

Abstract
Although carbon dioxide (CO2) is highly abundant, its low reactivity has limited its use in chemical synthesis. In particular, methods for carbon–carbon bond formation generally rely on two-electron mechanisms for CO2 activation and require highly activated reaction partners. Alternatively, radical pathways accessed via photoredox catalysis could provide new reactivity under milder conditions. Here we demonstrate the direct coupling of CO2 and amines via the single-electron reduction of CO2 for the photoredox-catalyzed, continuous flow synthesis of α-amino acids. By leveraging advantages for utilizing gases and photochemistry in flow, a commercially available organic photoredox catalyst effects the selective α-carboxylation of amines bearing various functional groups and heterocycles. Preliminary mechanistic studies support CO2 activation and carbon–carbon bond formation via single-electron pathways, and we expect that this strategy will inspire new perspectives on using this feedstock chemical in organic synthesis.
Atmospheric carbon dioxide (CO2) is an attractive one-carbon building block in chemical synthesis due to its abundance, availability, and sustainability1–3. To date, most examples of carbon–carbon bond formation with CO2 have relied on two-electron mechanisms4. The low reactivity and high stability of CO2 generally limits these transformations to couplings with activated partners such as extended π systems or organometallic reagents (Fig. 1a)5,6, including work from our group7. Herein we apply the single-electron reduction of CO2 for the synthesis of α-amino acids via the direct, photoredox-catalyzed α-carboxylation of amines in continuous flow (Fig. 1b). Relying on advantages for utilizing gases and photochemistry in flow, this protocol provides a procedure for accessing α-amino acids8 by combining an amine and CO2 in the presence of light and a commercially available organic photoredox catalyst. Furthermore, this work demonstrates that single-electron reduction of CO2 is a productive strategy for carbon–carbon bond formation with this stable feedstock chemical.
Figure 1. Design plan for α-carboxylation of amines with CO2.
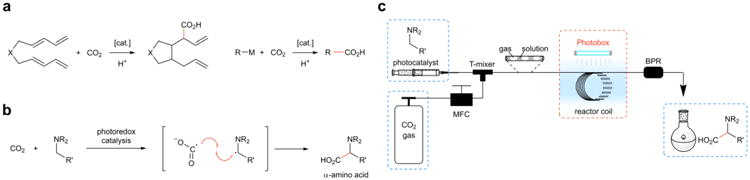
a, Carbon–carbon bond formation with CO2 has generally relied on two-electron reaction pathways with an extended π system or a strong nucleophile. cat., catalyst. b, Single-electron reduction of carbon dioxide and its reaction with an α-amino radical to provide α-amino acid. c, Continuous flow setup for the photoredox-catalyzed synthesis of α-amino acids. The reactants were introduced via a gas-tight syringe containing a solution of the amine substrate, base, and catalyst. CO2 gas was metered into the system by a MFC. These two streams were joined by a T-mixer before irradiation under a UV lamp. The pressure of CO2 is controlled by a BPR. MFC, mass flow controller; BPR, back pressure regulator.
Although the single-electron reduction of CO2 has been demonstrated electrochemically and photochemically, the CO2 radical anion has not shown broad utility as a reactive intermediate in organic synthesis9,10. Under the operationally mild conditions of photoredox catalysis11-16, we envisioned that the productive coupling of this radical anion with another radical would provide the mechanistic framework for novel transformations. The main challenge for this mode of CO2 activation is the high reduction potential [E0 = −2.21 V versus saturated calomel electrode (SCE) in N,N-dimethylformamide (DMF)], which is commonly supplemented by an overpotential of 0.1 to 0.6 V17,18. The necessary reduction potential is therefore outside the photoreducing capabilities of the ruthenium and iridium catalysts that are commonly used in visible light photoredox catalysis (the highest reduction potential available for this class of catalysts is for tris[2-phenylpyridinato-C2, N]iridium(III), or Ir(ppy)3, E1/2red = –2.19 V vs SCE in acetonitrile)12. Although oligophenylenes have not been widely applied as photoredox catalysts in organic synthesis, we expected that para-terphenyl (E0 = –2.63 V vs SCE in DMF) could overcome this limitation since this organic photoredox catalyst has been studied by Yanagida and co-workers for the reduction of CO2 to formic acid under UV light19.
From the outset, we recognized that this transformation would operate ideally in continuous flow, a field which has recently emerged as an important design tool for organic synthesis20,21. Figure 1c illustrates our continuous flow setup, in which a liquid solution containing the amine substrate and catalyst are mixed with CO2 gas in line. Irradiation of this segmented flow mixture with a UV lamp provides the amino acid product. Importantly, our setup allows for control of the amount and pressure of gas, and gas-liquid mixing is generally superior in flow than in batch22. The short path length of light also provides enhancements for photochemistry, and flow reactors remove scale limitations encountered with batch photoreactors23.
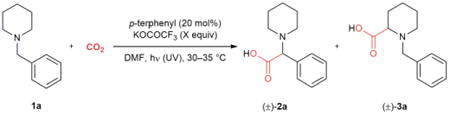
We began our investigations using N-benzylpiperidine (1a) as a model substrate (Table 1). The presence of a benzylic C–H bond was expected to enhance both the reactivity and regioselectivity of the carboxylation reaction. Under conditions similar to those utilized by Yanagida and co-workers19, p-terphenyl catalyzed the formation of α-amino acids 2a and 3a in 21% combined yield with 6.6:1 regioselectivity in favor of carboxylation at the benzylic position (entry 1). As in other recent reports of photoredox-catalyzed radical-radical couplings24, we hypothesized that the inclusion of an exogenous base would increase the yield by promoting deprotonation of an amine radical cation to produce the α-amino radical. Although our choice of base was somewhat limited by the constraint of a homogeneous reaction mixture for our continuous flow setup (see Supplementary Table 1), potassium trifluoroacetate (KOCOCF3) provided the highest yield, with a concomitant increase in regioselectivity, of the organic and inorganic bases investigated (entry 2). Further optimization by increasing the amount of KOCOCF3 from one to three equivalents, decreasing the pressure from 0.69 MPa to 0.34 MPa, and using a 4-minute residence time provided the maximum yield (78%) without a UV filter (entry 3). Operating at this pressure limited side reactions such as the formation of carbamate salts25, but further pressure reductions were detrimental to yield (see Supplementary Table 2).
Table 1.
Optimization of α-carboxylation of N-benzylpiperidine in continuous flow.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Entry* | Equiv KOCOCF3 | Pressure† (MPa) | tR (min) | UV filter | Yield 2a+3a (%)‡ | Regioselectivity (2a/3a)‡ |
| 1 | 0 | 0.69 | 5 | none | 21 | 6.6:1 |
| 2 | 1 | 0.69 | 5 | none | 45 | 33:1 |
| 3 | 3 | 0.34 | 4 | none | 78 | 30:1 |
| 4 | 3 | 0.34 | 10 | > 280 nm | 92 | 52:1 |
Reactions were carried out using the original photochemistry system (entry 1–3) or using Beeler's photochemistry system (entry 4). See Supplementary Fig. 1 and 2 for details of the setup.
Pressure of BPR. 0.69 MPa is equivalent to 7.3 equiv of CO2 and 0.34 MPa to 3.6 equiv.
Calculated by gas chromatography (GC) after esterification with (trimethylsilyl)diazomethane, using methyl benzoate as an internal standard.
DMF, N,N-dimethylformamide; tR, residence time.
In further optimizing this transformation, we reasoned that short wavelength UV light could lead to other unproductive reactions. We therefore investigated filters for UV light and found that the system recently reported by Beeler and co-workers was ideal for easily testing an array of commercial filters (see Supplementary Fig. 2)26. Using a long-pass filter with a 280 nm cut-on wavelength (p-terphenyl excitation λmax = 283 nm), the desired product was obtained in 92% yield with almost exclusive regioselectivity in favor of 2a (entry 4). In comparison, the batch reaction conducted with continuously bubbling CO2 provided 2a in poor yield (30%), even after prolonged reaction time (see Supplementary Table 4). These results highlight the advantages of reaction development in continuous flow, as it provides the opportunity to vary parameters that are difficult to adjust in traditional batch optimizations.
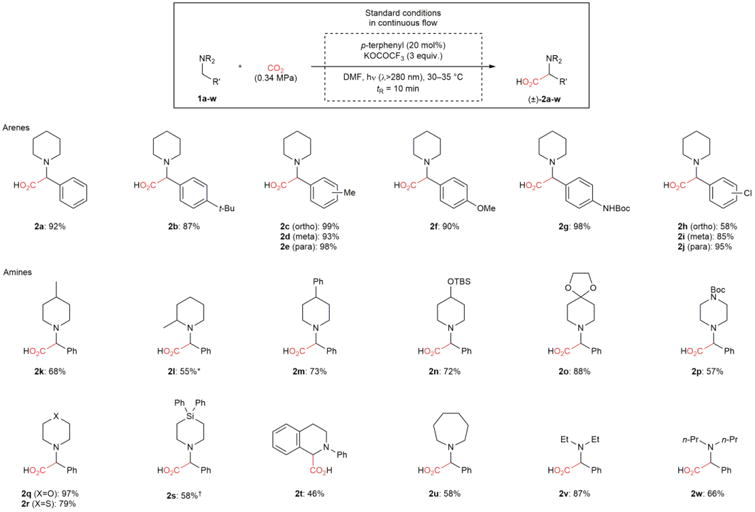
Our optimized conditions were applicable to the synthesis of a variety of α-amino acids (Table 2). In all cases, >20:1 regioselectivity in favor of carboxylation at the benzylic position was observed. Initially, we investigated reactions of tertiary N-benzylpiperidines bearing a range of electron-neutral and electron-rich arenes. Substrates bearing arenes with ortho-, meta-, and para-alkyl substituents provided the desired amino acids in high yield (2b–2e). Although para-methoxybenzylamines can be deprotected by single electron oxidants such as 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ)27, our conditions provided 2f in 90% yield without C–N bond cleavage. As demonstrated by the synthesis of 2g, protic functional groups such as N–H bonds are also tolerated by these conditions. Substrates containing ortho-, meta-, and para-chloroarenes provide good to excellent yields of 2h, 2i, and 2j with handles for further elaboration via cross-coupling. N-Benzylamines with electron-poor arenes are not suitable substrates for α-carboxylation under these conditions.
Table 2.
Substrate scope for the α-carboxylation of amines with CO2.
|
Various amino acids are synthesized in high yields and excellent regioselectivities (>20:1) with a short residence time. This protocol utilizes readily available starting materials and a commercially available, inexpensive organic catalyst. Products were isolated as trifluoroacetate salts on 0.7 mmol scale. (2s and 2t were isolated as free amines)
1.3:1 diastereomeric ratio was observed by GC analysis.
Reaction was carried out under atmospheric pressure of CO2 (1.1 equiv CO2) with tR = 5 min. Boc, tert-butyloxycarbonyl; TBS, tert-butyldimethylsilyl.
Next, other variations to the amine structure were investigated. 4- and 2-methylpiperidine derivatives provided the corresponding amino acids 2k and 2l in good yields. Non-activated benzylic C–H bonds, such as those found in 1m, are not carboxylated under these conditions. Although unprotected alcohols and ketones are not tolerated, silyl ether 2n and ketal 2o are produced in 72% and 88% yield, respectively, and can serve as masked versions of these functional groups. Amino acids containing a number of other heterocycles, including piperazine (2p), morpholine (2q), thiomorpholine (2r), and silapiperidine (2s) derivatives, can also be synthesized. Fused rings and different ring sizes, as exemplified by tetrahydroisoquinoline 2t and azepane 2u, are accommodated by this protocol. Importantly, amino acids bearing acyclic amines (2v and 2w) can be produced in good yields.
We next sought to expand the scope of this methodology to other substrate classes (Fig. 2a). Ticlopidine (4), a marketed antiplatelet agent, was regioselectively derivatized to provide 5 as a single isomer despite the presence of two activated sites. This result points to potential applications of this method for late stage C–H functionalization of complex structures. We also expected that this reactivity would apply to non-benzylic amines with less activated alkyl C–H bonds adjacent to nitrogen. Accordingly, N-cyclohexylpiperidine (6) underwent regioselective carboxylation of a secondary, rather than tertiary, C–H bond to produce amino acid 7. Despite the moderate yield (43%), we are encouraged that substrates beyond N-benzylamines undergo photoredox-catalyzed carboxylation and that considerations other than electronic effects can influence regioselectivity. Without applying a UV filter, the product yields in Table 2 and Figure 2a were significantly lower; this comparison highlights the importance of this modification to realize broad substrate scope.
Figure 2. Expanding the scope of the α-carboxylation protocol.
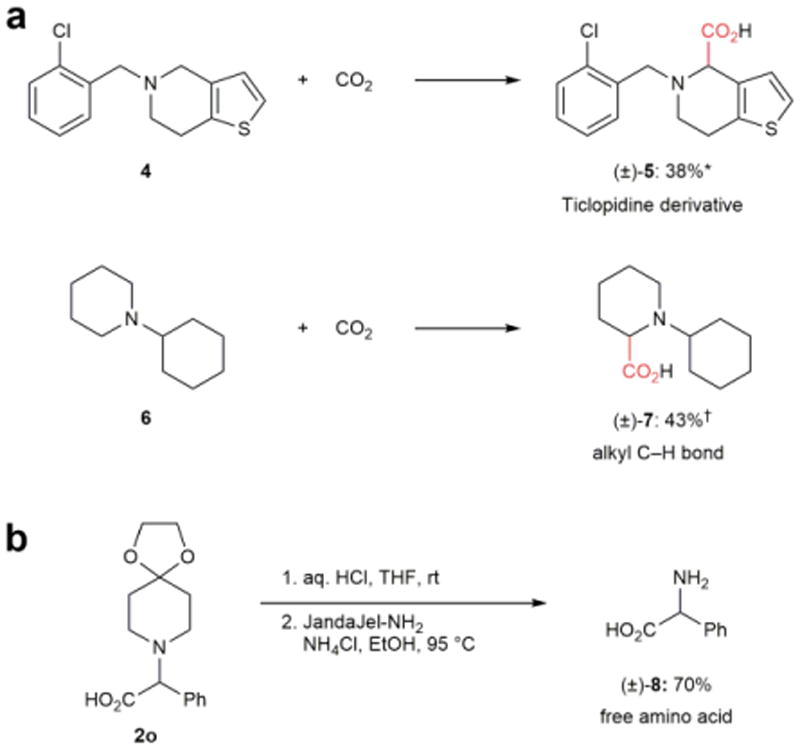
a, The carboxylation protocol can be applied to ticlopidine (an active pharmaceutical ingredient containing a heterocycle) and a substrate without a benzylic C–H bond. *Under standard conditions (see Table 2) †Under standard conditions, with atmospheric pressure of CO2 and tR = 6 min. b, Synthesis of a free amino acid can be achieved via deprotection of a piperidone derivative. THF, tetrahydrofuran.
Lastly, we recognized that the synthesis of a free amino acid via the combination of an amine and CO2 would provide access to this important motif (Fig. 2b). Primary amines were not suitable as substrates, but a screen of amine bis-protecting groups identified 4-piperidone analogues (e.g., 1o) as optimal for our α-carboxylation conditions. Cleavage of the 4-piperidone protecting group was accomplished under neutral conditions using a polymer-supported amine scavenger to afford free amino acid 828. Although the current protocol provides a racemic mixture, chiral resolution of such amino acids has been commonly utilized to isolate single enantiomers29,30.
In view of expanding the scope of this method and investigating further applications of this reactivity, preliminary studies were performed to understand the reaction mechanism. Our proposed catalytic cycle (Fig. 3) combines features of the mechanisms proposed by Yanagida19 and MacMillan24 for CO2 reduction and photoredox-catalyzed radical-radical couplings, respectively. Irradiation of organic photoredox catalyst p-terphenyl (9) with UV light produces the excited singlet state of p-terphenyl (10), which undergoes single-electron transfer (SET) with a tertiary amine (11) to provide the strongly reducing p-terphenyl radical anion (12) and the corresponding amine radical cation (13). In support of this step, Stern-Volmer plots illustrate that the luminescence of the excited state of p-terphenyl (9) is quenched by N-benzylpiperidine but not CO2 (see Supplementary Fig. 4). Strong reductant 12 then donates an electron to CO2 to form the CO2 radical anion (14). Concurrently, deprotonation of amine radical cation 13 affords neutral α-amino radical 15. The key bond-forming step occurs by radical-radical coupling of 14 and 15 to produce the desired α-amino acid (16). Although the fluorescence quenching experiments indicate that base does not appear to be directly involved in the photoredox catalytic cycle (see Supplementary Fig. 4 and 5), the nature of the exogenous base may limit product decomposition by stabilization as a salt. Results from control reactions and radical quenching experiments also agree with this preliminary mechanistic proposal (see Supplementary Table 5 and 6).
Figure 3. Proposed mechanism for photoredox catalytic α-carboxylation of amines with CO2.
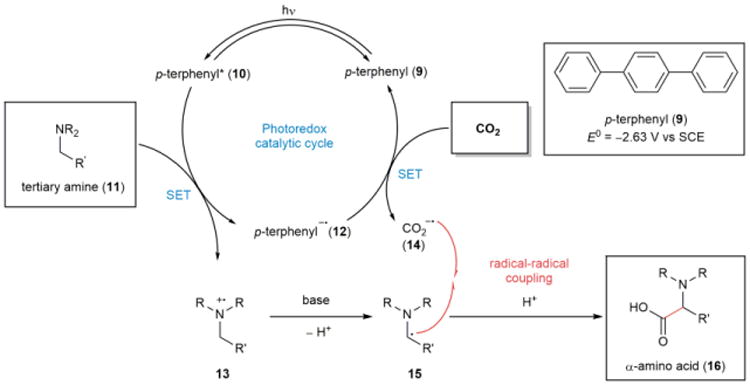
The excited singlet state of the photoredox catalyst p-terphenyl (10) is quenched by tertiary amine 11, and the radical anion of p-terphenyl (12) reduces CO2 to its radical anion (14). After the deprotonation of amine radical cation 13 to afford α-amino radical 15, radical-radical coupling of 14 and 15 provides α-amino acid 16.
In summary, we have developed a novel photoredox catalytic system for the α-carboxylation of amines with CO2 in continuous flow. In this high-yielding protocol, carbon–carbon bond formation occurs via a single-electron pathway for the conversion of two stable, unactivated reactants to α-amino acids. We anticipate that the single-electron activation of CO2 via the system described herein will result in the development of additional transformations with other reaction partners that provide new avenues for utilizing this abundant single carbon synthon.
Supplementary Material
Acknowledgments
We thank the Novartis-MIT Center for Continuous Manufacturing for financial support and several Novartis colleagues for suggestions, in particular, Berthold Schenkel, Benjamin Martin, Jörg Sedelmeier, Gerhard Penn, Francesco Venturoni, and Julien Haber. H.S. is grateful for a graduate fellowship from the Korean Government Scholarship Program for Study Overseas, and M.H.K. was supported by a postdoctoral fellowship from the NIGMS (F32GM108181). The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Prof. A. B. Beeler for guidance in setting up the flow photochemistry system, Prof. T. M. Swager for use of a spectrophotometer and L. Li for mass spectral data (conducted on an instrument purchased with the assistance of NSF Grant CHE-0234877).
Footnotes
Supplementary Information: is available in the online version of the paper.
Author Contributions: H.S. and M.H.K. performed the experiments: M.H.K. and T.F.J. conceived the idea: H.S., M.H.K. and T.F.J. designed the research and wrote the manuscript: All authors commented on the final draft of the manuscript and contributed to the analysis and interpretation of the data.
The authors declare competing financial interests: details accompany the full-text HTML version of the paper.
Readers are welcome to comment on the online version of the paper.
Competing financial interests: T.F.J. is a cofounder of Snapdragon Chemistry, Inc., and a scientific adviser for Zaiput Flow Technologies, Continuus Pharmaceuticals, Paraza Pharmaceuticals, and Asymchem.
References
- 1.Arakawa H, et al. Catalysis Research of Relevance to Carbon Management: Progress, Challenges, and Opportunities. Chem Rev. 2001;101:953–996. doi: 10.1021/cr000018s. [DOI] [PubMed] [Google Scholar]
- 2.Appel AM, et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem Rev. 2013;113:6621–6658. doi: 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Q, Wu L, Jackstell R, Beller M. Using carbon dioxide as a building block in organic synthesis. Nat Commun. 2015;6 doi: 10.1038/ncomms6933. [DOI] [PubMed] [Google Scholar]
- 4.Sakakura T, Choi JC, Yasuda H. Transformation of Carbon Dioxide. Chem Rev. 2007;107:2365–2387. doi: 10.1021/cr068357u. [DOI] [PubMed] [Google Scholar]
- 5.Aresta M. In: Activation of small molecules: organometallic and bioinorganic perspectives. Tolman WB, editor. Ch 1. Wiley-VCH; 2006. pp. 1–41. [Google Scholar]
- 6.Omae I. Recent developments in carbon dioxide utilization for the production of organic chemicals. Coord Chem Rev. 2012;256:1384–1405. [Google Scholar]
- 7.Wu J, Yang X, He Z, Mao X, Hatton TA, Jamison TF. Continuous Flow Synthesis of Ketones from Carbon Dioxide and Organolithium or Grignard Reagents. Angew Chem Int Ed. 2014;53:8416–8420. doi: 10.1002/anie.201405014. [DOI] [PubMed] [Google Scholar]
- 8.Jones J. Amino acid and peptide synthesis. Oxford University Press; 2002. [Google Scholar]
- 9.Otero MD, Batanero B, Barba F. CO2 anion–radical in organic carboxylations. Tetrahedron Lett. 2006;47:2171–2173. [Google Scholar]
- 10.Morgenstern DA, Wittrig RE, Fanwick PE, Kubiak CP. Photoreduction of carbon dioxide to its radical anion by nickel cluster [Ni3(μ3-I)2(dppm)3]: formation of two carbon-carbon bonds via addition of carbon dioxide radical anion to cyclohexene. J Am Chem Soc. 1993;115:6470–6471. [Google Scholar]
- 11.Fagnoni M, Dondi D, Ravelli D, Albini A. Photocatalysis for the Formation of the C–C Bond. Chem Rev. 2007;107:2725–2756. doi: 10.1021/cr068352x. [DOI] [PubMed] [Google Scholar]
- 12.Prier CK, Rankic DA, MacMillan DWC. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicewicz DA, Nguyen TM. Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis. ACS Catal. 2014;4:355–360. [Google Scholar]
- 14.Schultz DM, Yoon TP. Solar Synthesis: Prospects in Visible Light Photocatalysis. Science. 2014;343:1239176. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beatty JW, Stephenson CRJ. Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc Chem Res. 2015;48:1474–1484. doi: 10.1021/acs.accounts.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Merging photoredox with nickel catalysis: Coupling of α-carboxyl sp3-carbons with aryl halides. Science. 2014;345:437–440. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamy E, Nadjo L, Saveant JM. Standard potential and kinetic parameters of the electrochemical reduction of carbon dioxide in dimethylformamide. J Electroanal Chem Interfacial Electrochem. 1977;78:403–407. [Google Scholar]
- 18.Fujita E, Brunschwig BS. In: Electron Transfer in Chemistry. Balzani V, editor. Ch 3. Wiley-VCH Verlag GmbH; 2001. pp. 88–126. [Google Scholar]
- 19.Matsuoka S, Kohzuki T, Pac C, Ishida A, Takamuku S, Kusaba M, Nakashima N, Yanagida S. Photocatalysis of oligo(p-phenylenes): photochemical reduction of carbon dioxide with triethylamine. J Phys Chem. 1992;96:4437–4442. [Google Scholar]
- 20.McQuade DT, Seeberger PH. Applying Flow Chemistry: Methods, Materials, and Multistep Synthesis. J Org Chem. 2013;78:6384–6389. doi: 10.1021/jo400583m. [DOI] [PubMed] [Google Scholar]
- 21.Ley SV, Fitzpatrick DE, Myers RM, Battilocchio C, Ingham RJ. Machine-Assisted Organic Synthesis. Angew Chem Int Ed. 2015;54:10122–10136. doi: 10.1002/anie.201501618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mallia CJ, Baxendale IR. The Use of Gases in Flow Synthesis. Org Process Res Dev. 2016;20:327–360. [Google Scholar]
- 23.Cambié D, Bottecchia C, Straathof NJW, Hessel V, Noël T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem Rev. 2016 doi: 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
- 24.McNally A, Prier CK, MacMillan DWC. Discovery of an α-Amino C-H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science. 2011;334:1114–1117. doi: 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chowdhury FA, Yamada H, Higashii T, Goto K, Onoda M. CO2 Capture by Tertiary Amine Absorbents: A Performance Comparison Study. Ind Eng Chem Res. 2013;52:8323–8331. [Google Scholar]
- 26.Telmesani R, Park SH, Lynch-Colameta T, Beeler AB. [2+2] Photocycloaddition of Cinnamates in Flow and Development of a Thiourea Catalyst. Angew Chem Int Ed. 2015;54:11521–11525. doi: 10.1002/anie.201504454. [DOI] [PubMed] [Google Scholar]
- 27.Singh SB. Total synthesis of flutimide, a novel endonuclease inhibitor of influenza virus. Tetrahedron Lett. 1995;36:2009–2012. [Google Scholar]
- 28.Aschwanden P, Stephenson CRJ, Carreira EM. Highly Enantioselective Access to Primary Propargylamines: 4-Piperidinone as a Convenient Protecting Group. Org Lett. 2006;8:2437–2440. doi: 10.1021/ol060876w. [DOI] [PubMed] [Google Scholar]
- 29.Clark JC, Phillipps GH, Steer MR, Stephenson L, Cooksey AR. Resolution of esters of phenylglycine with (+)-tartaric acid. J Chem Soc, Perkin Trans. 1976;1:471. [PubMed] [Google Scholar]
- 30.Wegman MA, Hacking MAPJ, Rops J, Pereira P, van Rantwijk F, Sheldon RA. Dynamic kinetic resolution of phenylglycine esters via lipase-catalysed ammonolysis. Tetrahedron: Asymmetry. 1999;10:1739–1750. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.