

Abstract
Ischemia-reperfusion injury (IRI) is a common cause of acute kidney injury leading to an induction of oxidative stress, cellular dysfunction, and loss of renal function. DNA damage, including oxidative base modifications and physical DNA strand breaks, is a consequence of renal IRI. Like many other organs in the body, a redundant and highly conserved set of endogenous repair pathways have evolved to selectively recognize the various types of cellular DNA damage and combat its negative effects on cell viability. Severe damage to the DNA, however, can trigger cell death and elimination of the injured tubular epithelial cells. In this minireview, we summarize the state of the current field of DNA damage and repair in the kidney and provide some expected and, in some cases, unexpected effects of IRI on DNA damage and repair in the kidney. These findings may be applicable to other forms of acute kidney injury and could provide new opportunities for renal research.
Keywords: DNA repair, TRIP13, acute kidney injury, tubular epithelial cells
acute kidney injury (AKI) remains a major problem, affecting up to two-thirds of intensive care unit patients (16), and can result in increased hospitalization time, increased risk of developing chronic kidney disease, and possibly death (5, 7, 19, 25). Ischemia-reperfusion injury (IRI), a common cause of AKI, can exert varying degrees of energetic stress on tubular epithelial cells, particularly the proximal tubules and thick ascending limb of Henle (3, 4). In the face of sublethal stress, tubular epithelia are highly adaptable and compensate for the cellular damage by dedifferentiating to a primordial state, where repair and proliferative pathways are activated in an attempt to reestablish normal structure (3, 4, 11, 24, 46, 53). Some tubular cells are unable to cope with the biological injury and progress toward cell death. This can lead to partial or, in some cases, complete loss of renal function (3, 4). The mechanisms involved in determining the fate of the cell following IRI remain to be fully understood.
In the past few years, there has been increased interest in the role of DNA repair pathways and their influence on cell viability following IRI. Renal IRI produces copious amounts of reactive oxygen and nitrogen species, which can modify the composition and structure of an individual base or a string of bases or possibly, generate DNA strand breaks in both the nucleus and mitochondria (28, 45, 49, 54). Preservation of DNA integrity is of paramount importance to the cell, since perturbations in the proper structure and function of the DNA can initiate abnormal cellular pathologies (21, 37). Similar DNA repair systems have been identified in the mitochondria and nucleus (29), but little is known about the mitochondrial DNA repair system during IRI. Therefore, we will summarize the repair pathways involved in the restoration of nuclear DNA damage and their influence on tubular epithelial cell viability following ischemic AKI.
DNA Damage Response in Renal Tubules
In general, the kidney is an organ undergoing low levels of mitosis. Tubular epithelial cells are predominantly in a semipermanent G0 phase, where only 1% of the cells enter a state of active proliferation (44). Upon exposure to a genotoxic stress, such as IRI, cellular DNA can be modified in the nucleus or the mitochondria of the injured tubular epithelia. In the nucleus, the DNA damage response (DDR) system is activated, and unidirectional entry into the cell cycle is enacted by the injured epithelia. In fact, a majority (>70%) of the recovering tubular epithelial cells progress to the DNA synthesis (S) phase to prepare for the proliferative stage (17, 18).
Cell cycle progression is controlled by the DDR system, a sophisticated system consisting essentially of two phases: a surveillance, or sensor, phase and an effector phase (48). Upstream sensor kinases (e.g., ataxia telangiectasia-mutated, ataxia telangiectasia- and Rad3-related, and DNA protein kinase) are activated selectively, depending on the type and severity of the DNA damage to the kidney (see below) (15, 28, 30, 32, 33). Subsequently, the sensor information relays the state of the DNA to downstream effector kinases, such as checkpoint kinases 1 and 2. These effector kinases regulate other downstream effectors, such as p53 (35) or cell division cycle 25 (50), to halt the cell cycle at specific checkpoints to ensure that the fidelity of the DNA remains intact before permitting DNA replication (S phase) and cell division (M phase) (20, 43). Tubular cells arrested in the G1 phase exhibited a protective effect following AKI (9, 11). However, blockade of cell cycle progression at the G2/M checkpoint led to augmented activation of apoptosis through a JNK-dependent pathway and also promoted profibrotic TGFβ signaling (11, 45, 51, 54).
These findings demonstrate that the cell cycle status regulated by the DDR system plays an intricate part in the recovery of the kidney by providing sufficient time for injured cells to restore their normal architecture and organ function and also to remove damaged cells that cannot recover from the injurious stimuli (12, 30, 41, 43).
Mechanisms of DNA Damage Repair System in AKI
As shown in Fig. 1, biological insults lead to a variety of nuclear modifications that require correction through a number of excision repair systems, including nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR). Physical DNA breaks, such as single- or double-stranded breaks, which are more severe forms of DNA damage, are repaired by homologous recombination (HR) or nonhomologous end joining (NHEJ) (20). Examples of each of these types of repair mechanisms with respect to ischemic AKI are discussed below.
Fig. 1.
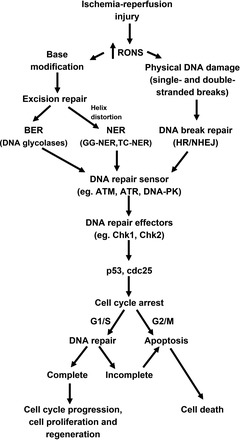
DNA repair mechanisms in ischemic acute kidney injury. Basic overview displays mechanisms by which ischemia-reperfusion injury produces reactive oxygen and nitrogen species (RONS), leading to DNA damage in the renal tubular epithelial cells. Various forms of DNA repair mechanisms are involved in simple base changes with [nuclear excision repair (NER)] or without [base excision repair (BER)] helix distortion or more lethal physical strand breaks [homologous recombination (HR)/nonhomologous end joining (NHEJ)]. Depending on the type of DNA damage, sensor kinases, including ataxia telangiectasia-mutated (ATM), ataxia telangiectasia- and Rad3-related (ATR), and DNA protein kinase (DNA-PK), can initiate a cascade of signaling pathways that activate effector systems, checkpoint kinase 1/2 (Chk1/2), to control cell cycle progression through the activity of p53 or cell division cycle 25 (cdc25). Arrest in the cell cycle allows the cell to determine that the damaged DNA can be fully repaired and proceed to cellular recovery or that cell death needs to occur because DNA damage is severe and DNA repair cannot be completed. GG-NER, global genome NER; TC-NER, transcription-coupled NER.
DNA excision repair.
Of the three types of excision repair, only BER and NER have been described as coping mechanisms for DNA damage in renal cells following IRI. No published studies have investigated the role of MMR in IRI.
In IRI, the most prevalent forms of DNA damage are hydrolytic, alkylated, or oxidative base modifications mediated by free radicals and other reactive species (20). These types of DNA modifications are primarily repaired by a distinct group of DNA glycolases in the BER pathway that can selectively recognize and remove individual bases or short stretches of lesioned DNA throughout the cell cycle (20). As an example, the accretion of 8-oxo-2-deoxyguanosine (8-oxo-dG), the most common type of oxidative base modification, was readily detected in kidneys following IRI (49). This was concomitantly associated with increased expression of 8-oxoguanine DNA glycolase (OGG1), which selectively removes 8-oxo-dG (49). From these observations, one might infer that DNA glycolases, such as OGG1, have a positive influence on tubular recovery by repairing the damaged DNA. However, mice deficient in another DNA glycolase, alkyladenine DNA glycosylase (AAG), which removes a broader range of modified nucleotides, actually demonstrated an unexpectedly reduced level of damage to the renal tubular epithelia following IRI compared with AAG+/+ mice (10). In light of these findings, our view on the role of DNA glycolases in the BER pathway to positively influence cell recovery needs to be revisited.
The NER pathway involves the activation of two distinct modes of excision repair when distortions in the DNA helical structure due to the appearance of modified nucleotides are detected. The first mode is global genome (GG) NER, which repairs bulky DNA lesions located throughout the entire genome, even in intervening sequences (13). The second mode is transcription-coupled (TC) NER, which fixes damaged DNA where the replication fork has collapsed or RNA polymerase-directed transcription has stalled (31). TC-NER operates to restore DNA integrity at the damaged site by recruiting proteins, including Cockayne syndrome A (Csa) and B (Csb), to aid in the repair process and enable RNA polymerase II to proceed (31). Using a congenital mouse model of NER insufficiency, which is deficient in Csa or Csb, Susa et al. (47) reported reduced tubular injury from the damaging effects of renal IRI. Similar to AAG deficiency, TC-NER appears to play a deleterious role during tubular epithelial cell recovery.
The reasons for the divergent effects mediated by the various excision pathways to either promote beneficial recovery of the injured tubular epithelia or activate death pathways are not fully known. During IRI, imbalanced repair may occur because of the inability of some BER enzymes to meet the increased DNA repair capacity due to the increased accumulation of modified bases. DNA glycolases, such as AAG, generate toxic DNA intermediates during the repair process and, therefore, depending on the efficiency of removal of these intermediates, would lead to differences in the extent of tissue damage (6). Alternatively, competition by multiple repair pathways could affect the efficiency in correcting the DNA damage and influence the viability of the damaged cells (14). In NER, the unwinding of DNA is ATP-dependent, which may not be favorable in injured tubular epithelial cells, where energy could be diverted to other survival pathways, leading to incomplete repair and increased cellular damage (43). There is a clear need to further study the importance of each excision pathway and the role of these pathways in determining the fate of the damaged tubular epithelia.
DNA break repair.
Physical breakage of DNA, a more toxic form of damage, can occur during the initial injury phase following exposure to free radicals (29, 35). In addition, physical breaks in double-stranded DNA are part of the normal biological process involved in DNA replication and chromosome segregation before cytokinesis of newly formed epithelial cells. As mentioned above, the primary modes available to repair physically broken DNA strands are HR and NHEJ (15, 30, 32, 33, 43). Of the two mechanisms, HR resolves double-stranded DNA breaks (DSB) at a higher fidelity than in the case of NHEJ; HR functions primarily during the S and G2 phases of the cell cycle, whereas NHEJ repairs the majority of DNA breaks during the G1 phase (20).
Mechanistically, single- or double-stranded DNA break repair is initiated at the site of DNA damage by the formation of protein complexes consisting of Mre11, Rad50, and Nbs1 (MRN) (39). This key protein complex recognizes DSB leading to cell cycle arrest, so either NHEJ or HR mechanisms could commence repair of the damaged DNA (8). There is a lack of empirical data regarding the role of the MRN complex during renal IRI, but there is some evidence for its role in another common type of AKI, i.e., toxic chemical exposure with cisplatin (23). Pharmacological inhibition of the MRN complex hampered cell viability by reducing cell proliferation (22). Moreover, in mice, NAD(P)H:quinone oxidoreductase 1 deficiency, which demonstrated reduced expression of the MRN complex, was associated with increased tubular damage following cisplatin administration (22). In addition, the MRN complex functions in concert with thyroid receptor-interacting protein 13 (TRIP13), which is a regulator of double-stranded break repair and spindle apparatus checkpoint, to promote cell survival in nonrenal cells (38, 42). Our laboratory has identified increased transcript levels of Trip13 following renal IRI (40), and overexpression of TRIP13 exaggerated DNA repair through the activation of DNA-PK in the NHEJ pathway, resulting in aberrant oncogenic cell proliferation (2). In the kidney, lack of TRIP13 production following renal IRI led to persistent tubular damage (40), which could be associated as observed in non-renal cells with a failure to complete mitosis or meiosis due to increased accumulation of DSB (26, 27, 34, 36, 52). Unlike the varying effects mediated by the excision repair systems on cell survival, these studies illustrate that the completion of DNA strand break repair, particularly DSB, is essential to maintain cell viability following DNA damage. However, further investigation is needed to better understand the role of single- and double-stranded DNA damage pathways in the kidney, particularly in the context of IRI.
Conclusion and Perspectives on the Field
Modifications to the genetic code are a normal part of life for all living organisms, and all cells have evolved DNA repair systems to provide checks and balances to maintain DNA integrity. In some cases, however, a fully intact set of DNA repair mechanisms was not beneficial to recovery of the damaged tubular epithelia following renal IRI. It is probable that the damaged cells during IRI have a compromised ability to properly complete the DNA repair process, which could lead to the manifestation of other renal pathologies, including chronic kidney disease (1). Our current level of knowledge regarding DNA damage repair in kidney disease remains at a nascent stage, so further investigation is clearly necessary to fully elucidate the importance of each distinct DNA repair pathway in terms of timing, specificity of action, and biological response on the fate of the tubular epithelia following injury. Exploiting the benefits of DNA repair pathways may help uncover new therapeutic targets in the treatment of ischemic and other types of AKI.
GRANTS
This work is funded by National Institute of Diabetes and Digestive and Kidney Diseases Grant RO1 DK-90123.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D.P. and F.P. conceived and designed the research; J.D.P. and F.P. analyzed the data; J.D.P. and F.P. prepared the figure; J.D.P. and F.P. drafted the manuscript; J.D.P. and F.P. edited and revised the manuscript; J.D.P. and F.P. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Sean J. Jansen for assistance with manuscript preparation.
REFERENCES
- 1.Aamann MD, Norregaard R, Kristensen ML, Stevnsner T, Frokiaer J. Unilateral ureteral obstruction induces DNA repair by APE1. Am J Physiol Renal Physiol. 310: F763–F776, 2015. doi: 10.1152/ajprenal.00613.2014 . [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R, Russo N, Liu M, Basrur V, Bellile E, Palanisamy N, Scanlon CS, van Tubergen E, Inglehart RC, Metwally T, Mani RS, Yocum A, Nyati MK, Castilho RM, Varambally S, Chinnaiyan AM, D’Silva NJ. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nat Commun 5: 4527, 2014. doi: 10.1038/ncomms5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353, 2012. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger K, Moeller MJ. Mechanisms of epithelial repair and regeneration after acute kidney injury. Semin Nephrol 34: 394–403, 2014. doi: 10.1016/j.semnephrol.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Bhandari S, Turney JH. Survivors of acute renal failure who do not recover renal function. QJM 89: 415–421, 1996. doi: 10.1093/qjmed/89.6.415. [DOI] [PubMed] [Google Scholar]
- 6.Çağlayan M, Wilson SH. Oxidant and environmental toxicant-induced effects compromise DNA ligation during base excision DNA repair. DNA Repair (Amst) 35: 85–89, 2015. doi: 10.1016/j.dnarep.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 8.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, Ferguson DO. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol 16: 808–813, 2009. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiRocco DP, Bisi J, Roberts P, Strum J, Wong KK, Sharpless N, Humphreys BD. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am J Physiol Renal Physiol 306: F379–F388, 2014. doi: 10.1152/ajprenal.00475.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebrahimkhani MR, Daneshmand A, Mazumder A, Allocca M, Calvo JA, Abolhassani N, Jhun I, Muthupalani S, Ayata C, Samson LD. Aag-initiated base excision repair promotes ischemia reperfusion injury in liver, brain, and kidney. Proc Natl Acad Sci USA 111: E4878–E4886, 2014. doi: 10.1073/pnas.1413582111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 36: 133–140, 2011. doi: 10.1016/j.tibs.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillet LC, Schärer OD. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem Rev 106: 253–276, 2006. doi: 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- 14.Guo J, Hanawalt PC, Spivak G. Comet-FISH with strand-specific probes reveals transcription-coupled repair of 8-oxoguanine in human cells. Nucleic Acids Res 41: 7700–7712, 2013. doi: 10.1093/nar/gkt524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heijink AM, Krajewska M, van Vugt MA. The DNA damage response during mitosis. Mutat Res 750: 45–55, 2013. doi: 10.1016/j.mrfmmm.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Hoste EA, Schurgers M. Epidemiology of acute kidney injury: how big is the problem? Crit Care Med 36, Suppl: S146–S151, 2008. doi: 10.1097/CCM.0b013e318168c590. [DOI] [PubMed] [Google Scholar]
- 17.Humphreys BD, Czerniak S, DiRocco DP, Hasnain W, Cheema R, Bonventre JV. Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci USA 108: 9226–9231, 2011. doi: 10.1073/pnas.1100629108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphreys BD, Valerius MT, Kobayashi A, Mugford JW, Soeung S, Duffield JS, McMahon AP, Bonventre JV. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2: 284–291, 2008. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 19.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 20: 223–228, 2009. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyama T, Wilson DM 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 12: 620–636, 2013. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 461: 1071–1078, 2009. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim YJ, Kim TW, Park SR, Kim HT, Jung DY, Ryu SY, Jung JY. Deletion of NAD(P)H:quinone oxidoreductase 1 represses Mre11-Rad50-Nbs1 complex protein expression in cisplatin-induced nephrotoxicity. Toxicol Lett 243: 22–30, 2016. doi: 10.1016/j.toxlet.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Kim YJ, Kim TW, Park SR, Kim HT, Ryu SY, Jung JY. Expression of the Mre11-Rad50-Nbs1 complex in cisplatin nephrotoxicity. Environ Toxicol Pharmacol 40: 12–17, 2015. doi: 10.1016/j.etap.2015.04.018. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, Liu J, McMahon AP. Defining the acute kidney injury and repair transcriptome. Semin Nephrol 34: 404–417, 2014. doi: 10.1016/j.semnephrol.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levy EM, Viscoli CM, Horwitz RI. The effect of acute renal failure on mortality. A cohort analysis. JAMA 275: 1489–1494, 1996. doi: 10.1001/jama.1996.03530430033035. [DOI] [PubMed] [Google Scholar]
- 26.Li XC, Schimenti JC. Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS Genet 3: e130, 2007. doi: 10.1371/journal.pgen.0030130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma HT, Poon RY. TRIP13 regulates both the activation and inactivation of the spindle-assembly checkpoint. Cell Rep 14: 1086–1099, 2016. doi: 10.1016/j.celrep.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Ma Z, Wei Q, Dong G, Huo Y, Dong Z. DNA damage response in renal ischemia-reperfusion and ATP-depletion injury of renal tubular cells. Biochim Biophys Acta 1842: 1088–1096, 2014. doi: 10.1016/j.bbadis.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin SA. Mitochondrial DNA repair. In: DNA Repair—On the Pathways to Fixing DNA Damage and Errors (Storici F, editor). Rijeka, Croatia: Intech, 2011, chapt 15, doi: 10.5772/871. [DOI] [Google Scholar]
- 30.McGowan CH, Russell P. The DNA damage response: sensing and signaling. Curr Opin Cell Biol 16: 629–633, 2004. doi: 10.1016/j.ceb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Melis JP, van Steeg H, Luijten M. Oxidative DNA damage and nucleotide excision repair. Antioxid Redox Signal 18: 2409–2419, 2013. doi: 10.1089/ars.2012.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 14: 237–245, 2002. doi: 10.1016/S0955-0674(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 33.Mermershtain I, Glover JN. Structural mechanisms underlying signaling in the cellular response to DNA double strand breaks. Mutat Res 750: 15–22, 2013. doi: 10.1016/j.mrfmmm.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miniowitz-Shemtov S, Eytan E, Kaisari S, Sitry-Shevah D, Hershko A. Mode of interaction of TRIP13 AAA-ATPase with the Mad2-binding protein p31comet and with mitotic checkpoint complexes. Proc Natl Acad Sci USA 112: 11536–11540, 2015. doi: 10.1073/pnas.1515358112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan SE, Kastan MB. p53 and ATM: cell cycle, cell death, and cancer. Adv Cancer Res 71: 1–25, 1997. doi: 10.1016/S0065-230X(08)60095-0. [DOI] [PubMed] [Google Scholar]
- 36.Nelson CR, Hwang T, Chen PH, Bhalla N. TRIP13PCH-2 promotes Mad2 localization to unattached kinetochores in the spindle checkpoint response. J Cell Biol 211: 503–516, 2015. doi: 10.1083/jcb.201505114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Driscoll M. Diseases associated with defective responses to DNA damage. Cold Spring Harb Perspect Biol 4: a012773, 2012. doi: 10.1101/cshperspect.a012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pacheco S, Marcet-Ortega M, Lange J, Jasin M, Keeney S, Roig I. The ATM signaling cascade promotes recombination-dependent pachytene arrest in mouse spermatocytes. PLoS Genet 11: e1005017, 2015. doi: 10.1371/journal.pgen.1005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porter-Goff ME, Rhind N. The role of MRN in the S-phase DNA damage checkpoint is independent of its Ctp1-dependent roles in double-strand break repair and checkpoint signaling. Mol Biol Cell 20: 2096–2107, 2009. doi: 10.1091/mbc.E08-09-0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pressly JD, Hama T, Regner KR, Park F. Reduced levels of thyroid receptor interacting protein 13 prevents recovery of tubular epithelial cells following renal ischemia-reperfusion injury (Abstract). FASEB J 30 Suppl 1217.11, 2016. [Google Scholar]
- 41.Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int 76: 604–613, 2009. doi: 10.1038/ki.2009.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roig I, Dowdle JA, Toth A, de Rooij DG, Jasin M, Keeney S. Mouse TRIP13/PCH2 is required for recombination and normal higher-order chromosome structure during meiosis. PLoS Genet 6: e1001062, 2010. doi: 10.1371/journal.pgen.1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73: 39–85, 2004. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 44.Schmitt R, Cantley LG. The impact of aging on kidney repair. Am J Physiol Renal Physiol 294: F1265–F1272, 2008. doi: 10.1152/ajprenal.00543.2007. [DOI] [PubMed] [Google Scholar]
- 45.Schumer M, Colombel MC, Sawczuk IS, Gobé G, Connor J, O’Toole KM, Olsson CA, Wise GJ, Buttyan R. Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am J Pathol 140: 831–838, 1992. [PMC free article] [PubMed] [Google Scholar]
- 46.Srisawat N, Murugan R, Kellum JA. Repair or progression after AKI: a role for biomarkers? Nephron Clin Pract 127: 185–189, 2014. doi: 10.1159/000363254. [DOI] [PubMed] [Google Scholar]
- 47.Susa D, Mitchell JR, Verweij M, van de Ven M, Roest H, van den Engel S, Bajema I, Mangundap K, Ijzermans JN, Hoeijmakers JH, de Bruin RW. Congenital DNA repair deficiency results in protection against renal ischemia reperfusion injury in mice. Aging Cell 8: 192–200, 2009. doi: 10.1111/j.1474-9726.2009.00463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomasova D, Anders HJ. Cell cycle control in the kidney. Nephrol Dial Transplant 30: 1622–1630, 2015. doi: 10.1093/ndt/gfu395. [DOI] [PubMed] [Google Scholar]
- 49.Tsuruya K, Furuichi M, Tominaga Y, Shinozaki M, Tokumoto M, Yoshimitsu T, Fukuda K, Kanai H, Hirakata H, Iida M, Nakabeppu Y. Accumulation of 8-oxoguanine in the cellular DNA and the alteration of the OGG1 expression during ischemia-reperfusion injury in the rat kidney. DNA Repair (Amst) 2: 211–229, 2003. doi: 10.1016/S1568-7864(02)00214-8. [DOI] [PubMed] [Google Scholar]
- 50.Xiao G, Kue P, Bhosle R, Bargonetti J. Decarbamoyl mitomycin C (DMC) activates p53-independent ataxia telangiectasia and rad3 related protein (ATR) chromatin eviction. Cell Cycle 14: 744–754, 2015. doi: 10.1080/15384101.2014.997517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye Q, Rosenberg SC, Moeller A, Speir JA, Su TY, Corbett KD. TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. eLife 4: e07367, 2015. doi: 10.7554/eLife.07367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshida M, Honma S. Regeneration of injured renal tubules. J Pharmacol Sci 124: 117–122, 2014. doi: 10.1254/jphs.13R12CP. [DOI] [PubMed] [Google Scholar]
- 54.Yoshida T, Shimizu A, Masuda Y, Mii A, Fujita E, Yoshizaki K, Higo S, Kanzaki G, Kajimoto Y, Takano H, Fukuda Y. Caspase-3-independent internucleosomal DNA fragmentation in ischemic acute kidney injury. Nephron Exp Nephrol 120: e103–e113, 2012. doi: 10.1159/000337358. [DOI] [PubMed] [Google Scholar]