

Abstract
Background:
Epilepsy is a disorder of the central nervous system characterized by recurrent seizures. It is a very common disease in which approximately 30% of patients do not respond favourably to treatment with anticonvulsants. Oxidative stress is associated with neuronal damage arising from epileptic seizures. The present study investigated the effects of naringenin in pilocarpine-induced epilepsy in mice. Naringenin, one of the most frequently occurring flavanone in citrus fruits, was evaluated for its shielding effect against the pilocarpine induced behavioural, oxidative and histopathological alterations in rodent model of epilepsy.
Methodology:
Epilepsy was induced by giving pilocarpine (300mg/kg) and sodium valproate (300mg/kg) was given as standard anti-epileptic drug Pilocarpine was administered (300 mg /kg body weight) intraperitoneally to the mice on 15th day while naringenin was administered orally (20 and 40 mg/kg body weight) for 15 days prior to administration of pilocarpine.
Results:
The intraperitoneal administration of pilocarpine enhanced lipid peroxidation, caused reduction in antioxidant enzymes, viz., catalase, superoxide dismutase and glutathione reductase. Treatment of mice orally with naringenin (20 mg/kg body weight and 40 mg/kg body weight) resulted in a significant decrease in lipid peroxidation. There was significant recovery of glutathione content and all the antioxidant enzymes studied. Also in case of behavioural parameters studied, naringenin showed decrease in seizure severity. All these changes were supported by histological observations, which revealed excellent improvement in neuronal damage.
Conclusion:
The higher dose of naringenin was more potent in our study and was comparable to the standard drug (sodium valproate) in effectiveness.
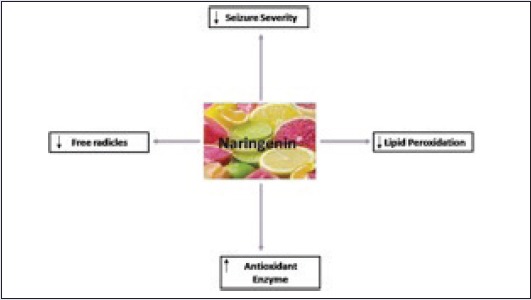
SUMMARY
Naringenin ameliorated the development of ROS formation in hippocamus.
Naringenin helped in recovery of antioxidant enzymes.
Naringenin decreased seizure severity.
Naringenin treatment reduced lipid peroxidation.
Abbreviations used: 6-OHDA: 6-hydroxydopamine, AED: Anti epileptic drugs, AIDS: Acquired immune deficiency syndrome, ANOVA: Analysis of variance, ATP: Adenosine triphosphate, CA: Cornu ammonis, CAT: Catalase, DG: Dentate gyrus, EDTA: Ethylenediamine tetra acetic acid, GR: Glutathione reductase, GSH: Glutathione reduced, HCl: Hydrochloric acid, IL-1β: Interleukin 1 beta, LPO: Lipid peroxidation, MDA: Malondialdehyde, NADPH: Nicotinamide adenine dinucleotide phosphate, PMS: post mitochondrial supernatant, SE: Status epilepticus, SEM: Standard error of the mean, SOD Superoxide dismutase, TBA: Thiobarbituric acid, TBARS: Thiobarbituric acid reactive substance, TLE: Temporal lobe epilepsy, TNF-α: Tumor necrosis factor alpha
Keywords: Antioxidant, anticonvulsants, epilepsy, naringenin
INTRODUCTION
Epilepsy is a common neurologic illness marked by the manifestation of spontaneous seizures as a result of unexpected neuronal activity in the brain. It has afflicted human beings since the dawn of our species and has been recognized since the earliest medical writings. Only when a person has two or more unprovoked seizures, he/she is diagnosed with epilepsy.[1] Robert et al.[2] defined epilepsy as a disorder of the brain identified by a persisting susceptibility to generate epileptic seizures and by the neurobiologic, psychologic, cognitive, and social consequences of this condition.
Usually, the cause of epilepsy is unknown, but sometimes the origin of epilepsy may be associated with stroke, brain injury, brain tumors, or infections (such as meningitis, brain abscess, AIDS). Exposure to some particular drugs, peculiar sodium/glucose levels in blood, or hyperpyrexia can be sometimes linked to impermanent cause of seizures. A person is not considered as epileptic if seizures fail to persist after the prime cause is rectified.
The customary type of epilepsy in humans is temporal lobe epilepsy (TLE) consisting of partial seizures that originate from the hippocampus, entorhinal cortex, or amygdala. TLE can be replicated by administration of glutaminergic (kainic acid) or cholinergic (pilocarpine) agonists in rodents[3] in laboratory model. Till date, no experimental model has been able to reproduce all features of TLE, but some models have been utilized in the preceding decades owing to the fact their influential level of resemblance with human epilepsy. Among these is the pilocarpine model, emerging as tremendously isomorphic with human epilepsy, therefore used in many laboratories since its introductory explanation a quarter of a century ago.[4,5]
Oxidative and nitrosative stress are looked on as the potential processes in epileptic pathogenesis. Until now, studies have proved that status epilepticus modifies redox potential and cuts down the level of ATP, which can induce a collapse in brain energy production and supply. Oxidative injury to vulnerable targets is caused by continuous seizures (status epilepticus).[6,7] Various genetic studies and animal models have validated an increase in mitochondrial oxygen and nitrosative stress, followed by cell damage after persistent seizures.
Flavonoids (or bioflavonoids) are the class of plant secondary metabolites, which have been found to be of substantial significance as antioxidants.[8] More than 4000 flavonoids have been discovered and naringenin is one of them. Naringenin is present in citrus fruits like grapes as “naringin” in inactive form and is broken down into an active aglycone form “naringenin” by the action of intestinal bacterial enzymes. Chemically, it is 4,5,7-trihydroxyflavanone. Renowned for its diverse biologic effects on animal as well as well-being, naringenin has been found to exhibit the properties and characteristics of being used as hepatoprotective,[9] anticarcinogenic,[10] anti-oxidant,[11] antidiabetic,[12] anti-atherogenic,[13] and anti-inflammatory.[14] Further, neuroprotective effect of naringenin has also been reported previously in animal model of amnesia,[15] Alzheimer's disease,[16] and 6-OHDA model of Parkinson's disease.[17] Also, naringenin supplementation has shown to restore expression of choline acetyl transferase and improvement in learning and memory.[16,18] Cytokine regulation and inflammation control by naringenin in brain via TNF-α and IL-1β have also been reported.[19]
No major research work has been carried out till date regarding the preventive effects of naringenin on epilepsy. Therefore, the present study was designed to investigate the efficacy of naringenin against pilocarpine-induced epilepsy in mice model.
MATERIALS AND METHODS
Animals
Adult Swiss albino mice were procured from the Central Animal House Facility IIIM, Jammu. The animals were housed in clean polypropylene cages under natural light/dark cycle and controlled conditions of temperature and humidity (25 ± 5°C). Standard rodent chow and water ad libitum was provided to animals. At least a week before performing the experiments, mice were acclimatized to the laboratory conditions. Animals were divided randomly into five groups of 10 mice in each group. Male and female mice were kept in separate cages. The weights of the animals were in the range of 20–30 g. The experiments were conducted between 09:00 and 16:00 h. All the experiments were carried out after proper approval of the Institutional Animal Ethics Committee (IAEC) of PG department of pharm sciences (Ethical clearance no: F (IAEC-Approval) KU/2015/03).
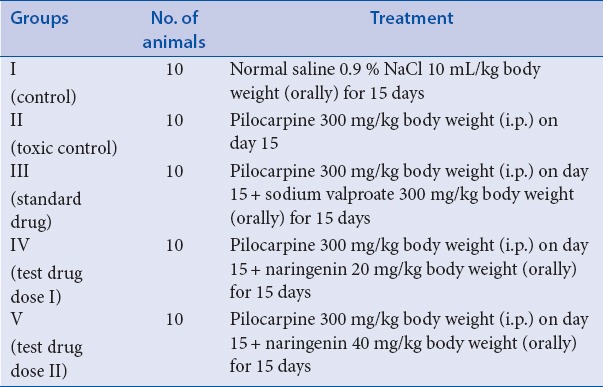
Group I received normal saline 0.9 % NaCl 10 mL/kg body weight (orally) for 15 days. Group II was treated with single dose of pilocarpine on day 15 at the dose of 300 mg/kg. Group III received sodium valproate 300 mg/kg body weight (orally) for 15 days and pilocarpine 300 mg/kg body weight (i.p.) on day 15. Group IV received naringenin 20 mg/kg body weight (orally) for 15 days and pilocarpine 300 mg/kg body weight (i.p.) on day 15, while group V received naringenin 40 mg/kg body weight (orally) for more than 15 days and pilocarpine 300 mg/kg body weight (i.p.) on day 15.
The dose of pilocarpine was standardized before the start of actual experiment. For the purpose of standardization, six groups of five mice each were selected and pilocarpine was injected with 280, 290, 300, 310, 340 mg/kg body weight. Doses below 300 mg/kg body weight showed little or no effect, while doses above 300 mg/kg body weight showed enhanced mortality. We observed 300 mg/kg body weight to be optimum dose for mice-based studies with around 30–40% mortality only.
Mortality percentage after pilocarpine treatment groups is as follows. Group II: 40%, group III: 20%, group IV: 30%, and group V: 20%.
Drugs and doses
Epilepsy was induced by giving pilocarpine (300 mg/kg) and sodium valproate (300 mg/kg) was given as standard antiepileptic drug. Pilocarpine {(3S,4R)-3-ethyl-4-((1-methyl-1H-imidazol5yl)methyl)dihydrofuran-2(3H)-one} was obtained from HiMedia. It was prepared freshly by dissolving in distilled water and injected i.p. at a dose of 300 mg/kg to mice for induction of status epilepticus.[20] Control animals were treated with an equivalent volume of distilled water (10 mL/kg body weight). Sodium valproate (sodium 2-propylpentanoate) was obtained from Unicure Remedies Pvt. Ltd. It was prepared freshly by dissolving in distilled water and given orally to mice at a dose of 300 mg/kg[21] for 15 days prior to administration of pilocarpine. Naringenin was obtained from Sigma–Aldrich (India). It was freshly prepared by dissolving in normal saline and given orally at dose of 20 and 40 mg/kg body weight. For treatment schedule refer to [Table 1].
Table 1.
Treatment schedule
After administering pilocarpine, the alterations in seizure activity in different groups were recorded for a period of 1 h as per the following Racine classification.[22]
Stage I: mouth and facial movements
Stage II: head nodding
Stage III: forelimb clonus, chewing
Stage IV: rearing and forelimb clonus
Stage V: rearing and falling with forelimb clonus
Tissue preparation for histopathology and morphologic analysis of neuronal degeneration
Animals from each group were deeply anesthetized with diethyl ether, brains removed from the skull, and then cleaned of blood in normal saline and preserved in formalin solution. Histopathologic studies of hippocampus were carried out at SKUAST-K, Srinagar. Hematoxylin-eosin staining was used to assess neuronal, cellular, and terminal degeneration.
Tissue homogenization
After the 15 days treatment schedule, the animals were sacrificed, the hippocampus blotted dry, weighed (10% w/v), and homogenized in 0.01 mM phosphate buffered saline pH 7.4. Homogenates were centrifuged to obtain postmitochondrial fluid (PMF/PMS) of hippocampus, which was subjected to biochemical estimations.
Protein
Protein concentration in all the samples was determined by the method of Lowry et al.[23] using bovine serum albumin as the standard.
Assay for superoxide dismutase
Superoxide dismutase (SOD) activity was measured by the method of Marklund and Marklund.[24] The reaction mixture consisted of 2.875 mL Tris-HCl buffer (50 mM, pH 8.5), pyrogallol (24 mM in 10 mM HCl) and 100 mL PMS, in a total volume of 3 mL. Enzyme activity was measured at 420 nm and was expressed as units/mg protein. One unit of enzyme is defined as the enzyme activity that inhibits the auto-oxidation of pyrogallol by 50%.
Assay for catalase
Catalase (CAT) activity was assessed by the method of Claiborne.[25] In short, the reaction mixture consisted of 0.05 mL PMS, 1.0 mL of H2O2 (0.019 M), 1.95 mL phosphate buffer (0.1 M, pH 7.4), in a total volume of 3 mL. Changes in absorbance were recorded at 240 nm and the change in absorbance was calculated as nmol H2O2 consumed/min per mg protein.
Estimation of thiobarbutric acid reactive substance (TBARS)
The assay of lipid peroxidation (LPO) was done according to the method of Wright et al.[26] The reaction mixture consisted of 0.58 mL phosphate buffer (0.1 M, pH 7.4), 0.2 mL microsome, 0.2 mL ascorbic acid (100 mM), and 0.02 mL ferric chloride (100 mM) in a total volume of 1 mL. This reaction mixture was then incubated at 378°C in a shaking water bath for 1 h. The reaction was stopped by the addition of 1 mL trichloroacetic acid (10%). Following the addition of 1.0 mL thiobarbutric acid (TBA) (0.67 %), all the tubes were placed in a boiling water bath for a period of 20 min. The tubes were shifted to an ice bath and then centrifuged at 2500g for 10 min. The amount of malondialdehyde (MDA) formed in each of the samples was assessed by measuring the optical density of the supernatant at 535 nm. The results were expressed as nmol TBA formed/h per g tissue at 37°C by using a molar extinction coefficient of 1.56 × 105 M–1 per cm.
Estimation of glutathione reductase
Glutathione reductase (GR) activity was determined by the method of Carlberg and Manevrick.[27] The reaction mixture consisted of 1.65 mL phosphate buffer (0.1 M, pH 7.6), 0.1 mL EDTA (0.5 mM), 0.05 mL GSH (1 mM), 0.1 mL NADPH (0.1 mM), and 0.1 mL of 10% PMS, in a total volume of 2 mL. Enzyme activity was quantified at 258°C by measuring the disappearance of NADPH at 340 nm and was calculated as nmol NADPH oxidized/min per mg protein using a molar extinction coefficient of 6.22 × 103 M–1 per cm.
Statistical analysis
The data from individual groups are presented as the mean ± standard error of the mean (SEM). Differences between groups were analyzed by using one-way analysis of variance (ANOVA) followed by Tukey–Kramer multiple comparisons test and minimum criterion for statistical significance was set at P less than 0.05 for all comparisons.
RESULTS
Effect of naringenin and sodium valproate on seizure activity against pilocarpine induced epilepsy.
Naringenin administration at the doses of 20 and 40 mg/kg body weight to mice for a period of 15 days, followed by pilocarpine administration on the last day of drug treatment for induction of epilepsy in mice revealed the following results.
Effect on seizure severity
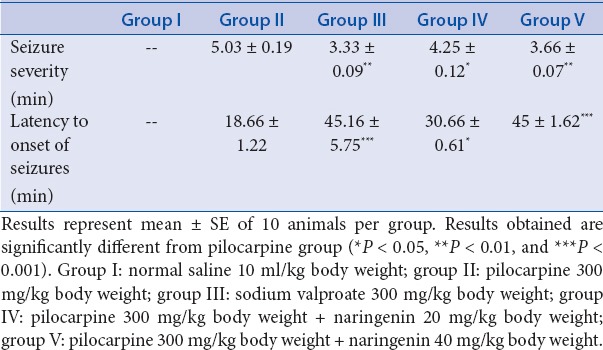
The seizure severity score reduced significantly (P < 0.01) in valproate pretreatment mice as compared with that observed in toxic control (receiving pilocarpine only) group. Pretreatment with naringenin at 20 and 40 mg/kg also reduced seizure severity (P < 0.05 and P < 0.01), respectively, compared with pilocarpine control group [Table 2].
Table 2.
Effect of treatment of naringenin and sodium valproate on seizure severity and latency to onset of status epilepticus in pilocarpine-induced epilepsy in mice
Effect on latency to onset of seizures
The latency to seizure onset score revealed extremely significant (P < 0.001) rise in mice of group III as compared with that of score observed in toxic control that received pilocarpine only. Pretreatment with naringenin at 20 mg/kg to mice of group IV showed significant (P < 0.05) rise in the latency to seizure onset score, while the mice of group V that received 40 mg/kg body weight of naringenin revealed extremely significant (***P < 0.001) rise in the score [Table 2].
Effect of naringenin and sodium valproate on different biochemical parameters against pilocarpine-induced epilepsy
SOD activity
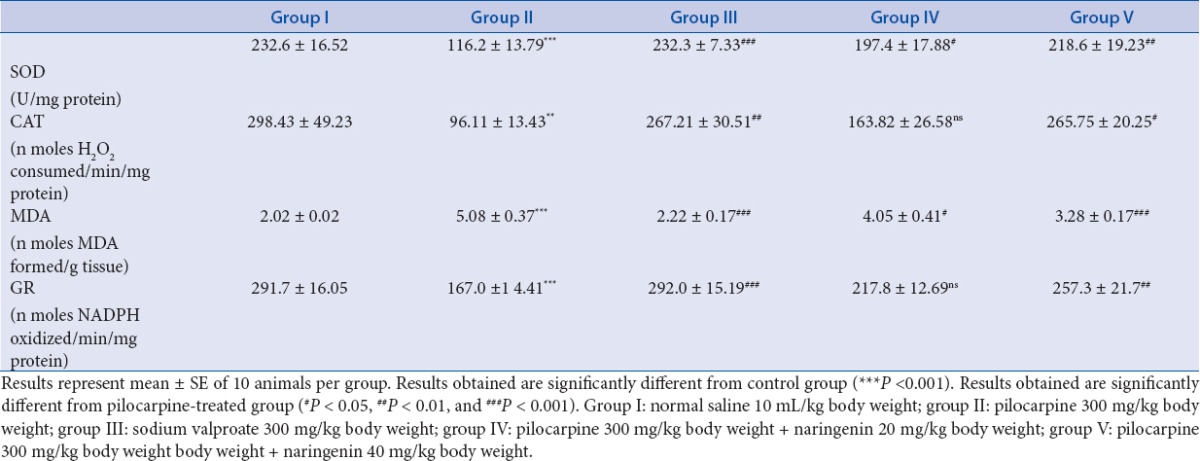
Significant (***P < 0.001) fall in SOD levels was observed in mice of group II as compared with the levels in control mice (group I). A significant (*P < 0.05) increase in levels was observed in the mice of group IV that had received naringenin (20 mg/kg), while a highly significant (**P < 0.01) rise was seen in mice administered naringenin (40 mg/kg) compared with levels in toxic group that had received pilocarpine only. Sodium valproate, the standard antiepileptic drug administered to mice of group III displayed extremely significant (***P < 0.001) rise in SOD levels as compared with group II [Table 3].
Table 3.
Effect of treatment of naringenin on the antioxidant enzymes and LPO on pilocarpine-induced status epilepticus in experimental mice
CAT activity
A highly significant decrease occurred in CAT levels in mice of group II as compared with the levels in control mice (group I). A nonsignificant (ns) rise in the levels was observed in the mice of group IV that had received naringenin (20 mg/kg), while a significant (*P < 0.05) rise was seen in mice administered naringenin (40 mg/kg) compared with levels in toxic group that had received pilocarpine only. Sodium valproate, the standard antiepileptic drug administered to mice of group III showed highly significant (**P < 0.01) rise in CAT levels as compared with group II [Table 3].
Lipid peroxidation (MDA)
We observed a significant (***P < 0.001) rise in levels of MDA in mice of group II as compared with the levels in control mice (group I). A significant (*P < 0.05) decrease was observed in the mice of group IV that had received naringenin (20 mg/kg), while an extremely significant (***P < 0.001) fall in levels of MDA was seen in mice administered naringenin (40 mg/kg) compared with levels in toxic group that had received pilocarpine only. Sodium valproate, the standard antiepileptic drug administered to mice of group III exhibited extremely significant (***P < 0.001) fall in MDA levels as compared with group II [Table 3].
GR levels
An extremely significant (***P < 0.001) fall occurred in GR levels in mice of group II as compared with the levels in control mice (group I). A nonsignificant (ns) rise was observed in the mice of group IV that had received naringenin (20 mg/kg), while a highly significant (**P < 0.01) rise in the levels was seen in mice administered naringenin (40 mg/kg) compared with levels in toxic mice. Sodium valproate, the standard antiepileptic drug administered to mice of group III, revealed significant (***P < 0.001) increase in these levels [Table 3].
Histopathologic analysis
Histologic examination of hippocampal sections revealed normal morphology of neurons of group I animals. No deformity in dentate gyrus (DG), CA1, CA2, and CA3 sections of normal mice was observed. Neuronal condensation, necrosis, and nuclear degeneration in the DG, CA1, CA2, and CA3 regions were observed in hippocampus of mice of group II animals that had received pilocarpine only. Hippocampus of mice of group III that had received sodium valproate revealed almost normal morphology in CA1, CA2, and DG regions, while CA3 revealed decrease in neuronal number and neuronal degeneration at few spots. Mild condensation and degeneration of neurons was observed in hippocampus of mice of group IV administered 20 mg/kg body weight of naringenin, while almost normal morphology was observed in hippocampal sections of mice of group V that had been administered 40 mg/kg body weight of naringenin [Figure 1].
Figure 1.
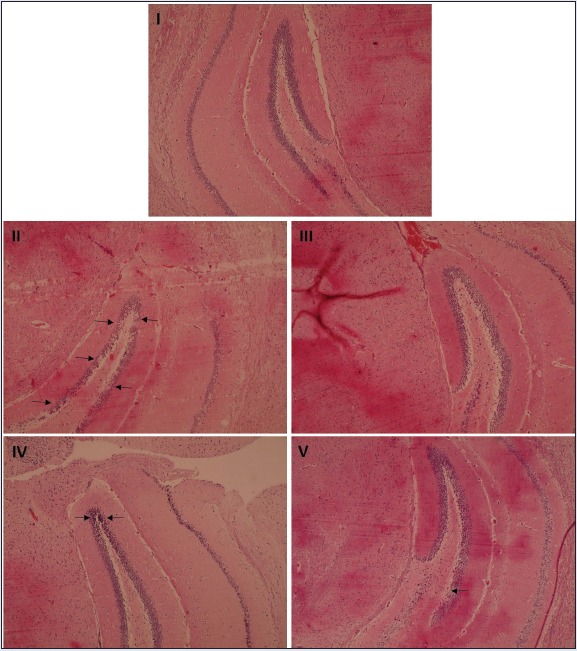
The histologic sections of control group showed normal histoarchitecture, while pilocarpine-treated group (group II) exhibited condensation of pyramidal neurons with increased perineural space. In group III, normal morphology of DG was preserved by sodium valproate treatment. In group IV, histologic sections showed that treatment with pilocarpine and lower dose of naringenin resulted in mild condensation and degeneration of neurons in DG. Group V sections showed that treatment with pilocarpine and naringenin revealed almost normal neuronal morphology in the DG.
DISCUSSION
The present study was designed to investigate the anticonvulsant effect of naringenin using the pilocarpine model of epilepsy. Naringenin could suppress onset and duration of seizure in pilocarpine model and we observed that this effect increased dose dependently. Also, naringenin treatment restored the antioxidant status and reduced LPO in hippocampus of epileptic mice.
Epilepsy is one of the most common serious neurologic conditions. In contemporary society, the frequency and importance of epilepsy can hardly be overstated from the epidemiologic studies. However, in most studies, the overall incidence of epilepsy in developed societies has been found to be around 50 cases per 100,000 persons per year, and it rises sharply in elder people.[28] The current therapeutic treatment of epilepsy with modern antiepileptic drugs (AEDs) is associated with dose-related side effects, chronic toxicity, and teratogenic effects. Approximately 30% of the patients continue to have seizures with current AEDs therapy.[29]
In recent past, a number of evidences have demonstrated that natural products from folk therapies have contributed significantly in the discovery of modern drugs. Herbal medicines are often considered to be a gentle and safe alternative to synthetic drugs. More than half of the medically important pharmaceutical drugs are either natural products or derivatives of natural products.[30,31,32] Additionally, numerous herbal medicines are active on central nervous system, and they have at least a hypothetical potential to affect chronic conditions that do not respond well to conventional treatments.[33,34]
Development and introduction of new drugs with greater safety and efficacy is essential to enhance treatment of epilepsy. The search for less toxic alternatives resulted in decreased use of synthetic substances and the introduction of natural products. This notion is supported by previous studies showing that some natural products, such as phytol,[35] isopulegol,[36] thymoquinone,[37] citronellol,[38] and α-β,-epoxy carvone,[39] possess anticonvulsant activity.[40]
Pilocarpine disrupts the blood–brain barrier and impairs brain functions by inducing free radicals,[41] which leads to the development of seizure activity in animals,[42] followed by neuronal death. The hyperexcitability of brain neurons and production of free radicals by pilocarpine administration are major factors responsible for subsequent epileptogenesis in animal models of epilepsy.[43]
In pilocarpine-induced epilepsy, the increase in the number of free radicals in the brain neurons interferes with respiratory chain in the mitochondria, destabilizes the lysosomal membranes, and lowers the convulsion threshold.[44,45] Neuronal firing associated with prolonged epileptic discharges and seizures may lead to a number of neurochemical changes and cascades of events at the cellular and molecular level. This, in turn, results in mitochondrial dysfunction, increased reactive oxygen species that precede neuronal degeneration and death with possible subsequent epileptogenesis and cortical epileptization – secondary epileptogenesis – that may result in cognitive function impairments and with chronic intractable epilepsy.
Known to play an important part in resolving the pathophysiology of epilepsy and development of AED, animal models of TLE have been widely used ever since their introduction. The pilocarpine model of TLE finds place in those groups of animal models, which recreate the overall sequence of epileptic episodes as observed in humans. In the pilocarpine model, systemic administration of pilocarpine is carried out, which leads to induction of status epilepticus possibly through activation of M1 muscarinic receptors.[44] Pilocarpine-induced seizures were rated using Racine scale.[22] In pilocarpine (convulsant) treated mice, significant increase in seizure severity was spotted as compared with naringenin-treated animals. The latency to onset of SE determines the extent to which apoptotic or necrotic mechanisms contribute to the degeneration following SE.[46] In our studies, we observed an extremely significant decrease in latency to onset of seizure score in pilocarpine-treated mice as compared with treatment groups.
Oxidative stress and antioxidant enzymes are integral to epileptic phenomena and reactive oxygen species have been implicated in seizure-induced neurodegeneration.[47] Using the epilepsy model obtained by systemic administration of pilocarpine in mice, LPO (TBARS), GR, SOD, and CAT activities were investigated in the hippocampus of mice.
LPO assay is a measure of damage caused by free radicals produced as a consequence of recurring seizures. As an index of LPO, the formation of TBARS was used, which is widely adopted as a sensitive method for the measurement of LPO.[48] There was a significant rise in LPO level in hippocampus of epileptic mice. These data are reflected by increase in TBARS concentrations, which is related to its intermediate free radicals formation during seizures. The enhanced oxidative stress condition results in a series of changes in the cellular structure and function.[49] Oxidative stress can drastically affect membrane properties through LPO, which is one of the most biologically relevant free radicals reactions.[50] Phytochemicals are well-known potent-free radical scavengers and it has also been reported that the naringenin tends to reverse the changes in LPO and damage to cells.[51] The treatment with naringenin considerably decreased TBARS levels, indicating reduced generation of free radicals, suggesting naringenin as a potent antioxidant; our results are in agreement with previously published report of Lee et al.[52]
The biologic effects of free radicals are controlled in vivo by a wide range of antioxidants such as GR, SOD, glutathione peroxidase, and CAT. We assessed the activity of GR, SOD, and CAT in order to analyze the modifications in antioxidant activity in hippocampus of mice. Oxidative stress mimics the epileptic condition by severely altering the antioxidant system.[53] A significant decrease in GR, SOD, and CAT activity in the hippocampus of pilocarpine induced epileptic mice was observed as compared with control mice. Similar kind of alteration in the activity of antioxidant enzymes like GR, SOD, and CAT has been reported and explained to be due to oxidative deactivation of antioxidant enzymes.[54] Previous investigators have reported enhanced GR, SOD, and CAT activity after treatment with naringenin.[52,53,54,55,56] We observed that treatment with naringenin in epileptic mice has resulted in increased activity of GR, SOD, and CAT, indicating supplemented antioxidant system. These results are in agreement with published reports of Annadurai[55] and Chtourou.[56]
The hippocampus, one of the important parts of brain consists of complex interfolded layers of DG and cornu ammonis (CA).[57] Each region of the hippocampus includes the various subfields and lamellae, which play a role in the storage and retrieval of episodic memory.[58] The main target of hippocampal damage by pilocarpine is CA3 and CA1 region of the dorsal hippocampus. In the present study, the complete destruction of neurons along with neuronal condensation and necrosis in DG, CA1, CA2, and CA3 was observed in the mice treated with pilocarpine alone as compared with normal mice, while as administration of naringenin (40 mg/kg) and sodium valproate exhibited antiepileptic effect, which was evident from almost normal morphology of neurons and less number of shrunken neurons in mice in the regions observed.
CONCLUSION
Summing up, the data obtained through our study indicate that naringenin possesses a defensive effect against pilocarpine-induced epilepsy. Pilocarpine, a cholinergic agonist is thought to induce seizures via formation of free radicals, while the antioxidant activity of naringenin is responsible for its protective role against seizures instigated by pilocarpine. The results of our work supported by strong published literature reports suggest that this naturally occurring flavanone (naringenin) is a potential candidate for prevention of pilocarpine-induced epilepsy, since it restrains several biomarkers of oxidative stress in animal model. These properties make naringenin studied preclinically useful. Further detailed mechanistic studies are necessary to divulge the beneficial effect of naringenin before proceeding for clinical trials.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
The authors acknowledge Department of Science and Technology (DST), Govt of India for providing facilities under FIST scheme. The authors are also highly thankful to Prof. Shayiab Kamil and Dr. Masood Saleem for guidance, inputs, and analysis of histopathologic studies.
REFERENCES
- 1.Patricia O, Shafer RNMN. Epilepsy Foundation; About epilepsy. About epilepsy. [Retrieved May 1, 2015]. A from http://www. epilepsyfoundation.org/aboutepilepsy/index.cfm/
- 2.Robert SF, Carlos A, Alexis A, Alicia B, Helen CJ, Christian EE, et al. A practical clinical definition of epilepsy. Epilepsia. 2014;55:475–82. doi: 10.1111/epi.12550. [DOI] [PubMed] [Google Scholar]
- 3.Pitkanen A, Schwartzkroin P, Moshe S. The rat modles of seizures and epilepsy. San Diego: Elsevier Academic Press; 2005. [Google Scholar]
- 4.Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: behavioural, electroencephalographic and neuropathological study. Behav Brain Res. 1983;9:315–35. doi: 10.1016/0166-4328(83)90136-5. [DOI] [PubMed] [Google Scholar]
- 5.Turski WA, Czuczwar SJ, Kleinrok Z, Turski L. Cholinomimetics produce seizures and brain damage in rats. Experientia. 1983;39:1408–11. doi: 10.1007/BF01990130. [DOI] [PubMed] [Google Scholar]
- 6.Anovadiya AP, Sanmukhani JJ, Tripathi CB. Epilepsy: novel therapeutic targets. J Pharmacol Pharmacother. 2012;3:112–7. doi: 10.4103/0976-500X.95505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang LP, Patel M. Seizure-induced changes in mitochondrial redox status. Free Rad Biol Med. 2006;40:316–22. doi: 10.1016/j.freeradbiomed.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 8.Katrin S. Cytotoxicity of dietary flavonoids on different human cancer types. Pharmacogsy Rev. 2014;8:122–46. doi: 10.4103/0973-7847.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renugadevi J, Prabu SM. Naringenin protects against cadmium-induced oxidative renal dysfunction in rats. Toxicology. 2009;256:128–34. doi: 10.1016/j.tox.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 10.Ekambaram G, Rajendran P, Magesh V, Sakthisekaran D. Naringenin reduces tumor size and weight lost in N-methyl-N’-nitro-N-nitrosoguanidine-induced gastric carcinogenesis in rats. Nutr Res. 2008;28:106–12. doi: 10.1016/j.nutres.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Santos KF, Oliveria TT, Nagem TJ, Pinto AS, Oliveria MG. Hypolipidamic effects of naringenin, rutin nicotinic acid and their association. Pharmacol Res. 1999;40:493–6. doi: 10.1006/phrs.1999.0556. [DOI] [PubMed] [Google Scholar]
- 12.Rayidi S, Pari L. Effect of naringenin on carbohydrate metabolism in streptozotocin- nicotinamide induced diabetic rats. Biomirror. 2011;2:12–9. [Google Scholar]
- 13.Goldwasser J, Pazit YC, Wenu L, Danny K, Patrick B, Stephen JP, et al. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J Hepatol. 2011;55:963–71. doi: 10.1016/j.jhep.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinho-Ribeiro FA, Zarpelon AC, Fattori V, Manchope MF, Mizokami SS, Casagrande R, et al. Naringenin reduces inflammatory pain in mice. Neuropharmacology. 2016;105:508–19. doi: 10.1016/j.neuropharm.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 15.Zaki HF, May A, Amina SA. Naringenin protects against scopolamine-induced dementia in rats.Bull. Faculty Pharm. 2014;52:15–25. [Google Scholar]
- 16.Khan MB, Khan MM, Khan A, Ahmed ME, Ishrat T, Tabassum R, et al. Naringenin ameliorates Alzheimer's disease (AD)-type neurodegeneration with cognitive impairment (AD-TNDCI) caused by the intracerebroventricular-streptozotocin in rat model. Neurochem Int. 2012;61:1081–93. doi: 10.1016/j.neuint.2012.07.025. [DOI] [PubMed] [Google Scholar]
- 17.Zbarsky V, Datla KP, Parkar S, Rai DK, Aruoma OI, Dexter DT. Neuroprotective properties of the natural phenolic antioxidants curcumin and naringenin but not quercetin and fisetin in a 6-OHDA model of Parkinson's disease. Free Radic Res. 2005;39:1119–25. doi: 10.1080/10715760500233113. [DOI] [PubMed] [Google Scholar]
- 18.Muthaiah VK, Venkitasamy L, Michael FM, Chandrasekar K, Venkatachalam S. Neuroprotective role of naringenin on carbaryl induced neurotoxicity in mouse neuroblastoma cells. J Pharmacol Pharmacother. 2013;4:192–7. doi: 10.4103/0976-500X.114599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunot S, Dugas N, Faucheux B, Hartmann A, Tardieu M, Debre P, et al. Fcepsilon RII/CD23 is expressed in Parkinson's disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J Neurosci. 1999;19:3440–7. doi: 10.1523/JNEUROSCI.19-09-03440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Nagao T, Desjardins GC, Gloor P, Avoli M. Quantitative evaluation of neuronal loss in the dorsal hippocampus in rats with long-term pilocarpine seizures. Epilepsy Res. 1994;17:237–47. doi: 10.1016/0920-1211(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 21.Pinder RM, Brogden RN, Speight TM, Avery GS. Sodium valproate: a review of its pharmacological properties and therapeutic efficacy in epilepsy.Drugs. 1997;13:81–123. doi: 10.2165/00003495-197713020-00001. [DOI] [PubMed] [Google Scholar]
- 22.Racine RJ. Modification of seizure activity by electrical stimulation II motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–94. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 23.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent J Biol chem. 1951;1:193–265. [PubMed] [Google Scholar]
- 24.Marklund S, Marklund G. Involvement of superoxide anion radical in the auto oxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974;47:469–74. doi: 10.1111/j.1432-1033.1974.tb03714.x. [DOI] [PubMed] [Google Scholar]
- 25.Claiborne A. Catalase activity. In: Greenwald RA, editor. Handbook of methods for oxygen radical research. Boca Raton, FL: 1985. p. 283.p. 3. [Google Scholar]
- 26.Wright JR, Colby HD, Miles PR. Cytosolic factors which affect microsomal lipid peroxidation in lung and liver. Arch Biochem Biophys. 1981;206:296–304. doi: 10.1016/0003-9861(81)90095-3. [DOI] [PubMed] [Google Scholar]
- 27.Carlberg I, Mannervik B. Glutathione reductase levels in rat brain. J Biol Chem. 1975;250:5475–9. [PubMed] [Google Scholar]
- 28.Ropper AH, Brown RH. Epilepsy and other seizures disorder. In: Ropper AH, Brown RH, editors. Adams and Victor's principles of neurology. 8 ed. New York: McGraw-Hill; 2005. pp. 271–97. [Google Scholar]
- 29.Poole K, Moran N, Bell G, Solomon J, Kendall S, McCarthy M, et al. Patients’ perspectives on services for epilepsy: a survey of patient satisfaction, preferences and information provision in 2394 people with epilepsy. Seizure. 2000;9:551–8. doi: 10.1053/seiz.2000.0450. [DOI] [PubMed] [Google Scholar]
- 30.Koehn FE, Carter GT. The evolving role of natural products in drug discovery. Nat Rev Drug Discov. 2005;4:206–20. doi: 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- 31.Sucher NJ. Insights from molecular investigations of traditional Chinese herbal stroke medicines: implications for neuroprotective epilepsy therapy. Epilepsy Behav. 2006;8:350–62. doi: 10.1016/j.yebeh.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 32.Okoli CO, Ezike AC, Agwagah OC, Akah PA. Anticonvulsant and anxiolytic evaluation of leaf extracts of Ocimum gratissimum, a culinary herb. Pharmacognosy Res. 2010;2:36–40. doi: 10.4103/0974-8490.60580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlini EA. Plants and the central nervous system. Pharmacol Biochem Behav. 2003;3:501–12. doi: 10.1016/s0091-3057(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 34.Campelo LML, Feitosa CM, Tome AR, Freitas RM. Evaluation of neuroprotective potential of Citrus limon essential oil in hippocampus and striatum of mice after pilocarpine-induced seizures. Boletin Latinoamericano y del Caribe de Plantas Medicinales y Aromaticas. 2010;10:116–26. [Google Scholar]
- 35.Costaa JP, Ferreiraa PB, De Sousab DP, Jordanc J, Freitas RM. Anticonvulsant effect of phytol in a pilocarpine model in mice: Neurosci Lett. 2012;523:115–8. doi: 10.1016/j.neulet.2012.06.055. [DOI] [PubMed] [Google Scholar]
- 36.Silva MI, Silva MA, Aquino MR, Moura BA, Sousa HL, Lavor EP, et al. Effects of isopulegol on pentylenetetrazol-induced convulsions in mice: possible involvement of GABAergic system and antioxidant activity. Fitoterapia. 2009;80:506–13. doi: 10.1016/j.fitote.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 37.Hosseinzadeh H, Parvardeh S, Nassiri AM, Mansouri MT. Intracerebroventricular administration of thymoquinone, the major constituent of Nigella sativa seeds, suppress epileptic seizures in rats. Med Sci Monitor. 2005;11:106–10. [PubMed] [Google Scholar]
- 38.Sousa DP, Gonc JCR¸ Alves L, Quintans J, Cruz JS, Araújo DAM, et al. Study of anticonvulsant effect of citronellol, a monoterpene alcohol, in rodents. Neurosci Lett. 2006;401:231–5. doi: 10.1016/j.neulet.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 39.De Almeida RN, De Sousa DP, Nóbrega FFF, Claudino FS, Araújo DAM, Leite JR, et al. Anticonvulsant effect of a natural compound, epoxycarvone and its action on the nerve excitability. Neurosci Lett. 2008;443:51–5. doi: 10.1016/j.neulet.2008.07.037. [DOI] [PubMed] [Google Scholar]
- 40.Kumar GP, Khanum F. Neuroprotective potential of phytochemicals. Pharmacognosy Rev. 2012;6:81–90. doi: 10.4103/0973-7847.99898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uzum G, Diler AS, Bahcekapili N, Ziylan Z. Erythropoietin prevents the increase in blood brain barrier permeability during pentylenetetrazole induced seizures. Life Sci. 2006;78:2571–6. doi: 10.1016/j.lfs.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 42.Eun-Joo S, Ji HJ, Yoon HC, Won-Ki K, Kwang-Ho K, Jae-Hyung B, et al. Role of oxidative stress in epileptic seizures. Neurochem Int. 2011;59:122–37. doi: 10.1016/j.neuint.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frantseva MV, Perez VJL, Tsoraklidis G, Mendonca AJ, Adamchik Y, Mills LR, et al. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neurosciences. 2000;97:431–5. doi: 10.1016/s0306-4522(00)00041-5. [DOI] [PubMed] [Google Scholar]
- 44.Waldbaum S, Patel M. Mitochondrial dysfunction and oxidative stress: a contributing link to acquired epilepsy. J Bioenerg Biomembr. 2010;42:449–55. doi: 10.1007/s10863-010-9320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamilton SE, Loose MD, Qi M, Levey AI, Hille B, McKnight GS. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc Natl Acad Sci USA. 1997;94:13311–6. doi: 10.1073/pnas.94.24.13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kondratyev A, Gale K. Latency to onset of status epilepticus determines molecular mechanisms of seizure-induced cell death. Brain Res Mol Brain Res. 2004;5:86–94. doi: 10.1016/j.molbrainres.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 47.Freitas RM, Sousa FC, Vasconcelos SM, Viana GS, Fonteles MM. Pilocarpine-induced status epilepticus in rats: lipid peroxidation level, nitrite formation, GABAergic and glutamatergic receptor alterations in the hippocampus, striatum and frontal cortex. Pharmacol Biochem Behav. 2004;78:327–32. doi: 10.1016/j.pbb.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 48.Manmohan KP, Purnima M, Maheshwari PK. The lipid peroxidation product as a marker of oxidative stress in epilepsy. J Clin Diagn Res. 2012;6:590–2. [Google Scholar]
- 49.Chuang YC. Mitochondrial dysfunction and oxidative stress in seizure-induced neuronal cell death. Acta Neurol Taiwan. 2010;19:3–15. [PubMed] [Google Scholar]
- 50.Uchida A. Activation of the stress signalling pathways by the end products of lipid peroxidation. J Biol Chem. 2000;274:2234–42. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- 51.Dhuley JN. Effect of Ashwagandha on lipid peroxidation in stress-induced animals. J Ethnopharmacol. 1998;60:173–8. doi: 10.1016/s0378-8741(97)00151-7. [DOI] [PubMed] [Google Scholar]
- 52.Lee MH, Yoon S, Moon JK. The flavonoid naringenin inhibits dimethyl nitro samine-induced liver damage in rats. Biol Pharm Bull. 2004;27:72–6. doi: 10.1248/bpb.27.72. [DOI] [PubMed] [Google Scholar]
- 53.Oliver CN, Starke RPE, Stadtman ER, Liu GJ, Carney JM, Floyd RA. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc Natl Acad Sci USA. 1990;87:5144–7. doi: 10.1073/pnas.87.13.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halliwell B, Gutteridge JM. Free radicals in biology and medicine. 3rd ed. New York: Oxford University Press; 1999. [Google Scholar]
- 55.Annadurai T, Thomas PA, Geraldine P. Ameliorative effect of naringenin on hyperglycemia-mediated inflammation in hepatic and pancreatic tissues of Wistar rats with streptozotocin-nicotinamide-induced experimental diabetes mellitus. Free Radic Res. 2013;47:793–803. doi: 10.3109/10715762.2013.823643. [DOI] [PubMed] [Google Scholar]
- 56.Chtourou Y, Fetoui H, Jemai R, Slima AB, Makni M, Gdoura R. Naringenin reduces cholesterol-induced hepatic inflammation in rats by modulating matrix metalloproteinases-2,9 via inhibition of nuclear factor κB pathway. Eur J Pharmacol. 2015;746:96–105. doi: 10.1016/j.ejphar.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 57.Williams PL, Warwick R. Gray's anatomy. 36th ed. London: Longman; [Google Scholar]
- 58.Samsonovich AV, Ascoli GA. A simple neural network model of hippocampus suggesting its path finding role in episodic memory retrieval. Learn Memory. 2005;12:193–208. doi: 10.1101/lm.85205. [DOI] [PMC free article] [PubMed] [Google Scholar]