/j.bbagrm.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97(12):6658–63. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocklein W, Piepersberg W. Binding of Cycloheximide to Ribosomes from Wild-Type and Mutant Strains of S. cerevisiae. Antimicrob Agents Chemother. 1980;18(6):863–867. doi: 10.1128/aac.18.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero A, Sternglanz R, Reinberg D. NAD+-dependent deacetylation of H4 Lys 16 by class III HDACs. Oncogene. 2007;26(37):5505–20. doi: 10.1038/sj.onc.1210617. [DOI] [PubMed] [Google Scholar]
- Xie Y, et al. Enhanced Enzyme Kinetic Stability by Increasing Rigidity within the Active Site. J Biol Chem. 2014;289(11):7994–8006. doi: 10.1074/jbc.M113.536045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.