

Abstract
Mice transgenic for genomic segments harboring PHAII (pseudohypoaldosteronism type II) mutant Wnk4 (with-No-Lysine kinase 4) (TgWnk4PHAII) have hyperkalemia which is currently believed to be the result of high activity of Na-Cl cotransporter (NCC). This leads to decreasing Na+ delivery to the distal nephron segment including late distal convoluted tubule (DCT) and connecting tubule (CNT). Since epithelial Na+ channel (ENaC) and renal outer medullary K+ channel (ROMK or Kir4.1) are expressed in the late DCT and play an important role in mediating K+ secretion, the aim of the present study is to test whether ROMK and ENaC activity in the DCT/CNT are also compromised in the mice expressing PHAII mutant Wnk4. Western blot analysis shows that the expression of βENaC and γENaC subunits but not αENaC subunit was lower in TgWnk4PHAII mice than that in wild-type (WT) and TgWnk4WT mice. Patch-clamp experiments detected amiloride-sensitive Na+ currents and TPNQ-sensitive K+ currents in DCT2/CNT, suggesting the activity of ENaC and ROMK. However, both Na+ and ROMK currents in DCT2/CNT of TgWnk4PHAII mice were significantly smaller than those in WT and TgWnk4WT mice. In contrast, the basolateral K+ currents in the DCT were similar among three groups, despite higher NCC expression in TgWnk4PHAII mice than those of WT and TgWnk4WTmice. An increase in dietary K+ intake significantly increased both ENaC and ROMK currents in the DCT2/CNT of all three groups. However, high-K+ (HK) intake-induced stimulation of Na+ and K+ currents was smaller in TgWnk4PHAII mice than those in WT and TgWnk4WT mice. We conclude that ENaC and ROMK channel activity in DCT2/CNT are inhibited in TgWnk4PHAII mice and that Wnk4PHAII-induced inhibition of ENaC and ROMK may contribute to the suppression of K+ secretion in the DCT2/CNT in addition to increased NCC activity.
Keywords: hyperkalemia, Kir4.1, Kir1.1, K secretion, pseudohypoaldosteronism type II
mutations in with no-lysine kinase 1 (Wnk1) and Wnk4 cause pseudohypoaldosteronism type II (PHAII), characterized by hypertension and hyperkalemia (28). The mouse model for PHAII generated by introducing genomic segments harboring PHAII Wnk4 into the mouse genome (TgWnk4PhAII) developed typical PHAII phenotypes including hypertension and hyperkalemia (11, 28). In contrast, the mice with introduction of wild-type (WT) Wnk4 (TgWnk4WT) had a lower blood pressure than that of WT and TgWnk4PHAII mice and a normal plasma K+ concentration. While the hypertension was caused by an excessive Na+ absorption through Na-Cl cotransporter (NCC) in the distal convoluted tubule (DCT), hyperkalemia was currently believed to be induced by a low-Na+ delivery to the distal nephron due to high NCC activity. Because Wnk4 has been reported to inhibit ROMK and epithelial Na+ channel (ENaC) by stimulating endocytosis (10, 16, 29), it raises the possibility that ROMK and ENaC activity may be compromised in TgWnk4PHAII mice. Thus, the present study explores the role of ENaC and ROMK in causing hyperkalemia by examining their activity in the late DCT (DCT2)/connecting tubule (CNT). The DCT2 is functionally different from the early part of DCT (DCT1) because ROMK and ENaC are expressed in the apical membrane of DCT2 (1, 2, 25). Electrophysiological studies have demonstrated that amiloride-sensitive Na+ currents were significantly larger in the DCT2/CNT than that in the cortical collecting duct (CCD) under normal conditions (13). Since high-ENaC activity is expected to enhance K+ excretion, it is conceivable that DCT2/CNT should play an important role in mediating K+ secretion. Thus, the aim of the present study is to test whether ENaC and ROMK activity are also suppressed in the DCT2/CNT of TgWnk4PhAII mice thereby impairing K+ secretion.
METHODS
Animals and tubule preparation.
C57BL/6 mice (either sex, 4–6 wk old) were purchased from the Jackson Laboratory (Bar Harbor, ME) and were used as WT control. We obtained TgWnk4WT and TgWnk4PHAII mice (either sex, 4–6 wk old) from Dr. Lifton’s laboratory at Yale University (11). The mice were fed with the control diet (1% KCl) or HK diet (5% KCl) for 7 days and had free access to water. HK diet (catalog no. TD.110866) was purchased from Harlan Laboratory (Madison, WI). After mice were euthanized by cervical dislocation, we perfused the left kidney with 5 ml collagenase type 2 (1 mg/1 ml) containing L-15 medium (Life Technology). The collagenase-perfused kidney was removed and the renal cortex was cut with a sharp razor. The animal use protocol was approved by an independent IACUC at both Yale University and New York Medical College.
Patch-clamp experiment.
For the single channel recording, an Axon200B patch-clamp amplifier was used to record the channel current. Borosilicate glass (1.7-mm OD) was used to make the patch-clamp pipettes using a Narishige electrode puller. The pipette solution contained (in mM) 140 KCl, 1.8 MgCl2, and 10 HEPES (pH 7.4) and the bath solution was composed of 140 NaCl, 5 KCl, 1.8 CaCl2, 1.5 MgCl2, 10 HEPES (pH 7.4). The currents were low-pass filtered at 1 kHz and digitized by an Axon interface with sampling rate of 4 kHz. The channel open probability (Po) was calculated from the channel number (N) and NPo (a product of channel number and open probability) which was calculated from data samples of 60-s duration in the steady state as follows:
where ti is the fractional open time spent at each of the observed current levels. The channel conductance was determined by measuring the current amplitudes over several voltages.
For the measurement of whole cell Na+ or K+ currents with the perforated patch recording, we used an Axon 200A amplifier. The tip of the pipette was filled with pipette solution containing 125 mM K+-gluconate, 15 mM KCl, 2 mM MgATP, 1 mM EGTA 10 mM HEPES (pH 7.4) for Na+ currents or 140 mM KCl, 2 mM MgCl2, 1 mM EGTA, and 5 mM HEPES (pH 7.4) for K+ currents. The pipette was then back-filled with amphotericin B (20 μg/0.1 ml) containing the pipette solution. The bath solution contains 130 mM Na+-gluconate, 10 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 10 mM HEPES (pH 7.4) for Na+ currents or symmetrical 140 mM KCl solution as used in the pipette for K+ currents. After a high-resistance seal (>2 GΩ) was formed, the membrane capacitance was monitored until the whole cell patch configuration was formed. The currents were low-pass filtered at 1 kHz and digitized by an Axon interface with 4 kHz sampling rate (Digidata 1440A). Data were analyzed using the pClamp software system 9.0 (Axon).
Preparation of protein samples and Western blot.
The tissue of renal cortex was homogenized in an ice-cold solution containing 250 mM sucrose, 50 mM Tris·HCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1% protease, and phosphatase inhibitor cocktails (Sigma) titrated to pH 7.6. After homogenization, the sample was subjected to centrifugation at 2,000 rpm for 15 min at 4°C and the protein concentration was measured using the DC Protein Assay Kit (Bio-Rad, Hercules, CA). The proteins were separated by electrophoresis on 4–20% gradient gels and transferred to nitrocellulose membrane. The membranes were blocked with LI-COR blocking buffer (PBS). An Odyssey infrared imaging system (LI-COR, Lincoln, NE) was used to scan the membrane at a wavelength of 680 or 800 nM. For Western blot, ENaC-α, ENaC-β, and ENaC-γ antibody were diluted at 1:1,000.
Experimental materials and statistics.
We used αENaC antibody from StressMarq (catalog no. SPC-403, Victoria, Canada) and it is raised against a synthetic peptide mapping to the NH2 terminal (amino acids 46–68) of rat αENaC. We obtained βENaC antibody from Sigma (catalog no. SAB5200106, St. Louis, MO) and it is produced against amino acids 617–638 of the rat sequence. The antibody of γENaC is also from StressMarq (catalog no. SPC-405) and it is generated against the COOH-terminal tail (amino acids 629–650) of rat γENaC. NCC antibody was a gift from Dr. Ellison’s laboratory at Oregon Health & Science University. The data are presented as means ± SE. We used one-way ANOVA to determine the statistical significance. If the P value is <0.05, the results were considered to be significant.
RESULTS
We followed the method described previously to prepare the DCT/CNT for the experiments (31, 32). Figure 1A is an image of an isolated tubule segment corresponding to DCT, CNT, and CCD. Figure 1B shows a magnified image of the split-open DCT2/CNT and Fig. 1C shows that a patch-pipette (Pip) was placed in the lumen of a split-open DCT2/CNT. Although DCT2 is morphologically different from the CNT (the diameter of the CNT is smaller than that of the DCT2), it is hard to determine the exact transition between DCT2 and CNT, especially for the tubule of TgWnk4PHAII mice due to the hypertrophic DCT and CNT (Fig. 1D). Thus we refer that the patch-clamp experiments were performed in the DCT2/CNT.
Fig. 1.

Distal convoluted tubule (DCT) preparation for the patch-clamp recording. A: tubule image showing the location of DCT1, DCT2, connecting tubule (CNT), and cortical collecting duct (CCD). B: tubule image showing the split-open DCT2 and CNT. C: image showing a patch-clamp pipette (Pip) was placed in the apical membrane of a split open DCT2/CNT. The pipette position pointed by an arrow is also indicated in A. D: images of isolated DCT/CNT from wild-type (WT) and TgWnk4PhAII mouse. Note that DCT/CNT tubule from TgWnk4PHAII mouse is hypertrophic in comparison to WT.
After forming whole cell recording configuration with GΩ resistance, we clamped the membrane potential at −60 mV with 140 mM Na+-5 mM K+ in the bath and 140 mM K+ in the pipette (intracellular solution). Under such experimental conditions, an inward current can be observed and addition of 0.1 mM amiloride largely abolished the inward current. We defined the amiloride-sensitive cation current as ENaC activity. Figure 2A shows a set of recordings illustrating amiloride-sensitive Na+ currents measured with the perforated whole cell recording in wild-type (WT), TgWnk4WT, and TgWnk4PHAII mice. The expression of TgWnk4WT has no significant effect on ENaC activity in the DCT2/CNT in comparison with that in WT and amiloride-sensitive Na+ currents were 213 ± 9 pA (WT) and 208 ± 25 pA (TgWnk4WT), respectively (Fig. 2B). In contrast, amiloride-sensitive Na+ currents decreased significantly in TgWnk4PHAII mice (42 ± 10 pA). We also used Western blot to examine the expression of α−, β−, and γENaC in WT, TgWnk4WT, and TgWnk4PHAII mice (Fig. 3A). While the expression of uncleaved αENaC was similar in all three groups (n = 4 mice), the expression of βENaC (20 ± 10% control value), uncleaved γENaC (30 ± 10% control value), and cleaved γENaC (50 ± 15% control value) subunit was significantly lower in TgWnk4PHAII mice than those in WT and TgWnk4WT mice (Fig. 3B). The observation that the expression of βENaC subunit was lower in TgWnk4PHAII mice than those in WT and TgWnk4WT mice was a little different from the previous report that βENaC subunit expression was the same among three groups (11). However, this conclusion was based on immunostaining using βENaC antibody rather than Western blot.
Fig. 2.

Epithelial Na+ channel (ENaC) is inhibited in TgWNK4PHAII mice. A: set of amiloride-sensitive Na+ current traces measured with the perforated whole cell recording in WT, TgWnk4WT, and TgWnk4PHAII mice. The pipette solution contains 125 mM K-gluconate, 15 mM KCl, 2 mM MgATP, 1 mM EGTA, and 10 mM HEPES (pH 7.4). The bath solution contains130 mM Na-gluconate, 10 mM NaCl, 5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 10 mM HEPES (pH 7.4). For the measurement of whole cell amiloride-sensitive Na+ currents, the membrane potential was clamped at −60 mV. Addition and washout of amiloride (0.1 mM) are indicated by arrows. B: bar graph summarizes the results of Na+ currents measured at −60 mV from 5 to 7 experiments. C: bar graph summarizes the effect of high dietary K+ intake (5%) for 7 days on amiloride-sensitive Na+ currents in the DCT2/CNT of WT, TgWnk4WT, and TgWnk4PHAII mice. The amiloride-sensitive Na+ currents were measured at −60 mV. The statistical significance is indicated by *P < 0.05 or **P < 0.01.
Fig. 3.

Expression of α-, β-, and γENaC in WT, TgWnk4WT, and TgWnk4PHAII mice. A: Western blot showing the expression of α-, β-, and γENaC in TgWnk4WT, WT, and TgWnk4PHAII mice on normal-K+ (NK) diet or on a high-K+ (HK) diet, respectively. Arrows indicate the full-length (f) γENaC and cleaved (c) γENaC subunit, respectively. B: bar graph shows the normalized band density of α-, β-, γ- and cleaved γENaC (C-γENaC) subunits. *Significant difference.
We next examined whether HK intake was still able to stimulate ENaC activity in DCT2/CNT of TgWnk4PHAII mice. WT, TgWnk4WT, and TgWnk4PHAII mice were maintained on a HK (5%) diet for 7 days and the patch-clamp experiments were performed in the split-open DCT2/CNT as described above. Results from 5 to 7 experiments are summarized in Fig. 2C showing that HK increased amiloride-sensitive Na+ currents in all three groups (WT, 339 ± 20 pA; TgWnk4WT, 380 ± 30 pA; TgWnk4PHAII, 160 ± 21 pA), suggesting that HK intake is still able to stimulate ENaC in TgWnk4PHAII mice. However, amiloride-sensitive Na+ currents in DCT2/CNT of TgWnk4PHAII mice on a HK diet were significantly lower than those in both WT and TgWnk4WT mice, suggesting a compromised ENaC activity in the DCT2/CNT of TgWnk4PHAII mice. The notion that HK intake is able to stimulate ENaC in TgWnk4PHAII mice is also supported by immunoblotting (Fig. 3, A and B). A HK intake significantly stimulates the expression of βENaC and cleaved γENaC subunit in TgWnk4PHAII mice by 190 ± 25 and by 90 ± 15% in comparison with those on a normal-K+ diet, respectively.
We and others demonstrated that Wnk4 inhibited ROMK channels in vitro (7, 10, 12, 30). Because ROMK plays a key role in mediating K+ secretion in aldosterone-sensitive distal nephron (6, 8), we examined ROMK channel activity by measuring TPNQ-sensitive K+ currents in the DCT2/CNT with perforated whole cell recording (5). After a whole cell recording configuration was formed, we clamped the membrane potential at −40 mV with 140 mM K+ in the bath and in the pipette and observed inward current. Figure 4A is a set of typical traces showing TPNQ-sensitive K+ currents in DCT2/CNT of WT, TgWnk4WT, and TgWnk4PHAII mice, respectively. Adding 400 nM TPNQ decreased inward currents, presumably ROMK currents and further application of 0.1 mM Ba2+ decreased inward currents near zero. We refer that TPNQ-sensitive cation currents represent ROMK activity, whereas Ba2+-sensitive K+ currents correspond to the basolateral Kir4.1/5.1 currents. From the inspection of Fig. 4B, it is apparent that the expression of TgWnk4WT has no significant effect on ROMK activity in the DCT2/CNT in comparison with that in WT since TPNQ-sensitive K+ currents were 1,198 ± 117 pA (WT) and 1,041 ± 137 pA (TgWnk4WT), respectively. However, TPNQ-sensitive K+ current decreased significantly in the DCT2/CNT of TgWnk4PHAII mice (410 ± 45 pA), suggesting that ROMK channel activity in the DCT2/CNT was inhibited in TgWnK4PHAII mice. This notion is also confirmed by the single channel recording (Fig. 4C) and it shows that the ROMK channel open probability in the DCT2/CNT of TgWnk4PHAII mice was 0.45 ± 0.1 (n = 4), a value significantly lower than 0.95 ± 0.1 in WT mice (n = 6; Fig. 4D) and those reported previously (3, 26). Moreover, the probability of finding ROMK in DCT2/CNT (4 patches with ROMK from total 39 patches) was lower in TgWnk4PhAII mice than those of WT mice (6 patches with ROMK from total 22 patches). Thus, decreased K+ currents in the DCT2/CNT of TgWnk4PHAII mice were at least partially the result of low-channel open probability.
Fig. 4.

ROMK activity was inhibited in TgWnk4PHAII mice. A: set of TPNQ-sensitive K+ current traces measured with the perforated whole cell recording in WT, TgWnk4WT, and TgWnk4PHAII mice. The pipette solution was composed of 140 mM KCl, 2 mM MgCl2, 1mM EGTA, and 5 mM HEPES (pH 7.4). The membrane potential was clamped at −40 mV for the measurement of K+ currents. Addition and washout of TPNQ (400 nM) are indicated by arrows. B: bar graph summarizes the results measured at −40 mV. C: single channel recording demonstrates diminished ROMK channel activity in the apical membrane of DCT2 of TgWnk4PHAII mice. D: single channel recording demonstrates ROMK channel activity in the apical membrane of DCT2 in WT mouse. The patch-clamp experiments were performed in a cell-attached patch and the channel closed level is indicated by “C.” The holding potential is indicated in the top of each trace.
We next examined whether HK intake was still able to stimulate ROMK channels in DCT2/CNT of TgWnk4PHAII mice. As described above, we measured TPNQ-sensitive K+ currents at −40 mV in the DCT2/CNT of WT, TgWnk4WT, and TgWnk4PHAII mice on a HK diet for 7 days. Results from 5 to 7 experiments were summarized in Fig. 5 showing that HK increased TPNQ-sensitive K+ currents in all three groups (WT, 1,900 ± 200pA; TgWnk4WT, 1,750 ± 150pA; TgWnk4PHAII, 1,150 ± 125 pA). This finding is also consistent with a previous report that HK intake stimulates ROMK expression in all three groups (11). However, TPNQ-sensitive ROMK currents in TgWnk4PHAII mice were significantly lower than those in both WT and TgWnk4WT mice, suggesting a compromised ROMK activity in the DCT2/CNT of TgWNK4PHAII mice. The observation that HK intake was still able to stimulate ROMK in the DCT2/CNT of TgWnk4PHAII mice suggests that the expression of PHAII mutant Wnk4 did not completely prevent HK-induced stimulation of ROMK. However, the activity of ROMK in the DCT2/CNT was still smaller in TgWnk4PHAII mice than in WT mice, indicating an enhanced inhibition of ROMK by PHAII mutant Wnk4. Thus both ENaC and ROMK activity in the DCT2/CNT are lower in TgWnk4PHAII mice than in WT mice. It is conceivable that low ENaC and ROMK activity in the DCT2/CNT should impair K+ secretion thereby contributing to hyperkalemia in TgWnk4PHAII mice.
Fig. 5.
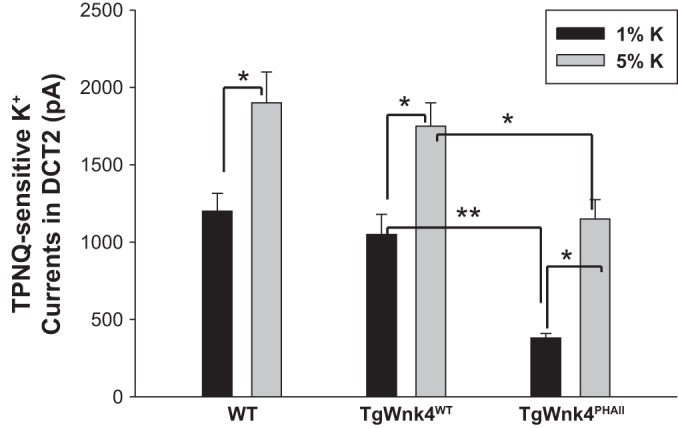
High-K+ intake stimulates ROMK in DCT2/CNT. A bar graph summarizes the effect of high dietary K intake (5%) for 7 days on TPNQ-sensitive K+ currents in the DCT2/CNT of WT, TgWnk4WT, and TgWnk4PHAII mice. TPNQ (400 nM)-sensitive ROMK currents in DCT2/CNT were measured at −40 mV. The statistical significance is indicated by *P < 0.05 or **P < 0.01.
We next examined whether the expression of PHAII mutant Wnk4 has an effect on the basolateral K+ channel activity. Because the expression of PHAII mutant Wnk4 increased NCC expression (11), it is expected that a high NCC activity should stimulate Na+-K+-ATPase activity. Consequently, the stimulation of Na+-K+-ATPase should augment K+ recycling across the basolateral membrane via a pump-leaky mechanism (19). Thus, we used the whole cell recording to measure the basolateral K+ conductance in the DCT1. Because the only type K+ channel expressed in the DCT1 is Kir4.1/Kir5.1 heterotetramer in the basolateral membrane (31, 32), the whole cell K+ conductance in the DCT1 represents the basolateral K+ channel activity. We confirmed that TgWnk4PHAII transgenic mice have higher NCC expression than WT or TgWnk4WT transgenic mice (Fig. 6, A and B) (11). However, increased NCC expression in TgWnk4PHAII mice did not augment the basolateral K+ conductance in the DCT1 in comparison to that of WT and TgWnk4WT transgenic mice. Figure 6C is a trace showing Ba2+-sensitive K+ currents measured with whole cell recording using ramp protocol at a range from −100 to 100 mV in the DCT1 of both WT and TgWnk4PHAII transgenic mice, and Fig. 6D summarizes the results measured at −60 mV from 4 to 6 experiments. It is apparent that basolateral K+ channel activity is similar in the DCT1 of all three groups (WT, 1,400 ± 90 pA; TgWnk4WT, 1,350 ± 120 pA and TgWnk4PHAII, 1,290 ± 180 pA). The results suggest that high NCC activity in TgWnk4PHAII mice did not stimulate the basolateral K+ channel activity.
Fig. 6.

High Na-Cl cotransporter (NCC) expression does not affect the basolateral Kir4.1/5.1 activity in the DCT. A: Western blot showing the expression of NCC in WT, TgWnk4WT, and TgWnk4PHAII mice. B: normalized results are summarized in a bar graph. #Significant difference from other bars. C: trace showing Ba2+-sensitive K+ currents measured with whole cell recording using ramp protocol at a range from −100 to 100 mV in the DCT1 of both WT and TgWnk4PHAII transgenic mice. D: bar graph summarizes the results measured at −60 mV.
DISCUSSION
The aldosterone-sensitive distal nephron including the DCT2, CNT, and CCD is responsible for K+ secretion (6, 8). The classic mechanism for the K+ secretion is that K+ ions are secreted to the lumen of the CNT and CCD via ROMK and Ca2+-activated big-conductance K+ (BK) channels along a favorable electrochemical gradient created by Na+ absorption via ENaC (6, 9, 14, 15, 18, 27). Recent paradigm-shifting discoveries concerning the control of systemic K+ balance have identified the thiazide-sensitive NCC of the DCT (4, 17, 22, 24). The role of NCC in regulating renal K+ secretion and K+ homeostasis is also convincingly established by human genetic and clinical studies demonstrating that an abnormal NCC activity is responsible for causing hyperkalemia or hypokalemia. For instance, PHAII or familial hyperkalemic hypertension (FHHt) is caused by high activity of NCC (11, 20, 23), whereas hypokalemia in patients with Gitelman syndrome is due to the loss-function mutations of NCC (21). The role of NCC in causing hyperkalemia in FHHt is also convincingly demonstrated in the PHAII mouse model by introducing genomic segments harboring PHAII Wnk4 into the mouse genome (11). However, it has been suggested that hyperkalemia in the TgWnk4PHAII mouse model is solely induced by high NCC activity without the involvement of ENaC and ROMK. This conclusion was based on the following observations (11): 1) the downregulation of NCC corrected hyperkalemia; 2) immunostaining images showed that the expressions of ENaC and ROMK were similar among WT, TgWnk4WT, and TgWnk4PHAII mice. But, the present experiments have demonstrated that both ENaC and ROMK activity in DCT2/CNT were lower in TgWnk4PHAII mice on a normal or a HK diet than in WT and TgWnk4WT mice. Moreover, the decrease in ENaC activity in TgWnk4PHAII mice is partially due to the low expression of βENaC and γENaC subunits. Since ENaC and ROMK activity in the DCT2/CNT are critically involved in mediating K+ secretion, it is conceivable that a diminished ROMK and ENaC activity should contribute to hyperkalemia in TgWnk4PHAII mice. Also, the observation that HK intake-induced stimulation of ENaC and ROMK in the DCT2/CNT is also compromised in TgWnk4PHAII mice is consistent with the finding that the mice had more severe hyperkalemia than those of WT mice during increasing dietary K+ intake (11). The main argument of the previous study to exclude the role of ENaC and ROMK in causing hyperkalemia in TgWnk4PHAII mice is that the depletion of NCC could correct hyperkalemia (11). However, this effect may be due to high volume delivery induced by the depletion of NCC to the collecting duct thereby stimulating BK channel-dependent K+ secretion. Thus a flow-stimulated BK-dependent K secretion may compensate the compromised function of ENaC and ROMK. We conclude that Wnk4 with PHAII mutations inhibits ENaC and ROMK and that a low ENaC and ROMK activity in the DCT2/CNT should contribute to hyperkalemia in PHAII syndrome.
GRANTS
The work is supported by National Institutes of Health Grant DK54983 (to W. H. Wang) and Dr. Chengbiao Zhang is supported by National Science Foundation of China Grant 31571187.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.Z., L.W., X.-T.S., J.Z., and D.-H.L. performed experiments; C.Z., L.W., X.-T.S., D.-H.L., and W.-H.W. analyzed data; C.Z., L.W., X.-T.S., and W.-H.W. interpreted results of experiments; C.Z., L.W., X.-T.S., and W.-H.W. prepared figures; C.Z., L.W., X.-T.S., J.Z., and W.-H.W. edited and revised manuscript; C.Z., L.W., X.-T.S., J.Z., D.-H.L., and W.-H.W. approved final version of manuscript; W.-H.W. drafted manuscript.
REFERENCES
- 1.Bachmann S, Velázquez H, Obermüller N, Reilly RF, Moser D, Ellison DH. Expression of the thiazide-sensitive Na-Cl cotransporter by rabbit distal convoluted tubule cells. J Clin Invest 96: 2510–2514, 1995. doi: 10.1172/JCI118311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellison DH, Velázquez H, Wright FS. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am J Physiol Renal Fluid Electrolyte Physiol 253: F546–F554, 1987. [DOI] [PubMed] [Google Scholar]
- 3.Frindt G, Palmer LG. Low-conductance K channels in apical membrane of rat cortical collecting tubule. Am J Physiol Renal Fluid Electrolyte Physiol 256: F143–F151, 1989. [DOI] [PubMed] [Google Scholar]
- 4.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299: F890–F897, 2010. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frindt G, Shah A, Edvinsson J, Palmer LG. Dietary K regulates ROMK channels in connecting tubule and cortical collecting duct of rat kidney. Am J Physiol Renal Physiol 296: F347–F354, 2009. doi: 10.1152/ajprenal.90527.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giebisch G, Hebert SC, Wang WH. New aspects of renal potassium transport. Pflügers Arch 446: 289–297, 2003. doi: 10.1007/s00424-003-1029-8. [DOI] [PubMed] [Google Scholar]
- 7.He G, Wang HR, Huang SK, Huang C-L. Intersectin links WNK kinases to endocytosis of ROMK1. J Clin Invest 117: 1078–1087, 2007. doi: 10.1172/JCI30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev 85: 319–371, 2005. doi: 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362: 31–38, 1993. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- 10.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet 35: 372–376, 2003. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 11.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 12.Lin DH, Yue P, Yarborough O III, Scholl UI, Giebisch G, Lifton RP, Rinehart J, Wang WH. Src-family protein tyrosine kinase phosphorylates WNK4 and modulates its inhibitory effect on KCNJ1 (ROMK). Proc Natl Acad Sci USA 112: 4495–4500, 2015. doi: 10.1073/pnas.1503437112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol 303: F1289–F1299, 2012. doi: 10.1152/ajprenal.00247.2012. [DOI] [PubMed] [Google Scholar]
- 14.Palmer LG, Frindt G. Aldosterone and potassium secretion by the cortical collecting duct. Kidney Int 57: 1324–1328, 2000. doi: 10.1046/j.1523-1755.2000.00970.x. [DOI] [PubMed] [Google Scholar]
- 15.Pluznick JL, Sansom SC. BK channels in the kidney: role in K+ secretion and localization of molecular components. Am J Physiol Renal Physiol 291: F517–F529, 2006. doi: 10.1152/ajprenal.00118.2006. [DOI] [PubMed] [Google Scholar]
- 16.Ring AM, Cheng SX, Leng Q, Kahle KT, Rinehart J, Lalioti MD, Volkman HM, Wilson FH, Hebert SC, Lifton RP. WNK4 regulates activity of the epithelial Na+ channel in vitro and in vivo. Proc Natl Acad Sci USA 104: 4020–4024, 2007. doi: 10.1073/pnas.0611727104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandberg MB, Maunsbach AB, McDonough AA. Redistribution of distal tubule Na+-Cl− cotransporter (NCC) in response to a high-salt diet. Am J Physiol Renal Physiol 291: F503–F508, 2006. doi: 10.1152/ajprenal.00482.2005. [DOI] [PubMed] [Google Scholar]
- 18.Satlin LM. Developmental regulation of expression of renal potassium secretory channels. Curr Opin Nephrol Hypertens 13: 445–450, 2004. doi: 10.1097/01.mnh.0000133979.17311.21. [DOI] [PubMed] [Google Scholar]
- 19.Schultz SG. Homocellular regulatory mechanisms in sodium-transporting epithelia: avoidance of extinction by “flush-through.” Am J Physiol Renal Fluid Electrolyte Physiol 241: F579–F590, 1981. [DOI] [PubMed] [Google Scholar]
- 20.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP. Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci USA 110: 7838–7843, 2013. doi: 10.1073/pnas.1304592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 22.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- 23.Take C, Ikeda K, Kurasawa T, Kurokawa K. Increased chloride reabsorption as an inherited renal tubular defect in familial type II pseudohypoaldosteronism. N Engl J Med 324: 472–476, 1991. doi: 10.1056/NEJM199102143240707. [DOI] [PubMed] [Google Scholar]
- 24.van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AHJ, Fenton RA, Zietse R, Hoorn EJ. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl− cotransporter. Am J Physiol Renal Physiol 305: F1177–F1188, 2013. doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- 25.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA. Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 300: F1385–F1393, 2011. doi: 10.1152/ajprenal.00592.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang WH, Schwab A, Giebisch G. Regulation of small-conductance K+ channel in apical membrane of rat cortical collecting tubule. Am J Physiol Renal Fluid Electrolyte Physiol 259: F494–F502, 1990. [DOI] [PubMed] [Google Scholar]
- 27.Wang WH. View of K+ secretion through the apical K channel of cortical collecting duct. Kidney Int 48: 1024–1030, 1995. doi: 10.1038/ki.1995.385. [DOI] [PubMed] [Google Scholar]
- 28.Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G, Lifton RP. Molecular pathogenesis of inherited hypertension with hyperkalemia: the Na-Cl cotransporter is inhibited by wild-type but not mutant WNK4. Proc Natl Acad Sci USA 100: 680–684, 2003. doi: 10.1073/pnas.242735399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu L, Cai H, Yue Q, Alli AA, Wang D, Al-Khalili O, Bao HF, Eaton DC. WNK4 inhibition of ENaC is independent of Nedd4-2-mediated ENaC ubiquitination. Am J Physiol Renal Physiol 305: F31–F41, 2013. doi: 10.1152/ajprenal.00652.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue P, Lin DH, Pan CY, Leng Q, Giebisch G, Lifton RP, Wang WH. Src family protein tyrosine kinase (PTK) modulates the effect of SGK1 and WNK4 on ROMK channels. Proc Natl Acad Sci USA 106: 15061–15066, 2009. doi: 10.1073/pnas.0907855106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang C, Wang L, Thomas S, Wang K, Lin DH, Rinehart J, Wang WH. Src family protein tyrosine kinase regulates the basolateral K channel in the distal convoluted tubule (DCT) by phosphorylation of KCNJ10 protein. J Biol Chem 288: 26135–26146, 2013. doi: 10.1074/jbc.M113.478453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C, Wang L, Zhang J, Su X-T, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 111: 11864–11869, 2014. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]