

The governing bodies for international apnea competition, the Association Internationale pour le Développment de l’Apnée and La Confédération Mondaile des Activités Subaquatiques, have banned the use of β-blockers based on anecdotal reports that they improve apnea duration. Using a randomized placebo-controlled trial, we are the first to empirically confirm that β-blockade improves apnea duration. This improvement in apnea duration coincided with a reduced myocardial oxygen consumption.
Keywords: esmolol, rate pressure product, breath-hold diving, cerebral blood flow
Abstract
We hypothesized that the cardioselective β1-adrenoreceptor antagonist esmolol would improve maximal apnea duration in elite breath-hold divers. In elite national-level divers (n = 9), maximal apneas were performed in a randomized and counterbalanced order while receiving either iv esmolol (150 μg·kg−1·min−1) or volume-matched saline (placebo). During apnea, heart rate (ECG), beat-by-beat blood pressure, stroke volume (SV), cardiac output (CO), and total peripheral resistance (TPR) were measured (finger photoplethysmography). Myocardial oxygen consumption (MV̇o2) was estimated from rate pressure product. Cerebral blood flow through the internal carotid (ICA) and vertebral arteries (VA) was assessed using Duplex ultrasound. Apnea duration improved in the esmolol trial when compared with placebo (356 ± 57 vs. 323 ± 61 s, P < 0.01) despite similar end-apnea peripheral oxyhemoglobin saturation (71.8 ± 10.3 vs. 74.9 ± 9.5%, P = 0.10). The HR response to apnea was reduced by esmolol at 10–30% of apnea duration, whereas MAP was unaffected. Esmolol reduced SV (main effect, P < 0.05) and CO (main effect; P < 0.05) and increased TPR (main effect, P < 0.05) throughout apnea. Esmolol also reduced MV̇o2 throughout apnea (main effect, P < 0.05). Cerebral blood flow through the ICA and VA was unchanged by esmolol at baseline and the last 30 s of apnea; however, global cerebral blood flow was reduced in the esmolol trial at end-apnea (P < 0.05). Our findings demonstrate that, in elite breath-hold divers, apnea breakpoint is improved by β1-blockade, likely owing to an improved total body oxygen sparring through increased centralization of blood volume (↑TPR) and reduced MV̇o2.
NEW & NOTEWORTHY The governing bodies for international apnea competition, the Association Internationale pour le Développment de l’Apnée and La Confédération Mondaile des Activités Subaquatiques, have banned the use of β-blockers based on anecdotal reports that they improve apnea duration. Using a randomized placebo-controlled trial, we are the first to empirically confirm that β-blockade improves apnea duration. This improvement in apnea duration coincided with a reduced myocardial oxygen consumption.
voluntary apnea elicits an integrative circulatory and neural response to maintain adequate perfusion of vital organs (e.g., the brain) while simultaneously reducing both flow to nonvital organs (e.g., skin and skeletal muscle) and total body oxygen consumption (9, 10, 20). Bradycardia, peripheral vasoconstriction, and centralization of blood volume are the primary cardiovascular responses to voluntary apnea, which together represent the mammalian diving reflex (20). During the initial phase (~25%) of a maximal apnea the extreme lung volumes mechanically compress the heart, whereas high intrathoracic pressures impede venous return, resulting in an abrupt drop in stroke volume (SV), cardiac output (CO), and consequently mean arterial pressure (MAP) and cerebral blood flow (CBF) (5, 8, 26). Concomitant unloading of the baroreceptors causes a transient increase in muscle sympathetic nerve activity (7), heart rate (22), and total peripheral resistance (TPR). Following this initial phase, bradycardia (via reduced baroreflex output in addition to increases in vagal tone; see Ref. 18) occurs alongside progressive increases in CO, MAP, TPR (5, 25), and CBF (23, 31). These elevations in CO, MAP, and TPR are thought to be mediated via marked elevations in sympathetic nerve activity (10- to 20-fold) (14, 29). Along with hypoxemic- and hypercapnic-mediated dilation of cerebral blood vessels, this increase in MAP aids in the augmenting the CBF response to maintain cerebral oxygen delivery throughout the apnea (31).
The precise mechanism(s) for apnea break point remains poorly understood (24) but in the motivated and elite breath-hold diver is likely related in part to chemoreflex stress (3, 4) and a critical oxygen level required to maintain consciousness (4, 31). Additional mechanisms have been theorized to contribute to break point in the untrained breath-holder, such as diaphragmatic afference (24); however, how this translates to the motivated and elite breath-holder is unknown.
β-Adrenoreceptor antagonists (β-blockers) have been used commonly in elite apnea competitions due to anecdotal reports of improved maximal apnea duration (personal correspondence, Drvis I, Croatian National Apnea Coach). As a result, the Association Internationale pour le Développment de l’Apnée (AIDA; https://www.aidainternational.org) and La Confédération Mondaile des Activités Subaquatiques (CMAS; http://www.cmas.org) prohibit the use of β-blockers in and out of competition. Given that recent work in elite apnea divers supports the presence of a critical oxygen tension as a major contributor to apnea break point (4), the present theory is that β-blockade improves maximal apnea duration via myocardial oxygen sparing, yet there are no published data demonstrating this effect. In the first randomized and placebo-controlled study of its kind, we determined the effects of β1-blockade on maximal apnea duration and the circulatory parameters that are characteristic of extreme apnea. It was hypothesized that β1-blockade would prolong apnea duration, owing to a reduced heart rate throughout the apnea and a consequently reduced rate of myocardial oxygen consumption (MV̇o2). We further reasoned that the effects of β1-blockade would be reflected in the cerebral vasculature through a lower CBF during baseline and end apnea compared with the placebo control due to reduced CO and MAP.
METHODS
Subjects
Elite breath-hold divers (n = 9; 1 female) were recruited from the National Croatian Apnea team to participate in this study. The divers were 29 ± 9 yr old, weighed 80 ± 13 kg, and were 184 ± 8 cm tall (body mass index = 23.5 ± 2.6). Personal record static apnea time was on average 394 s (range: 296–496), whereas divers had a mean of 4 yr (range: 2–11) of experience in competitive apnea training and a forced vital capacity (FVC) of 6.9 liters (range: 5.0–7.8). Following written informed consent, divers visited the laboratory on one occasion for the experimental session. Subjects arrived at either 8 AM or 12 PM, having abstained from alcohol, caffeine, and exercise for 24 h before arrival. All subjects were free from any cardiovascular, respiratory, and cerebrovascular disease at the time of study, as assessed by a screening questionnaire and spirometry. This study was approved by the University of British Columbia Clinical Research Ethics Board and by the Ethics Committee of the University of Split School of Medicine and conformed to the Declaration of Helsinki.
Experimental Design
Upon arrival to the laboratory, subjects first performed a spirometry test (Quark PFT; Cosmed, Rome, Italy) in the upright position. Subjects voided their bladders and were then instructed to rest supine, at which time a 20-gauge intravenous catheter was placed into an antecubital vein under local anesthesia (lidocaine 1.0%). Following cannulation, subjects rested for ≥20 min during the setup of experimental monitoring equipment.
This study implemented two experimental conditions: 1) cardiac-specific β1-adrenergic receptor blockade (Esmolol, 10 mg/ml; Esmocard) and 2) a volume-matched placebo (saline; 0.9% NaCl) condition. Esmolol was first infused at a rate of 500 μg·kg−1·min−1 for 1 min, followed by 50 μg·kg−1·min−1 for 5 min. This was repeated two additional times before a steady maintenance infusion of 150μg·kg−1·min−1 for the remainder of the drug condition. These doses have previously been established to effectively reduce HR, systolic blood pressure, rate pressure product (RPP), and both left and right ventricular ejection fraction during rest and exercise (16). The order of infusions was randomized in a counterbalanced manner and separated by >5 half-lives (60 min) (30). The participants and apnea coach were blinded to the experimental condition.
All experimental breath holds (maximal breath holds) were completed in the presence of the breath hold divers’ coach, the Croatian national apnea coach. The coach was present to ensure that all divers were attaining a true maximal apnea. Each maximal breath hold was preceded by two practice breath holds. The preparatory breath holds included one at functional residual capacity lasting until seven involuntary breathing movements (IBMs) and, after two minutes of rest, a second breath hold at total lung capacity lasting until 10 IBMs. Subjects then rested quietly for 6 min in preparation for their maximal breath hold. The experimental breath hold was performed at TLC, whereas the extent of glossopharyngeal insufflation (lung packing) performed was based upon the individual capacity of each subject but standardized between trials.
Experimental Measures
Cardiovascular measures.
All cardiovascular and respiratory variables were sampled continuously at 1,000 Hz via an analog to digital acquisition system (Powerlab; ADInstruments, Colarado Springs, CO). Finger photoplethysmography (Finometer, Finapres Medical Systems, Amsterdam, The Netherlands) was used to measure beat-by-beat blood pressure and estimate cardiac output (CO), stroke volume (SV), and total peripheral resistance (TPR), whereas standard three-lead electrocardiogram (ECG) was used to measure heart rate. Peripheral oxyhemoglobin saturation () was measured during baseline and throughout the apneas (Poet II; Criticare). The IBM onset was recorded in real time during the apnea by the apnea coach and verified from a chest plethysmography belt. All data were interfaced with LabChart (version 7; ADInstruments) on a laboratory computer and stored for offline analysis.
Myocardial oxygen consumption.
For each trial, MV̇o2 of the left ventricle was estimated from RPP (the product of systolic blood pressure and HR) and left ventricular mass. To calculate MV̇o2 per 100 g tissue, we used the equation reported by Gobel et al. (12):
| (1) |
Left ventricular mass was estimated at baseline using echocardiography from a parasternal short-axis view, in accordance with the recommendations of the American Society of Echocardiography (19) using the following equation:
| (2) |
where IVS and PWT are the diameters of the ventricular septum and inferolateral wall during diastole, respectively, and LVID is the internal diameter of the LV during diastole.
To then calculate absolute MV̇o2 for each subject, left ventricular mass was incorporated into the following equation on an individual basis:
| (3) |
The cumulative myocardial oxygen consumption throughout apnea was then calculated by estimating MV̇o2 from RPP on a beat-by-beat basis. The volume cost of oxygen per beat was calculated by dividing beat-by-beat MV̇o2 by the R-R interval for each heart-beat. Total myocardial oxygen consumption for the entire apnea was calculated as the area under the curve of the beat-to-beat O2 cost.
Cerebrovascular measures.
Blood velocity in the right middle cerebral artery (MCAv) and left posterior cerebral artery (PCAv) were measured using a 2M-Hz transcranial Doppler ultrasound (TCD; Spencer Technologies, Seattle, WA). The TCD probes were attached bilaterally to a specialized commercial headband (model M600 bilateral head frame; Spencer Technologies) and secured in place. Insonation of the MCA and PCA was performed through the transtemporal window using previously described location and standardization techniques (32).
Blood flow through extracranial cerebral vessels was measured using a 10-MHz, multifrequency, linear array vascular ultrasound (Terason T3200; Teratech, Burlington, MA). The internal carotid (ICA) and vertebral artery (VA) were insonated ipsilateral to the MCA and PCA, respectively. Arterial diameter was measured via B-mode imaging, whereas peak blood velocity was simultaneously measured with pulse-wave mode. Measurements of ICA diameter and velocity (ICAv) were acquired ~1.5 cm distal to the common carotid bifurcation, with no evidence of turbulent or retrograde flow present during recording. Vertebral artery diameter and velocity (VAv) were acquired between either the C4–C5 or C5–C6 vertebral segment. This location was determined on an individual basis in an attempt to select the most reproducible measures, with the same location repeated within subjects. Measurement location and insonation angle were standardized within subjects and between the esmolol and placebo conditions. An angle correction of 60° was used for all subjects, whereas the velocity sample volume was placed in the center of the vessel and adjusted to span across the entire luminal diameter. Baseline recordings were made in all subjects (n = 9); however, the sample size for VA measures throughout an entire breath hold was reduced (n = 6) due to strict exclusion criteria such as obvious angle changes.
Ultrasound images of the extracranial vessels were recorded as video files at 30 Hz and stored for offline analysis. Concurrent values for arterial diameter and peak blood velocity were acquired at 30 Hz, using customized edge detection software designed to mitigate observer bias (33). Blood flow was subsequently calculated for each vessel using the following formula:
| (4) |
where QICA represents ICA blood flow and QVA represents VA blood flow. For the maximal apneas, QICA and QVA were averaged for a 1-min baseline and the last 30 s of apnea. Furthermore, values were also calculated on an individual basis to represent 10% increments in total apnea duration. No-flow calculation included <12 consecutive cardiac cycles, and where possible it included the entire averaging period. Once individual vessel flow was calculated, global cerebral blood flow (gCBF) was estimated for each apnea time point using the following formula:
| (5) |
During the latter half of each maximal apnea (i.e., struggle phase), IBMs resulted in large diaphragmatic movements and neck muscle recruitment (i.e., sternocleidomastoid contraction). As such, reliable velocity traces were no longer attainable. Once measures of QICA and QVA were no longer attainable due to poor velocity traces, flow was estimated using the following formulas in subsequent fashion for the remainder of the apnea:
| (6) |
| (7) |
where changes in MCAv and PCAv were used to calculate an estimated ICA (eICAv) and VA velocity (eVAv), respectively (Eq. 3). The term “pre-IBM ICAv or VAv” represents the average velocity value of the last 30 s before the IBMs that precluded the measurement of velocity occurred, and “%ΔMCAv or %ΔPCAv” represents the relative change in MCAv and PCAv from the same pre-IBM stage. As such, this multiplication gives a reliable estimate of neck vessel blood velocity during the struggle phase of apnea in the event that changes in downstream vessel (MCA and PCA) velocity represent upstream vessel (ICA and VA) velocity changes. As reliable diameter measurements (d) were still attainable throughout IBMs, changes in vessel diameter were accounted for in our estimation of volumetric blood flow (Eq. 4). Volumetric flow through the neck vessels (QICA and QVA) was then calculated from the estimated velocity values and vessel cross-sectional area . To exemplify the feasibility of this estimation, we ran a Pearson’s correlation between the percent changes in ICAv and VAv with those of MCAv and PCAv, respectively, throughout apnea until velocity measures were unattainable. The average within-subject r2 values between ICAv and MCAv were 0.88 ± 0.12 and 0.90 ± 0.10 during the placebo and esmolol trials, respectively. For VAv and PCAv, the r2 average was 0.88 ± 0.09 and 0.95 ± 0.03 for the placebo and esmolol trials, respectively. The strong correlation between blood velocity of confluent intra- and extracranial blood vessels supports the accuracy of our estimation. Furthermore, we have demonstrated previously that MCAv and ICAv reactivity do not differ statistically over a wide range of end-tidal CO2 (rest to +9 mmHg) (15), whereas work in our laboratory also indicates that ICAv and VAv reactivity do not differ from MCAv and PCAv reactivity, respectively, during hypoxia at an SaO2 of 98, 90, 80, and 70% (14a).
Statistical Analyses
Differences in apnea duration and time to IBM onset between esmolol and placebo were compared using two-tailed paired t-tests. To analyze the effect of esmolol over the progression of apnea, apnea duration was normalized to 100%, with comparisons being made between placebo and esmolol conditions at 10% relative increments in apnea duration using a two-way repeated-measures ANOVA (factors: esmolol and apnea duration). When significant F-ratios were detected, post hoc comparisons were made using Tukey’s honest significant difference test. End-apnea variables (final 30 s) in the placebo and esmolol trials were compared using two-tailed paired t-tests (Table 1). Data are presented as means ± SD.
Table 1.
End-apnea hemodynamic variables
| Units | Placebo | Esmolol |
|---|---|---|
| HR, beats/min | 54.2 ± 16.9 | 52.2 ± 13.3 |
| MAP, mmHg | 139.0 ± 18.6 | 129.6 ± 19.1 |
| SV, ml | 92.7 ± 21.8 | 78.5 ± 21.3† |
| CO, l/min | 5.4 ± 2.4 | 4.4 ± 1.3 |
| RPP, mmHg·beats−1·min−1 | 13,192 ± 3,882 | 10,124 ± 3,318† |
| Systolic BP, mmHg | 247.9 ± 37.6 | 195.7 ± 25.8† |
| , % | 74.9 ± 9.5 | 71.8 ± 10.3 |
Values are means ± SE. HR, heart rate; MAP, mean arterial pressure; SV, stroke volume; CO, cardiac output; RPP, rate pressure product; BP, blood pressure; , peripheral oxyhemoglobin saturation.
Significant difference between placebo and esmolol, P < 0.05.
RESULTS
Apnea Duration
Apnea duration increased 10 ± 10% from 323.4 ± 60.6 s during the placebo trial to 355.9 ± 56.8 s after β1-blockade with esmolol (P < 0.01; Fig. 1). The time to IBM onset was delayed by 21 ± 15% (from 160.4 ± 41.3 s during the placebo trial to 192.0 ± 45.3 s after treatment with esmolol, P < 0.01). There was no relationship between the absolute (r2 = 0.06, P = 0.53) or percent (r2 = 0.21, P = 0.22) change in IBM onset and apnea duration. Despite the prolonged apnea duration, during the last 30 s of apnea was not significantly different between placebo (74.9 ± 9.5%) and esmolol trials (71.8 ± 10.3%) (P = 0.10).
Fig. 1.
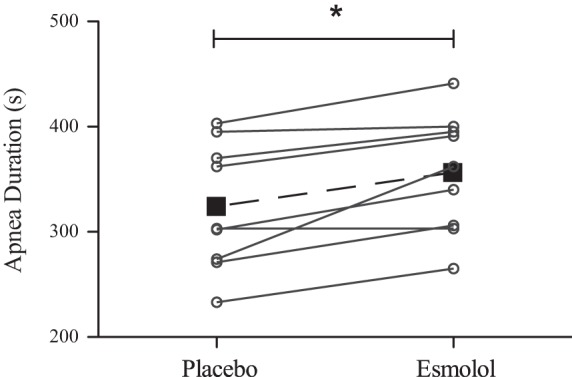
Maximal apnea duration during placebo and esmolol trials. Individual data are represented by ○ and solid lines, whereas mean data are represented by ■ and the dashed line. Overall, there was a 32 ± 25-s increase in apnea duration with esmolol treatment. *Significant difference in apnea duration (P < 0.01).
Hemodynamics
Values and statistical output for HR, MAP, SV, CO, and TPR during each relative time point throughout apnea are presented in Fig. 2. During the initial phase of apnea, HR was elevated in both placebo and esmolol trials, whereas esmolol reduced HR compared with placebo at 10–30% of apnea duration. In both trials, MAP was elevated from baseline. Throughout apnea, SV and CO were reduced from baseline and lower (main effect: both P < 0.01) during the esmolol compared with the placebo trial from 40% to max apnea and 20% to max apnea, respectively. End-apnea (final 30 s; Table 1) HR and MAP were not different between esmolol and placebo, whereas SV and CO were reduced by 15 and 18%, respectively, in the esmolol trial; however, the difference in CO did not reach statistical significance (P = 0.06). Esmolol increased TPR (main effect: P = 0.04) compared with placebo, whereas within-condition TPR was elevated above baseline from 20 to 100% of apnea duration.
Fig. 2.
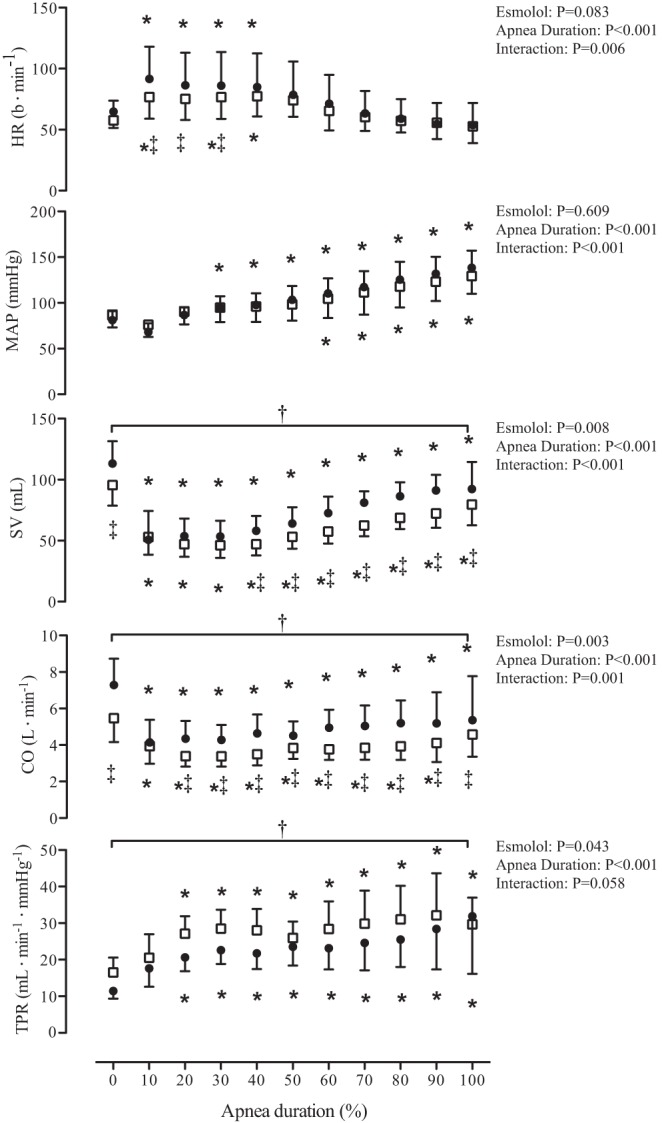
The hemodynamic response to maximal apnea during placebo and esmolol infusion. Data are normalized within subjects to represent 10% increments in apnea duration. ●, Data from the placebo trial; □, data from the esmolol trial. Data were analyzed using a 2-way repeated-measures ANOVA with the factors of apnea duration and esmolol treatment. *Significant difference from baseline P < 0.05; †main effect of treatment, P < 0.05; ‡interaction between apnea time and treatment. HR, heart rate; MAP, mean arterial pressure; SV, stroke volume; CO, cardiac output; TPR, total peripheral resistance.
There was a main effect of esmolol on MV̇o2 (P < 0.01; Fig. 3), which was lower in the esmolol trial. In both trials MV̇o2 was elevated from baseline at 30, 40, and 90%. There was a 16.2 ± 4.2% reduction in MV̇o2 across all stages (range: 8.0–21.6%) in the esmolol trial. However, there was no difference in the cumulative myocardial oxygen consumption between the placebo (76.3 ± 19.9 ml) and esmolol (70.2 ± 21.7 ml) trials (P = 0.27; Fig. 3B) due to the prolonged duration of the esmolol apnea.
Fig. 3.

Myocardial oxygen consumption (MV̇o2) during maximal apnea in elite breath-hold divers. A: data are normalized within subjects to represent 10% increments in apnea duration. ●, Data from the placebo trial; □, data from the esmolol trial. Data were analyzed using a 2-way repeated-measures ANOVA with the factors of apnea duration and esmolol treatment. *Significant difference from baseline, P < 0.05; †main effect of treatment, P < 0.05. B: total MV̇o2 throughout apnea was unaltered between placebo and esmolol trials. Individual data are represented by ○ and solid lines, whereas mean data are represented by ■ and the dashed line.
Cerebral Blood Flow
Measurements of QICA, QVA, gCBF, MCAv, and PCAv were not different between placebo and esmolol at baseline (Table 2). At the end of apnea (final 30 s), the sample sizes for gCBF and QVA were reduced to six participants. In these six participants, gCBF was lower at the end of apnea during the esmolol trial compared with placebo (Table 2).
Table 2.
Baseline and end-apnea cerebrovascular variables
| Baseline |
Final 30 s |
|||
|---|---|---|---|---|
| Units | Placebo | Esmolol | Placebo | Esmolol |
| QICA, ml/min | 222.9 ± 33.5 | 201.5 ± 33.0 | 384.8 ± 77.1 | 347.6 ± 66.3 |
| QVA, ml/min | 74.3 ± 35.5 | 72.6 ± 38.7 | 144.5 ± 61.9 | 133.5 ± 57.3 (n = 6) |
| gCBF, ml/min | 583.6 ± 99.8 | 531.1 ± 88.2 | 935.2 ± 415.0 (n = 6) | 808.8 ± 373.1 (n = 6)† |
| MCAv, cm/s | 57.3 ± 11.5 | 54.8 ± 10.3 | 94.8 ± 11.8 | 92.2 ± 12.0 |
| PCAv, cm/s | 41.0 ± 13.4 (n = 8) | 39.2 ± 6.3 (n = 8) | 68.4 ± 17.6 (n = 8) | 64.6 ± 15.5 (n = 8) |
Values are means ± SE. QICA, internal carotid artery blood flow; ICAv, internal carotid artery blood velocity; DICA, internal carotid artery diameter; QVA, vertebral artery blood flow; VAv, vertebral artery blood velocity; DVA, vertebral artery diameter; gCBF, global cerebral blood flow; MCAv, middle cerebral artery blood velocity; PCAv, posterior artery blood velocity. As noted in methods, the values for QICA, QVA, and gCBF are based off of estimated velocity values combined with real diameter measurements.
Significant difference between placebo and esmolol, P < 0.05; n = 9 unless otherwise specified.
DISCUSSION
The primary findings of this study are as follows: 1) β1-blockade with esmolol improves maximal apnea duration in elite breath-hold divers by ~10%; 2) esmolol reduces MV̇o2 at each time point throughout maximal apnea by ~16%, whereas the cumulative myocardial oxygen consumption is unaltered; 3) there are marked changes in the hemodynamic response (TPR and CO) to apnea between the esmolol and placebo trials; and 4) end-apnea was not different despite the prolonged apnea in the esmolol trial. Collectively, these data support the hypothesis that apnea break point is influenced by β1-blockade but ultimately governed by a critical oxygen tension.
β1-Blockade and Apnea Duration
It is now recognized that β-blocker drugs act as an ergogenic aid, and therefore, they are banned from international apnea competition by the AIDA. In support of this rule, using a placebo-controlled and blinded design, this study is the first to empirically demonstrate the increased maximal apnea time with β1-blockade in elite breath-hold divers.
Hemodynamic Response to Apnea
β1-blockade affects hemodynamics, and thus CO, through chronotropic and inotropic means (2, 16). In the present study, we observed a predominantly negative inotropic influence of β1-blockade with esmolol on CO (reduced SV), with only slight chronotropic changes. With the exception of a lower HR during the initial hypotensive phase of apnea (first 10–30% apnea duration), esmolol did not have an appreciable effect on HR; MAP was not different from placebo at any relative time point during the esmolol trial apnea. However, esmolol did cause a reduction in SV throughout the apnea (likely through reduced contractility, i.e., negative inotropy), which caused a concomitant reduction in CO compared with the placebo trial. This reduction in CO would likely have acted to further baroreceptor unloading and augment the reflex increase in sympathetic output (and TPR) throughout the apnea (14) to maintain the MAP response. Despite a reduced CO, increased centralization of blood volume (due to ↑TPR) in the esmolol trial likely reduced nonvital organ perfusion while aiding in the selective maintenance of oxygen delivery to vital organs.
Esmolol reduced left ventricular MV̇o2 throughout apnea when compared with the placebo trial, indicating that whole heart MV̇o2 would likely have been reduced (Fig. 3). Reduced MV̇o2 would further spare oxygen and also contribute in part to the prolonged apnea duration associated with β1-blockade. Despite a lower MV̇o2 throughout apnea, the cumulative myocardial oxygen consumption was not different between trials, owing to the prolonged apnea in the esmolol trial.
Mechanisms Governing Apnea Termination
The physiological apnea break point (i.e., cessation of breath hold) in untrained breath holders, which is observed as IBM onset in elite breath-hold divers, is governed largely by chemoreceptor afferents (6, 11). Breskovic et al. (6) suggested that IBM onset is related predominantly to a threshold level of 6.5 ± 0.5 kPa (48.8 mmHg). A similar threshold for IBM onset has been reported in most (1, 21) but not all (4) earlier studies, highlighting the importance of in initiating the physiological break point. Nevertheless, the prevailing oxygen tension will impact the level of at which IBM onset occurs (6). These chemoreflex interactions, along with a modulatory role from lung afferents (reviewed in Ref. 24), indicate that it is the integration of signals (i.e., peripheral and central chemoafference) that collectively govern the physiological break point.
Concurrent decreases in O2 depletion and CO2 production, both of which are a consequence of a reduced metabolism, will act to attenuate arterial blood gas changes and hence, chemoreflex stimulation. Indeed, our findings show that IBM onset was delayed by ~32 s but nevertheless occurred at the same level of between trials (placebo = 97.8 ± 1.3%; esmolol = 96.4 ± 2.2%). Under the assumption that the respiratory exchange ratio was unaltered between trials, a similar reduction in signifies that metabolic CO2 production likely did not differ and was also similar between trials at IBM onset. Therefore, despite the delayed IBM onset with esmolol treatment, IBMs likely occurred at the same level of chemoreflex stress with no influence from altered chemosensitivity (13) or CO2 washout in the brainstem (i.e., QVA, an index of brainstem flow, was similar between trials). Therefore, it is likely that the delay in reaching a chemoreflex threshold was responsible for delayed IBM onset (or physiological break point). In contrast to the physiological break point, the actual apnea termination in elite breath-hold divers appears to be only modestly affected by suppression of peripheral chemoreflex drive (4).
Typically, it is accepted that man is incapable of sustaining a voluntary apnea to the point of losing consciousness (27). However, many elite apnea divers are able to suppress their ventilatory drive during an apnea to the loss of consciousness (unpublished observations). This latter point is reflected in that AIDA includes loss of consciousness as a criterion for disqualification during apnea competition. At this point, the apnea “break point” is governed to a potentially large extent by a threshold oxygen tension to maintain cerebral functioning and, therefore, consciousness. Current literature indicates this threshold occurs at around 25–35 mmHg (4, 31), which coincides with P50. Despite a prolonged apnea duration following esmolol in the current study, was not significantly different between trials, indicating similar end-apnea hypoxic stress. Based upon the commonly used Severinghaus equation (28) and our pulse-oximetry measurements, it appears that end-apnea would have been ~35–40 mmHg in both the placebo and esmolol trials. Although the achieved hypoxic stimulus does not appear as severe as that of a previous study indicating the potential of an oxygen threshold for apnea termination (4), the similar end-apnea indicates some role of hypoxia in mediating apnea break point in both trials. Of note is that data from our previous study (4), where concurrent (finger pulse-oximetry) and (radial artery blood draw) measurements were collected, indicate that overestimates by a mean value of ~5% and that this over estimation is largest below an of 70% (Fig. 4). Therefore, it is likely that the end-apnea hypoxic stimulus was similar to our previous studies in elite breath-hold divers (4, 31). These data are similar to our previous findings in elite breath-hold divers of similar end-apnea despite moderate prolongation (but in some cases reductions) in maximal apnea time following peripheral chemoreflex blunting with low-dose dopamine (4).
Fig. 4.
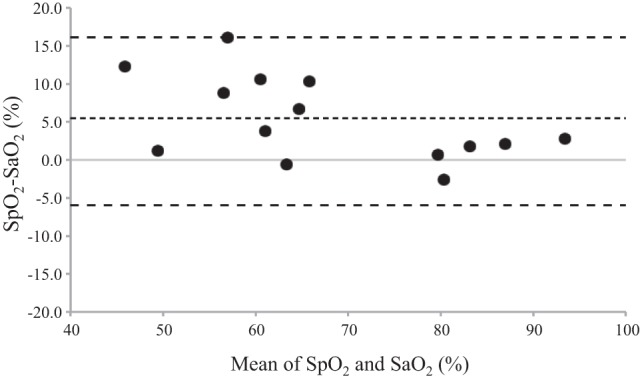
A Bland-Altman plot of and at the end of a maximal apnea. This figure demonstrates an overestimation of (radial blood draw) by (finger pulse-oximetry) at the end of apnea in elite breath-hold divers using data from our previous study (4). Dotted line represents the mean difference (5.3), whereas the dashed lines represent the upper and lower limits of agreement (16.1 and −5.6, respectively). Of note is that the overestimation of by is most evident below a of 70%. Each data point represents an individual subject; n = 14.
In the current study, centralization of blood volume and, therefore, a reduction of nonvital organ perfusion but maintenance of vital organ oxygen delivery, in combination with myocardial oxygen sparring, likely reduced both the hypoxic and acidotic stresses’ characteristic of extreme apnea. Together, these factors likely underscore the increased apnea duration with esmolol.
Methodological Considerations
In the current study, we have used RPP and echocardiographic assessment of left ventricular mass to estimate left ventricular MV̇o2 from the equation reported by Gobel et al. (12). Although RPP has been shown to be an excellent (r = 0.85) correlate of MV̇o2 during exercise pre- and post-β-blockade (propranolol) (17), we acknowledge that this validation has not been made during the specific conditions of prolonged apnea. Despite this, it is unlikely that our estimates of RPP and MV̇o2 would differ between conditions and discredit our findings. Furthermore, although we used doses comparable with those in other studies (16), we must acknowledge that we did not assess the completeness of our β1-blockade. However, the presence of an incomplete block would have led to an underestimation of the magnitude by which esmolol impacts apnea duration.
We used to quantify the hypoxic stimulus during prolonged breath hold during placebo (saline) and esmolol infusion. As demonstrated in Fig. 4, overestimates substantially below a of ~70%. However, given the overestimation of , our data should be interpreted as presenting end-apnea hypoxic stress similar to that in previous investigations by our group (4, 31).
Conclusion
This study contributes to the recent body of literature (4, 31) supporting the emerging hypothesis that apnea break point in elite breath-hold divers is governed to a large extent by an oxygen threshold for the maintenance of consciousness. Cardiac-specific β1-blockade with esmolol improved apnea duration in the current study and thus acts as an effective ergogenic aid in apnea competition. The likely mechanisms by which esmolol increased apnea duration are through increased centralization of blood flow (due to ↑TPR), maintenance of vital organ perfusion, and a reduced MV̇o2. Together, these two factors would contribute to improved total body oxygen sparring and reduced metabolic CO2 production.
GRANTS
This study was funded through a Canadian Research Chair and NSERC Discovery grant held by P. N. Ainslie. Z. Dujic, O. Barak, and P. N. Ainslie were also funded through the Croatian Science Foundation (Grant No. IP-2014-09-1937).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.L.H., P.N.A., A.R.B., D.B.M., M.S., I.D., D.M., O.F.B., D.M.M., and Z.D. performed experiments; R.L.H., P.N.A., and A.R.B. analyzed data; R.L.H., P.N.A., and A.R.B. interpreted results of experiments; R.L.H. prepared figures; R.L.H. drafted manuscript; R.L.H., P.N.A., A.R.B., D.B.M., M.S., I.D., D.M., O.F.B., D.M.M., and Z.D. edited and revised manuscript; R.L.H., P.N.A., A.R.B., D.B.M., M.S., I.D., D.M., O.F.B., D.M.M., and Z.D. approved final version of manuscript; P.N.A., A.R.B., D.B.M., I.D., and Z.D. conception and design of research.
ACKNOWLEDGMENTS
We acknowledge the apnea divers from the Croatia National Apnea team for their participation.
REFERENCES
- 1.Agostoni E. Diaphragm activity during breath holding: factors related to its onset. J Appl Physiol 18: 30–36, 1963. [DOI] [PubMed] [Google Scholar]
- 2.Askenazi J, MacCosbe PE, Hoff J, Turlapaty P, Hua TA, Laddu A. Hemodynamic effects of esmolol, an ultrashort-acting beta blocker. J Clin Pharmacol 27: 567–573, 1987. doi: 10.1002/j.1552-4604.1987.tb03068.x. [DOI] [PubMed] [Google Scholar]
- 3.Bain AR, Ainslie PN, Hoiland RL, Willie CK, MacLeod DB, Madden D, Maslov PZ, Drviš I, Dujić Ž. Role of cerebral blood flow in extreme breath holding. Transl Neurosci 7: 12–16, 2016. doi: 10.1515/tnsci-2016-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bain AR, Dujic Z, Hoiland RL, Barak OF, Madden D, Drvis I, Stembridge M, MacLeod DB, MacLeod DM, Ainslie PN. Peripheral chemoreflex inhibition with low-dose dopamine: new insight into mechanisms of extreme apnea. Am J Physiol Regul Integr Comp Physiol 309: R1162–R1171, 2015. doi: 10.1152/ajpregu.00271.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batinic T, Utz W, Breskovic T, Jordan J, Schulz-Menger J, Jankovic S, Dujic Z, Tank J. Cardiac magnetic resonance imaging during pulmonary hyperinflation in apnea divers. Med Sci Sports Exerc 43: 2095–2101, 2011. doi: 10.1249/MSS.0b013e31821ff294. [DOI] [PubMed] [Google Scholar]
- 6.Breskovic T, Lojpur M, Maslov PZ, Cross TJ, Kraljevic J, Ljubkovic M, Marinovic J, Ivancev V, Johnson BD, Dujic Z. The influence of varying inspired fractions of O2 and CO2 on the development of involuntary breathing movements during maximal apnoea. Respir Physiol Neurobiol 181: 228–233, 2012. doi: 10.1016/j.resp.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Breskovic T, Steinback CD, Salmanpour A, Shoemaker JK, Dujic Z. Recruitment pattern of sympathetic neurons during breath-holding at different lung volumes in apnea divers and controls. Auton Neurosci 164: 74–81, 2011. doi: 10.1016/j.autneu.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Cross TJ, Kavanagh JJ, Breskovic T, Zubin Maslov P, Lojpur M, Johnson BD, Dujic Z. The Effects of Involuntary Respiratory Contractions on Cerebral Blood Flow during Maximal Apnoea in Trained Divers. PLoS One 8: e66950, 2013. doi: 10.1371/journal.pone.0066950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dujic Z, Breskovic T, Bakovic D. Breath-hold diving as a brain survival response. Transl Neurosci 4: 302–313, 2013. doi: 10.2478/s13380-013-0130-5. 25370857 [DOI] [Google Scholar]
- 10.Dujic Z, Breskovic T. Impact of breath holding on cardiovascular respiratory and cerebrovascular health. Sports Med 42: 459–472, 2012. doi: 10.2165/11599260-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.Feiner JR, Bickler PE, Severinghaus JW. Hypoxic ventilatory response predicts the extent of maximal breath-holds in man. Respir Physiol 100: 213–222, 1995. doi: 10.1016/0034-5687(94)00132-J. [DOI] [PubMed] [Google Scholar]
- 12.Gobel FL, Norstrom LA, Nelson RR, Jorgensen CR, Wang Y. The rate-pressure product as an index of myocardial oxygen consumption during exercise in patients with angina pectoris. Circulation 57: 549–556, 1978. doi: 10.1161/01.CIR.57.3.549. [DOI] [PubMed] [Google Scholar]
- 13.Gonsalves SF, Smith EJ, Nolan WF, Dutton RE. beta-Adrenoceptor blockade spares chemoreceptor responsiveness to hypoxia. Brain Res 324: 349–353, 1984. doi: 10.1016/0006-8993(84)90047-7. [DOI] [PubMed] [Google Scholar]
- 14.Heusser K, Dzamonja G, Tank J, Palada I, Valic Z, Bakovic D, Obad A, Ivancev V, Breskovic T, Diedrich A, Joyner MJ, Luft FC, Jordan J, Dujic Z. Cardiovascular regulation during apnea in elite divers. Hypertension 53: 719–724, 2009. doi: 10.1161/HYPERTENSIONAHA.108.127530. [DOI] [PubMed] [Google Scholar]
- 14a.Hoiland RL, Bain AR, Tymko MM, Rieger MG, Howe CA, Willie CK, Hansen AB, Flück D, Wildfong KW, Stembridge M, Subedi P, Anholm JD, Ainslie PN. Adenosine receptor dependent signaling is not obligatory for normobaric and hypobaric hypoxia-induced cerebral vasodilation in humans. J Appl Physiol. doi: 10.1152/japplphysiol.00840.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoiland RL, Tymko MM, Bain AR, Wildfong KW, Monteleone B, Ainslie PN. Carbon dioxide-mediated vasomotion of extra-cranial cerebral arteries in humans: a role for prostaglandins? J Physiol 594: 3463–3481, 2016. doi: 10.1113/JP272012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iskandrian AS, Hakki AH, Laddu A. Effects of esmolol on cardiac function: evaluation by noninvasive techniques. Am J Cardiol 56: F27–F32, 1985. doi: 10.1016/0002-9149(85)90913-0. [DOI] [PubMed] [Google Scholar]
- 17.Jorgensen CR, Wang K, Wang Y, Gobel FL, Nelson RR, Taylor H, Gams FR, Vilandre JE. Effect of propranolol on myocardial oxygen consumption and its hemodynamic correlates during upright exercise. Circulation 48: 1173–1182, 1973. doi: 10.1161/01.CIR.48.6.1173. [DOI] [PubMed] [Google Scholar]
- 18.Kiviniemi AM, Breskovic T, Uglesic L, Kuch B, Maslov PZ, Sieber A, Seppänen T, Tulppo MP, Dujic Z. Heart rate variability during static and dynamic breath-hold dives in elite divers. Auton Neurosci 169: 95–101, 2012. doi: 10.1016/j.autneu.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, Lancellotti P, Muraru D, Picard MH, Rietzschel ER, Rudski L, Spencer KT, Tsang W, Voigt JU. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 28: 1–39.e14, 2015. doi: 10.1016/j.echo.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Lin YC. Breath-hold diving in terrestrial mammals. Exerc Sport Sci Rev 10: 270–307, 1982. doi: 10.1249/00003677-198201000-00009. [DOI] [PubMed] [Google Scholar]
- 21.Lin YC, Lally DA, Moore TO, Hong SK. Physiological and conventional breath-hold breaking points. J Appl Physiol 37: 291–296, 1974. [DOI] [PubMed] [Google Scholar]
- 22.Overgaard K, Friis S, Pedersen RB, Lykkeboe G. Influence of lung volume, glossopharyngeal inhalation and P(ET) O2 and P(ET) CO2 on apnea performance in trained breath-hold divers. Eur J Appl Physiol 97: 158–164, 2006. doi: 10.1007/s00421-006-0156-2. [DOI] [PubMed] [Google Scholar]
- 23.Palada I, Obad A, Bakovic D, Valic Z, Ivancev V, Dujic Z. Cerebral and peripheral hemodynamics and oxygenation during maximal dry breath-holds. Respir Physiol Neurobiol 157: 374–381, 2007. doi: 10.1016/j.resp.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Parkes MJ. Breath-holding and its breakpoint. Exp Physiol 91: 1–15, 2006. doi: 10.1113/expphysiol.2005.031625. [DOI] [PubMed] [Google Scholar]
- 25.Pingitore A, Gemignani A, Menicucci D, Di Bella G, De Marchi D, Passera M, Bedini R, Ghelarducci B, L’Abbate A. Cardiovascular response to acute hypoxemia induced by prolonged breath holding in air. Am J Physiol Heart Circ Physiol 294: H449–H455, 2008. doi: 10.1152/ajpheart.00607.2007. [DOI] [PubMed] [Google Scholar]
- 26.Potkin R, Cheng V, Siegel R. Effects of glossopharyngeal insufflation on cardiac function: an echocardiographic study in elite breath-hold divers. J Appl Physiol (1985) 103: 823–827, 2007. doi: 10.1152/japplphysiol.00125.2007. [DOI] [PubMed] [Google Scholar]
- 27.Schneider EC. Observations on holding the breath. Am J Physiol 94: 464–470, 1930. [Google Scholar]
- 28.Severinghaus JW. Simple, accurate equations for human blood O2 dissociation computations. J Appl Physiol Respir Environ Exerc Physiol 46: 599–602, 1979. [DOI] [PubMed] [Google Scholar]
- 29.Steinback CD, Salmanpour A, Breskovic T, Dujic Z, Shoemaker JK. Sympathetic neural activation: an ordered affair. J Physiol 588: 4825–4836, 2010. doi: 10.1113/jphysiol.2010.195941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiest DB, Haney JS. Clinical pharmacokinetics and therapeutic efficacy of esmolol. Clin Pharmacokinet 51: 347–356, 2012. doi: 10.2165/11631590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 31.Willie CK, Ainslie PN, Drvis I, MacLeod DB, Bain AR, Madden D, Maslov PZ, Dujic Z. Regulation of brain blood flow and oxygen delivery in elite breath-hold divers. J Cereb Blood Flow Metab 35: 66–73, 2015. doi: 10.1038/jcbfm.2014.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willie CK, Colino FL, Bailey DM, Tzeng YC, Binsted G, Jones LW, Haykowsky MJ, Bellapart J, Ogoh S, Smith KJ, Smirl JD, Day TA, Lucas SJ, Eller LK, Ainslie PN. Utility of transcranial Doppler ultrasound for the integrative assessment of cerebrovascular function. J Neurosci Methods 196: 221–237, 2011. doi: 10.1016/j.jneumeth.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 33.Woodman RJ, Playford DA, Watts GF, Cheetham C, Reed C, Taylor RR, Puddey IB, Beilin LJ, Burke V, Mori TA, Green D. Improved analysis of brachial artery ultrasound using a novel edge-detection software system. J Appl Physiol (1985) 91: 929–937, 2001. [DOI] [PubMed] [Google Scholar]