

This study identifies the calcium-regulated transcription factor nuclear factor of activated T cells as a novel regulator of cystathionine γ-lyase (CSE). This pathway is basally active in mesenteric artery endothelial cells, but, after exposure to intermittent hypoxia to mimic sleep apnea, nuclear factor of activated T cells c3 nuclear translocation and CSE expression are decreased, concomitant with decreased CSE-dependent vasodilation.
Keywords: hydrogen sulfide, vasodilation, mesenteric arteries, sleep apnea, nuclear factor of activated T cells c3, calcineurin
Abstract
Sleep apnea is a risk factor for cardiovascular disease, and intermittent hypoxia (IH, 20 episodes/h of 5% O2-5% CO2 for 7 h/day) to mimic sleep apnea increases blood pressure and impairs hydrogen sulfide (H2S)-induced vasodilation in rats. The enzyme that produces H2S, cystathionine γ-lyase (CSE), is decreased in rat mesenteric artery endothelial cells (EC) following in vivo IH exposure. In silico analysis identified putative nuclear factor of activated T cell (NFAT) binding sites in the CSE promoter. Therefore, we hypothesized that IH exposure reduces Ca2+ concentration ([Ca2+]) activation of calcineurin/NFAT to lower CSE expression and impair vasodilation. In cultured rat aortic EC, inhibiting calcineurin with cyclosporine A reduced CSE mRNA, CSE protein, and luciferase activity driven by a full-length but not a truncated CSE promoter. In male rats exposed to sham or IH conditions for 2 wk, [Ca2+] in EC in small mesenteric arteries from IH rats was lower than in EC from sham rat arteries (Δfura 2 ratio of fluorescence at 340 to 380 nm from Ca2+ free: IH = 0.05 ± 0.02, sham = 0.17 ± 0.03, P < 0.05), and fewer EC were NFATc3 nuclear positive in IH rat arteries than in sham rat arteries (IH = 13 ± 3, sham = 59 ± 11%, P < 0.05). H2S production was also lower in mesenteric tissue from IH rats vs. sham rats. Endothelium-dependent vasodilation to acetylcholine (ACh) was lower in mesenteric arteries from IH rats than in arteries from sham rats, and inhibiting CSE with β-cyanoalanine diminished ACh-induced vasodilation in arteries from sham but not IH rats but did not affect dilation to the H2S donor NaHS. Thus, IH lowers EC [Ca2+], NFAT activity, CSE expression and activity, and H2S production while inhibiting NFAT activation lowers CSE expression. The observations that IH exposure decreases NFATc3 activation and CSE-dependent vasodilation support a role for NFAT in regulating endothelial H2S production.
NEW & NOTEWORTHY This study identifies the calcium-regulated transcription factor nuclear factor of activated T cells as a novel regulator of cystathionine γ-lyase (CSE). This pathway is basally active in mesenteric artery endothelial cells, but, after exposure to intermittent hypoxia to mimic sleep apnea, nuclear factor of activated T cells c3 nuclear translocation and CSE expression are decreased, concomitant with decreased CSE-dependent vasodilation.
hydrogen sulfide (H2S) is a recently described vasodilator that is generated from the amino acid cysteine by the enzyme cystathionine γ-lyase (CSE) in the vasculature (17, 43). H2S levels and expression and activity of CSE are decreased in many cardiovascular diseases (1, 3, 5, 22, 38), including sleep apnea (22, 27). Sleep apnea, characterized by intermittent cessation of breathing during sleep, affects up to 20% of adults (36) and is a risk factor for cardiovascular disease (2, 31, 37, 39). Recent results from our laboratory using an intermittent hypoxia (IH) rat model of sleep apnea demonstrated that vasodilation to the CSE substrate cysteine requires the endothelium and is impaired following IH exposure (20). Conversely, vasodilation in response to the exogenous H2S donor NaHS is similar between control and IH groups. Furthermore, expression of CSE is decreased in the endothelium of arteries from IH rats as is the production of H2S (21). Taken together, these findings suggest that arteries from IH-exposed rats dilate to H2S but may have a deficit in the ability to produce H2S endogenously because of decreased enzyme expression. Previous findings from our laboratory suggest endothelial function is compromised in arteries from IH rats. For example, myogenic or pressure-induced tone is increased in mesenteric arteries from IH rats. Inhibition of CSE or removal of the endothelium increases myogenic tone in arteries from control rats but has no effect in the IH group (21), suggesting that endothelial-derived H2S normally restrains myogenic tone, but this mechanism is impaired after IH exposure. Although there is evidence of decreased CSE expression and H2S production following IH exposure, the mechanisms of transcriptional regulation of CSE under control or IH conditions are not clearly defined. To identify potential transcriptional regulators of CSE, we conducted an in silico analysis of the CSE promoter, the sequence of which was reported by Ishii and colleagues (19), and identified several putative binding sites for the transcription factor nuclear factor of activated T cells (NFAT, Fig. 1). The NFAT family of transcription factors is activated through Ca2+ activation of calcineurin, which dephosphorylates cytosolic NFAT, triggering its translocation to the nucleus (reviewed in Ref. 16). The role of endothelial NFAT activity following IH exposure has not been previously evaluated; therefore, we hypothesized that IH exposure reduces endothelial Ca2+ levels and NFATc3 activity to decrease CSE expression and impair H2S-dependent vasodilation.
Fig. 1.

Cystathionine γ-lyase promoter sequence adapted from Ref. 10. Regions in underlined gray indicate putative binding sites for the transcription factor nuclear factor of activated T cells (NFAT). Binding sites were predicted by in silico analysis with Patch 1.0 and AliBaba 2.1 public software.
MATERIALS AND METHODS
Rodent model of sleep apnea.
Male Sprague-Dawley rats (275–300 g) were obtained from Harlan Laboratories. Rats were exposed to sham or IH conditions as previously described (23). IH exposure consisted of cycling between room air (21% O2-0% CO2) and hypoxia (5% O2-5% CO2), with 20 hypoxic episodes/h for 7 h/day for 2 wk during the sleep cycle. Rats in the sham group were housed in identical Plexiglas containers, with exposure to the same air flow and sound conditions, but were maintained in room air only. A lethal concentration of pentobarbital sodium (200 mg/kg ip) was used to euthanize rats at the end of the 2-wk cycling period. The Institutional Animal Care and Use Committee of the University of New Mexico School of Medicine reviewed and approved all animal protocols. All protocols conform to the National Institutes of Health guidelines for animal use.
In silico analysis of CSE promoter.
Patch 1.0 and AliBaba 2.1 public software were used to predict transcription factor binding sites over the sequence of the CSE promoter, as identified by Ishii and colleagues (19).
CSE promoter activity assay.
CSE promoter activity was measured in rat aortic endothelial cells (RAEC; Cell Applications) under conditions in which NFAT activity was inhibited. Cells were grown in rat endothelial cell basal medium with rat endothelial cell growth supplement (Cell Applications). Cells were maintained in 5% CO2 with controlled humidity at 37°C. RAEC were electroporated (using Nucleofector; Lonza) with a luciferase reporter plasmid containing a 3.5-kb segment upstream of the CSE transcriptional start site (CSE-1) or a truncated construct that did not include the putative NFAT binding sites (CSE-10) [kindly provided by Dr. I. Ishii (National Institute of Neuroscience, Tokyo, Japan)] (19) and a Renilla reporter plasmid (Promega) that was used to normalize differences in transfection efficiency. NFAT transcriptional activity was determined using a 9× NFATc-luciferase reporter plasmid and normalized to Renilla luciferase. The 9× NFATc-luciferase plasmid has nine NFATc binding sites upstream of the luciferase gene [kindly provided by Dr. J. D. Molkentin (Department of Pediatrics, Children’s Hospital Medical Center, Cincinnati, OH)]. RAEC were treated for 48 h with vehicle (ethanol) or cyclosporine A (CsA; 1 µM), an inhibitor of calcineurin. Luciferase and Renilla activity were detected simultaneously using a Dual-Luciferase Reporter Assay System (Promega). Data are expressed as relative luciferase units compared with relative Renilla units.
mRNA expression.
RAEC were used to evaluate NFAT regulation of CSE transcription. Cells were treated with vehicle (ethanol) or CsA (1 µM) for 48 h. Total RNA was extracted using the RNeasy Mini Kit (Qiagen) and reverse transcribed to cDNA using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies). In other studies, total RNA was collected from mesenteric arteries isolated in RNA later (Ambion) to evaluate in vivo NFATc3 and NFATc2 mRNA levels. In all studies, the Roche Universal Probe Library System was used for real-time detection of CSE in cells (5′-cgaagacctgggtcaagc, 3′-agaaggtctggccccttg, probe 121), cyclooxygenase 2 (COX2) in cells (5′-ctacaccagggcccttcc, 3′-tccagaacttcttttgaatcagg, probe 5), NFATc2 in arteries, (5′-accagacttacctggatgacg, 3′-tgatttcgggagggaggt, probe 12), NFATc3 in arteries (5′-tggtggccatcctgttgt, 3′-catctgtagatttattggcttttcac, probe 128), and cyclophillin B (5′-accaatggctcccagttctt; 3′-ctccaccttccgtaccacat, probe 79) and/or β-actin (5′-cccgcgagtacaaccttct, 3′-cgtcatccatggcgaact, probe 17) in both as a reference gene. The normalized gene expression method (2−ΔΔCT) was used for relative quantification of gene expression (30).
CSE protein expression.
RAEC were also used to evaluate NFAT regulation of CSE protein expression. Preliminary studies treating cells with CsA for 48, 60, 72, or 90 h showed that a 90-h treatment caused the greatest decrease in CSE expression, so cells were treated with vehicle (ethanol) or CsA (1 µM) for 90 h before collection with 1× Laemli sample buffer (Bio-Rad). This long stability of the CSE protein is in agreement with earlier observations by Madurga et al. (24). Samples were run in a 10% acrylamide gel, transferred to PVDF membrane, blocked with 5% milk in phosphate-buffered saline with 0.05% Tween 20 (PBS-T), and probed with a CSE polyclonal antibody in 0.05% gelatin in PBS-T (1:1,000; ProteinTech 12217–1), as previously characterized (13). Homogenate of RAEC overexpressing CSE was included in every gel as a positive control since the antibody bound to multiple protein targets. Protein was visualized using a horseradish peroxidase rabbit secondary (1:5,000; Bio-Rad), enhanced chemiluminescence reagent (Pierce Scientific), and photographic film. After CSE blot images were obtained, membranes were washed and reprobed for GAPDH (1:5,000; 2118S; Cell Signaling) and an anti-rabbit secondary (1:5000; Bio-Rad). Scanned images were analyzed using SigmaGel software.
Endothelial Ca2+ measurements.
Because NFAT is regulated by Ca2+, endothelial cell Ca2+ was measured in isolated pressurized mesenteric arteries from sham and IH rats. Mesenteric arterial cascades were isolated in ice-cold HEPES buffer (in mM: 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 0.026 EDTA, and 10 glucose). Fourth- or fifth-order endothelium-intact artery segments were cannulated, and the lumen was loaded with the ratiometric Ca2+ indicator fura 2-AM [2 µM in HEPES buffer with 0.05% pluronic acid (Life Technologies)] for 10 min in the dark. The lumen was washed with physiological salt solution (PSS; in mM: 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 0.43 NaH2PO4, 19 NaHCO3, 1.8 CaCl2, and 5.5 glucose), and the artery was equilibrated for 40 min at an intraluminal pressure of 60 mmHg and superfused with gassed (21% O2-6% CO2-N2 balance) and heated (37°C) PSS at a rate of 5 ml/min. Intraluminal pressure was increased to 100 mmHg, and artery diameter was allowed to stabilize. Arteries were then excited alternately at 340 and 380 nm by a dual-excitation light source (IonOptix Hyperswitch), and the 510-nm emission was collected with a photomultiplier tube. Basal endothelial cell Ca2+ is expressed as the ratio of the background-subtracted emission at 340 and 380 nm compared with the ratio in Ca2+-free conditions (in mM: 129.8 NaCl, 5.4 KCl, 0.83 MgSO4, 0.43 NaH2PO4, 19 NaHCO3, 5.5 glucose, and 4.5 EGTA).
H2S assay.
H2S levels were measured in mesenteric tissue using a modified version of the method of Stipanuk and Beck (33) in the presence or absence of β-cyano-l-alanine (BCA, 10−4 mol/l). Mesentery segments containing arterioles and surrounding tissue (19–26 mg tissue/reaction) were added to phosphate-buffered saline (PBS) containing pyridoxal 5′-phosphate (8 mmol/l), cysteine (10−5 mol/l), and complete protease inhibitor (Roche) in plastic center wells suspended in 25 ml Erlenmeyer flasks fitted with septum stoppers. The plastic center wells also contained small sections of filter paper wetted with PBS to trap evolved H2S. The flasks were sealed and flushed with N2. Reactions were initiated by placing the flasks in a 37°C incubator for 30 min. H2S was trapped by injection of ZnAc (1%) through the septum stopper in the center wells, and then reactions were immediately stopped by injection of trichloroacetic acid (10%). To detect the H2S, 67 μl of 20 mmol/l N,N-dimethyl-p-phenylenediamine sulfate in 7.2 mol/l HCl and 67 μl of 30 mmol/l FeCl3 in 1.2 mmol/l HCl were added. After 20 min of incubation, absorbance was measured at 670 nm, and sample concentration was calculated from a standard curve (10−7 to 10−4 mol/l NaHS) and normalized for tissue weight (mg) to yield nanomoles per minute per milligram tissue.
NFATc3 and NFATc2 nuclear localization.
To determine if the observed decrease in endothelial Ca2+ diminishes NFAT translocation in vivo, a section of the small intestine was collected from anesthetized sham and IH rats and immediately frozen in OCT Compound (Tissue Tek) without fixation. Cross sections (10 µm) were thaw mounted on slides and incubated with biotin-labeled mouse anti-rat CD31 antibody (BD Pharmigen) or rabbit anti-NFATc3 or rabbit anti-NFATc2 antibodies (Santa Cruz Biotechnology) followed by staining with Cy3-streptavidin (Jackson ImmunoResearch), anti-rabbit Dylight 649 (Thermo Scientific), and SYTOX Green (Life Technologies). Sections were imaged with a Leica TCS SP5 laser scanning confocal microscope with a ×63 objective. NFATc3- or NFATc2-positive endothelial cell nuclei were quantified in multiple fields from each artery using MetaMorph software (Molecular Devices). The software was programmed so that individual pixels appeared white instead of yellow if the green nucleic acid stain and red NFATc3 or NFATc2 stain colocalized. Thus, a cell was considered positive if colocalization (white) was uniformly distributed in the nucleus and negative if no colocalization (green only) or perinuclear colocalization was observed (4, 10, 11, 14). In cultured RAEC, translocation studies used the same antibodies with cells grown on cover slips that were fixed with paraformaldehyde before being stained and with the CD31 antibody omitted.
Vasodilation studies.
Fourth- and fifth-order mesenteric arteries from sham and IH rats were isolated, cannulated, and pressurized as described in Endothelial Ca2+ measurements. Arteries were incubated with the CSE inhibitor BCA (100 µM; Sigma-Aldrich) or vehicle (water) for 20 min and then preconstricted with phenylephrine to ~50% of their starting diameter. Cumulative concentrations (1 nM to 1 µM) of the endothelium-dependent vasodilator acetylcholine (ACh; Sigma-Aldrich) were administered. In another set of arteries from sham rats, arteries were pretreated with either BCA or vehicle for 20 min and then contracted to 50% resting diameter with phenylephrine as above. Vasodilation to the H2S donor NaHS (10 µM) was recorded to determine if CSE inhibition of dilation could be overcome by exogenous H2S. Vasodilation is expressed as the percentage change from phenylephrine preconstriction vs. the maximal change from preconstriction in Ca2+-free conditions. The average artery diameter in Ca2+-free buffer at a pressure of 100 mmHg was 132 µm.
Statistical analysis.
CSE promoter activity was analyzed by two-way ANOVA, and CSE mRNA and protein levels were compared by Student’s t-test. Basal endothelial cell Ca2+, dilation to NaHS, and endothelial NFATc3 or NFATc2 nuclear localization were compared between sham and IH by Student’s t-tests. Vasodilation to ACh was compared between sham and IH ± BCA by repeated-measures two-way ANOVA. All values are expressed as means ± SE. P values <0.05 were considered statistically significant.
RESULTS
Previous work from our laboratory has indicated impairment of endogenous H2S production in the endothelium of mesenteric arteries from rats exposed to the IH model of sleep apnea (20). In an attempt to identify potential transcriptional regulators of CSE, in silico analysis of the CSE promoter sequence was conducted, and potential transcription factor binding sites were uncovered. Analysis revealed 16 putative binding sites for the Ca2+-sensitive transcription factor NFAT, which has been previously implicated in IH-induced hypertension (8, 9). Putative NFAT binding sites are denoted in Fig. 1.
To determine if NFAT regulates CSE, promoter activity and mRNA levels were measured in RAEC with and without inhibition of NFAT activity. In cells expressing full-length CSE promoter (CSE-1), inhibiting calcineurin with CsA decreased CSE promoter activity as measured by luciferase activity (Fig. 2A). In RAEC transfected with a truncated version of the CSE promoter (CSE-10), which lacks the putative NFAT binding sites, basal CSE promoter activity was decreased, and NFAT inhibition had no effect. CSE mRNA (Fig. 2B) was lower in nontransfected RAEC treated with CsA, similar to the CsA-induced decrease in COX-2 mRNA (Fig. 2C), a prototypical NFAT-regulated endothelial gene (18, 26). As expected, CsA significantly decreased basal NFAT transcriptional activity in RAEC (Fig. 3A). Interestingly, NFATc3 was localized in the nucleus under basal conditions, whereas NFATc2 was cytosolic (Fig. 3B). The nonselective inhibition of NFAT by CsA decreased nuclear localization of NFATc3 but did not affect NFATc2 localization, supporting a role for NFATc3 in CSE regulation (Fig. 3B). Furthermore, CsA did not affect transcript levels of either NFATc3 or NFATc2 (Fig. 3C).
Fig. 2.
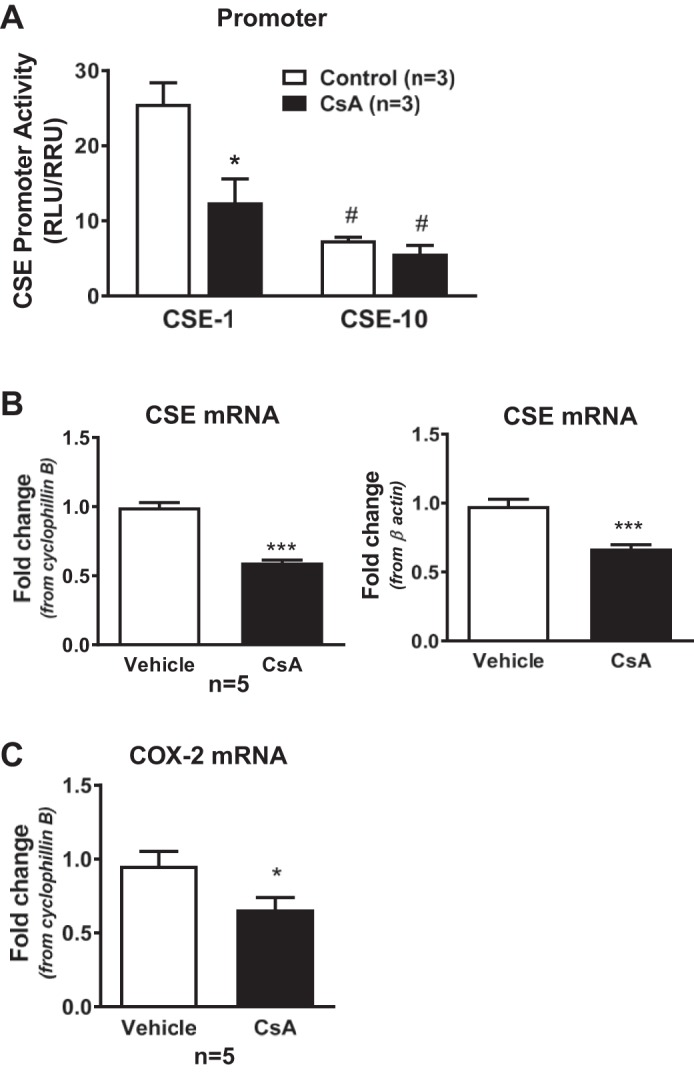
Cystathionine γ-lyase (CSE) promoter activity, CSE mRNA expression, and NFAT transcriptional activity in rat aortic endothelial cells (RAEC). A: cells expressing a full-length (CSE-1) or truncated version (CSE-10) of the CSE promoter upstream of luciferase were treated with vehicle (ethanol) or cyclosporine A (CsA; 1 µM) for 48 h. A Renilla construct was used to account for differences in transfection efficiencies, and data are expressed as the ratio of relative luciferase units (RLU) to relative Renilla units (RRU). B: CSE mRNA expression in RAEC treated with vehicle or CsA for 48 h using cyclophillin B or β-actin as endogenous control. C: cyclooxygenase 2 (COX2) mRNA expression in RAEC treated with vehicle or CsA for 48 h using cyclophillin B as endogenous control. P < 0.05 vs. control or vehicle (*) or CSE-1 (#) and ***P < 0.0001; n = 5–6 batches of RAEC from passage 2 to 5.
Fig. 3.

CsA decreases NFAT transcriptional activity and reduces NFATc3 nuclear localization in RAEC. A: cells expressing a 9× NFAT luciferase reporter construct treated with vehicle or CsA for 48 h. B: representative immunofluorescence confocal microscopic images of RAEC stained for NFATc2 and NFATc3. Nuclei were labeled with SYTOX green. C: NFATc2 and NFATc3 mRNA expression in RAEC treated with vehicle or CsA for 48 h using cyclophillin B as endogenous control. Values are means ± SE. *P < 0.05 vs. control or vehicle; n = 3–6 batches of RAEC from passage 2 to 5.
In Western blots for CSE, two bands were present that comigrated with the positive control (EC overexpressing CSE), and both were analyzed, normalized to GAPDH levels as a loading control. Bands from the CsA-treated cell homogenates had lower density than bands from vehicle-treated cell homogenates (Fig. 4). These data suggest that basal NFAT activity regulates CSE expression.
Fig. 4.
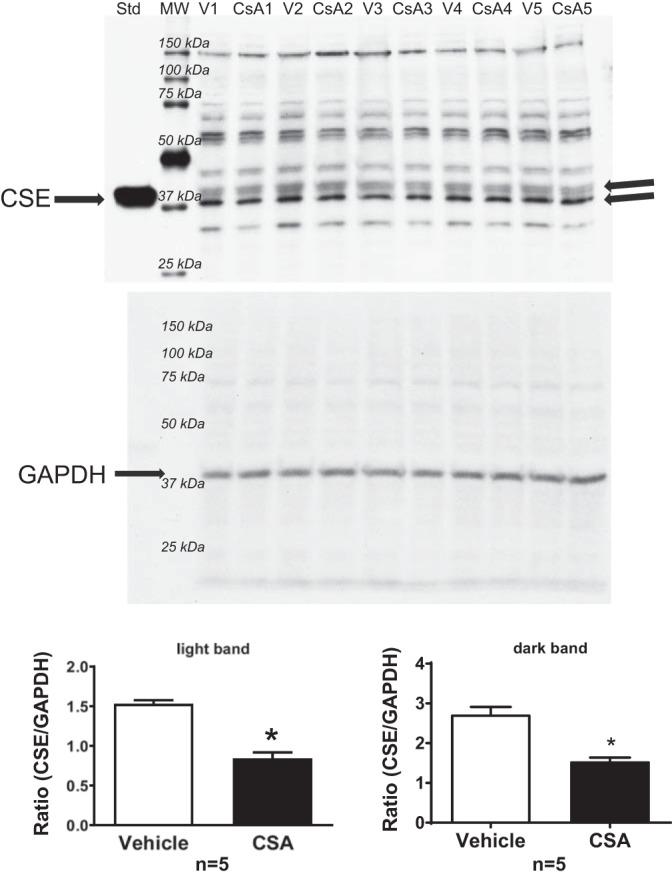
CSE protein expression in RAEC treated with vehicle or CsA (1 µM) for 90 h. A single blot was probed with either an antibody against CSE (top) or GAPDH (bottom), and then CSE expression was calculated as the ratio of CSE/GAPDH density to control for loading differences. Values are means ± SE. *P < 0.05 vs. vehicle; n = 5 batches of RAEC from passage 2 to 5.
Because NFAT requires Ca2+ for its activation, basal endothelial cell Ca2+ was measured in isolated pressurized mesenteric arteries from sham and IH rats to determine if IH exposure decreases Ca2+ levels in the endothelium. The fura 2 ratio in endothelial cells was lower in arteries from IH rats compared with the ratios in endothelial cells of arteries from sham rats (Fig. 5A), indicating that 2 wk of IH exposure decreases Ca2+ levels in the endothelium under unstimulated conditions. Consistent with loss of CSE (21), H2S production was also decreased in mesenteric tissue from IH rats compared with that in tissues from sham rats, and this production was inhibited by BCA only in the samples from the sham rats (Fig. 5B).
Fig. 5.
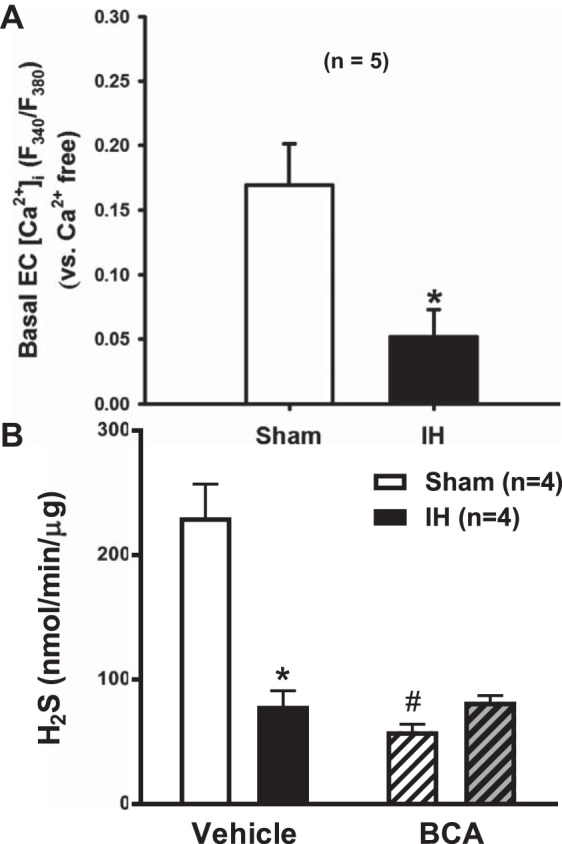
Basal endothelial cell Ca2+ in mesenteric arteries and hydrogen sulfide (H2S) production in mesenteric tissue from rats exposed to sham and intermittent hypoxia (IH) conditions. A: Ca2+ was measured in the endothelium of pressurized arteries with the ratiometric Ca2+ indicator fura 2-AM. Values are expressed as the difference between the fura 2 ratio in Ca2+-containing conditions and Ca2+-free conditions. Values are means ± SE. *P < 0.05 vs. sham; n = 5. B: H2S was measured in mesenteric tissue from rats exposed to sham and IH conditions using a modified version of the method developed by Stipanuk and Beck (33). Values are means ± SE and expressed as nmol·min−1·mg tissue−1. P < 0.05 vs. sham (*) or vehicle (#); n = 5.
The percentage of endothelial cell nuclei containing NFATc3 and NFATc2 was determined in cross sections of mesenteric arteries from sham and IH rats. Artery cross sections were triple fluorescently labeled for an endothelial cell marker (CD31), nuclei (SYTOX Green), and NFATc3 (Fig. 6A) or NFATc2 (data not shown). Nuclei were considered NFAT positive (indicated with white arrow) if NFAT staining colocalized with nuclear staining in CD31+ cells. There were significantly less NFATc3-positive nuclei in the endothelium of naïve arteries from IH rats compared with those in arteries of sham rats (Fig. 6B) but no difference in NFATc2-positive nuclei (Fig. 6C), suggesting NFATc3 activity is decreased under basal conditions following IH exposure. Again, this is consistent with the lower resting Ca2+ levels in the endothelial layer of IH mesenteric arteries.
Fig. 6.

Endothelial nuclear localization of NFATc3 and NFATc2 in mesenteric arteries from rats exposed to sham and IH conditions. A: cross sections of mesenteric arteries stained with antibodies for NFATc3 (red) and the endothelial marker CD31 (blue) as well as SYTOX Green for nuclei (green). White arrows indicate colocalization of NFATc3 and nuclei in CD31+ endothelial cells. B and C: quantification of the percentage of endothelial cell nuclei with NFATc3 (B) or NFATc2 (C) staining. Values are means ± SE. *P < 0.05 vs. sham; n = 13–27 arteries from at least 3 rats/group.
Because NFATc2 and NFATc3 activity increases in pulmonary arteries by transcriptional regulation (25, 28), NFATc2 and NFATc3 mRNA levels were determined in small mesenteric arteries from sham and IH-exposed rats. No significant difference was observed in transcript NFAT levels between groups (Fig. 7), suggesting that IH does not affect the expression of these NFAT isoforms but that the decreased CSE levels after IH are due to increased activity downstream of lower endothelial Ca2+ levels.
Fig. 7.
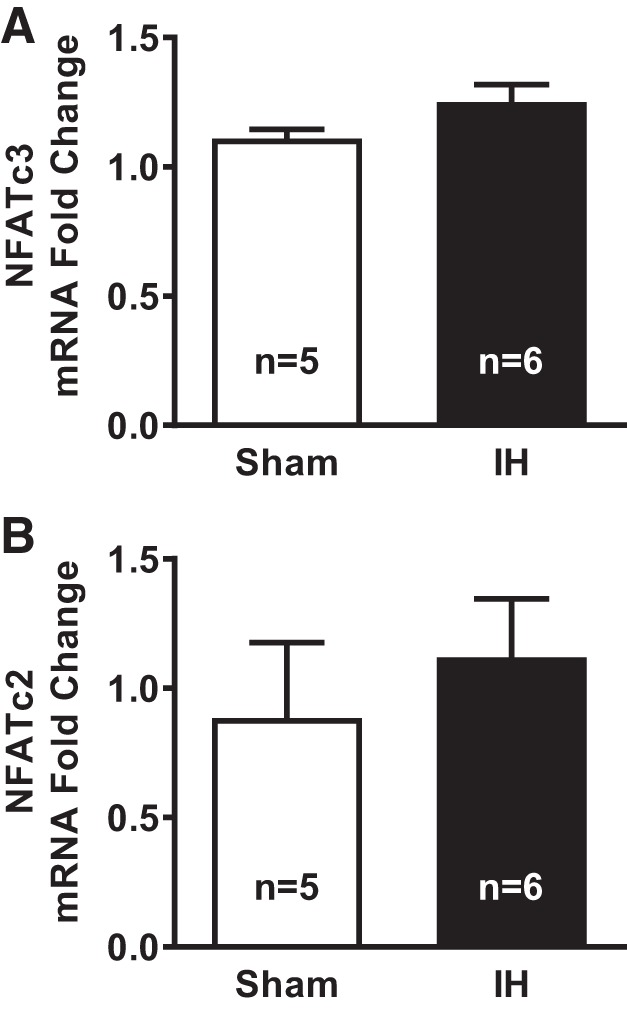
NFATc3 (A) and NFATc2 (B) mRNA levels in mesenteric arteries from rats exposed to sham and IH conditions. NFATc3 and NFATc2 transcripts were determined by real-time PCR in mesenteric arteries from rats exposed to sham or IH. Values are means ± SE; n = 5–6 rats/group.
Finally, to evaluate the functional impact on the endothelium of the IH-induced reduction in endothelial cell Ca2+ levels, NFATc3 nuclear localization, CSE expression, and H2S production, vasodilation to the endothelium-dependent dilator ACh was determined. Arteries from rats exposed to IH dilated less to ACh compared with arteries from sham rats (Fig. 8A). Inhibition of the H2S-producing enzyme CSE significantly decreased dilation in the sham group but did not further diminish dilation in the IH group. However, the CSE inhibitor did not affect vasodilator responses to the H2S donor NaHS, which was slightly but not significantly greater in arteries treated with the CSE inhibitor BCA (Fig. 8B). These findings suggest that, following IH exposure, basal endothelial cell Ca2+ and NFATc3 nuclear localization are decreased, and there is a reduced capacity for endothelium-derived H2S-dependent dilation that contributes to diminished endothelium-dependent vasodilation.
Fig. 8.

Acetylcholine (ACh)-mediated vasodilation in mesenteric arteries from rats exposed to sham and IH conditions. A: arteries were pretreated with vehicle (water) or the CSE inhibitor β-cyano-l-alanine (BCA; 100 µM) and then constricted to ~50% of their starting diameter with phenylephrine. Dilation to cumulative concentrations of ACh was measured. B: arteries were pretreated with vehicle (water) or the CSE inhibitor BCA (100 µM) and then constricted to ~50% of their starting diameter with phenylephrine. Dilation to 10 µM NaHS was measured. Values are means ± SE. *P < 0.05 vs. sham; n = 4–5 rats.
DISCUSSION
This study provides several novel observations: 1) NFAT is a potential transcriptional regulator of CSE in endothelial cells, 2), IH exposure decreases mesenteric artery endothelial cell Ca2+ and mesenteric H2S production under basal conditions, 3) NFATc3 nuclear localization is decreased in mesenteric artery endothelial cells following IH exposure, and 4) endothelium-dependent vasodilation of mesenteric arteries is impaired by IH exposure because of loss of H2S-induced dilation.
Ishii et al. (19) reported the sequence of the mouse CSE promoter, and subsequent studies demonstrate that transcriptional regulation of this H2S-producing enzyme is complex with regulation by multiple transcriptional factors. Studies by Yang and coworkers (40) using a luciferase promoter in HEK-293 and COS-7 cells or mRNA levels in smooth muscle cell lines observed a biphasic effect on CSE promoter activity in response to increases in Ca2+. Thus, Ca2+-sensitive transcription factors such as NFAT are good candidates for CSE regulation. Results from our in silico analysis of the CSE promoter identified putative NFAT binding sites in the promoter sequence, pointing to NFAT as a potential Ca2+-dependent regulator of CSE expression (11). Therefore, experiments were conducted in cultured cells to more directly assess NFAT regulation of CSE. NFAT nuclear translocation and binding to consensus sequences in the DNA require dephosphorylation by the Ca2+-activated phosphatase calcineurin (29). We observed that inhibiting NFAT activation with the calcineurin inhibitor CsA decreased CSE promoter activity in RAEC transfected with the full-length version of the CSE promoter. CsA also decreased NFAT transcriptional activity and decreased nuclear NFATc3 levels without affecting NFATc2 nuclear localization consistent with elevated basal NFAT activity in RAEC. CsA also decreased CSE mRNA and protein expression, further supporting that CsA-sensitive elements regulate CSE expression. Finally, when RAEC expressed a truncated version of the CSE promoter lacking the putative NFAT binding sites, CSE promoter activity was greatly diminished at baseline, and NFAT inhibition had no further effect (Fig. 2). These findings strongly support our hypothesis that NFAT is a transcriptional regulator of CSE expression.
Observations in mesenteric arteries further support that NFATc3 is an endogenous regulator of CSE. Both resting Ca2+ levels (Fig. 4A) and the nuclear location of NFATc3, but not of NFATc2, are lower in endothelial cells of arteries from IH rats compared with arteries from sham rats (Fig. 5). Both the current observation of reduced NFAT translocation and our previous report of lower CSE expression (21) are consistent with NFATc3 functioning as an activator of CSE expression under physiological conditions and that this activation is reduced after IH exposure. However, these data did not define a direct interaction of NFATc3 with the CSE promoter so that other members of the NFAT family of transcription factors could be implicated.
The novel observation that basal endothelial cell Ca2+ is reduced following IH exposure implies that multiple endothelial Ca2+ signaling pathways may be impaired. Yang et al. (41) reported that Ca2+-calmodulin acutely activates the H2S-producing enzyme CSE in vitro, and Jackson-Weaver and colleagues (21) observed that loss of endogenous H2S production enhances myogenic tone of small mesenteric arteries following IH exposure. Therefore, diminished endothelial Ca2+ could contribute to augmented myogenic tone in part through loss of H2S production in this rodent model of sleep apnea. Our data showing reduced nuclear localization of NFATc3 after IH exposure indicate that the reduced endothelial cell Ca2+ also limits activation of this Ca2+-sensitive transcription factor (Fig. 5), since nuclear localization is required for NFATc3 activity. These results indicate that IH exposure impairs NFATc3 activity in mesenteric artery endothelial cells and suggest that loss of NFAT-dependent activation of CSE expression contributes to IH-induced endothelial dysfunction. These results are in contrast to our previous findings in mouse mesenteric artery smooth muscle cells showing increased basal Ca2+ and NFATc3 nuclear localization and activity following IH exposure. Therefore, Ca2+ regulation and downstream signaling are differentially regulated in EC and smooth muscle cells in response to IH (9). The decrease in endothelial Ca2+ levels suggests multiple signaling pathways may be impaired after IH exposure. This would potentially include endothelial pathways under transcriptional regulation by NFAT such as CSE (32, 42) and enzymes regulated by intracellular Ca2+ that contribute to vasodilation such as endothelial nitric oxide synthase (12), prostacyclin (15), cytochrome P-450 products (6), and Ca2+-activated K+ channels (7, 34, 35).
Previous work from our laboratory (21) suggests that endothelial dysfunction in mesenteric arteries from IH rats is independent of loss of nitric oxide synthase, COX, or other known endothelial dilators. This is substantiated by the current observation that ACh-induced endothelium-dependent vasodilation in mesenteric arteries from IH rats is severely blunted compared with that in arteries from sham rats. Inhibiting CSE decreased ACh-mediated dilation in arteries from sham rats to the point that it was indistinguishable from dilation in the IH group. Thus, reduced levels of CSE after IH (21) appear to contribute to loss of both basal and agonist-stimulated endothelium-dependent dilation. However, the mechanism by which endothelial Ca2+ is reduced in arteries from IH rats to drive the loss of CSE expression is unclear and presents an interesting area for future investigations that may uncover additional vascular impairments in this model of sleep apnea-induced hypertension.
Taken together, these findings suggest that decreased endothelial cell Ca2+ and diminished NFAT activation of CSE transcription contribute to impaired endothelium-dependent vasodilation after exposure to IH.
GRANTS
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute (Grants HL-82799, HL-007736, and HL-088151), the American Heart Association (Grant 12POST8690008), and institutional funding from the University of New Mexico.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.V.G.B., J.M.O., O.J.-W., and N.L.K. conceived and designed research; L.V.G.B., J.M.O., W.G., C.E.P., J.L.R., O.J.-W., and N.L.K. performed experiments; L.V.G.B., J.M.O., W.G., C.E.P., J.L.R., O.J.-W., and N.L.K. analyzed data; L.V.G.B., J.M.O., and N.L.K. interpreted results of experiments; L.V.G.B., J.M.O., and N.L.K. prepared figures; L.V.G.B. and N.L.K. edited and revised manuscript; L.V.G.B., J.M.O., W.G., C.E.P., J.L.R., O.J.-W., and N.L.K. approved final version of manuscript; J.M.O. drafted manuscript.
REFERENCES
- 1.Aminzadeh MA, Vaziri ND. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol Dial Transplant 27: 498–504, 2012. doi: 10.1093/ndt/gfr560. [DOI] [PubMed] [Google Scholar]
- 2.Bagai K. Obstructive sleep apnea, stroke, and cardiovascular diseases. Neurologist 16: 329–339, 2010. doi: 10.1097/NRL.0b013e3181f097cb. [DOI] [PubMed] [Google Scholar]
- 3.Bełtowski J, Jamroz-Wiśniewska A, Tokarzewska D. Hydrogen sulfide and its modulation in arterial hypertension and atherosclerosis. Cardiovasc Hematol Agents Med Chem 8: 173–186, 2010. doi: 10.2174/187152510792481207. [DOI] [PubMed] [Google Scholar]
- 4.Bierer R, Nitta CH, Friedman J, Codianni S, de Frutos S, Dominguez-Bautista JA, Howard TA, Resta TC, Gonzalez Bosc LV. NFATc3 is required for chronic hypoxia-induced pulmonary hypertension in adult and neonatal mice. Am J Physiol Lung Cell Mol Physiol 301: L872–L880, 2011. doi: 10.1152/ajplung.00405.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brancaleone V, Roviezzo F, Vellecco V, De Gruttola L, Bucci M, Cirino G. Biosynthesis of H2S is impaired in non-obese diabetic (NOD) mice. Br J Pharmacol 155: 673–680, 2008. doi: 10.1038/bjp.2008.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch 459: 881–895, 2010. doi: 10.1007/s00424-010-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crane GJ, Garland CJ. Thromboxane receptor stimulation associated with loss of SKCa activity and reduced EDHF responses in the rat isolated mesenteric artery. Br J Pharmacol 142: 43–50, 2004. doi: 10.1038/sj.bjp.0705756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Frutos S, Caldwell E, Nitta CH, Kanagy NL, Wang J, Wang W, Walker MK, Gonzalez Bosc LV. NFATc3 contributes to intermittent hypoxia-induced arterial remodeling in mice. Am J Physiol Heart Circ Physiol 299: H356–H363, 2010. doi: 10.1152/ajpheart.00341.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Frutos S, Duling L, Alò D, Berry T, Jackson-Weaver O, Walker M, Kanagy N, González Bosc L. NFATc3 is required for intermittent hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol 294: H2382–H2390, 2008. doi: 10.1152/ajpheart.00132.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Frutos S, Spangler R, Alò D, Bosc LV. NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with alpha-actin up-regulation. J Biol Chem 282: 15081–15089, 2007. doi: 10.1074/jbc.M702679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doxey JC, Roach AG, Smith CF. Studies on RX 781094: a selective, potent and specific antagonist of alpha 2-adrenoceptors. Br J Pharmacol 78: 489–505, 1983. doi: 10.1111/j.1476-5381.1983.tb08809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleming I, Bauersachs J, Busse R. Calcium-dependent and calcium-independent activation of the endothelial NO synthase. J Vasc Res 34: 165–174, 1997. doi: 10.1159/000159220. [DOI] [PubMed] [Google Scholar]
- 13.Fu M, Zhang W, Yang G, Wang R. Is cystathionine gamma-lyase protein expressed in the heart? Biochem Biophys Res Commun 428: 469–474, 2012. doi: 10.1016/j.bbrc.2012.10.084. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez Bosc LV, Wilkerson MK, Bradley KN, Eckman DM, Hill-Eubanks DC, Nelson MT. Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation: role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase. J Biol Chem 279: 10702–10709, 2004. doi: 10.1074/jbc.M312920200. [DOI] [PubMed] [Google Scholar]
- 15.Gryglewski RJ, Chłopicki S, Uracz W, Marcinkiewicz E. Significance of endothelial prostacyclin and nitric oxide in peripheral and pulmonary circulation. Med Sci Monit 7: 1–16, 2001. [PubMed] [Google Scholar]
- 16.Hill-Eubanks DC, Gomez MF, Stevenson AS, Nelson MT. NFAT regulation in smooth muscle. Trends Cardiovasc Med 13: 56–62, 2003. doi: 10.1016/S1050-1738(02)00212-8. [DOI] [PubMed] [Google Scholar]
- 17.Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun 237: 527–531, 1997. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 18.Iñiguez MA, Martinez-Martinez S, Punzón C, Redondo JM, Fresno M. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J Biol Chem 275: 23627–23635, 2000. doi: 10.1074/jbc.M001381200. [DOI] [PubMed] [Google Scholar]
- 19.Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, Kimura H. Murine cystathionine gamma-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J 381: 113–123, 2004. doi: 10.1042/BJ20040243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson-Weaver O, Osmond JM, Riddle MA, Naik JS, Gonzalez Bosc LV, Walker BR, Kanagy NL. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca2+-activated K+ channels and smooth muscle Ca2+ sparks. Am J Physiol Heart Circ Physiol 304: H1446–H1454, 2013. doi: 10.1152/ajpheart.00506.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson-Weaver O, Paredes DA, Gonzalez Bosc LV, Walker BR, Kanagy NL. Intermittent hypoxia in rats increases myogenic tone through loss of hydrogen sulfide activation of large-conductance Ca(2+)-activated potassium channels. Circ Res 108: 1439–1447, 2011. doi: 10.1161/CIRCRESAHA.110.228999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jain SK, Kahlon G, Morehead L, Lieblong B, Stapleton T, Hoeldtke R, Bass PF 3rd, Levine SN. The effect of sleep apnea and insomnia on blood levels of leptin, insulin resistance, IP-10, and hydrogen sulfide in type 2 diabetic patients. Metab Syndr Relat Disord 10: 331–336, 2012. doi: 10.1089/met.2012.0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanagy NL, Walker BR, Nelin LD. Role of endothelin in intermittent hypoxia-induced hypertension. Hypertension 37: 511–515, 2001. doi: 10.1161/01.HYP.37.2.511. [DOI] [PubMed] [Google Scholar]
- 24.Madurga A, Golec A, Pozarska A, Ishii I, Mižíková I, Nardiello C, Vadász I, Herold S, Mayer K, Reichenberger F, Fehrenbach H, Seeger W, Morty RE. The H2S-generating enzymes cystathionine β-synthase and cystathionine γ-lyase play a role in vascular development during normal lung alveolarization. Am J Physiol Lung Cell Mol Physiol 309: L710–L724, 2015. doi: 10.1152/ajplung.00134.2015. [DOI] [PubMed] [Google Scholar]
- 25.Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, Chabot S, Ruffenach G, Henry S, Breuils-Bonnet S, Tremblay È, Nadeau V, Lambert C, Paradis R, Provencher S, Bonnet S. Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension. Circ Res 117: 525–535, 2015. doi: 10.1161/CIRCRESAHA.115.307004. [DOI] [PubMed] [Google Scholar]
- 26.Mena MP, Papiewska-Pajak I, Przygodzka P, Kozaczuk A, Boncela J, Cierniewski CS. NFAT2 regulates COX-2 expression and modulates the integrin repertoire in endothelial cells at the crossroads of angiogenesis and inflammation. Exp Cell Res 324: 124–136, 2014. doi: 10.1016/j.yexcr.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Olson KR, Whitfield NL, Bearden SE, St Leger J, Nilson E, Gao Y, Madden JA. Hypoxic pulmonary vasodilation: a paradigm shift with a hydrogen sulfide mechanism. Am J Physiol Regul Integr Comp Physiol 298: R51–R60, 2010. doi: 10.1152/ajpregu.00576.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulin R, Courboulin A, Meloche J, Mainguy V, Dumas de la Roque E, Saksouk N, Côté J, Provencher S, Sussman MA, Bonnet S. Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 123: 1205–1215, 2011. doi: 10.1161/CIRCULATIONAHA.110.963314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15: 707–747, 1997. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- 30.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 31.Selim B, Won C, Yaggi HK. Cardiovascular consequences of sleep apnea. Clin Chest Med 31: 203–220, 2010. doi: 10.1016/j.ccm.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Sharma-Walia N, George Paul A, Patel K, Chandran K, Ahmad W, Chandran B. NFAT and CREB regulate Kaposi’s sarcoma-associated herpesvirus-induced cyclooxygenase 2 (COX-2). J Virol 84: 12733–12753, 2010. doi: 10.1128/JVI.01065-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J 206: 267–277, 1982. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun D, Huang A, Koller A, Kaley G. Endothelial K(ca) channels mediate flow-dependent dilation of arterioles of skeletal muscle and mesentery. Microvasc Res 61: 179–186, 2001. doi: 10.1006/mvre.2000.2291. [DOI] [PubMed] [Google Scholar]
- 35.Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, Adelman JP, Nelson MT. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res 93: 124–131, 2003. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- 36.Tishler PV, Larkin EK, Schluchter MD, Redline S. Incidence of sleep-disordered breathing in an urban adult population: the relative importance of risk factors in the development of sleep-disordered breathing. JAMA 289: 2230–2237, 2003. doi: 10.1001/jama.289.17.2230. [DOI] [PubMed] [Google Scholar]
- 37.Turgut Celen Y, Peker Y. Cardiovascular consequences of sleep apnea: II-Cardiovascular mechanisms. Anadolu Kardiyol Derg 10: 168–175, 2010. doi: 10.5152/akd.2010.044. [DOI] [PubMed] [Google Scholar]
- 38.Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PW, Wang R, Gratacós E, Buhimschi IA, Buhimschi CS, Ahmed A. Dysregulation of hydrogen sulfide producing enzyme cystathionine γ-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 127: 2514–2522, 2013. doi: 10.1161/CIRCULATIONAHA.113.001631. [DOI] [PubMed] [Google Scholar]
- 39.Wattanakit K, Boland L, Punjabi NM, Shahar E. Relation of sleep-disordered breathing to carotid plaque and intima-media thickness. Atherosclerosis 197: 125–131, 2008. doi: 10.1016/j.atherosclerosis.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 40.Yang G, Pei Y, Cao Q, Wang R. MicroRNA-21 represses human cystathionine gamma-lyase expression by targeting at specificity protein-1 in smooth muscle cells. J Cell Physiol 227: 3192–3200, 2012. doi: 10.1002/jcp.24006. [DOI] [PubMed] [Google Scholar]
- 41.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322: 587–590, 2008. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zetterqvist AV, Blanco F, Öhman J, Kotova O, Berglund LM, de Frutos Garcia S, Al-Naemi R, Wigren M, McGuire PG, Gonzalez Bosc LV, Gomez MF. Nuclear factor of activated T cells is activated in the endothelium of retinal microvessels in diabetic mice. J Diabetes Res 2015: 428473, 2015. doi: 10.1155/2015/428473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J 20: 6008–6016, 2001. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]