

Data presented here demonstrate novel signaling to sarcomeric proteins by chronic alterations in fatty acid metabolism induced by PPARα. The mechanism involves modifications of key myofilament regulatory proteins modifying cross-bridge dynamics with differential effects in controls and hearts stressed by pressure overload.
Keywords: metabolism, sarcomere mechanics, calcium, cMyBP-C, phosphorylation
Abstract
Although alterations in fatty acid (FA) metabolism have been shown to have a negative impact on contractility of the hypertrophied heart, the targets of action remain elusive. In this study we compared the function of skinned fiber bundles from transgenic (Tg) mice that overexpress a relatively low level of the peroxisome proliferator-activated receptor α (PPARα), and nontransgenic (NTg) littermates. The mice (NTg-T and Tg-T) were stressed by transverse aortic constriction (TAC) and compared with shams (NTg-S and Tg-S). There was an approximate 4-fold increase in PPARα expression in Tg-S compared with NTg-S, but Tg-T hearts showed the same PPARα expression as NTg-T. Expression of PPARα did not alter the hypertrophic response to TAC but did reduce ejection fraction (EF) in Tg-T hearts compared with other groups. The rate of actomyosin ATP hydrolysis was significantly higher in Tg-S skinned fiber bundles compared with all other groups. Tg-T hearts showed an increase in phosphorylation of specific sites on cardiac myosin binding protein-C (cMyBP-C) and β-myosin heavy chain isoform. These results advance our understanding of potential signaling to the myofilaments induced by altered FA metabolism under normal and pathological states. We demonstrate that chronic and transient PPARα activation during pathological stress alters myofilament response to Ca2+ through a mechanism that is possibly mediated by MyBP-C phosphorylation and myosin heavy chain isoforms.
NEW & NOTEWORTHY Data presented here demonstrate novel signaling to sarcomeric proteins by chronic alterations in fatty acid metabolism induced by PPARα. The mechanism involves modifications of key myofilament regulatory proteins modifying cross-bridge dynamics with differential effects in controls and hearts stressed by pressure overload.
heart failure (HF) is characterized by a depression of myocardial contractility in response to chronic stressors such as hypertension, which induce cardiac remodeling, resulting in altered Ca2+ homeostasis (3, 29), myofilament Ca2+ responsiveness (7, 16), altered substrate metabolism, and profound changes in lipid dynamics and nuclear receptor activation (14, 58). Although these modifications are often considered in isolation, there is evidence of reciprocal interactions among the remodeling processes, where, for example, a change in Ca2+ flux induced by altered substrate metabolism (45) may alter transcriptional regulation by Ca2+-calmodulin-dependent protein kinase, leading to maladaptation. Reciprocal signaling also occurs between myofilament Ca2+ response and metabolic remodeling. There is evidence that alteration of FA metabolism leads to altered cardiac contractility (1, 50, 54, 63) and myofilament Ca2+ response (27). Moreover, primary changes at the level of the sarcomeres are known to induce metabolic remodeling in the myocardium (3, 12, 17, 36). However, despite the evidence that contractility is negatively affected with metabolic remodeling and altered FA metabolism, the underlying mechanisms related to modifications at the level of the sarcomeres have not been investigated.
To explore the mechanisms by which metabolic remodeling affects sarcomere function, we employed a mouse model of cardiac-specific peroxisome proliferator-activated receptor α (PPARα) overexpression (lowest level line 404–4) (4, 25, 31). Pathological hypertrophy is characterized by a marked depression in the expression and activity of the PPARα (5). To determine whether a model of chronic PPARα upregulation would counter the fundamental contractile deficits induced by pathological remodeling, we compared nontransgenic (NTg-S) and transgenic (Tg-S) shams to mice (NTg-T and Tg-T) stressed by transverse aortic constriction (TAC). We measured the effects of the metabolic alterations induced by PPARα on sarcomere mechanics, ATP hydrolysis, and protein phosphorylation. Compared with NTg-S there was a ~4-fold increase in PPARα expression in Tg-S, which was associated with a significant increase in the maximum rate of ATP hydrolysis compared with all other groups. However Tg-T hearts expressed a level of PPARα not significantly different from NTg-T hearts. Never-the-less there was an increase in phosphorylation of cMyBP-C in the Tg-T group associated with a significant decrease in ejection fraction (EF) compared with other groups. Our results indicate that chronic and possibly transient upregulation of PPARα is not beneficial as a means to counter reduced PPARα activation in hypertrophied hearts. PPARα upregulation induces a metabolic inflexibility in the opposite polarity of that observed in hypertrophied hearts, and so induces deleterious effects on contractility. Potentially, the reduced PPAR activity in the hypertrophied hearts is an adaptive mechanism to preserve contractile activity in cardiac hypertrophy, but is offset by the already recognized maladaptive metabolic consequences (25).
MATERIALS AND METHODS
Animal model.
Three-month-old Tg male mice overexpressing PPARα, and NTg littermates were used in this study. The PPARα gene is driven by the cardiac-specific α-myosin heavy chain promoter (MHC-PPARα). As shown in a previous study (26), a high level of MHC-PPARα overexpression is associated with the development of cardiac dysfunction; therefore, we used a transgenic mouse line (404-4) overexpressing a lower level that does not show any cardiac dysfunction at this age. All experiments were performed according to guidelines instituted by, and with protocol review and approval from, the Animal Care and Use Committee at the University of Illinois at Chicago.
Chronic pressure-overload cardiac hypertrophy.
NTg and Tg male mice were randomly selected to undergo transverse aortic constriction (TAC) as previously described (21) to induce chronic pressure-overload cardiac hypertrophy. TAC was performed by placing a titanium metal microclip to constrict the transverse aorta (ID 0.4 mm). In control, sham-operated males, the aorta was exposed, but not constricted. At 8 and 16 wk postsurgery, in vivo cardiac function was determined by echocardiography as described previously (2, 28) with slight modifications. The Tg groups were studied at 8 wk and the NTg groups were studied at 16 wk due to the lethality of the TAC in the Tg groups, and the lack of hypertrophy in the NTg group at 8 wk. Therefore the NTg groups had to be extended an additional 8 wk for the TAC to develop similar hypertrophy as the Tg group. At these time points, mice were euthanized and hearts were isolated and used to either study the mechanical properties of the heart, or stored at −80°C for biochemical assessment.
Measurements of Ca2+-dependent activation of force, ATPase, and ktr.
Simultaneous measurements of isometric tension and ATPase rate of skinned left ventricular fiber bundles were performed using an experimental apparatus and methodology described previously (19, 20). Fiber bundles were dissected from the left ventricular papillary muscles and detergent extracted (skinned) in a high-relaxing (HR) buffer containing 1% Triton X-100. Fibers were mounted between a force transducer and displacement motor using aluminum T-clips, and the sarcomere length (SL) was set to 2.2 µm using He-Ne laser diffraction. SL remained stable throughout the experiment. The fiber bundle was moved sequentially into HR (pCa 10.0) containing 10 mM EGTA, 48.95 mM K-Prop, 100 mM BES, 1 mM free Mg2+, 5 mM MgATP2−, and 1 mM DTT, then into the preactivating solution (0.50 mM EGTA, 49.19 mM K-Prop, 100 mM BES, 1 mM free Mg2+, 5 mM MgATP2−, 9.50 mM HDTA, and 1 mM DTT), and finally into the experimental pCa solution. Solutions of varying pCa values were generated by mixing appropriate volumes of HR and maximal activating solution (pCa 4.5) containing 10 mM EGTA, 10.01 mM CaCl2, 29.06 mM K-Prop, 75 mM BES, 1 mM free Mg2+, 5 mM MgATP2−, and 1 mM DTT. Pyruvate kinase (final concentration of 1 mg/ml) and lactate dehydrogenase (final concentration of 0.1 mg/ml) were added to all three solutions before use. Solution compositions were calculated by customized software based upon binding constants described previously (23). After these parameters were measured, the fiber bundle was returned to the relaxing solution until force decreased to the preactivation level. Measurements of the rate of force redevelopment (ktr) were carried out as previously described (8–10) with minor modifications (13). The fiber bundle was activated quickly by transferring from a preactivating solution (pCa 9.0) to an activating solution containing various pCa values. After the steady state was reached a slack of 20% of the fiber length was rapidly induced at one end of the fiber, which decreased the force to zero within 1 ms. Then the fiber was immediately unloaded for 20 ms and restretched rapidly (1 ms) to the original length to detach remaining bound cross bridges and then tension redeveloped. The rate of tension redevelopment increased with Ca2+ concentration. Only fiber bundles that retained more than 75% of their initial maximum tension were included in the analysis. All mechanical experiments were performed at 20°C.
Immunoblotting.
Ventricular heart homogenates were prepared from NTg and Tg mouse hearts that underwent either Sham or TAC operations. About 25 mg of frozen tissue was homogenized with Duall homogenizers in 2x Laemmli sample buffer (Bio-Rad, Hercules, CA), containing protease (Sigma, St. Louis, MO) and phosphatase (Calbiochem, Darmstadt, Germany) inhibitors at a 1:100 dilution, and a final concentration of 1 µM calyculin A. The protein concentration of the samples was determined using an RCDC assay kit (Bio-Rad). Proteins were then separated on 12% SDS gels and transferred to a 0.2-µm polyvinylidene difluoride (PVDF) membrane for Western blot analysis (40, 43). For MLC-2 and cTnI phosphorylation studies, the proteins were separated on 12% SDS gels containing 50 µM Phos-Tag (37). We quantified MLC-2 and TnI phosphorylation by computing the ratio of the phosphorylated band to the total protein content for each phosphorylation site [i.e., P-MLC-2/(P-MLC-2 + MLC-2) × 100]. Membranes were blocked at room temperature with 5% nonfat dry milk and incubated overnight at 4°C in primary antibody: [cMyBPC (C0–C1), 1:25,000; phospho-cMyBP-C (Ser-273),1:500; phospho-cMyBP-C (Ser-282), 1:2,500; phosphor-cMyBP-C (Ser-302), 1:10,000; actin, 1:3,000; cTnI, 1:5,000; MLC-2, 1:2,000]. All MyBP-C antibodies were gifts from Dr. Sakthivel Sadayappan; actin was from Sigma, cTnI, Fitzgerald; and MLC-2, Enzo. Membranes were washed in Tris-buffer saline with 0.1% Tween-20 (TBS-T) and incubated for 1 h at room temperature in secondary antibody [goat anti-mouse (Sigma-Aldrich) 1:50,000 for actin, donkey anti-rabbit (GE Healthcare) 1:40,000 for all others], conjugated to horseradish peroxidase. Membranes were then washed in TBS-T for 30 min and developed by enhanced chemiluminescence using a Chemidoc XRS+ imager (Bio-Rad, Hercules, CA). Band densities were analyzed using ImageLab v5.0 software (Bio-Rad).
Assessment of β-myosin heavy chain expression.
Ventricular heart homogenates prepared as described above were used for the myosin heavy chain (MHC) isoform analysis in NTg and Tg mouse hearts with Sham or TAC surgeries. For control samples, the heart homogenates were prepared from liquid nitrogen frozen 1-day-old neonatal mouse heart tissue. Proteins were separated on a 6% SDS-PAGE gel as described previously (62), and run at constant amperage of 16 mA until the dye front ran off the gel. To visualize total protein, the gel was incubated with Coomassie blue stain for 30 min and then destained with a 10% methanol and 10% acetic acid solution. The gel was then imaged with a ChemiDoc XRS+ (Bio-Rad). Band densities were analyzed using ImageLab v5.0 software (Bio-Rad). The percent β-MHC was determined by ratio of band densities: β-MHC/(α-MHC+ β-MHC) × 100.
Echocardiography.
Echocardiography was performed on anesthetized mice (3.5% isoflurane with 100% oxygen induction with 1.5% isoflurane in 100% oxygen maintenance) using a VisualSonics’ Vevo 2100 ultrasound machine and MS550D (40 MHz) transducer as previously reported (2, 28). Transthoracic B-mode, M-mode, pulsed Doppler, and tissue Doppler images were obtained from the parasternal short-axis view (systolic and morphology parameters) and the apical view (diastolic parameters).
Data and statistical analysis.
Tension-pCa, ATPase-pCa, and ktr-pCa relations were fit to a modified four-parameter Hill equation with GraphPad Prism 6.0. The equation is defined as: Y = bottom + (top − bottom)/[1 + 10(pCa50−pCa)·HillSlope], where bottom is the Y value at the bottom plateau; top is the Y-value at the top plateau; pCa50 is the pCa value at which 50% of maximum value top is reached; and Hill slope describes the steepness of the curve and is also known as Hill coefficient. ATPase-tension, and ktr-relative tension (P/Po) relationships where fit with linear curves.
All values are presented as means ± SE. Data were analyzed using two-way ANOVA with Tukey’s post hoc test using GraphPad Prism 6.0, with a level of statistical significance set at P < 0.05.
RESULTS
TAC-induced cardiac hypertrophy in NTg and Tg hearts.
Measurements of the heart weight-to-tibia length indicated a similar development of cardiac hypertrophy at 8 and 16 wk post-TAC in both Tg and NTg hearts (Fig. 1A). The hypertrophic remodeling in response to TAC was also accompanied with an increase in the β-MHC expression in both groups (Fig. 1C), in agreement with previous reports (42). The β-MHC expression was not significantly different between Tg-T and NTg-T; however there were significant differences between NTg-S and Tg-S vs. Tg-T (Fig. 1C). No detectable β-MHC was observed in the respective control sham groups.
Fig. 1.
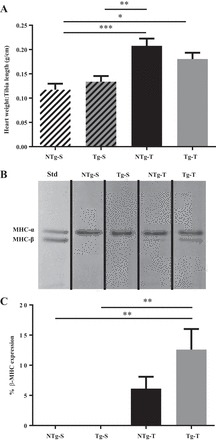
Transaortic constriction induced cardiac hypertrophy in both NTg and Tg mice. A: heart weight-to-tibia length ratio (n = 5–10 hearts/group). B: representative 6% SDS-PAGE gel image showing-myosin heavy chain isoform (α and β) expression (n = 6 hearts/group). Std is neonatal mouse heart expressing both myosin-heavy chain isoforms. Black lines demarcate region of interest spliced from the same image. C: quantification of myosin heavy chain isoform expression in NTg-S, Tg-S, NTg-T, and Tg-T whole homogenate mouse hearts. Data are represented as means ± SE. Statistically significant result based on Tukey’s post hoc test: *P < 0.05, **P < 0.01, ***P < 0.001.
Functional data obtained by echocardiography showed a significant decrease in ejection fraction (EF) and fractional shortening (FS) in TAC hearts compared with shams as shown in Fig. 2. Moreover, the data indicate that EF was significantly lower in Tg-T hearts compared with NTg-T (Fig. 2A). However, although there was a trend, FS (Fig. 2B) of the Tg-T hearts was not significantly different from NTg-T most likely due to a relatively large SE.
Fig. 2.
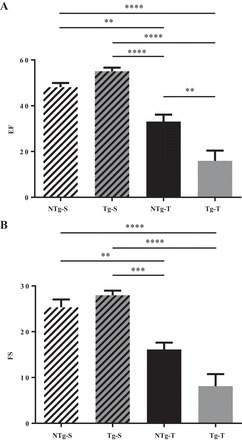
Effects of transaortic constriction are more pronounced in Tg mice. Ejection fraction (A) and fractional shortening (B) were recorded with echocardiography. The recordings were done at 16 wk for NTg-S (n = 11) and 8 wk for Tg-S (n = 8) mice. The recordings were done at 16 wk for NTg-T (n = 5) and 8 wk for Tg-T (n = 4–5). Data are represented as means ± SE. Statistically significant result based on Tukey’s post hoc test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
PPARα expression altered the chemomechanical properties of skinned fiber bundles.
To determine the functional effect of PPARα expression on sarcomere dynamics, we studied the Ca2+ dependency of tension, ATPase activity, and ktr of skinned fiber bundles isolated from NTg and Tg hearts with TAC, and their sham controls. Results are summarized in Table 1. The pCa-tension relation revealed no change in maximum tension or in Hill coefficient among the groups (Fig. 3A). However, compared with NTg-S myofilaments, there was a slight but significant increase in the Ca2+ sensitivity of tension in Tg-T (Table 1). Interestingly, the expression of PPARα resulted in a significant increase in the rate of ATP hydrolysis in the Tg-S group (Fig. 3B). Moreover, and in agreement with the results from the tension measurements, there was a small but significant increase in pCa50 of ATPase activity in Tg-T when compared with NTg-S (Table 1). As expected, the high rate of ATP hydrolysis observed in the Tg-S group was associated with an increase in the slope of the tension-ATPase relation, i.e., tension cost an increase in unit ATP hydrolysis per unit tension development (Fig. 3C and Table 1).
Table 1.
Mechanical properties of papillary fiber bundles isolated from the left ventricles of NTg and Tg hearts with TAC and their Sham controls
| Parameters | NTg-S | Tg-S | NTg-T | Tg-T |
|---|---|---|---|---|
| Maximum tension, mN/mm2 | 40.42 ± 1.12 | 44.49 ± 0.69 | 44.38 ± 0.7 | 42.10 ± 0.92 |
| pCa50 of tension | 5.75 ± 0.01 | 5.79 ± 0.01 | 5.77 ± 0.01 | 5.80 ± 0.01* |
| Hill slope of tension | 7.57 ± 1.24 | 7.41 ± 0.67 | 8.03 ± 0.74 | 6.78 ± 0.87 |
| Max ATPase, pmol·s−1·mm−3 | 451.3 ± 12.71 | 549.4 ± 13.21*† | 461.2 ± 10.83 | 398.7 ± 7.95§ |
| pCa50 of ATPase | 5.78 ± 0.01 | 5.80 ± 0.01 | 5.79 ± 0.01 | 5.81 ± 0.01* |
| Hill slope of ATPase | 8.46 ± 1.62 | 6.43 ± 0.99† | 9.22 ± 1.55 | 7.72 ± 1.01 |
| Tension cost | 9.77 ± 0.65 | 10.91 ± 0.56† | 9.39 ± 0.53 | 8.96 ± 0.33‡ |
| Maximum ktr, s−1 | 14.70 ± 0.81 | 18.59 ± 1.84 | 16.31 ± 0.86 | 16.19 ± 0.62 |
Values are means ± SE; n = 8–15 from 5 to 6 hearts. pCa50, −log10 of the [Ca2+] required to develop 50% of the maximal tension or ATPase; tension cost is estimated as the slope of the linear relationship between tension and ATPase activity; ktr, rate of tension redevelopment. *P < 0.05 vs. NTg-S;
P < 0.05 vs. NTg-T;
P < 0.05 and
P < 0.001 vs. Tg-S.
Fig. 3.
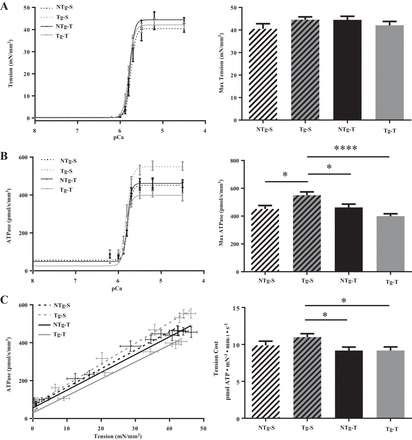
pCa-tension and pCa-ATPase relation in mouse cardiac skinned fiber bundles isolated from NTg-S, NTg-T, Tg-S, and Tg-T mice. A: tension-pCa relationship. B: ATPase-pCa relationship. C: tension cost. Data are presented as means ± SE; n = 8–15 fibers from 5 to 6 hearts per group. Tension cost is estimated as the slope of the linear relationship between tension (A) and ATPase activity (B). Data on right are represented as means ± SE. Statistically significant result based on Tukey’s post hoc test: *P < 0.05; ****P < 0.0001.
Based on additional measurements we interpret this increase in tension cost to largely reflect an increase in cross-bridge kinetics dominated by an increased rate of cross-bridge detachment (g) from the thin filaments (61). In the additional measurement and in the same experimental set-up, we subjected the fiber bundles to a rapid release restretch maneuver at each pCa value, which results in the detachment, then the reattachment of cross-bridges. Table 1 summarized the data as the maximum ktr (s−1) as previously described (32, 49). We found no change in ktr among the groups. The change in tension cost is dominated by a slow kinetic step in cross-bridge turnover kinetics as cross-bridges exit the force-generating state in association with the release of ADP from myosin heads and binding of ATP. This step in the cycle is referred to as gapp as originally described by Huxley in his 1957 model (35), whereas the rate of entry of cross-bridges into the force-generating state is referred to as fapp. To evaluate whether there was a change in fapp among the turnover kinetics of cross-bridge, in the same experiments in which we measured tension and ATPase, we subjected the fiber bundles to a quick release/restretch protocol, which provides a way of determining cross-bridge turnover kinetics as originally described by Brenner (8, 9). Fiber bundles at various levels of activation were quickly released inducing a detachment of force-generating cross-bridges, and then restretched to the original length, permitting force-generating cross-bridges to reattach. In the nomenclature used by Brenner (8, 9), based on Huxley’s 1957 model (35), the rate constant of tension recovery (ktr or k-redeveloped) represents turnover kinetics of force-generating cross-bridges, and is described by the sum of fapp (rate of entry of cross-bridges into the force-generating state) and gapp (rate of cross-bridge return to the non-force-generating state). As described in detail by Brenner (8, 9), the ATPase rate is related to fapp and gapp as follows: ATPase = (fapp) (gapp)/(fapp) + (gapp). As illustrated in the studies of Brenner (8, 9), in the case of ktr, the values are dominated by fapp. As there were no changes in ktr among the groups, the alteration in tension cost was not due to a change in rate of entry of cross-bridges into the force-generating state, but rather the exit of cross-bridges from the force-generating state.
Combination of PPARα expression and TAC altered myofilament protein phosphorylation.
Alterations in cross-bridge mechanics and in the response of the sarcomere to Ca2+ are usually associated with posttranslational modifications of the myofilament proteins (7, 56, 57). We therefore studied protein phosphorylation in NTg and Tg hearts with and without TAC. We first determined the levels of PPARα expression in the four groups studied; as illustrated in Fig. 4 both bands (doublet) were used in the quantitation of PPARα. The PPARα doublet may be due to a small terminal cleavage, as the expected molecular weight of the protein is correct. The PPARα expression was significantly higher in the Tg-S vs. NTg-S group as expected. However, with TAC there was a depression in PPARα expression resulting in a level not significantly different from the NTg-T. Previous studies have demonstrated that TAC is able to depress PPARα expression induced by a high-fat diet (41). Figures 5 and 6 show the quantification of phosphorylated proteins. In the case of cMyBP-C protein, our results show a remarkable increase in the phosphorylation of Ser-273 and Ser-302 in the Tg-T group relative to the NTg-T group. However, Ser-282 phosphorylation did not change (Fig. 5). Phosphorylation of cTnI and MLC-2 was assessed using a Phos-tag probe that was shown to bind to phosphorylated proteins and to slow their migration on the gel relative to unphosphorylated proteins (37). As shown in Fig. 6A, cTnI was separated into un- (0P), mono- (1P), and bis- (2P) phosphorylated species, with 2P containing cTnI species that have the highest number of phosphorylated sites (44). Also, MLC-2 was separated into unphosphorylated (MLC-2) and phosphorylated species (P-MLC-2) (Fig. 6B). Quantification of phosphorylated proteins indicated that, compared with all the other groups, there was a significant decrease in the 0P species of cTnI in Tg-T along with a nonsignificant increase in the 2P species phosphorylation. In the case of MLC-2, we could not detect any significant changes in phosphorylation.
Fig. 4.
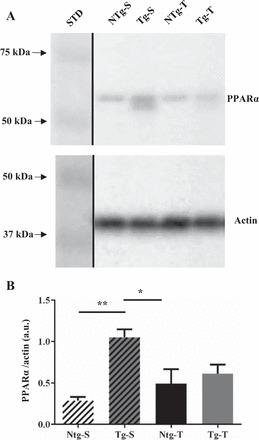
Immunoblotting for PPARα in whole heart homogenates from NTg-S, NTg-T, Tg-S, and Tg-T mice. A: representative images for PPARα (top) and actin loading control (bottom). Black lines demarcate region of interest spliced from the same image. B: densitometric analysis of PPARα expression. Data are represented as means ± SE; n = 6. Statistically significant result based on Tukey’s post hoc test: *P < 0.05, **P < 0.01. STD is molecular weight standard.
Fig. 5.

Site-specific immunoblotting for cMyBP-C phosphorylation. Left: representative images from Western blots using cMyBP-C phospho-specific antibodies against Ser-273 (A), Ser-282 (B), and Ser-302 (C) in whole heart homogenates from NTg-S, NTg-T, Tg-S, and Tg-T mice. STD is molecular weight standard. Right: quantification of cMyBP-C phosphorylation levels at: Ser-273 (A); Ser-282 (B), and Ser-302 (C). Data are represented as means ± SE; n = 6 hearts/group. Statistically significant result based on Tukey’s post hoc test: *P < 0.05. All samples were derived and processed at the same time and in parallel. Black lines demarcate spliced regions of interest.
Fig. 6.

cTnI and MLC2 phosphorylation. Phosphorylated and unphosphorylated proteins in whole heart homogenates from NTg-S, NTg-T, Tg-S, and Tg-T mice were separated using 12% SDS gels with Phos-tag. A, left: representative image of Western blots using a total cTnI antibody. A, right: quantification of cTnI phosphorylation. B, left: representative image of Western blots using a total MLC2 antibody. B, right: quantification of MLC2 phosphorylation. Data are represented as means ± SE; n = 6 hearts/group. Statistically significant result based on Tukey’s post hoc test: *P < 0.05 vs. all other 0P groups. Black lines demarcate region of interest spliced from the same image.
DISCUSSION
A major conclusion from our studies is that altered lipid metabolism induces signaling to sarcomeric proteins that modifies function. Thus our data indicate the importance of including these effects in thinking about the influence of altered lipid metabolism on cardiac function. Major findings supporting this conclusion include our data demonstrating that expression of PPARα promoted an increase in Ca2+ sensitivity of myofilaments from Tg stressed hearts compared with NTg controls. Yet, in view of the very small increase in myofilament Ca-sensitivity, we do not think this had a major functional effect in the hearts. On the other hand our results show that, compared with other groups, there was an increase in the maximum rate of ATP hydrolysis in the Tg-S group with no change in Ca2+ sensitivity of the myofilaments. However, when the Tg hearts were stressed with TAC (Tg-T) for 8 wk, an increased Ca2+ responsiveness of the myofilaments was observed, with a decrease in the rate of ATP hydrolysis and tension cost compared with Tg-S. The mechanism for these changes is likely to be associated with two significant modifications of the myofilaments: 1) an increase in expression of β-myosin heavy chain in the isoform population in Tg-T hearts and 2) site-specific increases in the MyBP-C phosphorylation in Tg-T hearts. Interestingly, compared with Tg and NTg sham controls, TAC decreased ATPase max and tension cost in the Tg-T mice but not the NTg-T mice. Moreover, ktr did not change. We speculate that the increased β-myosin heavy chain expression in the Tg-T, which would slow cross-bridge kinetics, together with the increase in MyBP-C phosphorylation, which would enhance cross-bridge kinetics, resulted in no changes in ktr.
Our data showing a difference in EF in Tg-T hearts compared with NTg-T indicate that, in the hypertrophied heart, altering FA metabolism by PPARα expression exacerbates cardiac dysfunction in the presence of stress. These results are consistent with the notion of impaired metabolic plasticity due to chronic activation of PPARα and a prior observation that administration of an agonist to counter reduced PPARα activity during pathological stress in the heart has deleterious consequences to mechanical function (64). Our result also suggests that even a transient change in expression of PPARα may significantly affect cardiac function by alterations at the level of the sarcomeres. They also support the hypothesis that there is a synergistic relation between substrate metabolism and control of cardiac contractility at the level of the sarcomeres.
Our findings are relevant to current concepts of the role of PPARα in HF. It is well established that, in the normal heart, long chain FA are the preferred substrates for oxidative ATP production by mitochondria with a very dynamic lipid metabolism; however, the failing myocardium displays a dramatic reduction in fatty acid oxidation and a general reduction in the intracellular lipid dynamics (14, 47, 58). The loss of triglyceride turnover impairs the lipolytic release of ligand for nuclear receptor activation, such as for PPARα that regulates genes involved in FA β-oxidation in mitochondria (6, 51, 52)). In the absence of a robust lipid metabolism, production of potentially lipotoxic, acyl-intermediates form within the cardiomyocyte. Included among the deleterious intermediates that accumulate are the long-chain ceramides that have been highlighted as lipotoxic (48). Although not part of the altered lipid profile of the pathological heart, a short-chain species of ceramide has recently been shown to alter myocyte contractility and myofilament protein phosphorylation (55).
Our data also support the hypothesis for the existence of reciprocal signaling between the contractile and regulatory state of the myofilaments and metabolic remodeling. There is ample evidence that modifications at the level of the sarcomere are able to influence metabolic remodeling of the myocardium. The metabolic phenotype of the pressure overloaded, decompensated heart has long been characterized as energy deprived, and thus adaptive responses in the mechanoenergetic coupling of the sarcomere are to be expected (11, 22, 38).
Familial hypertrophic cardiomyopathy (FHC)-linked mutations, that commonly occur at the level of sarcomeric regulatory proteins, also negatively impact the energy metabolism of the diseased myocardium (3). In this regard, the FHC-linked mutation R92Q in cardiac troponin (cTnT) not only decreased contractility but also reduced the metabolic efficiency of the myocardium as shown by a drop in creatine phosphate levels, with high concentrations of ADP and Pi (36). Furthermore, and in contrast to disease-causing mutations, alanine (Ala) substitution with histidine (A164H) in cTnI was shown to be protective against the decline in cardiac energetics observed in HF (17).
Interestingly, this histidine residue is present in slow skeletal TnI (ssTnI), the fetal isoform of cTnI. We have previously reported that, in a pressure-overload model of HF (CHF), substitution of cTnI with ssTnI maintained a normal cardiac function and increased the energy yield through reduction of anaplerotic flux that is usually enhanced in CHF (12). This synergy between sarcomere activity and metabolism is also emphasized by other studies pointing to maladaptive metabolic remodeling as a major cause of contractile dysfunction (1, 50, 54, 63). The mechanism underlying the effects of metabolic derangement on contractility is elucidated by evidence demonstrating that overexpression of the FA transport protein in the myocardium resulted in diastolic dysfunction that was likely caused by an increased sensitivity of the myofilaments to Ca2+ (27). Furthermore, mice expressing low levels of PPARα showed altered cardiac function when subjected to a high-fat diet (31). It has been shown previously that pharmacological activation of the PPARα gene aggravated cardiac dysfunction in rats stressed with TAC (64), which is in line with our findings in the Tg-T mice.
Results of our investigation of phosphorylation demonstrated that, compared with all other groups, in myofilaments from Tg-T hearts, there was a significant increase in phosphorylation serine (Ser)-302 of cMyBP-C. Also, a significant increase in Ser-273 was observed in the same group compared with Tg-S. However, phosphorylation of Ser-282 was the same among all the studied groups. Wang et al. (60) compared cross-bridge kinetics in controls to myofilaments regulated by phospho-mimetic cMyBP-C –S273D, S282A, and S302D, and reported that these phospho-mimetic myofilaments had altered cross-bridge kinetics at specific steps as revealed by sinusoidal analysis. Moreover, they reported a significant depression in maximum tension with the set of phosphorylation mimetics. Serendipitously, Tg-T myofilaments showed a similar charge pattern but with sites authentically phosphorylated. However, our functional results did not show reduced maximum tension in Tg-T vs. other groups. We attribute this difference to the presence of sites dynamically phosphorylated and to the presence of a difference in myosin isoform population. This dynamic phosphorylation and myosin isoform shift was induced by altered lipid signaling coupled with TAC stress. The change in cMyBP-C phosphorylation at Ser-273 and Ser-302, both phosphorylated by protein kinase C (PKC) (46), may be especially significant in view of the importance of PKC activation in heart failure (7). Despite the fact that cMyBP-C phosphorylation is essential for a normal heart function (53), discrepancy exists regarding its role in the modulation of cardiac mechanics (15, 30, 53, 60).
In conclusion, the results reported here indicate that in cardiac hypertrophy, PPARα alters sarcomere dynamics and may lead to contractile dysfunction. These effects seem to be indirectly mediated by the restricted metabolic remodeling caused by chronic PPARα activation. Possible mechanisms by which PPARα may be related to an increase in MyBP-C phosphorylation are through an associated increase in protein kinase C activity driven by a metabolic mechanism including generation of sphingolipids such as ceramide or by induction of altered redox state (7, 55). A tempting extension of these findings is that the reduced PPARα activity in the hypertrophied heart is a compensatory mechanism to maintain cardiac contractility. However, the actual balance between compensatory and decompensatory mechanisms in the pathogenesis of heart failure is complex and incompletely catalogued. Indeed, endogenous restoration of PPARα activity in cardiac hypertrophy, via lipolytic signaling, improves cardiac contractility (41). Thus the restricted metabolic phenotype induced by PPARα overexpression may be too far upstream for compensatory effects to outweigh decompensatory effects, and, as shown here, compromises the contractile response to pathological stress. Nevertheless, this current study demonstrates the profound regulatory role of PPARα on myofilament activity that are central to the impaired sarcomere function of the failing heart that for the first time are shown to be linked to MyBP-C phosphorylation.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) Grant POI-HL-062426 (R. J. Solaro, E. D. Lewandowski) and Core C (C. M. Warren), and NHLBI Grant RO1-HL-113057 (E. D. Lewandowski).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
C.N.K., C.M.W., M.H., and N.H.B. performed experiments; C.N.K., C.M.W., M.H., N.H.B., E.D.L., and R.J.S. analyzed data; C.N.K., C.M.W., M.H., N.H.B., E.D.L., and R.J.S. interpreted results of experiments; C.N.K., C.M.W., M.H., N.H.B., and R.J.S. prepared figures; C.N.K., C.M.W., M.H., N.H.B., E.D.L., and R.J.S. drafted manuscript; C.N.K., C.M.W., M.H., N.H.B., E.D.L., and R.J.S. edited and revised manuscript; C.N.K., C.M.W., M.H., N.H.B., E.D.L., and R.J.S. approved final version of manuscript.
REFERENCES
- 1.Abdurrachim D, Ciapaite J, Wessels B, Nabben M, Luiken JJ, Nicolay K, Prompers JJ. Cardiac diastolic dysfunction in high-fat diet fed mice is associated with lipotoxicity without impairment of cardiac energetics in vivo. Biochim Biophys Acta 1842: 1525–1537, 2014. doi: 10.1016/j.bbalip.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 2.Alves ML, Dias FA, Gaffin RD, Simon JN, Montminy EM, Biesiadecki BJ, Hinken AC, Warren CM, Utter MS, Davis RT III, Sadayappan S, Robbins J, Wieczorek DF, Solaro RJ, Wolska BM. Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ Cardiovasc Genet 7: 132–143, 2014. doi: 10.1161/CIRCGENETICS.113.000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashrafian H, McKenna WJ, Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res 109: 86–96, 2011. doi: 10.1161/CIRCRESAHA.111.242974. [DOI] [PubMed] [Google Scholar]
- 4.Banke NH, Wende AR, Leone TC, O’Donnell JM, Abel ED, Kelly DP, Lewandowski ED. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ Res 107: 233–241, 2010. doi: 10.1161/CIRCRESAHA.110.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest 105: 1723–1730, 2000. doi: 10.1172/JCI9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med 10: 238–245, 2000. doi: 10.1016/S1050-1738(00)00077-3. [DOI] [PubMed] [Google Scholar]
- 7.Belin RJ, Sumandea MP, Sievert GA, Harvey LA, Geenen DL, Solaro RJ, de Tombe PP. Interventricular differences in myofilament function in experimental congestive heart failure. Pflugers Arch 462: 795–809, 2011. doi: 10.1007/s00424-011-1024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenner B. The cross-bridge cycle in muscle. Mechanical, biochemical, and structural studies on single skinned rabbit psoas fibers to characterize cross-bridge kinetics in muscle for correlation with the actomyosin-ATPase in solution. Basic Res Cardiol 81, Suppl 1: 1–15, 1986. [DOI] [PubMed] [Google Scholar]
- 9.Brenner B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci USA 85: 3265–3269, 1988. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brenner B, Eisenberg E. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc Natl Acad Sci USA 83: 3542–3546, 1986. doi: 10.1073/pnas.83.10.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carley AN, Taegtmeyer H, Lewandowski ED. Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart. Circ Res 114: 717–729, 2014. doi: 10.1161/CIRCRESAHA.114.301863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carley AN, Taglieri DM, Bi J, Solaro RJ, Lewandowski ED. Metabolic efficiency promotes protection from pressure overload in hearts expressing slow skeletal troponin I. Circ Heart Fail 8: 119–127, 2015. doi: 10.1161/CIRCHEARTFAILURE.114.001496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandra M, Rundell VL, Tardiff JC, Leinwand LA, De Tombe PP, Solaro RJ. Ca2+ activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. Am J Physiol Heart Circ Physiol 280: H705–H713, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Christe ME, Rodgers RL. Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J Mol Cell Cardiol 26: 1371–1375, 1994. doi: 10.1006/jmcc.1994.1155. [DOI] [PubMed] [Google Scholar]
- 15.Copeland O, Sadayappan S, Messer AE, Steinen GJ, van der Velden J, Marston SB. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J Mol Cell Cardiol 49: 1003–1011, 2010. doi: 10.1016/j.yjmcc.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 16.Daniels MC, Naya T, Rundell VL, de Tombe PP. Development of contractile dysfunction in rat heart failure: hierarchy of cellular events. Am J Physiol Regul Integr Comp Physiol 293: R284–R292, 2007. doi: 10.1152/ajpregu.00880.2006. [DOI] [PubMed] [Google Scholar]
- 17.Day SM, Westfall MV, Fomicheva EV, Hoyer K, Yasuda S, La Cross NC, D’Alecy LG, Ingwall JS, Metzger JM. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat Med 12: 181–189, 2006. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- 18.de Tombe PP. Altered contractile function in heart failure. Cardiovasc Res 37: 367–380, 1998. doi: 10.1016/S0008-6363(97)00275-7. [DOI] [PubMed] [Google Scholar]
- 19.de Tombe PP, Stienen GJ. Impact of temperature on cross-bridge cycling kinetics in rat myocardium. J Physiol 584: 591–600, 2007. doi: 10.1113/jphysiol.2007.138693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Tombe PP, Stienen GJ. Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circ Res 76: 734–741, 1995. doi: 10.1161/01.RES.76.5.734. [DOI] [PubMed] [Google Scholar]
- 21.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, Lewandowski ED, Hajjar RJ. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation 104: 1424–1429, 2001. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 113: 709–724, 2013. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol 157: 378–417, 1988. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 24.Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol 517: 143–157, 1999. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 109: 121–130, 2002. doi: 10.1172/JCI0214080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 109: 121–130, 2002. doi: 10.1172/JCI0214080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flagg TP, Cazorla O, Remedi MS, Haim TE, Tones MA, Bahinski A, Numann RE, Kovacs A, Schaffer JE, Nichols CG, Nerbonne JM. Ca2+-independent alterations in diastolic sarcomere length and relaxation kinetics in a mouse model of lipotoxic diabetic cardiomyopathy. Circ Res 104: 95–103, 2009. doi: 10.1161/CIRCRESAHA.108.186809. [DOI] [PubMed] [Google Scholar]
- 28.Gaffin RD, Chowdhury SA, Alves MS, Dias FA, Ribeiro CT, Fogaca RT, Wieczorek DF, Wolska BM. Effects of nicotine administration in a mouse model of familial hypertrophic cardiomyopathy, α-tropomyosin D175N. Am J Physiol Heart Circ Physiol 301: H1646–H1655, 2011. doi: 10.1152/ajpheart.00277.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guinto PJ, Haim TE, Dowell-Martino CC, Sibinga N, Tardiff JC. Temporal and mutation-specific alterations in Ca2+ homeostasis differentially determine the progression of cTnT-related cardiomyopathies in murine models. Am J Physiol Heart Circ Physiol 297: H614–H626, 2009. doi: 10.1152/ajpheart.01143.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta MK, Robbins J. Post-translational control of cardiac hemodynamics through myosin binding protein C. Pflugers Arch 466: 231–236, 2014. doi: 10.1007/s00424-013-1377-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hankiewicz JH, Banke NH, Farjah M, Lewandowski ED. Early impairment of transmural principal strains in the left ventricular wall after short-term, high-fat feeding of mice predisposed to cardiac steatosis. Circ Cardiovasc Imaging 3: 710–717, 2010. doi: 10.1161/CIRCIMAGING.110.959098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henze M, Patrick SE, Hinken A, Scruggs SB, Goldspink P, de Tombe PP, Kobayashi M, Ping P, Kobayashi T, Solaro RJ. New insights into the functional significance of the acidic region of the unique N-terminal extension of cardiac troponin I. Biochim Biophys Acta 1833: 823–832, 2013. doi: 10.1016/j.bbamcr.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinken AC, Hanft LM, Scruggs SB, Sadayappan S, Robbins J, Solaro RJ, McDonald KS. Protein kinase C depresses cardiac myocyte power output and attenuates myofilament responses induced by protein kinase A. J Muscle Res Cell Motil 33: 439–448, 2012. doi: 10.1007/s10974-012-9294-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hofmann PA, Lange JH III. Effects of phosphorylation of troponin I and C protein on isometric tension and velocity of unloaded shortening in skinned single cardiac myocytes from rats. Circ Res 74: 718–726, 1994. doi: 10.1161/01.RES.74.4.718. [DOI] [PubMed] [Google Scholar]
- 35.Huxley AF. Muscle structure and theories of contraction. Prog Biophys Biophys Chem 7: 255–318, 1957. [PubMed] [Google Scholar]
- 36.Javadpour MM, Tardiff JC, Pinz I, Ingwall JS. Decreased energetics in murine hearts bearing the R92Q mutation in cardiac troponin T. J Clin Invest 112: 768–775, 2003. doi: 10.1172/JCI15967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5: 749–757, 2006. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113: 603–616, 2013. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Konhilas JP, Irving TC, Wolska BM, Jweied EE, Martin AF, Solaro RJ, de Tombe PP. Troponin I in the murine myocardium: influence on length-dependent activation and interfilament spacing. J Physiol 547: 951–961, 2003. doi: 10.1113/jphysiol.2002.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685, 1970. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 41.Lahey R, Wang X, Carley AN, Lewandowski ED. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation 130: 1790–1799, 2014. doi: 10.1161/CIRCULATIONAHA.114.011687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.López JE, Myagmar BE, Swigart PM, Montgomery MD, Haynam S, Bigos M, Rodrigo MC, Simpson PC. β-Myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circ Res 109: 629–638, 2011. doi: 10.1161/CIRCRESAHA.111.243410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsudaira P. Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J Biol Chem 262: 10035–10038, 1987. [PubMed] [Google Scholar]
- 44.Messer AE, Gallon CE, McKenna WJ, Dos Remedios CG, Marston SB. The use of phosphate-affinity SDS-PAGE to measure the cardiac troponin I phosphorylation site distribution in human heart muscle. Proteomics Clin Appl 3: 1371–1382, 2009. doi: 10.1002/prca.200900071. [DOI] [PubMed] [Google Scholar]
- 45.Mishra S, Ling H, Grimm M, Zhang T, Bers DM, Brown JH. Cardiac hypertrophy and heart failure development through Gq and CaM kinase II signaling. J Cardiovasc Pharmacol 56: 598–603, 2010. doi: 10.1097/FJC.0b013e3181e1d263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohamed AS, Dignam JD, Schlender KK. Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch Biochem Biophys 358: 313–319, 1998. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 47.O’Donnell JM, Fields AD, Sorokina N, Lewandowski ED. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J Mol Cell Cardiol 44: 315–322, 2008. doi: 10.1016/j.yjmcc.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 49: 2101–2112, 2008. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel JR, Fitzsimons DP, Buck SH, Muthuchamy M, Wieczorek DF, Moss RL. PKA accelerates rate of force development in murine skinned myocardium expressing alpha- or beta-tropomyosin. Am J Physiol Heart Circ Physiol 280: H2732–H2739, 2001. [DOI] [PubMed] [Google Scholar]
- 50.Pellieux C, Montessuit C, Papageorgiou I, Pedrazzini T, Lerch R. Differential effects of high-fat diet on myocardial lipid metabolism in failing and nonfailing hearts with angiotensin II-mediated cardiac remodeling in mice. Am J Physiol Heart Circ Physiol 302: H1795–H1805, 2012. doi: 10.1152/ajpheart.01023.2011. [DOI] [PubMed] [Google Scholar]
- 51.Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci USA 94: 6438–6443, 1997. doi: 10.1073/pnas.94.12.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94: 2837–2842, 1996. doi: 10.1161/01.CIR.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 53.Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ Res 109: 141–150, 2011. doi: 10.1161/CIRCRESAHA.111.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 18: 1692–1700, 2004. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 55.Simon JN, Chowdhury SA, Warren CM, Sadayappan S, Wieczorek DF, Solaro RJ, Wolska BM. Ceramide-mediated depression in cardiomyocyte contractility through PKC activation and modulation of myofilament protein phosphorylation. Basic Res Cardiol 109: 445, 2014. doi: 10.1007/s00395-014-0445-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res 112: 355–366, 2013. doi: 10.1161/CIRCRESAHA.112.268672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Solaro RJ, Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem 286: 9935–9940, 2011. doi: 10.1074/jbc.R110.197731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sorokina N, O’Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, Ballal K, Taegtmeyer H, Buttrick PM, Lewandowski ED. Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation 115: 2033–2041, 2007. doi: 10.1161/CIRCULATIONAHA.106.668665. [DOI] [PubMed] [Google Scholar]
- 59.Venema RC, Kuo JF. Protein kinase C-mediated phosphorylation of troponin I and C-protein in isolated myocardial cells is associated with inhibition of myofibrillar actomyosin MgATPase. J Biol Chem 268: 2705–2711, 1993. [PubMed] [Google Scholar]
- 60.Wang L, Ji X, Barefield D, Sadayappan S, Kawai M. Phosphorylation of cMyBP-C affects contractile mechanisms in a site-specific manner. Biophys J 106: 1112–1122, 2014. doi: 10.1016/j.bpj.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wannenburg T, Janssen PM, Fan D, de Tombe PP. The Frank-Starling mechanism is not mediated by changes in rate of cross-bridge detachment. Am J Physiol Heart Circ Physiol 273: H2428–H2435, 1997. [DOI] [PubMed] [Google Scholar]
- 62.Warren CM, Greaser ML. Method for cardiac myosin heavy chain separation by sodium dodecyl sulfate gel electrophoresis. Anal Biochem 320: 149–151, 2003. doi: 10.1016/S0003-2697(03)00350-6. [DOI] [PubMed] [Google Scholar]
- 63.Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes 51: 2587–2595, 2002. doi: 10.2337/diabetes.51.8.2587. [DOI] [PubMed] [Google Scholar]
- 64.Young ME, Laws FA, Goodwin GW, Taegtmeyer H. Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J Biol Chem 276: 44390–44395, 2001. doi: 10.1074/jbc.M103826200. [DOI] [PubMed] [Google Scholar]