

Western diet abolished protective effect of sex against cardiovascular disease (CVD) development in premenopausal animals with metabolic syndrome. Western diet accelerates progression of CVD in male and female animals with preexisting metabolic syndrome but not normal animals. Exacerbation of baseline oxidative stress correlates with accelerated progression of CVD in metabolic syndrome animals on Western diet.
Keywords: metabolic syndrome, Western diet, cardiovascular disease, sex differences, oxidative stress
Abstract
Thirty percent of the world population is diagnosed with metabolic syndrome. High-fat/high-sucrose (HF/HS) diet (Western diet) correlates with metabolic syndrome prevalence. We characterized effects of the HF/HS diet on vascular (arterial stiffness, vasoreactivity, and coronary collateral development) and cardiac (echocardiography) function, oxidative stress, and inflammation in a rat model of metabolic syndrome (JCR rats). Furthermore, we determined whether male versus female animals were affected differentially by the Western diet. Cardiovascular function in JCR male rats was impaired versus normal Sprague-Dawley (SD) rats. HF/HS diet compromised cardiovascular (dys)function in JCR but not SD male rats. In contrast, cardiovascular function was minimally impaired in JCR female rats on normal chow. However, cardiovascular function in JCR female rats on the HF/HS diet deteriorated to levels comparable to JCR male rats on the HF/HS diet. Similarly, oxidative stress was markedly increased in male but not female JCR rats on normal chow but was equally exacerbated by the HF/HS diet in male and female JCR rats. These results indicate that the Western diet enhances oxidative stress and cardiovascular dysfunction in metabolic syndrome and eliminates the protective effect of female sex on cardiovascular function, implying that both males and females with metabolic syndrome are at equal risk for cardiovascular disease.
NEW & NOTEWORTHY Western diet abolished protective effect of sex against cardiovascular disease (CVD) development in premenopausal animals with metabolic syndrome. Western diet accelerates progression of CVD in male and female animals with preexisting metabolic syndrome but not normal animals. Exacerbation of baseline oxidative stress correlates with accelerated progression of CVD in metabolic syndrome animals on Western diet.
metabolic syndrome is the combination of risk factors that enhance the risk of developing cardiovascular disease (CVD) and noninsulin-dependent diabetes mellitus (1, 14, 17). It is defined as concurrent visceral obesity, insulin resistance, dyslipidemia [elevated low-density lipoprotein (LDL) and triglycerides (TGs) and decreased high-density lipoprotein (HDL)], enhanced oxidative stress, and hypertension (1, 14).
Metabolic syndrome has been implicated in abnormal cardiovascular function and alterations in macro- and microvasculature function (vasculopathy) (17). With respect to the heart, a diagnosis of metabolic syndrome increases the risk of pathological cardiac hypertrophy, ischemic heart disease, myocardial infarction, and heart failure by two- to threefold (13). In the vasculature, the macrovascular changes include advanced atherosclerosis leading to peripheral ischemia and large artery stiffness leading to systolic hypertension. Large artery stiffness is likely the single most important determinant of systolic hypertension. It has been shown that systolic blood pressure is independently associated with pulse wave velocity, which is a direct measure of arterial stiffness. Furthermore, pulse wave velocity is increased even in very early stages of hypertension, suggesting that it may be a causal factor (42). Microvascular dysfunction, manifested as impaired vasodilation, results in increased total peripheral resistance and elevated systemic blood pressure (41). A high degree of coronary collateral development has been associated with decreased incidence of myocardial infarction (30). However, coronary collateral growth (CCG) is severely impaired in metabolic syndrome (27).
There are several rat models of metabolic syndrome: the Zucker fatty rat, the spontaneously hypertensive obese rat, the high-fat/high-sucrose (HF/HS)-fed rat, and the Russell rat (JCR:LA-cp or JCR) (2, 32, 40). While all models exhibit the metabolic dysfunction associated with metabolic syndrome as well as increased inflammation, oxidative stress, and endothelial dysfunction, only the JCR rat exhibits the full range of cardiovascular dysfunction associated with metabolic syndrome in humans: neointimal lesions, stroke, myocardial infarction, cardiac and vascular hypertrophy, and heart failure (40).
The Western diet has been implicated in the induction and advancement of the metabolic syndrome. The Western diet consists of high amounts of processed/complex sugars, saturated fats, and cholesterol and is responsible for the increases in obesity over the previous 50 yr (8, 11, 15, 44). This diet, the HF/HS diet, has been used to induce metabolic syndrome-like effects in animal models not known to be predisposed to the metabolic syndrome, which also developed evidence of vascular dysfunction associated with metabolic syndrome (15, 33, 35, 45). However, the effect of this type of diet on animals with preexisting metabolic syndrome has never been evaluated.
Furthermore, whether this diet has disparate effects on male versus female animals has never been studied. Traditionally, premenopausal females are considered to have protection from CVD development. However, in recent years, the incidence and prevalence of CVD in premenopausal women is increasing, concomitant with the increase in metabolic syndrome and consumption of the Western diet worldwide (13). However, a firm correlation between these three variables has not yet been established. Therefore, to establish these important correlations, the objective of this study was to investigate the effect of the HF/HS diet on cardiovascular function in both male and female normal and metabolic syndrome (JCR) rats. Since oxidative stress is a characteristic of metabolic syndrome and CVD, we also examined whether oxidative stress was a mechanism which may mediate these effects of the HF/HS diet.
METHODS
Animals.
Twelve-week-old male and female JCR:LA-cp (JCR; ~450-g male, ~300-g female; J.C. Russell and S. Proctor, University of Alberta, Edmonton, AB, Canada) and Sprague-Dawley (SD) (350-g male and ~220-g female; Charles Rivers, Wilmington, MA) rats were used in all experiments. The JCR rat is a cross between the lean Zucker (LA/N) and spontaneously hypertensive obese rat developed in the laboratory of Dr. Carl Hansen at the National Institutes of Health and sent to Drs. James C. Russell and Spencer Proctor (University of Alberta) (39). By 8 wk of age, JCR rats develop obesity with nonalcoholic fatty liver disease and, secondary to obesity, hypertension, insulin resistance with glucose intolerance, and complex dyslipidemia (low HDL, high LDL and VLDL) (Table 1). Unique to rodent models of metabolic syndrome, by 12 wk of age, JCR rats develop severe vasculopathy characterized by decreased endothelium-dependent and -independent vasodilation, neointimal hyperplasia, and intimal lesions morphologically identical to early atherosclerotic lesions in humans as well as left ventricular (LV) hypertrophy and myocardial and cerebral (micro)infarctions. At 16+ wk, the rats become prone to stroke and myocardial infarction, and at 18+ wk, they develop heart failure (6, 40). Like the development of metabolic syndrome and CVD in humans, the apparent complexity of the cardiometabolic phenotype exhibited by the JCR rats is suspected to be multifactorial and polygenetic in etiology. SD rats were used as control to mimic the normal heterogeneous human population. JCR lean (P. Rocic, unpublished observations), LA/N (16), and Wistar-Kyoto rats (29), which are proper genetic controls for JCR:LA-cp rats, are inbred strains with an increased propensity for the development of some of the component pathologies of metabolic syndrome and are thus phenotypically inappropriate controls for this study.
Table 1.
Weight and metabolic parameters in SD and JCR rats fed normal and HF/HS diet
| SD Male Normal | SD Female Normal | SD Male HF/HS | SD Female HF/HS | JCR Male Normal | JCR Female Normal | JCR Male HF/HS | JCR Female HF/HS | |
|---|---|---|---|---|---|---|---|---|
| Weight start, g | 350 ± 12 | 226 ± 8 | 348 ± 6 | 227 ± 11 | 450 ± 14* | 302 ± 11*† | 453 ± 9* | 303 ± 9* |
| Weight end, g | 602 ± 15 | 445 ± 12 | 775 ± 10* | 645 ± 22* | 650 ± 13* | 500 ± 7*† | 807 ± 14*† | 701 ± 12*†§ |
| CRP, µg/ml | 18 ± 0.8 | 20 ± 1 | 48 ± 3* | 45.3 ± 5* | 43 ± 4* | 28 ± 2*† | 55 ± 4*† | 58 ± 3*†§ |
| IL-6, pg/ml | 50 ± 2 | 48 ± 9 | 180 ± 5* | 85 ± 9* | 126 ± 11* | 78 ± 8*† | 344 ± 12*† | 327 ± 10*§ |
| MABP, mmHg | 98 ± 6 | 102 ± 7 | 146 ± 9* | 151 ± 11* | 152 ± 6* | 144 ± 9* | 160 ± 12* | 168 ± 6*§ |
| LDL, mg/dl | 71 ± 3 | 74 ± 2 | 140 ± 5* | 139 ± 7* | 130 ± 6* | 126 ± 4* | 152 ± 6*† | 155 ± 4*§ |
| HDL, mg/dl | 42 ± 2 | 44 ± 2 | 39 ± 1 | 40 ± 4 | 32 ± 1* | 29 ± 3* | 26 ± 2*† | 27 ± 1*§ |
| TGs, mg/dl | 96 ± 7 | 101 ± 4 | 275 ± 7* | 269 ± 11* | 183 ± 6* | 176 ± 4* | 275 ± 13*† | 275 ± 10*§ |
| Glucose, mg/dl | 78 ± 4 | 81 ± 4 | 274 ± 9* | 271 ± 6* | 138 ± 6* | 120 ± 4*† | 267 ± 8*† | 278 ± 8*§ |
| Insulin, IU/l | 20 ± 1 | 19 ± 2 | 284 ± 29* | 271 ± 36* | 346 ± 23* | 298 ± 23* | 434 ± 48*† | 401 ± 22*§ |
Body weight was measured in normal Sprague-Dawley (SD) and metabolic syndrome (JCR) rats before (at 12 wk of age) and after (at 24 wk of age) 12 wk of normal or high-fat/high-sugar (HF/HS) diet as indicated. All other parameters were measured after 12 wk of normal or HF/HS diet as indicated. CRP, C-reactive protein; MABP, mean arterial blood pressure; TGs, triglycerides.
P < 0.05 vs. male or female SD rats;
P < 0.05 vs. male JCR rats;
P < 0.05 vs. female JCR rats.
All experiments involving animals were performed in accordance with the Animal Welfare Act and were approved by the Institutional Animal Care and Use Committee of New York Medical College.
HF/HS diet.
The HF/HS diet was a Teklad custom diet purchased from Envigo [TD.03584, 35% fat (lard) and 36% carbohydrate (mostly sucrose)]. SD and JCR rats were maintained on the HF/HS diet or regular rat chow [LabDiet Rodent Chow 5001, 5% fat and 48% carbohydrate (mostly starch)] for 12 wk before measurements.
Body weight was measured every week of the 12-wk feeding protocol in all groups, and C-reactive protein (CRP) and IL-6 levels were measured by ELISA (eBioscience, San Diego, CA) at the end of the 12 wk.
Pressure myography.
Freshly isolated first-order mesenteric arteries [internal diameter: ~250 µm (JCR) and ~350–400 µm (SD)] were mounted on glass capillary pipettes in a pressure myograph (JP Trading, Aarhus, Denmark) and equilibrated for 1 h in oxygenated (95% O2-5% CO2) Krebs buffer (1.2 mM KHCO3 and 1 mM Ca2+, 37°C). A passive pressure-diameter curve was generated by increasing intraluminal pressure stepwise from 4 to 140 mmHg, and left and right wall thickness and internal diameters were recorded at each pressure. This range of pressures encompasses the physiological range of mean arterial pressure in SD and JCR animals. Wall thickness and vessel diameter sizes are plotted in response to increasing intravascular pressure as an index of arterial compliance.
Wire myography.
Freshly isolated mesenteric arteries (~50 µM) were mounted on wires in chambers of a multivessel myograph (JP Trading) in oxygenated (95% O2-5% CO2) Krebs buffer (1.2 mM potassium bicarbonate and 10 mM Ca2+, 37°C). After the vessels were allowed to develop spontaneous tone in response to an intramural pressure of 60 mmHg, a concentration-response curve to ACh (1 × 10−9 to 5 × 10−5 M) was generated in the presence and absence of nitro-l-arginine methyl ester (L-NAME; 50 µM). At the end of each experiment, contraction to phenylephrine (PE; 1 µM) and then dilation to sodium nitroprusside (SNP; 1 µM) were measured.
Echocardiography.
Echocardiography was performed as previously described (37). All measurements were performed under sevofluorene (1–2%) anesthesia with continuous monitoring of body temperature, blood pressure, and heart rate. LV end-diastolic diameter and end-systolic diameter septal wall diastolic thickness as well as LV free (posterior) wall diastolic thickness were measured by two-dimensional guided M-mode echocardiography from the parasternal long-axis view using a 12- to 38-MHz vascular probe (Vevo 770, 1,000 frames/s, Visual Sonics, Toronto, ON, Canada). LV end-diastolic and systolic volumes (LVEDV and LVESV) and ejection fraction (EF) were calculated with Vevo software (Visual Sonics).
Rat model of CCG/repetitive ischemia.
A pneumatic occluder was implanted over the left anterior descending coronary artery (LAD) as previously described (20–22). The repetitive ischemia (RI) protocol consisted of short, repetitive and transient ischemic episodes: eight 40-s occlusions, once every 20 min (2 h, 20 min total), followed by a nonischemic (rest) period of 5 h, 40 min. This 8-h cycle was repeated 3 times/day for 10 days.
Myocardial and collateral-dependent blood flow measurements.
Blood flow measurements were performed as previously described (21, 37, 38). At day 0 of RI, 5 × 105 gold-labeled (red in color) color microspheres were injected into the lumen of the LV lumen during LAD occlusion. At day 10 of RI, 5 × 105 samarium-labeled (blackberry in color) microspheres were injected into the LV lumen during LAD occlusion. Because of their diameter (15 µm), the microspheres lodge in precapillary vessels and thus remain in the tissue. Heart tissue from the normal nonischemic zone (NZ) and the collateral-dependent ischemic zone (CZ), which is the LAD perfusion territory, as well as a reference blood sample (via a carotid catheter) were then collected, weighed, and sent to BioPal (Worcester, MA) for analysis. When neutron-activated, gold and samarium emit at different frequencies; thus, the signals from each type of microsphere can be measured separately in the same animal. This enables us to obtain measurements of coronary blood flow at day 0 and day 10 of RI in the same animal. Boundaries of the CZ were determined both visually at the time of initial surgery as blanching on LAD occlusion (occluder inflation) followed by reactive hyperemia on reperfusion (occluder deflation) and by fluorescent microspheres (15 µm), which were injected into the LV at the same time and in the same manner as the first set of color microspheres (gold). Blood flow in the NZ and CZ (in ml·min−1·g−1) was calculated as previously described according to the following formula: blood flow = [radioactive count in myocardial tissue) × (blood withdrawal rate)/radioactive count in blood)]/(myocardial tissue weight) (21, 37, 38). Results were expressed as the CZ-to-NZ (CZ/NZ) flow ratio at day 10 of RI. Since CZ and NZ flows are measured while the LAD is occluded, low CZ blood flow correlates with low CCG, whereas high CZ blood flow correlates with high CCG. Since NZ blood flow is constant, low CZ/NZ flow ratios indicate low CCG and high CZ/NZ flow ratios indicate low CCG.
Western blot analysis.
First-order mesenteric arteries were excised, cut into ~1-mm pieces, and subjected to citric acid extraction. The supernatant and pellet were then homogenized in lysis buffer containing 0.2% SDS and 2% Triton, and the supernatants from both were collected. Equal amounts of protein (100 µg) were separated by SDS-PAGE and transferred to Trans-Blot Turbo Mini PVDF membranes (Bio-Rad, Hercules, CA) and blocked with blocking buffer (Li-Cor, Lincoln, NE), followed by incubation with primary and secondary antibodies. Anti-collagen type I (Abcam, Cambridge, MA), collagen type III (Abcam), antielastin (Abcam), and goat anti-rabbit IRDye 800CW (Li-Cor) antibodies were used for Western blot analysis. Fluorescence-based immunodetection was performed with the Li-Cor Odyssey Infrared Imaging System (Li-Cor), and band densities were analyzed by using the Odyssey Application software (version 3.0.21). Data were normalized to β-tubulin (loading control). Experiments were n = 5 animals/group and were analyzed by two-way ANOVA followed by Bonferroni post hoc analysis. P < 0.05 determined statistical significance.
Oxidative stress measurements.
Superoxide (O2·−) production was evaluated using X-band electroparamagnetic resonance (EPR) as in our previous studies (38). A Bruker EMX spectrometer was used for X-Band EPR measurements of O2·− using 1-hydroxy-3-carboxy-pyrrolidine (CP-H; Alexis) as a spin trap. At the end of the 9-day RI protocol, animals underwent two consecutive periods of ischemia-reperfusion. Animals were euthanized, the heart was removed, and the CZ was separated from the NZ. Tissue was immediately homogenized by sonication on ice, the supernatant was collected, and CP-H (3µg/1 mg tissue) was added. Samples were incubated at 37°C for 30 min and spun at 10,000 rpm for 1 min at 4°C, and the supernatant collected and frozen in liquid nitrogen overnight. O2·− concentration was calculated from arbitrary units (AU) (3.4 × 106 AU/nM).
Statistical analysis.
All experiments were at least n = 8 animals/treatment group. Results were analyzed by two-way ANOVA followed by Bonferroni correction. P < 0.05 determined statistical significance.
RESULTS
Effect of the HF/HS diet on arterial compliance in male and female metabolic syndrome animals.
Arterial compliance, as a parameter of macrovascular function, was markedly decreased in male metabolic syndrome (JCR) versus normal, healthy (SD) rats (~66%) (Fig. 1A). In contrast, loss of arterial compliance was only mild (~25% decrease vs. SD female rats) in female JCR rats (Fig. 1A). However, when JCR rats were fed the HF/HS diet for 12 wk, arterial compliance decreased to a similar extent in both male (~88% vs. SD male rats on the HF/HS diet) and female (~90% vs. SD female rats on the HF/HS diet) animals. Of note, the relative negative effect of the HF/HS diet was approximately threefold greater in JCR female versus JCR male animals (Fig. 1A). Interestingly, the HF/HS diet did not have a notable effect on arterial compliance in either male or female SD rats. Wall thickness was similarly increased in both JCR male and female rats on normal chow but was not further increased by the high-fat/high-carbohydrate diet in either sex or in SD rats (Fig. 1B).
Fig. 1.
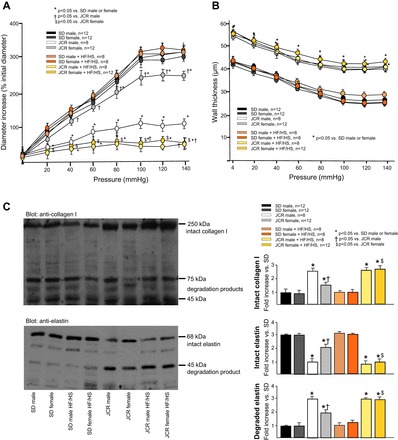
A and B: arterial compliance and changes in wall thickness were evaluated by pressure myography in large mesenteric arteries (250–400 µm) in normal Sprague-Dawley (SD) and metabolic syndrome (JCR) female and male rats fed normal chow or the high-fat/high-sugar (HF/HS) diet for 12 wk as indicated. A: increase in passive internal diameter plotted against increasing pressure. B: change in wall thickness plotted against increasing pressure. C: intact collagen type I and elastin content and their degradation were evaluated by Western blot analysis in large mesenteric arteries (250–400 µm) in SD and JCR female and male rats fed normal chow or the HF/HS diet for 12 wk as indicated. Representative Western blots (left) and cumulative data (right) are shown. *P < 0.05 vs. male or female SD rats; †P < 0.05 vs. male JCR rats; $P < 0.05 vs. female JCR rats.
These changes in compliance may be partially related to changes in total collagen content and elastin degradation. The approximately threefold increase in collagen type I expression correlated with decreased arterial compliance in JCR male animals on normal chow and in JCR male and female animals on the HF/HS diet (Fig. 1C). Collagen type III expression in these large arteries showed a nonsignificant trend toward a decrease in JCR male animals, but its expression in all groups was minimal (data not shown). Total elastin content was approximately threefold lower in large arteries of JCR male versus SD rats. Intact elastin was decreased in JCR female rats, but this decrease was lesser (~50%). The high-fat/high-carbohydrate diet also significantly increased elastin degradation in JCR male rats (~3-fold) and to a lesser extent (~2-fold) in JCR female versus JCR female and SD male and female rats (Fig. 1C), correlating with decreased compliance in these animals (Fig. 1A).
Effect of the HF/HS diet on endothelium-dependent vasodilation in male and female metabolic syndrome animals.
A similar trend was observed with respect to microvascular function as evaluated by endothelium-dependent vasodilation. While the ability to dilate in response to increasing concentrations of ACh was severely compromised in mesenteric microvessels from male JCR rats (~50%), it was only moderately decreased in mesenteric microvessels from female JCR rats (~25%) (Fig. 2). Twelve weeks of the HF/HS diet, however, significantly decreased ACh-dependent vasodilation in both male and female JCR rats, ~50% and 75%, respectively, and the negative effect of the HF/HS diet was approximately threefold greater in female than male metabolic syndrome animals (Fig. 2). As with macrovascular function, the HF/HS diet did not abrogate endothelium-dependent vasodilation in either male or female normal, healthy animals. L-NAME decreased ACh-dependent vasodilation in all SD rats, consistent with numerous studies showing that there is an endothelial nitric oxide (NO) synthase (eNOS)-derived NO-dependent component to endothelium-dependent vasodilation in normal animals (24). In agreement with a previous study (31), L-NAME inhibited ACh-mediated vasorelaxation in both male and female JCR rats on normal chow, but the magnitude of the response was attenuated in female compared with male animals (35% vs. 55% decrease at 10−5 log mol/l ACh, respectively). These data indicate that eNOS-derived NO may play a greater role in endothelial dysfunction in male than female JCR rats. L-NAME did not further reduce vasodilation in the presence of the HF/HS diet in either sex in JCR rats. This is likely due to the fact that ACh-induced vasodilation in the presence of HF/HS was already maximally impaired (>90%) and does not indicate a lack of involvement of eNOS-dependent NO in the regulation of endothelial-dependent vasodilation in these animals.
Fig. 2.
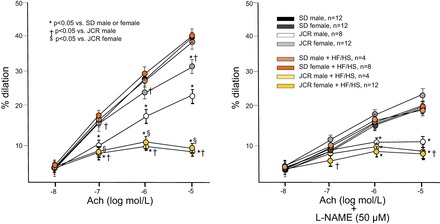
Endothelium-dependent vasodilation was measured by wire myography in mesenteric resistance arteries (~50 µm). Left: dilation to increasing doses of ACh in SD and JCR female and male rats fed normal chow or the HF/HS diet (Western diet) for 12 wk as indicated. Right: same as left except arteries were pretreated with 50 µM N-nitro-l-arginine methyl ester (l-NAME). *P < 0.05 vs. male or female SD rats; †P < 0.05 vs. male JCR rats; $P < 0.05 vs. female JCR rats.
Vasodilation was measured as increase in internal diameter from the starting internal diameter recorded at 60-mmHg transmural pressure, which was smaller in JCR (male and female average) than SD (male and female average) rats (48 ± 1.6 vs. 64 ± 1.4 µm), similar to large mesenteric arteries. There were no differences between the sexes, and the HF/HS diet did not affect the starting internal arterial diameter in any group.
Contraction to PE and dilation to SNP were assessed at the end of each experiment. As previously reported (31), both male and female JCR rats exhibit enhanced contraction in response to PE (14 ± 2.1 µm JCR vs. 38 ± 2.5 µm SD or ~70% vs. ~40% decrease vs. active diameter (48 ± 1.6 µm JCR vs. 64 ± 1.4 µm SD) and enhanced relaxation to SNP [~20% increase vs. ACh in JCR (male/female) vs. ~5% in SD (male/female)] compared with SD rats, which was not influenced by the sex of the animals. The HF/HS diet did not affect PE-dependent vasoconstriction in either phenotype; however, the effect of SNP was further accentuated by the HF/HS diet in female JCR rats (~35% increase vs. ACh).
Effect of the HF/HS diet on cardiac function in male and female metabolic syndrome animals.
Corresponding with vascular function, cardiac function [cardiac output (CO) and EF] was normal in normal, healthy (SD) animals regardless of diet (Fig. 3).
Fig. 3.
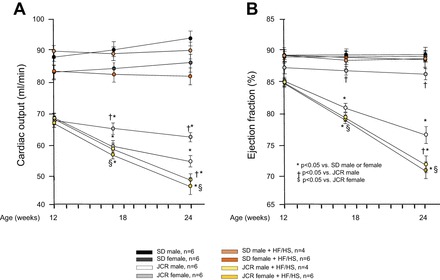
Cardiac function was measured by echocardiography (M-mode). Cardiac output (A) and ejection fraction (B) are shown in female and male SD and JCR rats fed normal chow or the HF/HS diet (Western diet) for 12 wk as indicated. *P < 0.05 vs. male or female SD rats; †P < 0.05 vs. male JCR rats; $P < 0.05 vs. female JCR rats.
JCR rats exhibited cardiac hypertrophy, secondary to hypertension, which accounted for the decreased CO in all JCR rats at baseline (at 12 wk of age before the onset of dietary treatment) (Fig. 3A). This reduction in all JCR rats at 12 wk of age and in JCR female rats on normal chow was a result of decreased stroke volume resulting from a similar decrease in LVEDV and LVESV (data not shown) and corresponding with preserved EF (Fig. 3B). In contrast, in male JCR rats on normal chow and male and female JCR rats that were fed the HF/HS diet for 12 wk, the decreasing CO over the subsequent 12 wk (Fig. 3A) was, however, the result of increasing LVESV, which was worsened by the HF/HS diet, especially in female JCR rats (Table 2), and resulted in the corresponding decrease in EF (Fig. 3B and Table 2).
Table 2.
Cardiac function in SD and JCR rats fed normal and HF/HS diet
| SD Male Normal | SD Female Normal | SD Male HF/HS | SD Female HF/HS | JCR Male Normal | JCR Female Normal | JCR Male HF/HS | JCR Female HF/HS | |
|---|---|---|---|---|---|---|---|---|
| MABP, mmHg | 98 ± 6 | 102 ± 7 | 146 ± 9 | 151 ± 11 | 152 ± 6 | 144 ± 9 | 160 ± 12 | 168 ± 6 |
| LVWT,d, mm | 1.70 ± 0.02 | 1.62 ± 0.02 | 1.70 ± 0.03 | 1.64 ± 0.03 | 2.64 ± 0.03* | 2.04 ± 0.03*† | 2.62 ± 0.03* | 2.58 ± 0.03*§ |
| IVSWT,d, mm | 1.61 ± 0.04 | 1.63 ± 0.03 | 1.65 ± 0.01 | 1.63 ± 0.02 | 2.11 ± 0.02* | 1.81 ± 0.02*† | 2.15 ± 0.02* | 2.11 ± 0.01*§ |
| LVEDD, mm | 7.7 ± 0.02 | 7.3 ± 0.02 | 7.7 ± 0.02 | 7.4 ± 0.06 | 5.2 ± 0.02* | 5.6 ± 0.02*† | 6.2 ± 0.02*† | 6.1 ± 0.04*†§ |
| LVESD, mm | 3.0 ± 0.01 | 2.8 ± 0.01 | 2.9 ± 0.01 | 2.8 ± 0.05 | 3.6 ± 0.04* | 2.2 ± 0.06*† | 5.4 ± 0.02*† | 5.6 ± 0.02*†§ |
| LVEDV, µl | 324 ± 6 | 308 ± 4 | 326 ± 3 | 307 ± 3 | 216 ± 4* | 229 ± 3*† | 258 ± 5*† | 253 ± 7*†§ |
| LVESV, µl | 39 ± 4 | 37 ± 2 | 36 ± 3 | 38 ± 2 | 49 ± 5* | 29 ± 3*† | 71 ± 2*† | 73 ± 2*†§ |
| CO, ml/min | 94 ± 3 | 87 ± 4 | 90 ± 2 | 82 ± 3 | 55 ± 1.5* | 63 ± 1*† | 49 ± 1.5*† | 46.5 ± 4*†§ |
| EF, % | 88 ± 5 | 88 ± 2.4 | 89 ± 3 | 88 ± 1 | 77 ± 2* | 86.7 ± 1*† | 72 ± 1.5*† | 71 ± 1.5*†§ |
LVWT, d, left ventricular (LV) wall thickness in diastole; IVSWT, d, intraventricular septum wall thickness at diastole; LVEDD and LVESD, LV end-diastolic and end-systolic diameter; LVEDV and LVESV, LV end-diastolic and end-systolic volume; CO, cardiac output; EF, ejection fraction.
P < 0.05 vs. male or female SD rats;
P < 0.05 vs. male JCR rats;
P < 0.05 vs. female JCR rats.
Effect of the HF/HS diet on coronary collateral growth in male and female metabolic syndrome animals.
We have previously shown that an RI protocol, which mimics stable angina, induced robust CCG in normal, healthy rats and that this adaptive response was severely compromised in male JCR rats (36). The results shown in Fig. 4 demonstrate that in response to RI, in contrast to male JCR rats, female JCR rats were able to develop a rather robust coronary collateral network, although to a lesser extent than their SD counterparts [CZ/NZ flow ratio was 0.56 ± 0.03 (JCR female RI) vs. 0.80 ± 0.05 (SD female RI)]. The HF/HS diet completely abolished the capacity for RI-induced CCG in female JCR rats, thereby eliminating any advantage afforded by the female sex [CZ/NZ flow ratio was 0.13 ± 0.02 (JCR female RI + HF/HS diet), 0.12 ± 0.01 (JCR male RI + HF/HS diet), and 0.15 ± 0.02 (JCR male RI + normal diet)] (Fig. 4).
Fig. 4.
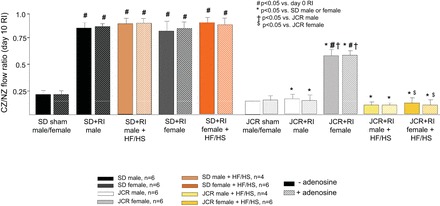
SD and JCR male and female rats were instrumented with the left anterior descending coronary artery (LAD) occluder and underwent 10 days of repetitive ischemia (RI) during the last 10 days of the 12-wk normal chow or HF/HS diet protocol as indicated. Coronary blood flow was measured in collateral-dependent ischemic zones (CZ) and normal nonischemic zones (NZ) using radioactive microspheres during LAD occlusion in the presence or absence of adenosine. Results are expressed as the ratio between CZ and NZ flow. *P < 0.05 vs. male or female SD rats; †P < 0.05 vs. male JCR rats; $P < 0.05 vs. female JCR rats.
We used a potent coronary vasodilator, adenosine (5 × 105 M), to determine if the observed changes in coronary blood flow could be explained by vasodilation; however, treatment with adenosine had no effect on coronary blood flow in any group, indicating that metabolic syndrome and the HF/HS diet changed coronary collateral structure (Fig. 4). This lack of response to adenosine in vivo should not be confused with changes in vasoreactivity in isolated vessels (Fig. 2). These in vivo measurements were obtained during total (>80%) occlusion of a major coronary artery. Under these conditions, the coronary circulation was already maximally dilated, i.e., the coronary flow reserve was zero (47).
Effect of the HF/HS diet on cardiac and vascular O2·− production in male and female metabolic syndrome animals.
The results shown in Fig. 5 demonstrate that myocardial O2·− production (Fig. 5B) was elevated approximately sixfold in male JCR vs. SD rats. Although O2·− production was significantly increased in female JCR versus SD rats, the increase was much smaller (~3-fold), correlating with pronounced cardiac dysfunction observed in male rats and only mild cardiac dysfunction observed in female JCR rats maintained on normal chow (Fig. 3). The HF/HS diet effectively abolished the difference between male and female JCR rats by eliciting equal and very high (~8-fold vs. SD male and female) levels of O2·− production in the heart (Fig. 5B), correlating with the effect of the HF/HS diet on cardiac function in JCR rats (Fig. 3). The HF/HS diet had no effect on O2·− production in male or female SD rats (Fig. 5B), correlating with its effect on cardiac function in SD rats (Fig. 3).
Fig. 5.
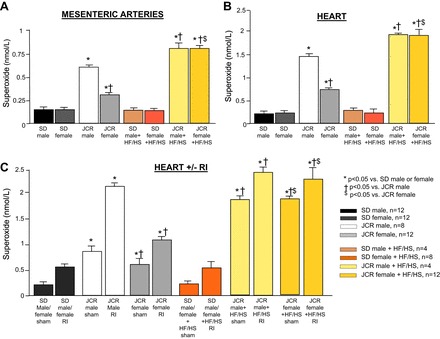
SD and JCR female and male rats were fed normal chow or the HF/HS diet (Western diet) for 12 wk, as indicated (A and B), and underwent 10 days or RI during the last 10 days of the 12-wk normal chow or HF/HS diet protocol, as indicated (C). O2·− production was measured by X-band electroparamagnetic resonance in mesenteric arteries (50–400 µm) (A), the whole heart (B), and the CZ (shown in C) and NZ (not shown). *P < 0.05 vs. male or female SD rats; †P < 0.05 vs. male JCR rats; $P < 0.05 vs. female JCR rats.
Similar results were observed with respect to O2·− production from mesenteric arteries. Although absolute amount of O2·− produced per gram of tissue was smaller, the trend was identical as that seen in the heart (Fig. 5A) and correlated with the exacerbated and equalized vascular dysfunction seen in male and female JCR rats on the HF/HS diet (Figs. 1 and 2). There was no difference between small (~50-µm internal diameter) and large (>250-µm internal diameter) mesenteric arteries; thus, data from both were combined to generate the results shown in Fig. 5A.
As reported in our previous studies, RI induced O2·− production in both SD and JCR male rats, but this increase was much greater (~4-fold vs. SD RI) in JCR rats (Fig. 5C) (38). The RI-induced increase in O2·− was comparatively smaller in female JCR rats (~2-fold vs. SD RI) (Fig. 5C), correlating with only partial inhibition of CCG in female JCR rats on normal chow (Fig. 4). The HF/HS diet had no effect on RI-induced O2·− production in SD rats but caused an even further and equal increase in O2·− production in both male and female JCR rats (~5-fold vs. SD RI) (Fig. 5C), correlating with complete inhibition of CCG not only in male but also female JCR rats (Fig. 4).
DISCUSSION
The major conclusions of our study are as follows: 1) macrovascular, microvascular, and cardiac function are severely compromised in male metabolic syndrome animals but relatively well preserved in female metabolic syndrome animals maintained on normal rat chow; 2) the HF/HS (Western) diet has no deleterious effect on cardiovascular function in normal animals (male or female) but has a major negative effect on cardiovascular function in both male and female metabolic syndrome animals; 3) the HF/HS diet abolishes the protective effect of the female sex with respect to CVD; and 4) the HF/HS diet exacerbates oxidative stress in the metabolic syndrome animals, which correlates with impaired cardiovascular function.
The observation that the HF/HS diet did not induce overt cardiovascular dysfunction in normal animals must be discussed within the context of the greater debate regarding whether this and similar diets, the high-fat or the high-sugar (usually fructose) diet, are capable of inducing cardiovascular dysfunction in normal, healthy rodents. Certain studies using phenotypically normal rodents have noted minimal, if any, cardiac dysfunction, yet others have shown comparable levels of dysfunction seen in genetic models of metabolic syndrome (9, 15, 33, 34). The key difference may lie with the differential use of SD versus Wistar rats, as SD rats have been known to be resistant to diet-induced obesity (22). However, through selective breeding and development of inbred strains, such as Wistar and Wistar-Kyoto rats, rats can develop susceptibility to metabolic syndrome (12, 29), giving further support to the polygenomic profile of obesity and metabolic syndrome predisposition. Additionally, even in studies that have demonstrated diet-induced metabolic syndrome in Wistar rats, none have shown convincing evidence of insulin resistance or type 2 diabetes, complicating their translational applicability (33). As such, these results may carry implications regarding a possible disparate effect of the Western diet depending on the genetic predisposition and/or the existence of parameters of metabolic syndrome in individuals exposed to the Western diet. Also, length of feeding and diet composition (HF/HS vs. high-fat or high-carbohydrate-only diets) play a critical role in metabolic syndrome development. With the exception of lower HDL levels, our HF/HS-fed SD rats developed all parameters of metabolic syndrome; some even match metabolic syndrome rats exposed to the HF/HS diet (absolute TGs and glucose and change in weight) (Table 1).
Our results demonstrating relatively preserved cardiovascular function in female rats despite metabolic syndrome are in agreement with clinical literature (13, 28). While we have not set out to determine a mechanistic explanation, several studies have pointed to estrogen as a potential source of protection in animals and humans (20, 26, 28, 43). Whether estrogen is directly responsible for this cardiovascular protection or if estrogens modulates critical components of metabolic syndrome have yet to be fully described.
Our observation that the Western diet abolishes the protective effect of the female sex with respect to CVD even in premenopausal animals is novel and has potentially important clinical as well as research implications. Current basic science and clinical metabolic syndrome research often excludes females since female experimental models of metabolic syndrome do not exhibit the extent of cardiovascular dysfunction seen in males. However, postmenopausal women are at an elevated risk for metabolic syndrome and CVD, and the incidence of metabolic syndrome among premenopausal women is now equal to that among men and higher in some populations (e.g., African Americans), correlating with increasing CVD prevalence and severity among premenopausal women (13, 28). Thus, future studies should incorporate Western-style diets, which, as our study shows, do induce equal cardiovascular dysfunction in males and females with metabolic syndrome.
Oxidative stress is a hallmark of obesity and metabolic syndrome. We have previously shown that myocardial O2·− production was elevated in male JCR versus SD rats (37, 38). The present study demonstrates that female metabolic syndrome animals are relatively protected from oxidative stress but that the HF/HS diet eliminates this protective effect of the female sex, correlating with deterioration of cardiovascular function in female animals. The apparent greater dependence of vasodilation on NO·− in male versus female metabolic syndrome animals on normal chow versus the particular improvement of vasodilation by a nitric oxide donor (SNP) in female but not male JCR rats on the HF/HS diet may further support this notion since NO·− bioavailability is in part regulated by its scavenging by superoxide. Although large clinical trials with mixed patient populations (metabolic syndrome and nonmetabolic syndrome) (HOPE, HOPE-TOO, and MRC/BHF Heart Protection Study) failed to see improvements in cardiovascular parameters with long-term antioxidant administration (19, 23), many smaller clinical trials using antioxidants specifically conducted in patients with metabolic syndrome had overwhelmingly positive effects on CVD (4, 5, 7). Moreover, drugs that have been shown to provide benefits in delay and prevention of CVD in metabolic syndrome patients, including ANG II-converting enzyme inhibitors, receptor blockers, metformin, and statins, have strong antioxidant properties (3, 10, 18, 25, 46). Thus, the HF/HS diet-mediated induction of oxidative stress in metabolic syndrome may provide a mechanistic intermediate that can be targeted to ameliorate the effects of the Western diet in metabolic syndrome patients.
In agreement with published data in animal models and humans, metabolic syndrome rats exhibited elevated levels of CRP and IL-6 compared with normal animals on normal chow. However, unlike with oxidative stress and cardiovascular dysfunction, similar increases in body weight precipitated by the HF/HS diet markedly elevated these markers of inflammation in both phenotypes, although the increase was more pronounced in metabolic syndrome. As with oxidative stress and cardiovascular dysfunction, the female sex did not provide any protection in either normal or metabolic syndrome phenotypes. These results support the idea that elevated inflammation is the earliest marker of metabolic syndrome, which alone is not sufficient to promote overt cardiovascular dysfunction but is instead an initiating step in the process.
A limitation of our study is that we did not measure metabolic parameters to demonstrate the induction of metabolic syndrome . However, in other studies, 5–8 wk of the HF/HS diet induced hyperglycemia, hyperinsulinemia and insulin resistance, and dyslipidemia (11, 33, 35).
Another potential limitation of the present study is the applicability of the feeding schedule to humans. The rats in our study were given both normal chow and HF/HS chow ad libitum. It may be unlikely that a human consumes a Western-type diet around the clock. As such, our feeding protocol may present an extreme scenario, so that regular and frequent consumption of meals consisting of the Western diet may result in similar albeit milder cardiovascular phenotype in humans.
Overall, our results support the notion that the deleterious consequences of metabolic syndrome are, in part, driven by additional environmental factors. While there is ample evidence of a correlation between environmental factors, especially diet and metabolic syndrome development, very few studies have focused on the effect of diet on pathologies associated with existing metabolic syndrome, especially CVD development. Second, our results demonstrate that the protection from CVD inherent to the female sex regardless of metabolic syndrome status can be overridden by environmental factors.
GRANTS
P. Rocic was supported by National Heart, Lung, and Blood Institute Grant R01-HL-093052.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
I.H., A.S., G.J., B.H., C.B., F.Z., B.J.P., and P.R. performed experiments; I.H., C.B., F.Z., B.J.P., and P.R. analyzed data; I.H. and P.R. prepared figures; I.H. drafted manuscript; I.H., A.S., G.J., B.H., C.B., F.Z., B.J.P., S.D.P., and P.R. approved final version of manuscript; S.D.P. and P.R. interpreted results of experiments; P.R. edited and revised manuscript.
REFERENCES
- 1.Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart J-C, James WPT, Loria CM, Smith SC Jr; International Diabetes Federation Task Force on Epidemiology and Prevention; Hational Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; International Association for the Study of Obesity . Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120: 1640–1645, 2009. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 2.Allen TJ, Cooper ME, Lan HY. Use of genetic mouse models in the study of diabetic nephropathy. Curr Atheroscler Rep 6: 197–202, 2004. doi: 10.1007/s11883-004-0032-7. [DOI] [PubMed] [Google Scholar]
- 3.Ansari JA, Bhandari U, Pillai KK, Haque SE. Effect of rosuvastatin on obesity-induced cardiac oxidative stress in Wistar rats—a preliminary study. Indian J Exp Biol 50: 216–222, 2012. [PubMed] [Google Scholar]
- 4.Basu A, Betts NM, Ortiz J, Simmons B, Wu M, Lyons TJ. Low-energy cranberry juice decreases lipid oxidation and increases plasma antioxidant capacity in women with metabolic syndrome. Nutr Res 31: 190–196, 2011. doi: 10.1016/j.nutres.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beydoun MA, Shroff MR, Chen X, Beydoun HA, Wang Y, Zonderman AB. Serum antioxidant status is associated with metabolic syndrome among U.S. adults in recent national surveys. J Nutr 141: 903–913, 2011. doi: 10.3945/jn.110.136580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunner F, Wölkart G, Pfeiffer S, Russell JC, Wascher TC. Vascular dysfunction and myocardial contractility in the JCR:LA-corpulent rat. Cardiovasc Res 47: 150–158, 2000. doi: 10.1016/S0008-6363(00)00056-0. [DOI] [PubMed] [Google Scholar]
- 7.Cangemi R, Angelico F, Loffredo L, Del Ben M, Pignatelli P, Martini A, Violi F. Oxidative stress-mediated arterial dysfunction in patients with metabolic syndrome: effect of ascorbic acid. Free Radic Biol Med 43: 853–859, 2007. doi: 10.1016/j.freeradbiomed.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Carrera-Bastos P, Fontes-Villalba M, O’Keefe JH, Lindeberg S, Cordain L. The western diet and lifestyle and diseases of civilization. Res Reports Clin Cardiol 2: 15–35, 2011. doi: 10.2147/RRCC.S16919. [DOI] [Google Scholar]
- 9.Carroll JF, Zenebe WJ, Strange TB. Cardiovascular function in a rat model of diet-induced obesity. Hypertension 48: 65–72, 2006. doi: 10.1161/01.HYP.0000224147.01024.77. [DOI] [PubMed] [Google Scholar]
- 10.Cingolani OH, Pérez NG, Ennis IL, Álvarez MC, Mosca SM, Schinella GR, Escudero EM, Cónsole G, Cingolani HE. In vivo key role of reactive oxygen species and NHE-1 activation in determining excessive cardiac hypertrophy. Pflugers Arch 462: 733–743, 2011. doi: 10.1007/s00424-011-1020-8. [DOI] [PubMed] [Google Scholar]
- 11.Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, O’Keefe JH, Brand-Miller J. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr 81: 341–354, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Couturier K, Batandier C, Awada M, Hininger-Favier I, Canini F, Anderson RA, Leverve X, Roussel AM. Cinnamon improves insulin sensitivity and alters the body composition in an animal model of the metabolic syndrome. Arch Biochem Biophys 501: 158–161, 2010. doi: 10.1016/j.abb.2010.05.032. [DOI] [PubMed] [Google Scholar]
- 13.Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol 28: 629–636, 2008. doi: 10.1161/ATVBAHA.107.151092. [DOI] [PubMed] [Google Scholar]
- 14.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC Jr, Spertus JA, Costa F; American Heart Association; National Heart, Lung, and Blood Institute . Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 112: 2735–2752, 2005. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 15.Heinonen I, Rinne P, Ruohonen ST, Ruohonen S, Ahotupa M, Savontaus E. The effects of equal caloric high fat and western diet on metabolic syndrome, oxidative stress and vascular endothelial function in mice. Acta Physiol (Oxf) 211: 515–527, 2014. doi: 10.1111/apha.12253. [DOI] [PubMed] [Google Scholar]
- 16.Henriksen EJ, Teachey MK, Lindborg KA, Diehl CJ, Beneze AN. The high-fat-fed lean Zucker rat: a spontaneous isocaloric model of fat-induced insulin resistance associated with muscle GSK-3 overactivity. Am J Physiol Regul Integr Comp Physiol 294: R1813–R1821, 2008. doi: 10.1152/ajpregu.00178.2008. [DOI] [PubMed] [Google Scholar]
- 17.Hutcheson R, Rocic P. The metabolic syndrome, oxidative stress, environment, and cardiovascular disease: the great exploration. Exp Diabetes Res 2012: 271028, 2012. doi: 10.1155/2012/271028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan BV, Sola S, Lauten WB, Natarajan R, Hooper WC, Menon RG, Lerakis S, Helmy T. Quinapril, an ACE inhibitor, reduces markers of oxidative stress in the metabolic syndrome. Diabetes Care 27: 1712–1715, 2004. doi: 10.2337/diacare.27.7.1712. [DOI] [PubMed] [Google Scholar]
- 19.Knekt P, Ritz J, Pereira MA, O’Reilly EJ, Augustsson K, Fraser GE, Goldbourt U, Heitmann BL, Hallmans G, Liu S, Pietinen P, Spiegelman D, Stevens J, Virtamo J, Willett WC, Rimm EB, Ascherio A. Antioxidant vitamins and coronary heart disease risk: a pooled analysis of 9 cohorts. Am J Clin Nutr 80: 1508–1520, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Koh KK. Effects of estrogen on the vascular wall: vasomotor function and inflammation. Cardiovasc Res 55: 714–726, 2002. doi: 10.1016/S0008-6363(02)00487-X. [DOI] [PubMed] [Google Scholar]
- 21.Leavesley SJ, Ledkins W, Rocic P. A device for performing automated balloon catheter inflation ischemia studies. PLoS One 9: e95823, 2014. doi: 10.1371/journal.pone.0095823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin BE, Dunn-Meynell AA, Balkan B, Keesey RE. Selective breeding for diet-induced obesity and resistance in Sprague-Dawley rats. Am J Physiol 273: R725–R730, 1997. [DOI] [PubMed] [Google Scholar]
- 23.Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, Ross C, Arnold A, Sleight P, Probstfield J, Dagenais GR. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA 293: 1338–1347, 2005. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 24.McCulloch AI, Randall MD. Sex differences in the relative contributions of nitric oxide and EDHF to agonist-stimulated endothelium-dependent relaxations in the rat isolated mesenteric arterial bed. Br J Pharmacol 123: 1700–1706, 1998. doi: 10.1038/sj.bjp.0701781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meaney E, Vela A, Samaniego V, Meaney A, Asbún J, Zempoalteca J-C, Elisa ZN, Emma MN, Guzman M, Hicks J, Ceballos G. Metformin, arterial function, intima-media thickness and nitroxidation in metabolic syndrome: the mefisto study. Clin Exp Pharmacol Physiol 35: 895–903, 2008. doi: 10.1111/j.1440-1681.2008.04920.x. [DOI] [PubMed] [Google Scholar]
- 26.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med 340: 1801–1811, 1999. doi: 10.1056/NEJM199906103402306. [DOI] [PubMed] [Google Scholar]
- 27.Mouquet F, Cuilleret F, Susen S, Sautière K, Marboeuf P, Ennezat PV, McFadden E, Pigny P, Richard F, Hennache B, Vantyghem MC, Bertrand M, Dallongeville J, Jude B, Van Belle E. Metabolic syndrome and collateral vessel formation in patients with documented occluded coronary arteries: association with hyperglycaemia, insulin-resistance, adiponectin and plasminogen activator inhibitor-1. Eur Heart J 30: 840–849, 2009. doi: 10.1093/eurheartj/ehn569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murase T, Hattori T, Ohtake M, Nakashima C, Takatsu M, Murohara T, Nagata K. Effects of estrogen on cardiovascular injury in ovariectomized female DahlS.Z-Lepr(fa)/Lepr(fa) rats as a new animal model of metabolic syndrome. Hypertension 59: 694–704, 2012. doi: 10.1161/HYPERTENSIONAHA.111.180976. [DOI] [PubMed] [Google Scholar]
- 29.Nascimento AR, Machado M, de Jesus N, Gomes F, Lessa MA, Bonomo IT, Tibiriçá E. Structural and functional microvascular alterations in a rat model of metabolic syndrome induced by a high-fat diet. Obesity (Silver Spring) 21: 2046–2054, 2013. doi: 10.1002/oby.20358. [DOI] [PubMed] [Google Scholar]
- 30.Nathoe HM, Koerselman J, Buskens E, van Dijk D, Stella PR, Plokker THW, Doevendans PA, Grobbee DE, de Jaegere PP; Octopus Study Group . Determinants and prognostic significance of collaterals in patients undergoing coronary revascularization. Am J Cardiol 98: 31–35, 2006. doi: 10.1016/j.amjcard.2006.01.050. [DOI] [PubMed] [Google Scholar]
- 31.O’Brien SF, Russell JC, Dolphin PJ, Davidge ST. Vascular wall function in insulin-resistant JCR:LA-cp rats: role of male and female sex. J Cardiovasc Pharmacol 36: 176–181, 2000. doi: 10.1097/00005344-200008000-00006. [DOI] [PubMed] [Google Scholar]
- 32.Panchal SK, Brown L. Rodent models for metabolic syndrome research. J Biomed Biotechnol 2011: 351982, 2011. doi: 10.1155/2011/351982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panchal SK, Poudyal H, Iyer A, Nazer R, Alam MA, Diwan V, Kauter K, Sernia C, Campbell F, Ward L, Gobe G, Fenning A, Brown L. High-carbohydrate, high-fat diet-induced metabolic syndrome and cardiovascular remodeling in rats. J Cardiovasc Pharmacol 57: 611–624, 2011. doi: 10.1097/FJC.0b013e3181feb90a. [DOI] [PubMed] [Google Scholar]
- 34.Pettersson US, Waldén TB, Carlsson P-O, Jansson L, Phillipson M. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS One 7: e46057, 2012. doi: 10.1371/journal.pone.0046057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poudyal H, Panchal SK, Ward LC, Waanders J, Brown L. Chronic high-carbohydrate, high-fat feeding in rats induces reversible metabolic, cardiovascular, and liver changes. Am J Physiol Endocrinol Metab 302: E1472–E1482, 2012. doi: 10.1152/ajpendo.00102.2012. [DOI] [PubMed] [Google Scholar]
- 36.Reed R, Kolz C, Potter B, Rocic P. The mechanistic basis for the disparate effects of angiotensin II on coronary collateral growth. Arterioscler Thromb Vasc Biol 28: 61–67, 2008. doi: 10.1161/ATVBAHA.107.154294. [DOI] [PubMed] [Google Scholar]
- 37.Reed R, Potter B, Smith E, Jadhav R, Villalta P, Jo H, Rocic P. Redox-sensitive Akt and Src regulate coronary collateral growth in metabolic syndrome. Am J Physiol Heart Circ Physiol 296: H1811–H1821, 2009. doi: 10.1152/ajpheart.00920.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rocic P, Kolz C, Reed R, Potter B, Chilian WM. Optimal reactive oxygen species concentration and p38 MAP kinase are required for coronary collateral growth. Am J Physiol Heart Circ Physiol 292: H2729–H2736, 2007. doi: 10.1152/ajpheart.01330.2006. [DOI] [PubMed] [Google Scholar]
- 39.Russell JC, Koeslag DG. Jcr:LA-corpulent rat: a strain with spontaneous vascular and myocardial disease. ILAR J 32: 27–32, 1990. doi: 10.1093/ilar.32.3.27. [DOI] [Google Scholar]
- 40.Russell JC, Proctor SD. Small animal models of cardiovascular disease: tools for the study of the roles of metabolic syndrome, dyslipidemia, and atherosclerosis. Cardiovasc Pathol 15: 318–330, 2006. doi: 10.1016/j.carpath.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Serné EH, de Jongh RT, Eringa EC, IJzerman RG, Stehouwer CDA. Microvascular dysfunction: a potential pathophysiological role in the metabolic syndrome. Hypertension 50: 204–211, 2007. doi: 10.1161/HYPERTENSIONAHA.107.089680. [DOI] [PubMed] [Google Scholar]
- 42.Smith ER, Tomlinson LA, Ford ML, McMahon LP, Rajkumar C, Holt SG. Elastin degradation is associated with progressive aortic stiffening and all-cause mortality in predialysis chronic kidney disease. Hypertension 59: 973–978, 2012. doi: 10.1161/HYPERTENSIONAHA.111.187807. [DOI] [PubMed] [Google Scholar]
- 43.Stier CT Jr, Chander PN, Rosenfeld L, Powers CA. Estrogen promotes microvascular pathology in female stroke-prone spontaneously hypertensive rats. Am J Physiol Endocrinol Metab 285: E232–E239, 2003. doi: 10.1152/ajpendo.00029.2003. [DOI] [PubMed] [Google Scholar]
- 44.Sweazea KL. Compounding evidence implicating Western diets in the development of metabolic syndrome. Acta Physiol (Oxf) 211: 471–473, 2014. doi: 10.1111/apha.12303. [DOI] [PubMed] [Google Scholar]
- 45.Varga O, Harangi M, Olsson IAS, Hansen AK. Contribution of animal models to the understanding of the metabolic syndrome: a systematic overview. Obes Rev 11: 792–807, 2010. doi: 10.1111/j.1467-789X.2009.00667.x. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida J, Yamamoto K, Mano T, Sakata Y, Nishikawa N, Nishio M, Ohtani T, Miwa T, Hori M, Masuyama T. AT1 receptor blocker added to ACE inhibitor provides benefits at advanced stage of hypertensive diastolic heart failure. Hypertension 43: 686–691, 2004. doi: 10.1161/01.HYP.0000118017.02160.fa. [DOI] [PubMed] [Google Scholar]
- 47.Yun J, Rocic P, Pung YF, Belmadani S, Carrao ACR, Ohanyan V, Chilian WM. Redox-dependent mechanisms in coronary collateral growth: the “redox window” hypothesis. Antioxid Redox Signal 11: 1961–1974, 2009. doi: 10.1089/ars.2009.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]