

Abstract
Androgens significantly alter muscle mass in part by shifting protein balance in favor of net protein accretion. During various atrophic conditions, the clinical impact of decreased production or bioavailability of androgens (termed hypogonadism) is important as a loss of muscle mass is intimately linked with survival outcome. While androgen replacement therapy increases muscle mass in part by restoring protein balance, this is not a comprehensive treatment option due to potential side effects. Therefore, an understanding of the mechanisms by which androgens alter protein balance is needed for the development of androgen-independent therapies. While the data in humans suggest androgens alter protein balance (both synthesis and breakdown) in the fasted metabolic state, a predominant molecular mechanism(s) behind this observation is still lacking. This failure is likely due in part to inconsistent experimental design between studies including failure to control nutrient/feeding status, the method of altering androgens, and the model systems utilized.
Keywords: Hypertrophy, Atrophy, Anabolic, Catabolic, Testosterone, Autophagy
3.1 INTRODUCTION
The importance of maintaining skeletal muscle mass during various catabolic conditions is becoming increasingly recognized since muscle wasting into older age is predictive of an unfavorable survival outcome (Martin, Birdsell et al., 2013). In males, reduced production or bioavailability of androgens, termed hypogonadism, directly contributes to muscle atrophy since androgens play a major role in the maintenance or restoration of muscle mass (Ferrando, Sheffield-Moore et al., 2003, Steiner, Fukuda et al., 2016, White, Gao et al., 2013, White, Puppa et al., 2013, Atkinson, Srinivas-Shankar et al., 2010). While a therapy such as resistance exercise is effective at increasing muscle mass during hypogonadal conditions (Sullivan, Roberson et al., 2005), there is also evidence that resistance exercise cannot increase mass to the same absolute value achieved by those with circulating androgen levels in the physiological range (Kvorning, Andersen et al., 2006). This highlights the important physiological role of androgens, in conjunction with other factors such as physical activity, in the overall maintenance of muscle mass.
Androgen-mediated changes in muscle mass are due in part to alterations in muscle protein balance with hypogonadism shifting this balance in favor of net protein breakdown (Ferrando et al., 2003, Ferrando, Tipton et al., 1998, Sheffield-Moore, Urban et al., 1999). Several studies have examined the molecular factors implicated in androgen-mediated changes in muscle size and protein metabolism (i.e. (Hughes D.C., 2012); however, numerous experimental inconsistencies preclude a definitive conclusion from being made about the predominant factors/pathways contributing to this change in protein balance. This is important because mimicking the effects of androgens pharmacologically to increase muscle mass is required for those individuals in which androgen replacement is not a treatment option due to potentially negative side effects (Atkinson et al., 2010, Bassil, Alkaade et al., 2009). For example, androgens may augment the growth of a cancer tumor, making androgen replacement a non-viable option for those with established cancer tumors and suffering from cancer cachexia (Huggins and Hodges, 2002, Amos-Landgraf, Heijmans et al., 2014)Therefore, the goal of this review is to critically discuss the molecular factors thought to contribute to the effects of androgens on skeletal muscle protein balance and to identify critical areas of future research required for the continual progression towards the development of androgen-independent therapies.
4.1 ANDROGENS
Androgens represent a class of hormones predominantly responsible for the development of male secondary sex characteristics including increased muscle mass (White et al., 2013, Guyton Ac, 2006). While females also synthesize androgens, circulating concentrations are much lower (Guyton AC, 2006), likely contributing to their smaller muscle mass. In males, androgens are synthesized in the Leydig cells of the testes using cholesterol as a precursor (Guyton AC, 2006). The adrenal cortex also produces androgen hormones, though the contribution of this alternative source to overall levels in males is thought to be negligible (Guyton AC, 2006). In contrast, this non-gonadal source in females accounts for a much larger portion of total androgen production (Guyton AC, 2006). Testosterone and its reduced metabolite, 5α-dihydrotestosterone (DHT), are the two most prominent anabolic androgens and can be produced locally in skeletal muscle from precursor androgens (i.e. Dehydroepiandrosterone; DHEA) via the enzymes 3β-hydroxy-steriod dehydrogenase, 17β-hydroxy-steriod dehydrogenase, or 5α-reductase (Sato, Iemitsu et al., 2008, Aizawa, Iemitsu et al., 2007). However, the in vitro concentrations of DHEA and/or testosterone precursors needed to induce this conversion were in the micromolar range and well above normal physiological concentrations, indicating that further in vivo studies are required to confirm whether this occurs when circulating concentrations are much lower than those used in vitro (Hopper and Yen, 1975, Velders and Diel, 2013). In general, circulating total testosterone values between 17 and 35 nmol/l are considered to be in the normal physiological range for males (Velders and Diel, 2013, Sader, Griffiths et al., 2003). Despite circulating concentrations of androgens being most frequently reported, concentrations within the tissues may also be important. For instance, evidence suggests that concentrations of androgens in skeletal muscle, rather than in circulation, is more predictive of strength and muscle cross sectional area at least in older men (Sato, Iemitsu et al., 2014). Further, while not conducted in muscle cells, intracellular androgen concentrations in cultured prostate cancer cells differ from those values observed in the surrounding culture media. When extrapolated to skeletal muscle, this suggests that measurement of hypogonadal or physiological concentrations of androgens in circulation may not be representative of those levels within skeletal muscle (Sedelaar and Isaacs, 2009, Wu, Godoy et al., 2013).
The most recognized androgen mechanism of action is through binding to the cytosolic androgen receptor (AR) (Guyton AC, 2006). Upon androgen binding, the AR translocates to the nucleus where it interacts with the androgen response element (ARE) of target genes to alter gene transcription (both positively and negatively) (Guyton AC, 2006). However, the role of this mechanism in vivo has been questioned since the dissociation constant (Kd) of testosterone or DHT for the androgen receptor was estimated to be ~2–5 nM (Wilson and French, 1976), which can be lower than androgen concentrations found in hypogonadal males (i.e. <17 nmol/l) (Velders and Diel, 2013). Thus, the receptor could be saturated even in a hypogonadal state, suggesting that alternative androgen-mediated mechanisms exist. Indeed, testosterone administration to L6 myoblasts in culture altered signaling events within 20 minutes of exposure (Wu, Bauman et al., 2010); a time frame which is likely to be shorter than the traditional AR-mediated changes in gene transcription; illustrating the presence of alternative mechanisms of action. However, these alternative mechanisms remain poorly defined and require further attention representing an avenue for pharmacological intervention.
5.1 REGULATION of PROTEIN BALANCE
Skeletal muscle mass is regulated in part by the coordinated balance between rates of muscle protein synthesis and muscle protein breakdown. In healthy individuals, where muscle mass is maintained, these two processes wax and wane throughout the diurnal cycle in response to anabolic (i.e. nutrient consumption) and catabolic (i.e. fasting) stimuli (Phillips, Glover et al., 2009). Conversely, a long-term shift in this balance favoring net protein synthesis results in muscle hypertrophy while a long-term shift favoring net protein breakdown results in muscle atrophy (Phillips et al., 2009). These concepts and the molecular regulation of each have been reviewed elsewhere and therefore are only briefly summarized (Gordon, Kelleher et al., 2013, Hornberger, 2011, Kimball and Jefferson, 2010, Laplante and Sabatini, 2009, Ma and Blenis, 2009, Milan, Romanello et al., 2015, Sandri, 2010, Sandri, 2013, Goodman and Hornberger, 2014).
In general, the increase in protein synthesis following anabolic stimuli requires signaling through the mechanistic target of rapamycin in complex 1 (mTORC1) (Dickinson, Fry et al., 2011, Drummond, Fry et al., 2009). Signaling through mTORC1 regulates mRNA translation initiation as well as peptide chain elongation through phosphorylation of at least two known substrates termed the 70 kD ribosomal protein S6 kinase 1 (p70S6K1) and the eukaryotic initiation factor 4E (eIF4) binding protein 1 (4E-BP1) (Kimball and Jefferson, 2010). Various upstream effectors regulate the magnitude of mTORC1 activity. Positive effectors include phosphorylation and activation of Akt (a.k.a. protein kinase b) by hormones such as insulin and IGF-1, which promotes mTORC1 activity through phosphorylation and subsequent inhibition of proteins including Tuberous Sclerosis 2 (TSC2; a.k.a Tuberin) and proline-rich Akt substrate of 40 kD (PRAS40) (Dennis, Baum et al., 2011, Dennis, Coleman et al., 2014). Amino acids also activate mTORC1 signaling through a mechanism that is distinct from Akt (Dennis et al., 2011, Sancak, Bar-Peled et al., 2010, Sancak, Peterson et al., 2008). Conversely, mTORC1 signaling is inhibited by activation of the 5′ AMP-activated protein kinase (AMPK) and expression of Regulated in Development and DNA Damage 1 (REDD1) (Sullivan et al., 2005, Dennis et al., 2014, Gordon, Steiner et al., 2014, Gordon, Williamson et al., 2015, Bolster, Crozier et al., 2002). While mTORC1 is required for the increase in protein synthesis following anabolic stimuli, its role in regulating protein synthesis during the basal, non-stimulated condition is less clear. For example, treating humans with rapamycin to inhibit mTORC1 did not alter global rates of muscle protein synthesis in the fasted metabolic condition (Dickinson, Drummond et al., 2013) though it was sufficient to completely block the nutrient-induced stimulation of this process (Dickinson et al., 2011). Evidence also indicates mTORC1-independent mechanisms are involved in the regulation of muscle protein synthesis following anabolic stimuli (West, Baehr et al., 2016), although this concept is less well-defined.
Protein breakdown encompasses two general processes; the selective protease-mediated breakdown of poly ubiquitylated proteins via the ubiquitin proteasome system (UPS), and the lysosomal-mediated breakdown of bulk or specific cellular components/proteins termed autophagy (Sandri, 2013). The activity of the UPS has been linked to changes in E3 ubiquitin ligase expression (Sandri, 2013). The two most prominent skeletal muscle specific E3 ligases, collectively termed the atrogenes, are Muscle RING-finger protein-1 (MuRF1) and Muscle Atrophy F-box (MAFbx/atrogin-1) (Bodine, Latres et al., 2001). Their expression is markedly increased during various atrophic conditions (White et al., 2013, Bodine et al., 2001, Kelleher, Gordon et al., 2014, Kelleher, Kimball et al., 2013), and deletion of these atrogenes can attenuate muscle atrophy (Bodine et al., 2001). However, expression of these atrogenes does not necessarily correlate with UPS activity as global deletion of MuRF1 increased UPS activity in the skeletal muscle of aged mice, suggesting that other factors contribute to its activation (Hwee, Baehr et al., 2014). Indeed, additional E3 ligases have recently been identified in muscle. For example, expression of Muscle ubiquitin ligase of SCF complex in atrophy 1 (MUSA1), FbxO21 (a.k.a. SMART), and FbxO31 accompanied the loss of skeletal muscle mass induced by fasting (Milan, Romanello et al., 2015). The expression of these E3 ligases, as well as atrogene expression, is regulated in part by the Forkhead box (FoxO) and Smad transcription factors (Sandri, Sandri et al., 2004, Sartori, Milan et al., 2009). FoxO signaling is inhibited by post translational modification (e.g. phosphorylation) from upstream effectors such as Akt (Sandri et al., 2004). Conversely, phosphorylation and subsequent activation of Smad transcription factors is increased following stimulation of the activin receptors by the transforming growth factor beta (TGFβ) family of cytokines (Goodman and Hornberger, 2014, Sartori et al., 2009). In particular, Myostatin, a TGFβ family member, is well known to negatively regulate muscle mass in mice, cattle, dogs, and humans (Lee, 2004) through upregulation of atrogene expression although other mechanisms have been suggested as the changes in atrogenes are inconsistent (Mcfarlane, Plummer et al., 2006, Trendelenburg, Meyer et al., 2009). In this regard, myostatin was shown to negatively affect muscle satellite cell number and activation, while muscle hypertrophy induced by myostatin inhibition occurred independent of myonuclear accretion (Welle, Mehta et al., 2011). Further details pertaining to the role of myostatin in skeletal muscle can be found elsewhere (Rodriguez, Vernus et al., 2014).
Initiation of autophagy is regulated by various signals including mTORC1 signaling, AMPK activation, and expression of regulatory proteins such as REDD1 and BCL2/Adenovirus E1B 19kDa Interacting Protein 3 (BNIP3) (Gordon et al., 2014, Kim, Kundu et al., 2011, Qiao, Dennis et al., 2015, Zhang, Xue et al., 2016). For instance, mTORC1 phosphorylates several proteins such as uncoordinated like kinase 1 (ULK1) to inhibit the initiation steps of autophagy (Kim et al., 2011). Conversely, AMPK phosphorylates ULK1 on different residues to promote the initiation of autophagy (Kim et al., 2011). Expression of REDD1 or BNIP3 also alters this metabolic process. For example, mouse embryonic fibroblasts (MEFs) lacking the REDD1 gene were resistant to autophagy induction in what appeared to be an mTORC1-independent manner (Qiao et al., 2015). Likewise, expression of BNIP3 is sufficient to induce the autophagic removal of mitochondria; a process termed mitophagy (Zhang et al., 2016).
6.1 ANDROGEN REGULATION OF MUSCLE PROTEIN SYNTHESIS
In humans, androgens alter protein synthesis when measured in the fasted, but not the fed, metabolic state. For example, short term (i.e. <5 days) and long term (i.e. 6 months) androgen treatment each increased rates of protein synthesis following an overnight fast in both young and aged male subjects (Ferrando et al., 1998, Sheffield-Moore et al., 1999, Griggs, Kingston et al., 1989, Urban, Bodenburg et al., 1995, Brodsky, Balagopal et al., 1996). However, no differences were observed following amino acid stimulation (Ferrando et al., 2003, Sheffield-Moore, Wolfe et al., 2000). Similarly, supraphysiological testosterone administration to post-menopausal women increased muscle protein synthesis following an overnight fast (Smith, Yoshino et al., 2014). This is not a universal phenomenon as treating aged males with androgens in which androgens were raised to the normal physiological range did not alter protein synthesis in the fasted state (Ferrando et al., 2003, Ferrando, Sheffield-Moore et al., 2002). A major factor promoting these discrepant findings related to protein synthetic rate may be due to the androgen concentrations prior to the measurement (hypogonadal vs. supraphysiological). For instance, synthetic rate was increased at 5–7 days following a testosterone enanthate (TE) injection even though testosterone levels were in the normal physiological range by the time of the synthetic measurement (Ferrando et al., 1998, Griggs et al., 1989, Urban et al., 1995). It is possible that the increased synthetic rate observed was a residual effect from when testosterone levels were in the supraphysiological range at earlier time points (i.e. 1–3 days post injection) (Snyder, 1984). Conversely, increasing androgen levels from the lower physiological/hypogondal range to the physiological range did not have this same effect (Ferrando et al., 2003, Ferrando et al., 2002), suggesting that only supraphysiological concentrations of androgens alter proteins synthesis or lead to a greater magnitude of change (Fig. 1). The androgen administered may have also contributed to the discordance in synthetic rate. For example, one study showed that testosterone cypionate (TC) increased muscle protein synthesis 14 days post injection while others using TE did not (Ferrando et al., 2003, Brodsky et al., 1996, Ferrando et al., 2002). Importantly, the circulating testosterone concentrations within these studies were all similar at the time of the synthetic measurement. This lone study reporting an increase in synthetic rate following TC administration may be an anomaly, or it may indicate that the cypionate ester in the testosterone molecule enhanced its anabolic effect since the pharmacokinetics of TE or TC is equivalent (Schultebeerbuhl and Nieschlag, 1980). In future studies, utilizing a consistent method of altering androgens such as implantable, timed release pellets, rather than a bolus method such as injections, will help resolve these issues.
FIGURE 1.
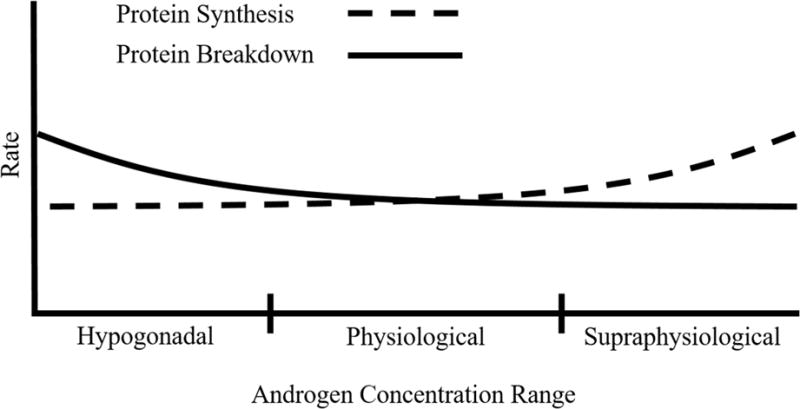
Theoretical model by which androgens alter muscle protein balance at differing androgen concentrations.
Studies in animal or ex vivo models have yielded conflicting results regarding androgen-mediated regulation of skeletal muscle protein synthesis. In one study, castration decreased rates of protein synthesis in the gastrocnemius of mice, while restoration of androgens through weekly injections of nandrolone decanoate normalized this measure (White et al., 2013). Likewise, ex vivo treatment of isolated extensor digitorum longus (EDL) and soleus muscle fiber bundles from elderly female mice (~700 days old) with 2 nM DHT increased rates of protein synthesis (Wendowski, Redshaw et al., 2016). Alternatively, castration did not change rates of protein synthesis in the tibialis anterior (TA) of mice or the gastrocnemius of rats following an overnight fast (Steiner et al., 2016, Jiao, Pruznak et al., 2009). Synthetic rates in the TA were also not different between castrated and sham mice 4 hr following refeeding, nor did castration affect the increase in protein synthesis following a bout of high frequency muscle contractions (Steiner et al., 2016). Conversely, castration prevented the leucine-induced stimulation of protein synthesis in the gastrocnemius of rats, suggesting mediation of the contributing pathways (Jiao et al., 2009). Several methodological issues may explain the discordant findings between animal studies including the feeding paradigm, model system employed, and muscle or animal species used. For example, the metabolic status of the mice at sacrifice was not specified in the study by (White et al., 2013) whereas the others utilized overnight fasting and timed refeeding (Steiner et al., 2016, Jiao et al., 2009). Thus, future studies should consider using timed refeeding or overnight fasting to bypass this likely contributing factor. Measuring protein synthesis ex vivo could have also contributed to this discrepancy as the Ringer’s solution in which the muscles were incubated may not have mimicked the in vivo cellular environment (Wendowski et al., 2016). Differences in the muscle analyzed (gastrocnemius vs. TA) or the species of the animals (rats vs. mice) may have further contributed. Regardless, the observation that castration did not alter muscle protein synthesis in the fasted state is consistent with the human studies in which global rates of muscle protein synthesis only appeared to be affected by supraphysiological androgen concentrations (Fig. 1). Thus, future studies should consider using timed refeeding or overnight fasting to ensure consistent findings across studies.
6.2 MOLECULAR REGULATION OF MUSCLE PROTEIN SYNTHESIS BY ANDROGENS
The prominent molecular mechanism(s) behind the androgen-mediated increase in protein synthesis remain undefined although increased signaling through mTORC1 via upstream effectors such as IGF-1/Akt and/or extracellular signal-regulated kinase 1/2 (ERK1/2) have been hypothesized to contribute. In humans, IGF-1 mRNA and protein content were increased in the muscle of humans following androgen administration while the mRNA content of IGF-1 binding protein 4 (IGF-1BP-4) was decreased (Urban et al., 1995, Ferrando et al., 2002). However, blocking the IGF-1 receptor did not impair the supraphysiological testosterone-induced increase in C2C12 myotube diameter (Hughes, Stewart et al., 2016). Conversely, AR blockade in these studies severely blunted the testosterone-induced increase in myotube formation and diameter in addition to reducing the mRNA content of the IGF-1 receptor, phosphorylation of Akt (Ser473) and phosphorylation of ERK1/2, suggesting that signaling through the AR precedes these downstream effects and is a more potent regulatory factor (Hughes et al., 2016). Additional indications of the importance of mTORC1 are garnered from animal and cell culture experiments including the finding that administration of supraphysiological concentrations of testosterone (100 nM) to primary rat myotubes increased myotube size in an mTORC1-dependent manner (Basualto-Alarcon, Jorquera et al., 2013). This report also showed that the androgen-mediated increase in mTORC1 signaling was preceded by activation of Akt (Basualto-Alarcon et al., 2013). Of interest, blocking the AR prior to testosterone treatment negated the increase in myotube size, but it was not determined whether Akt/mTORC1 signaling was affected by this blockade (Basualto-Alarcon et al., 2013). In further support of an mTORC1 dependent mechanism, administration of testosterone (100 nM) to L6 myoblasts increased cell diameter and protein content, and co-incubation with rapamycin negated this effect (Wu et al., 2010). ERK1/2 was implicated as an upstream activator as phosphorylation of ERK1/2 preceded the testosterone-induced mTORC1 activation (Wu et al., 2010). However, only inhibition of AR or phosphatidylinositol-3 kinase (PI3K), but not ERK1/2, prevented the testosterone induced increase in protein content. Additional support for a role of PI3K was also observed in control C2C12 myotubes and C2C12 myotubes that were subjected to population doubling to mimic “aged” skeletal muscle. Here, inhibition of PI3K reduced the testosterone (100 nM) induced increase in myotube differentiation and diameter in both cell types (Deane, Hughes et al., 2013). Despite these findings, the relationship between activation of either AR or PI3K and mTORC1 signaling remains elusive. Alternatively, the changes in mTORC1 signaling following androgen treatment may be due to enhanced availability of amino acids as ex vivo incubation of isolated soleus and EDL muscle fiber bundles from aged female mice with DHT increased the expression of the sodium-coupled neutral amino acid transporter (SNAT) 2 and L-type amino acid transporter (LAT) 2 channels (Wendowski et al., 2016). Collectively, these data support a role of mTORC1 in the regulation of muscle size and protein accretion by supraphysiological concentrations of androgens. However, protein synthesis and long-term changes in muscle mass were not measured in any of these mechanistic studies, which is important for defining the role of these molecular signals on androgen-mediated changes in muscle size. Furthermore, whether in vitro/ex vivo findings translate to humans is unclear considering that androgens appear to only increase protein synthetic rates under fasted conditions, which may not be accurately reproduced in vitro/ex vivo (Ferrando et al., 2003, Ferrando et al., 1998, Sheffield-Moore et al., 1999, Griggs et al., 1989, Urban et al., 1995, Brodsky et al., 1996, Sheffield-Moore et al., 2000, Smith et al., 2014, Ferrando et al., 2002).
Though shifting androgen levels from the physiological range to the hypogonadal range has yielded conflicting results regarding rates of protein synthesis, multiple studies show that mTORC1 signaling is reduced by this shift. For example, castration of mice reduced phosphorylation of mTOR (Ser2448), p70S6K1 (Thr389) and 4E-BP1 (Thr37/46) while androgen administration sufficiently restored these to sham levels (White et al., 2013). In this study, the changes in mTORC1 signaling were accompanied by corresponding changes in Akt and PRAS40 phosphorylation as well as REDD1 mRNA content, suggesting a regulatory role for these upstream factors (White et al., 2013). Of note, no change in the phosphorylation of AMPK (Thr172) was observed in this study following castration (White et al., 2013). In another study, castration of mice increased the phosphorylation of regulated associated protein of mTOR (Raptor) on Ser792 and decreased phosphorylation of TSC2 (Thr1462) in the levator ani and triceps brachii muscles (Serra, Sandor et al., 2013). Though these events would likely suppress mTORC1 signaling, no downstream measures of the activity of this signaling complex were assessed (e.g. phosphorylation of p70S6K1) (Serra et al., 2013). Using an overnight fasting model, phosphorylation of ribosomal protein s6 (rps6) on Ser235/236, a putative p70S6K1 substrate, was reduced in the gastrocnemius of castrated rats relative to sham values (Jiao et al., 2009). Notably, in this study, castration negated the leucine-induced stimulation of mTORC1 signaling, though a potential mechanism for this observation was not described (Jiao et al., 2009). Using a similar feeding paradigm, castration reduced the phosphorylation of mTORC1 substrates p70S6K1 (Thr389) and 4E-BP1 (Ser65) in the TA (Steiner et al., 2016). The protein content of REDD1 and phosphorylation of AMPK (Thr172) were also increased, likely contributing to the repression of mTORC1 signaling (Steiner et al., 2016). Interestingly, and in contrast to previous reports, the phosphorylation of Akt (Thr308) trended to increase (P = 0.06) in the muscle of castrated mice in the fasted state despite the repressed mTORC1 signaling (Steiner et al., 2016). This report went on to show that refeeding previously fasted mice negated the castration-induced repression in mTORC1 signaling (i.e. p70S6K1 (Thr389) and 4E-BP1 (Ser65)) as well as the increase in REDD1 protein content (Steiner et al., 2016). Collectively, these data suggest that mTORC1 signaling is sensitive to changes in androgen concentration, although alterations in the rate of muscle protein synthesis do not necessarily correspond (Table 1). Thus, the role of mTORC1 in the regulation of muscle protein synthesis, or one of its many other metabolic actions following changes in androgen levels, requires further investigation. Using genetic mouse models (i.e. gene knockout mice) or chemical inhibitors (i.e. rapamycin) in models of androgen administration will help define the role of mTORC1.
TABLE 1.
Summary of metabolic and molecular alterations following androgen manipulation.
| Ref | Species/Sex | Muscle Analyzed | Androgen Given | T Conc. at Protein Metabolic Measure (nmol/l) | Metabolic State at Protein Metabolic Measure | Protein Synthesis | Protein Breakdown | Molecular Events Associated with Change in Androgen |
|---|---|---|---|---|---|---|---|---|
| 67 | Human/Male | VL | TE | 17–28 | Fasted | ↔ | ↓ | ↑ IGF-1 protein |
| 65 | Human/Male | VL | Ox | Not Indicated | Fasted/AA Infusion | ↑ Fasted, ↔ AA Infusion | ↔ | N/A |
| 63 | Human/Male | VL | TE | 17.4–34.7 | Fasted | ↑ | Not Indicated | ↑ IGF-1 mRNA content; ↓ IGF-1BP-4 mRNA content |
| 2 | Human/Male | VL | TE | ~23 | Fasted/AA Infusion | ↔ | ↓ | N/A |
| 9 | Human/Male | VL | TE | ~33 | Fasted | ↑ | ↔ | N/A |
| 10 | Human/Male | VL | Ox | ~8 | Fasted | ↑ | ↔ | N/A |
| 64 | Human/Male | VL | TC | 10.4–13.9 | Fasted | ↑ | Not Indicated | N/A |
| 62 | Human/Male | VL | TE | >40 | Fasted | ↑ | Not Indicated | N/A |
| 66 | Human/Female | Quad | T | ~8.4 | Fasted | ↑ | Not Indicated | ↔ FOX03, MSTN, FST mRNA concentrations |
| 71 | Rat/Male | Gast | N/A | 0.1 | Fasted/Leucine Gavage | ↔ | ↑ | Fasted State: ↓ MuRF1 and MAFbx mRNA content, ↓ rps6 (Ser235/236). Following Leucine Gavage: ↓ rps6 (Ser235/236), 4E-BP1 phosphorylation, eIF4E bound to eIF4G; ↑ eIF4E bound to eIF4G |
| 81 | Rat/Male | LABC | TE/Tren | Not Indicated | Not Indicated | Not Indicated | Not Indicated | Castration: ↔ Myostatin Protein, Smad 2/3 phosphorylation (site not indicated); ↓ MyostaBn, AcBvin Receptor IIb mRNA Content. Castration + Testosterone Enanthate and Trenbolone: ↑ Myostatin Protein; ↑ MyostaBn, Activiin Receptor IIb mRNA Content ; ↔ Smad 2/3 phosphorylation |
| 76 | Rat/Male | LABC | TE | Not Indicated | Not Indicated | Not Indicated | Not Indicated | Castration: ↓ c-Myc, Nop56, Bop1, Ncl mRNA; ↓ 45S pre-rRNA. Castration + TE: Did not alter measures. |
| 78 | Rat/Male | LABC | TE/Tren | Not Indicated | Not Indicated | Not Indicated | Not Indicated | Castration: ↑ MuRF-1, MAFbx mRNA. Castration + TE or Tren: Normalized these measures. |
| 83 | Rat/Male | LA | TP | Not Indicated | Not Indicated | Not Indicated | Not Indicated | Castration: ↑ myostatin protein. Castration + Testosterone: ↓ myostatin protein |
| 4 | Mouse/Male | Gast | ND | Not Indicated | Not Indicated | ↓ by Castration ↑ by ND | Not Indicated | Castration: ↓ Akt (Ser473), mTOR (Ser2448), p70S6K (Thr389), 4E-BP1 (Thr37/46), PRAS40 (Thr246), Fox03A (Ser253); ↑ MuRF1, MAFbx, and REDD1 mRNA content. ND: Normalized all measures |
| 80 | Mouse/Male | Gast/Triceps Brachii | N/A | Not Indicated | Not Indicated | Not Indicated | Not Indicated | Castration: ↑Myostatin, Activin A, Activin B, Activin AG, GDF-11 in a transient manner throughout 2–10 week timecourse. ↑ Smad2 phosphorylation only at 4 week time point. |
| 75 | Mouse/Male | LABC | TP | Not Indicated | Not Indicated | Not Indicated | ↑ with Castration | Castration (Levator Ani): ↑MuRF1 and MAFbx mRNA; ↑LC3B, Bnip3, Beclin1, and Tfeb mRNA; ↓ Fox03A (Ser318/321), TSC2 (Thr1462); ↑ AMPKα (Thr172), Raptor (Ser792). Castration (Triceps Brachii): ↓ MuRF1 and ↔ MAFbx mRNA; ↑ LC3 II/I; ↑ Tfeb mRNA; ↓ Fox03A (Ser318/321), TSC2 (Thr1462); ↑ AMPKα (Thr172), Raptor (Ser792) |
| 3 | Mouse/Male | TA | N/A | <0.1 | Fasted/Refed | ↔ | ↑ | Fasted State: ↓ p70S6K1 (Thr389), 4E-BP1 (Ser65), ULK1 (Ser757); ↑ REDD1 protein content, AMPK (Thr172); ↔ MAFbx or MuRF-1 mRNA content. Refed State: ↔ p70S6K1 (Thr389), 4E-BP1 (Ser65), ULK1 (Ser757), REDD1 Protein Content |
| 79 | Mouse/Male | Soleus | ND | Not Indicated | Not Indicated | Not Indicated | Not Indicated | ↔ MuRF-1 mRNA Content |
| 86 | Mouse/Male | LA | T/DHT | Not Indicated | Not Indicated | Not Indicated | Not Indicated | AR KO and Castration: ↓ myostatin mRNA, ↑ activin IIB mRNA. |
| 82 | Mouse/Male | Gast-Plant and Soleus | T | Not Indicated | Not Indicated | Not Indicated | Not Indicated | Testosterone: ↓ myostatin expression in a dose dependent manner |
| 84 | Mouse/Male | Gast | T | 20–28 | Not Indicated | Not Indicated | Not Indicated | Testosterone: ↓ mature myostatin protein, ↑ Notch and PCNA protein |
| 85 | Mouse/Male | Gast | T | <0.1 | Not Indicated | Not Indicated | Not Indicated | Testosterone: ↑ Notch and PCNA protein |
| 77 | Mouse/Male | LA, Soleus, EDL | T | Not Indicated | Not Indicated | Not Indicated | Not Indicated | Castration: ↑ MuRF-1, MAFbx, myostatin mRNA. Castration + Testosterone: ↓ MuRF-1, MAFbx, myostatin mRNA |
| 72 | Mouse | C2C12 | T | N/A | DMEM + 2% Horse Serum | Not Indicated | Not Indicated | Testosterone: ↑ myotube diameter and myotube number in a n AR dependent manner. ↑ AR content |
| 74 | Mouse | C2C12 | T | N/A | DMEM + 2% Horse Serum | Not Indicated | Not Indicated | Testosterone: ↑ myotube diameter in a PI3K dependent manner. ↓ myostatin mRNA content |
| 25 | Rat | L6 Myoblasts | T | N/A | DMEM + 2% Horse Serum | Not Indicated | Not Measured | ↑ ERK1/2 (Thr202/Try204), p70S6K1 (Thr389), mTOR (Ser2481), rps6 (Ser235/236) |
| 73 | Rat/N/A | N/A | TE | N/A | DMEM-Ham’s F-12 without serum | Not Indicated | Not Measured | ↑ Phosphorylation of p70S6K1 (Thr412/389), ERK1/2 (Thr202/Try204), Akt (Ser473); ↔ MAFbx or MuRF-1 mRNA Content. |
| 70 | Mouse/Female | Soleus and EDL | DHT | N/A | Ringer’s Solution | ↑ | Not Measured | With Age: ↓ Protein Synthesis (both EDL and Soleus); ↓ SNAT2 and LAT2 protein in EDL only. With Age + DHT: ↑ Protein Synthesis (both EDL and Soleus), SNAT2 and LAT2 protein (both EDL and Soleus) |
T, Testosterone; TE, Testosterone Enanthate; TC, Testosterone Cypionate; ND, Nandrolone Decanoate; DHT, 5α-dihydrotestosterone; Gast, Gastrocnemius; Plant, Plantaris; EDL, Extensor Digitorum Longus; LABC, Levator Ani/Bulbocavernosus; N/A, Not Applicable; Quad, Quadriceps; Tren, Trenbolone; AA, Amino Acid; VL, Vastus Lateralis; Ox, Oxandrolone; TP, Testosterone Propionate; AR, Androgen Receptor; KO, Knockout; PCNA, Proliferating Cell Nuclear Antigen; PI3K, Phosphatidylinositol-3 Kinase
Lastly, there is evidence that androgens alter muscle translational capacity (i.e. ribosome content), which would impact protein balance. Here, castration of rats decreased the RNA content within the levator ani/bulbocavernosus muscle supporting a reduction in ribosome number. This paralleled reductions in markers of ribosome biogenesis including the content of the 47S pre-rRNA and the mRNA content of genes that regulate the transcription and processing of the pre-rRNA such as v-myc avian myelocytomatosis viral oncogene homolog (c-Myc), nuclear protein 56 (Nop56), block of proliferation1 (Bop1), and nucleolin (Ncl) (Mobley, Mumford et al., 2016). Interestingly, TE treatment restored the RNA content of the muscle to sham values, but the markers of ribosome biogenesis remained suppressed (Mobley et al., 2016), perhaps in an attempt to limit uncontrolled muscle growth.
7.1 ANDROGENS AND MUSCLE PROTEIN BREAKDOWN
In humans, the role of endogenous and exogenous androgens on muscle protein breakdown in the fasted state has yielded conflicting results. For example, TE administration in aged males reduced protein breakdown in two separate studies (Ferrando et al., 2003, Ferrando et al., 2002). Conversely, a single TE injection did not alter breakdown 5 days following administration in young males, though sufficient statistical power may have been lacking to measure the small change in breakdown that was observed (Ferrando et al., 1998). Further, 5 days of oxandrolone treatment to young males also failed to modulate rates of breakdown (Sheffield-Moore et al., 1999). Age, and thereby endogenous testosterone levels, may have contributed to the discrepant findings. For example, reduced protein breakdown occurred only in the aged (~67 years) subjects whose endogenous pretreatment testosterone levels were in the hypogonadal/lower physiological range (Ferrando et al., 2003, Ferrando et al., 2002). Meanwhile, endogenous pretreatment testosterone values in the young subjects were in the normal physiological range (Ferrando et al., 1998, Sheffield-Moore et al., 1999), suggesting that a shift in androgen levels from the hypogonadal range to the physiological range likely has a greater impact on protein breakdown compared to a shift from the physiological to the supraphysiological range (Fig. 1).
Androgen-mediated changes in markers of protein breakdown are also observed in animal models. For instance, 8 weeks of castration increased 20S proteasome activity within the gastrocnemius of rats following an overnight fast as well as in the levator ani muscle of mice 7 days post castration (Serra et al., 2013). Conversely, 20S proteasome activity was not increased in the triceps brachii at 50 days post castration surgery indicating a time and/or muscle dependent effect (Serra et al., 2013). Castration also reduced the content of ubiquitylated proteins in the TA of mice following an overnight fast relative to sham levels, while refeeding negated this effect (Steiner et al., 2016). Though speculative, it was concluded that the change in ubiquitylated proteins in this study following the overnight fast might have been reflective of an increase in UPS activity; however, UPS activity was not measured to confirm this possibility.
In addition to UPS activity, markers of autophagy activation are also sensitive to changes in androgen concentration. For example, activity of Cathepsin L and the microtubule-associated protein 1A/1B-light chain 3 (LC3) II/I ratio were elevated in the levator ani muscle within 7 days of castration, and administration of TE restored these markers (Serra et al., 2013). Similar findings were observed in the triceps brachii muscle in that Cathepsin L and the LC3 II/I ratio were increased in mice 50 days after castration surgery (Serra et al., 2013). However, at this time point, TE administration was unable to normalize the LC3 II/I ratio while Cathepsin L activity returned to sham values (Serra et al., 2013). In agreement with that study, the LC3 II/I ratio was increased and p62 protein content was decreased in the TA of castrated mice following an overnight fast relative to the values in sham mice (Steiner et al., 2016). Further, refeeding previously fasted castrated mice failed to restore these markers to levels observed in the refed sham mice (Steiner et al., 2016). The reason for the sustained elevation in autophagy markers in refed castrated mice is unknown, but it may indicate autophagic removal of specific muscle proteins or organelles.
7.2 MOLECULAR REGULATION OF MUSCLE PROTEIN DEGRADATION BY ANDROGENS
The prominent molecular mechanisms by which hypogonadism regulates protein breakdown are ill defined and based largely upon associative studies. In regards to the UPS, castration increased the mRNA content of both atrogenes (MuRF1 and MAFbx) within the levator ani, suggesting a role for the E3 ligases (Serra et al., 2013, De Naeyer, Lamon et al., 2014). A corresponding decrease in the phosphorylation of FoxO3a (Ser318/321) implied that the activation of this transcription factor promoted atrogene expression (Serra et al., 2013). These events were testosterone and AR sensitive as testosterone administration to castrated mice normalized these measures while AR blockade via flutamide negated the androgen-mediated restoration (Serra et al., 2013). In contrast, atrogene mRNA content was not increased 50 days post castration in the triceps brachii muscle, suggesting that the regulation of atrogene expression and UPS activity is either muscle type or time point specific (Serra et al., 2013). MuRF1 and MAFbx mRNA content were also increased in the levator ani/bulbocavernosus and gastrocnemius muscle of castrated rats and mice, respectively (White et al., 2013, Ye, Mccoy et al., 2014). Consistent with this change, the phosphorylation of FoxO3a (Ser253) was decreased in the gastrocnemius of castrated mice providing further support for this transcription factor in the atrogene expression during hypogonadism (White et al., 2013).
In contrast, reductions in atrogene mRNA content have also been reported following castration including a decrease in MuRF-1 and MAFbx in the gastrocnemius of castrated rats following an overnight fast compared to sham values (Jiao et al., 2009). Similarly, an overnight fast led to a non-significant decrease in atrogene mRNA content within the TA of castrated mice relative to sham values, and this was accompanied by a non-significant increase in FoxO3a (Ser253) phosphorylation (Steiner et al., 2016). Lastly, supraphysiological concentrations of testosterone (100 nM) failed to alter the mRNA content of MuRF-1 and MAFbx in primary rat myotubes while nandrolone decanoate treatment failed to alter atrogene expression in the soleus of mice (Basualto-Alarcon et al., 2013, Camerino, Desaphy et al., 2015). The reason(s) for the discrepant findings between animal studies is unknown, but like many of the other processes highlighted herein, it may be due to the feeding parameters and metabolic state of the animal when muscles were isolated. For instance, the later studies (i.e. (Steiner et al., 2016, Jiao et al., 2009) utilized overnight fasting to control the metabolic state at sacrifice while the others did not indicate the nutritional status of the animals (White et al., 2013, Serra et al., 2013). Additionally, the duration of androgen manipulation (7 days vs. 50 days) and/or the muscle analyzed (TA vs. Gastrocnemius vs. Triceps Brachii vs. Levator Ani) may also contribute. Future studies would benefit from using controlled feeding while also analyzing these molecular events in various muscle groups from the same animal.
Activin/Smad signaling also exhibited time-dependent effects following changes in androgen levels. Specifically, the content of myostatin, Activin A, Activin B, and Activin AB were all transiently increased throughout a 10-week castration time course in both the gastrocnemius and triceps brachii muscles (Pan, Singh et al., 2016). Further, the expression of growth differentiation factor 11 (GDF11), an activin receptor ligand, exhibited a transient expression pattern in these muscles following castration (Pan et al., 2016). Despite this transient pattern in TGFβ cytokine expression, phosphorylation of the downstream activin receptor substrate, Smad3 (Ser423/425), was only increased at the 4-week post castration time point (Pan et al., 2016). In contrast, another study showed phosphorylation of Smad2/3 and expression of mature myostatin protein were unaltered in the levator ani bulbocavernosus muscle 43 days following castration (Dalbo, Roberts et al., 2016). Administration of TE or trenbolone to previously castrated rats increased the expression of the mature myostatin protein and activin IIB mRNA content without altering Smad2/3 phosphorylation, suggesting that androgens may prevent Smad2/3 activation despite the increased myostatin expression (Dalbo et al., 2016). Despite these contradictory data, blocking activation of the activin receptors in castrated mice increased muscle mass to a value that was greater than those observed in sham mice (Pan et al., 2016). A caveat to this observation was that there was no mention of the effect of receptor blockade on the muscle mass of sham mice, precluding definitive conclusion(s) from being made.
While those studies show changes in activin/Smad signaling, other work has focused on androgen-mediated changes in the upstream mediator, myostatin. For example, myostatin expression in the gastrocnemius/plantaris complex and the soleus were repressed by testosterone in a dose dependent manner (Shigeo Kawada, 2006). Similarly, castration increased the expression of myostatin in the levator ani and EDL muscles, and this effect was reversed by testosterone administration (De Naeyer et al., 2014, Mendler, Baka et al., 2007). Testosterone administration to aged mice also decreased the expression of the mature myostatin peptide (Kovacheva, Hikim et al., 2010). This effect of testosterone on myostatin may be mediated via AR signaling inhibiting androgen binding to the AR increased myostatin mRNA content in both control and population doubled C2C12 myotubes (Hughes et al., 2016). The suppressive effect of androgens on myostatin may also be related to satellite cell function as expression of Notch and Proliferating Cell Nuclear Antigen (PCNA) were increased in the gastrocnemius of aged mice following testosterone administration (Kovacheva et al., 2010, Sinha, Sinha-Hikim et al., 2014).
In direct contrast, myostatin expression was decreased in the levator ani muscle 30 days post castration, and testosterone administration increased this measure back to sham levels (De Naeyer et al., 2014). In line with this discordant finding, the myostatin gene was identified as a direct target of the AR in skeletal muscle (Dubois, Laurent et al., 2014). However, contrary to the putative atrophic role of myostatin, this report showed that increased AR signaling enhanced myostatin expression (Dubois et al., 2014). While these studies were in murine and cell culture models, treating post-menopausal women with supraphysiolocial concentrations of testosterone failed to alter myostatin or follistatin mRNA expression in the fasted state (Smith et al., 2014). Collectively, the data indicate that myostatin expression is altered by androgens, although only one report has investigated the direct role of myostatin on androgen-mediated growth. In this regard, administration of supraphysiological concentrations of testosterone or DHT to previously castrated myostatin null mice appeared to enhance the growth promoting effects of the androgen compared to castrated, wild type mice (i.e. an interaction between genotype and androgens). While this finding is consistent with the previously described role of the AR in the promotion of myostatin expression, the actual changes in muscle weight observed in this study were not reported, but rather, muscle weight corrected for body weight was described, which may be why this outcome was observed. The reason(s) for discrepancies between studies in regards to myostatin expression and androgens are not clear, but it may be due in part to the failure to control for feeding as myostatin has been shown to be sensitive to nutrient consumption (Carneiro, Gonzalez et al., 2013), the difference in the muscle analyzed (levator ani vs. gastrocnemius), or the species analyzed (rodents vs. humans). Future androgen related studies using a controlled feeding experimental paradigm, while at the same time analyzing various muscle groups, will help to further define the role of the TGFβ/myostatin/activin/Smad signaling pathway.
Similar to the UPS, the molecular regulation of autophagy by androgens is also largely based upon associative studies. For example, castration increased phosphorylation of the autophagy activator, AMPK (Thr172), as well as the mRNA content of autophagy related genes including BNIP3, Beclin1, and Transcription factor EB (Tfeb) in the levator ani and triceps brachii of mice (Serra et al., 2013). Similarly, castration increased REDD1 protein content and phosphorylation of AMPK (Thr172) as well as decreased phosphorylation of ULK1 (Ser757) in the TA of castrated mice following an overnight fast (Steiner et al., 2016). However, in contrast to the previous study (i.e. (Serra et al., 2013)), castration did not alter BNIP3 protein content in the TA when measured in the fasted metabolic state (Steiner et al., 2016). While REDD1 protein content and phosphorylation of AMPK and ULK1 were altered in a manner consistent with elevated autophagy in the TA following an overnight fast, only REDD1 protein content was found to be significantly correlated with the LC3 II/I ratio autophagy marker, suggesting a predominant role of this protein in the fasting-induced regulation of autophagy (Steiner et al., 2016). Of note, refeeding castrated mice prevented the castration-induced changes in REDD1 protein content and ULK1 (Ser757) phosphorylation observed in the fasted state even though autophagy markers remained elevated, suggesting an unknown mediator(s) of autophagy contributed to this elevation (Steiner et al., 2016). Overall, the preponderance of data indicated that a lack of androgens altered various autophagy regulatory factors in favor of increased autophagy. As with other factors discussed herein, discrepant findings may be largely due to feeding paradigms utilized. However, a major shortcoming of this body of knowledge is the lack of mechanistic experiments utilizing the manipulation of the expression/activation of the proposed regulatory factors. Also, the use of autophagy inhibitors such as colchicine will help define the contribution of autophagy to the overall shift in protein balance following androgen deprivation.
8.1 CONCLUSION
It is well-accepted that androgens influence muscle mass, and this change is thought to occur in part through alterations in protein balance (Phillips et al., 2009). Accordingly, the data presented herein suggest that in the fasted metabolic state, supraphysiological concentrations of androgens increase muscle protein synthesis while hypogonadism increases protein breakdown (Fig. 1). Despite this concept being fairly well described in humans, the molecular factors thought to contribute to this effect (Fig. 2) are inconsistent between studies (both animal and cell culture). These inconsistencies are likely due in large part to the different nutrient/feeding paradigms utilized, especially as these methodological details were often not reported. As the human data overwhelmingly show that androgens alter muscle protein balance selectively in the fasted metabolic state (Sheffield-Moore et al., 2000), future mechanistic studies need to be cognizant of this concept. For example, the use of an overnight fast and/or refeeding paradigm will certainly help alleviate these discrepancies as this enables feeding status measurement at both ends of the metabolic spectrum (fasted vs. refed). However, this type of paradigm comes at the cost of making accurate muscle phenotype measurements (e.g. cross sectional area) due to the significant loss of body weight caused by the overnight fast. The translatability of cell culture systems to human models of either hypogonadism or androgen supplementation also needs to be addressed. For example, recapitulating the diurnal fluctuations in nutrient exposure that occur throughout the day in humans would be extremely time consuming and difficult in a cell-based system. Thus, it might be useful to perform serum or nutrient deprivation treatments prior to harvesting cells in this system in an attempt to more closely mimic the fasted metabolic state. Additionally, future work needs to be cognizant of how different androgen concentrations (i.e. hypogonadal vs. physiological vs. supraphysiological) appear to modulate protein balance (Fig. 1). While it is also important to initially identify molecular signals/events associated with androgen-mediated changes in protein synthesis and breakdown, future mechanistic work needs to delineate the role of these events by modifying their activation/expression. Using genetic knockout models (e.g. MuRF-1 and MAFbx null mice) or chemical inhibitors (e.g. rapamycin) would further our understanding of the androgen-mediated contribution of these factors/pathways in the regulation of muscle morphology and protein metabolism. Additionally, specificity of muscle groups/fiber types in response to androgen levels should also be determined including direct comparison of more androgen sensitive muscles, such as the levator ani, to less androgen sensitive muscles, such as the triceps brachii, within the same animal. Addressing these issues will help identify the most prominent mechanisms through which androgens regulate skeletal muscle protein balance, and therefore, expedite the development of new, androgen-independent therapies to offset muscle atrophy for those unable to undergo standard replacement therapy.
FIGURE 2.
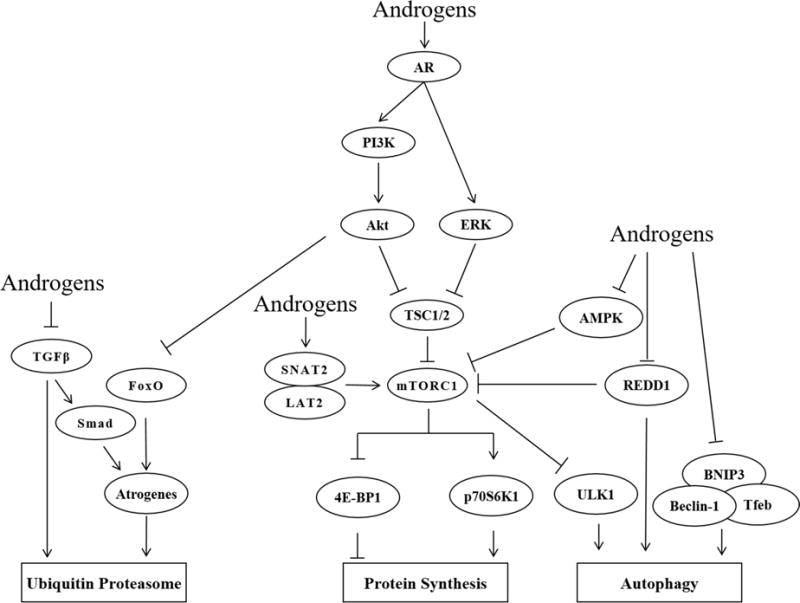
Summary of the proposed mechanisms by which androgens alter muscle protein balance.
Highlights.
In humans, androgens alter protein balance in the fasted metabolic state
Predominant molecular factors altering protein balance are inconclusive
Inconclusiveness likely due to many methodological differences between studies
Acknowledgments
FUNDING This review was supported in part by National Institutes of Health (Grant F32 AA-023422) to J.L. Steiner and a pilot feasibility grant from the University of Central Florida to B.S. Gordon. The funding agencies had no part in drafting the manuscript.
Abbreviations
- mTORC1
mechanistic target of rapamycin in complex 1
- AR
androgen receptor
- DHT
5α-dihydrotestosterone
- TE
testosterone enanthate
- TC
testosterone cypionate
- ARE
androgen response element
- p70S6K1
70 kD ribosomal protein S6 kinase 1
- 4E-BP1
eukaryotic initiation factor 4E binding protein 1
- TSC2
tuberous sclerosis 2/tuberin
- PRAS40
proline-rich Akt substrate of 40 kD
- AMPK
5′ AMP-activated protein kinase
- REDD1
regulated in development and DNA damage 1
- UPS
ubiquitin proteasome system
- BNIP3
BCL2/adenovirus E1B 19kDa interacting protein 3
- MuRF-1
muscle RING-finger protein-1
- MAFbx/atrogin-1
muscle Aatrophy F-box
- FoxO
forkhead box
- IGF-1BP-4
IGF-1 binding protein 4
- PI3K
phosphatidylinositol-3 kinase
- ERK1/2
extracellular signal-regulated kinase 1/2
- SNAT2
sodium-coupled neutral amino acid transporter 2
- LAT2
L-type amino acid transporter 2
- rps6
ribosomal protein s6
- EDL
extensor digitorum longus
- TA
tibialis anterior
- Raptor
regulated associated protein of mTOR
- c-myc
v-myc avian myelocytomatosis viral oncogene homolog
- Nop56
nuclear protein 56
- Bop1
block of proliferation1
- Ncl
nucleolin
- TGFβ
transforming growth factor beta
- GDF11
growth differentiation factor 11
- Tfeb
transcription factor EB
- LC3
microtubule-associated protein 1A/1B-light chain 3
- ULK1
uncoordinated like kinase 1
- PCNA
proliferating cell nuclear antigen
- DHEA
Dehydroepiandrosterone
- MUSA1
Muscle ubiquitin ligase of SCF complex in atrophy 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
There are no disclosures.
References
- Martin L, Birdsell L, Macdonald N, Reiman T, Clandinin MT, McCargar LJ, Murphy R, Ghosh S, Sawyer MB, Baracos VE. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J Clin Oncol. 2013;31:1539–47. doi: 10.1200/JCO.2012.45.2722. [DOI] [PubMed] [Google Scholar]
- Ferrando AA, Sheffield-Moore M, Paddon-Jones D, Wolfe RR, Urban RJ. Differential anabolic effects of testosterone and amino acid feeding in older men. J Clin Endocrinol Metab. 2003;88:358–62. doi: 10.1210/jc.2002-021041. [DOI] [PubMed] [Google Scholar]
- Steiner JL, Fukuda DH, Rossetti ML, Hoffman JR, Gordon BS. Castration Alters Protein Balance Following High Frequency Muscle Contraction. J Appl Physiol. 2016 doi: 10.1152/japplphysiol.00740.2016. 1985. jap 00740 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Gao S, Puppa MJ, Sato S, Welle SL, Carson JA. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol Cell Endocrinol. 2013;365:174–86. doi: 10.1016/j.mce.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Puppa MJ, Narsale A, Carson JA. Characterization of the male Apc>Min mouse as a hypogonadism model related to cancer cachexia. Biol Open. 2013;2:1346–1353. doi: 10.1242/bio.20136544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson RA, Srinivas-Shankar U, Roberts SA, Connolly MJ, Adams JE, Oldham JA, Wu FCW, Seynnes OR, Stewart CEH, Maganaris CN, Narici MV. Effects of Testosterone on Skeletal Muscle Architecture in Intermediate-Frail and Frail Elderly Men. J Gerontol A Biol Sci Med Sci. 2010;65:1215–1219. doi: 10.1093/gerona/glq118. [DOI] [PubMed] [Google Scholar]
- Sullivan DH, Roberson PK, Johnson LE, Bishara O, Evans WJ, Smith ES, Price JA. Effects of muscle strength training and testosterone in frail elderly males. Med Sci Sports Exerc. 2005;37:1664–72. doi: 10.1249/01.mss.0000181840.54860.8b. [DOI] [PubMed] [Google Scholar]
- Kvorning T, Andersen M, Brixen K, Madsen K. Suppression of endogenous testosterone production attenuates the response to strength training: a randomized, placebo-controlled, and blinded intervention study. Am J Physiol Endocrinol Metab. 2006;291:E1325–E1332. doi: 10.1152/ajpendo.00143.2006. [DOI] [PubMed] [Google Scholar]
- Ferrando AA, Tipton KD, Doyle D, Phillips SM, Cortiella J, Wolfe RR. Testosterone injection stimulates net protein synthesis but not tissue amino acid transport. Am J Physiol. 1998;275:E864–71. doi: 10.1152/ajpendo.1998.275.5.E864. [DOI] [PubMed] [Google Scholar]
- Sheffield-Moore M, Urban RJ, Wolf SE, Jiang J, Catlin DH, Herndon DN, Wolfe RR, Ferrando AA. Short-term oxandrolone administration stimulates net muscle protein synthesis in young men. J Clin Endocrinol Metab. 1999;84:2705–11. doi: 10.1210/jcem.84.8.5923. [DOI] [PubMed] [Google Scholar]
- Hughes DCSN, Sharples AP, Lewis MP. Perspectives on anabolic androgenic steroids (AAS) and doping in sport and health 2012 [Google Scholar]
- Bassil N, Alkaade S, Morley JE. The benefits and risks of testosterone replacement therapy: a review. Ther Clin Risk Manag. 2009;5:427–48. doi: 10.2147/tcrm.s3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins C, Hodges CV. Studies on prostatic cancer - I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. J Urol. 2002;168:9–12. doi: 10.1016/s0022-5347(05)64820-3. [DOI] [PubMed] [Google Scholar]
- Amos-Landgraf JM, Heijmans J, Wielenga MCB, Dunkin E, Krentz KJ, Clipson L, Ederveen AG, Groothuis PG, Mosselman S, Muncan V, Hommes DW, Shedlovsky A, Dove WF, van den Brink GR. Sex disparity in colonic adenomagenesis involves promotion by male hormones, not protection by female hormones. Proc Natl Acad Sci U S A. 2014;111:16514–16519. doi: 10.1073/pnas.1323064111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton ACHJ. Textbook of Medical Physiology. 11th. Elsvevier Inc; Philadelphia: 2006. [Google Scholar]
- Sato K, Iemitsu M, Aizawa K, Ajisaka R. Testosterone and DHEA activate the glucose metabolism-related signaling pathway in skeletal muscle. Am J Physiol Endocrinol Metab. 2008;294:E961–E968. doi: 10.1152/ajpendo.00678.2007. [DOI] [PubMed] [Google Scholar]
- Aizawa K, Iemitsu M, Maeda S, Jesmin S, Otsuki T, Mowa CN, Miyauchi T, Mesaki N. Expression of steroidogenic enzymes and synthesis of sex steroid hormones from DHEA in skeletal muscle of rats. Am J Physiol Endocrinol Metab. 2007;292:E577–E584. doi: 10.1152/ajpendo.00367.2006. [DOI] [PubMed] [Google Scholar]
- Hopper BR, Yen SSC. Circulating Concentrations of Dehydroepiandrosterone and Dehydroepiandrosterone Sulfate during Puberty. J Clin Endocrinol Metab. 1975;40:458–461. doi: 10.1210/jcem-40-3-458. [DOI] [PubMed] [Google Scholar]
- Velders M, Diel P. How Sex Hormones Promote Skeletal Muscle Regeneration. Sports Medicine. 2013;43:1089–1100. doi: 10.1007/s40279-013-0081-6. [DOI] [PubMed] [Google Scholar]
- Sader MA, Griffiths KA, Skilton MR, Wishart SM, Handelsman DJ, Celermajer DS. Physiological testosterone replacement and arterial endothelial function in men. Clin Endocrinol. 2003;59:62–67. doi: 10.1046/j.1365-2265.2003.01796.x. [DOI] [PubMed] [Google Scholar]
- Sato K, Iemitsu M, Matsutani K, Kurihara T, Hamaoka T, Fujita S. Resistance training restores muscle sex steroid hormone steroidogenesis in older men. FASEB J. 2014;28:1891–1897. doi: 10.1096/fj.13-245480. [DOI] [PubMed] [Google Scholar]
- Sedelaar JPM, Isaacs JT. Tissue Culture Media Supplemented With 10% Fetal Calf Serum Contains a Castrate Level of Testosterone. Prostate. 2009;69:1724–1729. doi: 10.1002/pros.21028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Godoy A, Azzouni F, Wilton JH, Ip C, Mohler JL. Prostate cancer cells differ in testosterone accumulation, dihydrotestosterone conversion, and androgen receptor signaling response to steroid 5 alpha-reductase inhibitors. Prostate. 2013;73:1470–1482. doi: 10.1002/pros.22694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EM, French FS. Binding properties of androgen receptors. Evidence for identical receptors in rat testis, epididymis, and prostate. J Biol Chem. 1976;251:5620–9. [PubMed] [Google Scholar]
- Wu Y, Bauman WA, Blitzer RD, Cardozo C. Testosterone-induced hypertrophy of L6 myoblasts is dependent upon Erk and mTOR. Biochem Biophys Res Commun. 2010;400:679–83. doi: 10.1016/j.bbrc.2010.08.127. [DOI] [PubMed] [Google Scholar]
- Phillips SM, Glover EI, Rennie MJ. Alterations of protein turnover underlying disuse atrophy in human skeletal muscle. J Appl Physiol. 2009;1985:107, 645–54. doi: 10.1152/japplphysiol.00452.2009. [DOI] [PubMed] [Google Scholar]
- Gordon BS, Kelleher AR, Kimball SR. Regulation of muscle protein synthesis and the effects of catabolic states. Int J Biochem Cell Biol. 2013;45:2147–57. doi: 10.1016/j.biocel.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornberger TA. Mechanotransduction and the regulation of mTORC1 signaling in skeletal muscle. Int J Biochem Cell Biol. 2011;43:1267–76. doi: 10.1016/j.biocel.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SR, Jefferson LS. Control of translation initiation through integration of signals generated by hormones, nutrients, and exercise. J Biol Chem. 2010;285:29027–32. doi: 10.1074/jbc.R110.137208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling at a glance. Journal of Cell Science. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XJM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Reviews Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA, Sandri M. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015;6:6670. doi: 10.1038/ncomms7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–6. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- Sandri M. Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol. 2013;45:2121–9. doi: 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CA, Hornberger TA. New roles for Smad signaling and phosphatidic acid in the regulation of skeletal muscle mass. F1000Prime Rep. 2014;6:20. doi: 10.12703/P6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson JM, Fry CS, Drummond MJ, Gundermann DM, Walker DK, Glynn EL, Timmerman KL, Dhanani S, Volpi E, Rasmussen BB. Mammalian target of rapamycin complex 1 activation is required for the stimulation of human skeletal muscle protein synthesis by essential amino acids. J Nutr. 2011;141:856–62. doi: 10.3945/jn.111.139485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond MJ, Fry CS, Glynn EL, Dreyer HC, Dhanani S, Timmerman KL, Volpi E, Rasmussen BB. Rapamycin administration in humans blocks the contraction-induced increase in skeletal muscle protein synthesis. J Physiol. 2009;587:1535–46. doi: 10.1113/jphysiol.2008.163816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MD, Baum JI, Kimball SR, Jefferson LS. Mechanisms involved in the coordinate regulation of mTORC1 by insulin and amino acids. J Biol Chem. 2011;286:8287–96. doi: 10.1074/jbc.M110.209171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MD, Coleman CS, Berg A, Jefferson LS, Kimball SR. REDD1 enhances protein phosphatase 2A-mediated dephosphorylation of Akt to repress mTORC1 signaling. Sci Signal. 2014;7:ra68. doi: 10.1126/scisignal.2005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BS, Steiner JL, Lang CH, Jefferson LS, Kimball SR. Reduced REDD1 expression contributes to activation of mTORC1 following electrically induced muscle contraction. Am J Physiol Endocrinol Metab. 2014;307:E703–11. doi: 10.1152/ajpendo.00250.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon BS, Williamson DL, Lang CH, Jefferson LS, Kimball SR. Nutrient-induced stimulation of protein synthesis in mouse skeletal muscle is limited by the mTORC1 repressor REDD1. J Nutr. 2015;145:708–13. doi: 10.3945/jn.114.207621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–80. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- Dickinson JM, Drummond MJ, Fry CS, Gundermann DM, Walker DK, Timmerman KL, Volpi E, Rasmussen BB. Rapamycin does not affect post-absorptive protein metabolism in human skeletal muscle. Metabolism. 2013;62:144–51. doi: 10.1016/j.metabol.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West DW, Baehr LM, Marcotte GR, Chason CM, Tolento L, Gomes AV, Bodine SC, Baar K. Acute resistance exercise activates rapamycin-sensitive and -insensitive mechanisms that control translational activity and capacity in skeletal muscle. J Physiol. 2016;594:453–68. doi: 10.1113/JP271365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–8. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Kelleher AR, Gordon BS, Kimball SR, Jefferson LS. Changes in REDD1, REDD2, and atrogene mRNA expression are prevented in skeletal muscle fixed in a stretched position during hindlimb immobilization. Physiol Rep. 2014;2:e00246. doi: 10.1002/phy2.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher AR, Kimball SR, Dennis MD, Schilder RJ, Jefferson LS. The mTORC1 signaling repressors REDD1/2 are rapidly induced and activation of p70S6K1 by leucine is defective in skeletal muscle of an immobilized rat hindlimb. Am J Physiol Endocrinol Metab. 2013;304:E229–36. doi: 10.1152/ajpendo.00409.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwee DT, Baehr LM, Philp A, Baar K, Bodine SC. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell. 2014;13:92–101. doi: 10.1111/acel.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao JH, Abraham R, Goldberg AL, Blaauw B, DePinho RA, Sandri M. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun. 2015;6 doi: 10.1038/ncomms7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. 2009;296:C1248–57. doi: 10.1152/ajpcell.00104.2009. [DOI] [PubMed] [Google Scholar]
- Lee SJ. Regulation of muscle mass by myostatin. Ann Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, Smith H, Sharma M, Kambadur R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappa B-independent, FoxO1-dependent mechanism. J Cell Physiol. 2006;209:501–514. doi: 10.1002/jcp.20757. [DOI] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- Welle S, Mehta S, Burgess K. Effect of postdevelopmental myostatin depletion on myofibrillar protein metabolism. Am J Physiol Endocrinol Metab. 2011;300:E993–E1001. doi: 10.1152/ajpendo.00509.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Vernus B, Chelh I, Cassar-Malek I, Gabillard JC, Sassi AH, Seiliez I, Picard B, Bonnieu A. Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell Mol Life Sci. 2014;71:4361–4371. doi: 10.1007/s00018-014-1689-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao S, Dennis M, Song X, Vadysirisack DD, Salunke D, Nash Z, Yang Z, Liesa M, Yoshioka J, Matsuzawa S, Shirihai OS, Lee RT, Reed JC, Ellisen LW. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6:7014. doi: 10.1038/ncomms8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R, Billiar TR, Tao WA, Zhang Z. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J Biol Chem. 2016;291:21616–21629. doi: 10.1074/jbc.M116.733410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griggs RC, Kingston W, Jozefowicz RF, Herr BE, Forbes G, Halliday D. Effect of testosterone on muscle mass and muscle protein synthesis. J Appl Physiol. 1989;1985:66, 498–503. doi: 10.1152/jappl.1989.66.1.498. [DOI] [PubMed] [Google Scholar]
- Urban RJ, Bodenburg YH, Gilkison C, Foxworth J, Coggan AR, Wolfe RR, Ferrando A. Testosterone Administration to Elderly Men Increases Skeletal-Muscle Strength and Protein-Synthesis. Am J Physiol Endocrinol Metab. 1995;269:E820–E826. doi: 10.1152/ajpendo.1995.269.5.E820. [DOI] [PubMed] [Google Scholar]
- Brodsky IG, Balagopal P, Nair KS. Effects of testosterone replacement on muscle mass and muscle protein synthesis in hypogonadal men–a clinical research center study. J Clin Endocrinol Metab. 1996;81:3469–75. doi: 10.1210/jcem.81.10.8855787. [DOI] [PubMed] [Google Scholar]
- Sheffield-Moore M, Wolfe RR, Gore DC, Wolf SE, Ferrer DM, Ferrando AA. Combined effects of hyperaminoacidemia and oxandrolone on skeletal muscle protein synthesis. Am J Physiol Endocrinol Metab. 2000;278:E273–E279. doi: 10.1152/ajpendo.2000.278.2.E273. [DOI] [PubMed] [Google Scholar]
- Smith GI, Yoshino J, Reeds DN, Bradley D, Burrows RE, Heisey HD, Moseley AC, Mittendorfer B. Testosterone and progesterone, but not estradiol, stimulate muscle protein synthesis in postmenopausal women. J Clin Endocrinol Metab. 2014;99:256–65. doi: 10.1210/jc.2013-2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, Lieberman SA, Tipton K, Wolfe RR, Urban RJ. Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am J Physiol Endocrinol Metab. 2002;282:E601–7. doi: 10.1152/ajpendo.00362.2001. [DOI] [PubMed] [Google Scholar]
- Snyder PJ. Clinical Use of Androgens. Annual Review of Medicine. 1984;35:207–217. doi: 10.1146/annurev.me.35.020184.001231. [DOI] [PubMed] [Google Scholar]
- Schultebeerbuhl M, Nieschlag E. Comparison of Testosterone, Dihydrotestosterone, Luteinizing-Hormone, and Follicle-Stimulating-Hormone in Serum after Injection of Testosterone Enanthate or Testosterone Cypionate. Fertility and Sterility. 1980;33:201–203. doi: 10.1016/s0015-0282(16)44543-7. [DOI] [PubMed] [Google Scholar]
- Wendowski O, Redshaw Z, Mutungi G. Dihydrotestosterone treatment rescues the decline in protein synthesis as a result of sarcopenia in isolated mouse skeletal muscle fibres. J Cachexia Sarcopenia Muscle. 2016 doi: 10.1002/jcsm.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao QN, Pruznak AM, Huber D, Vary TC, Lang CH. Castration differentially alters basal and leucine-stimulated tissue protein synthesis in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2009;297:E1222–E1232. doi: 10.1152/ajpendo.00473.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DC, Stewart CE, Sculthorpe N, Dugdale HF, Yousefian F, Lewis MP, Sharples AP. Testosterone enables growth and hypertrophy in fusion impaired myoblasts that display myotube atrophy: deciphering the role of androgen and IGF-I receptors. Biogerontology. 2016;17:619–639. doi: 10.1007/s10522-015-9621-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basualto-Alarcon C, Jorquera G, Altamirano F, Jaimovich E, Estrada M. Testosterone Signals through mTOR and Androgen Receptor to Induce Muscle Hypertrophy. Med Sci Sports Exerc. 2013;45:1712–1720. doi: 10.1249/MSS.0b013e31828cf5f3. [DOI] [PubMed] [Google Scholar]
- Deane CS, Hughes DC, Sculthorpe N, Lewis MP, Stewart CE, Sharples AP. Impaired hypertrophy in myoblasts is improved with testosterone administration. J Steroid Biochem Mol Biol. 2013;138:152–161. doi: 10.1016/j.jsbmb.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Serra C, Sandor NL, Jang H, Lee D, Toraldo G, Guarneri T, Wong S, Zhang A, Guo W, Jasuja R, Bhasin S. The effects of testosterone deprivation and supplementation on proteasomal and autophagy activity in the skeletal muscle of the male mouse: differential effects on high-androgen responder and low-androgen responder muscle groups. Endocrinology. 2013;154:4594–606. doi: 10.1210/en.2013-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley CB, Mumford PW, Kephart WC, Conover CF, Beggs LA, Balaez A, Yarrow JF, Borst SE, Beck DT, Roberts MD. Effects of testosterone treatment on markers of skeletal muscle ribosome biogenesis. Andrologia. 2016;48:967–977. doi: 10.1111/and.12539. [DOI] [PubMed] [Google Scholar]
- De Naeyer H, Lamon S, Russell AP, Everaert I, De Spaey A, Vanheel B, Taes Y, Derave W. Androgenic and estrogenic regulation of Atrogin-1, MuRF1 and myostatin expression in different muscle types of male mice. Eur J Appl Physiol. 2014;114:751–761. doi: 10.1007/s00421-013-2800-y. [DOI] [PubMed] [Google Scholar]
- Ye F, McCoy SC, Ross HH, Bernardo JA, Beharry AW, Senf SM, Judge AR, Beck DT, Conover CF, Cannady DF, Smith BK, Yarrow JF, Borst SE. Transcriptional regulation of myotrophic actions by testosterone and trenbolone on androgen-responsive muscle. Steroids. 2014;87:59–66. doi: 10.1016/j.steroids.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camerino GM, Desaphy JF, De Bellis M, Capogrosso RF, Cozzoli A, Dinardo MM, Caloiero R, Musaraj K, Fonzino A, Conte E, Jagerschmidt C, Namour F, Liantonio A, De Luca A, Conte Camerino D, Pierno S. Effects of Nandrolone in the Counteraction of Skeletal Muscle Atrophy in a Mouse Model of Muscle Disuse: Molecular Biology and Functional Evaluation. PLoS One. 2015;10:e0129686. doi: 10.1371/journal.pone.0129686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan CL, Singh S, Sahasrabudhe DM, Chakkalakal JV, Krolewski JJ, Nastiuk KL. TGF beta Superfamily Members Mediate Androgen Deprivation Therapy-Induced Obese Frailty in Male Mice. Endocrinology. 2016;157:4461–4472. doi: 10.1210/en.2016-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalbo VJ, Roberts MD, Mobley CB, Ballmann C, Kephart WC, Fox CD, Santucci VA, Conover CF, Beggs LA, Balaez A, Hoerr FJ, Yarrow JF, Borst SE, Beck DT. Testosterone and trenbolone enanthate increase mature myostatin protein expression despite increasing skeletal muscle hypertrophy and satellite cell number in rodent muscle. Andrologia. 2016 doi: 10.1111/and.12622. [DOI] [PubMed] [Google Scholar]
- Shigeo Kawada MO, Naokata Ishii. Testosterone Causes Decrease in the Content of Skeletal Muscle Myostatin. International Journal of Sport and Health Sciences. 2006;4:44–48. [Google Scholar]
- Mendler L, Baka Z, Kovacs-Simon A, Dux L. Androgens negatively regulate myostatin expression in an androgen-dependent skeletal muscle. Biochem Biophys Res Commun. 2007;361:237–242. doi: 10.1016/j.bbrc.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Kovacheva EL, Hikim APS, Shen RQ, Sinha I, Sinha-Hikim I. Testosterone Supplementation Reverses Sarcopenia in Aging through Regulation of Myostatin, c-Jun NH2-Terminal Kinase, Notch, and Akt Signaling Pathways. Endocrinology. 2010;151:628–638. doi: 10.1210/en.2009-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha I, Sinha-Hikim AP, Wagers AJ, Sinha-Hikim I. Testosterone is essential for skeletal muscle growth in aged mice in a heterochronic parabiosis model. Cell Tissue Res. 2014;357:815–821. doi: 10.1007/s00441-014-1900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois V, Laurent MR, Sinnesael M, Cielen N, Helsen C, Clinckemalie L, Spans L, Gayan-Ramirez G, Deldicque L, Hespel P, Carmeliet G, Vanderschueren D, Claessens F. A satellite cell-specific knockout of the androgen receptor reveals myostatin as a direct androgen target in skeletal muscle. FASEB J. 2014;28:2979–2994. doi: 10.1096/fj.14-249748. [DOI] [PubMed] [Google Scholar]
- Carneiro I, Gonzalez T, Lopez M, Senaris R, Devesa J, Arce VM. Myostatin expression is regulated by underfeeding and neonatal programming in rats. J Physiol Biochem. 2013;69:15–23. doi: 10.1007/s13105-012-0183-x. [DOI] [PubMed] [Google Scholar]