

Abstract
Cross-effect (CE) dynamic nuclear polarization (DNP) is a rapidly developing technique that enhances the signal intensities in magic angle spinning (MAS) NMR spectra. We report CE DNP experiments at 211, 600 and 800 MHz using a new series of biradical polarizing agents referred to as TEMTriPols, in which a nitroxide (TEMPO) and a trityl radical are chemically tethered. The TEMTriPol molecule with the optimal performance yields a record 1H NMR signal enhancement of 65 at 800 MHz at a concentration of 10 mM in a glycerol/water solvent matrix. The CE DNP enhancement for the TEMTriPol biradicals does not decrease as the magnetic field is increased in the manner usually observed for bis-nitroxides. Instead, the relatively strong exchange interaction between the trityl and nitroxide moieties determines the magnetic field at which the optimum enhancement is observed.
Keywords: solid-state NMR spectroscopy, dynamic nuclear polarization (DNP), cross-effect, polarizing agents, biradicals
Graphical abstract
A series of biradicals consisting of a nitroxide chemically tethered to a trityl are employed for cross-effect DNP at 211, 600 and 800 MHz. The relatively strong exchange interaction between the trityl and nitroxide moieties determines the field strength at which the enhancement is optimized, and yields a record 1H NMR signal enhancement of 65 at 800 MHz.
Over the last decade dynamic nuclear polarization (DNP) has emerged as a powerful method to enhance the sensitivity of magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy. Recent literature provides several elegant examples of new structural insights into complex chemical and biological systems that could not have been obtained without the extra sensitivity provided by DNP. [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] In these experiments, unpaired electrons (introduced into the NMR sample in the form of polarizing agents) are irradiated with microwaves at or near the electron Larmor frequency, and induce mutual electron-nuclear spin flips effecting a transfer of the electron spin polarization to the surrounding nuclei. Since its first application to MAS NMR at high field, [11] the efficiency of the DNP method has been increasing steadily through the development of dedicated high-power, high-frequency gyrotrons as microwave sources, specialized NMR/DNP cryo-probes, and optimization of sample preparation. Moreover, major increases in the enhancement factors have been derived from the chemical synthesis of improved polarizing agents.
The DNP mechanism that is currently most successful in high-field MAS NMR experiments is the cross-effect (CE). [12] [13] [14] [15] [16] [17] It requires two interacting electrons, one of which interacts with a nearby nucleus, commonly a proton. The difference between the electron Larmor frequencies of these two electrons must be equal to the 1H Larmor frequency, ω0e1 – ω0e2 = ±ω01H. At this simplified CE matching condition (vide infra), strong state mixing occurs and consequently there is a high probability that microwave radiation resonant with electron 1 will flip electron 2 and the coupled proton together. [18]
Initially, CE DNP experiments were performed with samples containing high concentrations of nitroxide monoradicals, up to 40 mM. [11a],[11b] The g-anisotropy of nitroxide radicals (gx = 2.0085, gy = 2.0061, gz = 2.0022) results in an electron paramagnetic resonance (EPR) frozen solution spectrum with a width of about three times the 1H Larmor frequency. Thus, within the frozen solution pairs of nitroxide radicals satisfy the CE matching condition. In 2004 Hu et al. realized that the most efficient way to introduce nitroxides into the MAS NMR sample is in pairs, in the form of biradicals. [19] A higher enhancement of the NMR signal, defined as the amplitude ratio of the DNP-enhanced and the non-enhanced signal, ε = Ion/Ioff - 1, could be accomplished at a lower radical concentration, typically 10 mM. This spawned a series of new nitroxide biradicals further optimized for CE DNP leading yet to higher enhancements. [20] [21] [22] [23] [24] [25] [26]
The most recent milestone is the nitroxide biradical AMUPol. [26] At a concentration of 10 mM in the DNP-optimized glycerol/water matrix of d8-glycerol:D2O:H2O 60:30:10 v:v:v (a.k.a. “DNP juice”) AMUPol gives enhancements on 1 M 13C-urea of 250 at 211 MHz, 235 at 400 MHz, 158 at 600 MHz, 60 at 700 MHz.1 At 800 MHz, where enhancements up to 15 have been reported for complex biomolecules,[10] this sample preparation results in an enhancement of 30. Clearly, enhancements decrease as the magnetic field is increased. In the literature this has been connected to broadening of the EPR spectrum of a frozen solution of nitroxide radicals by g-anisotropy, which decreases the fraction of the radicals in the sample that contributes to the CE roughly with 1/B0. [18a, 27]
In this communication we report CE DNP experiments at 211, 600 and 800 MHz with a new series of biradicals in which a nitroxide and a trityl radical are chemically tethered, hereafter referred to as TEMTriPols. This approach was inspired by Hu et al.,[28] who noted that the difference between gy of a nitroxide radical and the nearly isotropic g-value of trityl (gx = gy = 2.00319, gz = 2.00258) [29] satisfies the simplified CE 1H matching condition. They performed CE DNP experiments at 211 MHz on a sample containing a mixture of 20 mM TEMPO and 20 mM trityl and observed a 1H NMR enhancement of 160. [28] The structures of six TEMTriPol biradicals we studied for CE DNP are shown in Scheme 1. [30] DNP enhancements were measured on a sample containing 1 M 13C-urea in “DNP juice” doped with 10 mM of TEMTriPol at a temperature of 103 K via 13C-1H cross-polarization. Details on the experiments and instrumentation are contained in the Supporting Information. At 211 MHz the performance of all TEMTriPols we tested is inferior to AMUPol and the mixture of TEMPO and trityl, with a maximum ε = 75 observed with TEMTriPol-PPT. At 600 MHz the TEMTriPol biradicals perform surprisingly well, with TEMTriPol-1 yielding ε = 87, which is less than AMUPol, but higher than the mixture, ε = 38. At 800 MHz TEMTriPol-1 yields ε = 65, which is more than a factor of two higher than AMUPol for the same sample preparation. Thus, the TEMTriPol biradicals show that the CE DNP enhancement does not necessarily decrease at higher fields.
Scheme 1.

Molecular structures of the TEMTriPol biradicals tested for cross-effect 1H DNP. [30]
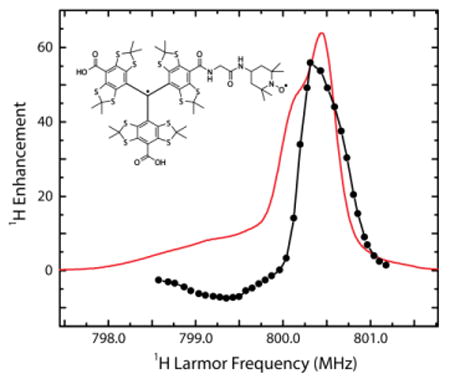
Figures 1(a-c) show the CE 1H DNP enhancement field profiles for TEMTriPol-1 at fields corresponding to 800, 600, and 211 MHz 1H Larmor frequencies. Figure 1(d) illustrates the echo-detected EPR spectrum of TEMTriPol-1 at 140 GHz. The narrow line between 4.990 and 4.995 T is due to the trityl moiety, while the broad feature spanning from 4.977→5.001 T is due to the nitroxide. Comparison of the EPR spectrum of a trityl monoradical (Figure S5) reveals a broadening of the trityl line due to interaction with the nitroxide in the TEMTriPol-1 biradical. The field profiles at 800 and 600 MHz differ drastically from the field profile at 211 MHz. The 211 MHz field profile resembles field profiles observed for bis-nitroxides. [20] [21] These profiles extend over the entire width of the nitroxide EPR spectrum (Figure 1d) and the profile is relatively symmetric (|εmax|/|εmin| is between 1 and 2). In contrast, the field profiles at 600 and 800 MHz show a narrow peak around 14.10 and 18.80 T, respectively, roughly at the position of the EPR resonance of the trityl radical. The ratio |εmax|/|εmin| is 5.8 at 600 and 8.1 at 800 MHz. A similar narrow peak and asymmetry appears in the 211 MHz field profile of the nitroxide-trityl mixture reported by Hu et al. [28] and arises from the CE between a trityl and a nitroxide. We conclude that at 211 MHz the enhancement for the TEMTriPol-1 mainly arises from an intermolecular nitroxide-nitroxide CE and that the contribution from the intramolecular trityl-nitroxide CE is small. At 600 and 800 MHz, however, the intramolecular trityl-nitroxide CE dominates and generates a large positive enhancement. The SI contains the field profiles of TEMTriPol-2, TEMTriPol-4, and TEMTriPol-PPT at 211 and at 600 MHz together with the 140 GHz EPR spectra. These field profiles show a similar rise of a narrow line associated with the trityl moiety and increased asymmetry when going from 211 to 600 MHz.
Figure 1.

Cross-effect 1H DNP field profiles of the TEMTriPol-1 at a) 800 MHz, gyrotron frequency 527.043 GHz, b) 600 MHz, gyrotron frequency 395.299 GHz, and c) 211 MHz, gyrotron frequency 139.60 GHz. In d) the echo-detected 140 GHz EPR spectrum of TEMTriPol-1 is shown, microwave frequency 139.997 GHz. The x-axes showing the magnetic field in the field profiles are scaled to match the 140 GHz EPR spectrum. Instrumentation and experimental details are given in the Supporting Information, as well as field profiles at 600 and 211 MHz and 140 GHz EPR spectra for the other TEMTriPol biradicals.
We believe that the remarkable field dependence of the CE efficiency for the TEMTriPol biradicals is related to the strength of the exchange interaction between the trityl and the nitroxide. Thus far the exchange interaction has played no role in biradical development for CE DNP. For nitroxide radicals the unpaired spin density is localized mainly on the N-O bond [32] and in a bis-nitroxide the exchange interaction between the nitroxides is smaller than the dipolar coupling, which is about 35 MHz for AMUPol. [26] However, the unpaired spin density in a trityl radical is highly delocalized, [33] [34] which means that in a TEMTriPol biradical the exchange interaction between the two radical moieties can easily be large. Using X-band EPR spectroscopy in solution at T = 279 K, Liu et al. determined the average strength of this interaction for the TEMTriPol biradicals (see Table 1). [30c] The observed exchange interaction depends on the length, flexibility, and rotational freedom of the chemical linker and can become as large as 820 MHz.
Table 1.
Results of cross-effect DNP NMR experiments at 211, 600 and 800 MHz with the TEMTriPol biradicals shown in Scheme 1, a mixture of monoradicals trityl and TEMPOL, and AMUPol. All DNP experiments were performed on samples containing 1 M 13C-urea in d8-glycerol:D2O:H2O 60:30:10 v:v:v and 10 mM biradical. The mixture contained 20 mM deuterated Finland trityl and 20 mM TEMPOL. The polarization build-up time, TB, nuclear T1 and T2 were measured for all biradical samples, as well as for an undoped sample. TB, T1, and T2 at 600 MHz are reported in the Supporting Information. The last column shows the strength of the exchange interaction, |J|, as estimated from solution EPR at X band at 279 K. [30c]
| (Bi)radical | 800 MHz / 527 GHz | 600 MHz / 395 GHz | 211 MHz / 140 GHz | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| εmax | TB (s) | T1 (s) | T2 (ms) | εmax | εmax | |J| (MHz) | |
|
| |||||||
| TEMTriPol-1 | 65[a] | 3.7 | 3.8 | 25 | 87 | 50 | 73 |
| TEMTriPol-2 | 27 | 2.4 | 2.5 | 14 | 43 | 27 | 89 |
| TEMTriPol-4 | 12 | 4.1 | 4.4 | 24 | 23 | 35 | 46 |
| TEMTriPol-PPT | 46 | 4.2 | 4.6 | 24 | 65 | 75 | 3, 36[b] |
| TEMTriPol-0 | 6 | 3.0 | 3.4 | 25 | - | 7 | 441 |
| ProxTriPol | 0 | 4.2 | 5.4 | - | - | 6 | 820 |
| Trityl-TEMPOL | - | - | - | - | 38 | 135 | - |
| AMUPol | 30 | 4.8 | 5.2 | - | 158 | 250 | 20[c] |
| Undoped | - | - | 92 | 40 | - | - | - |
Enhancements increase as the temperature is lowered. The maximum enhancement of 65 was obtained at a for this spectrometer minimum sample temperature of 100 K. At 103 K the enhancement was 61.
Two conformations were observed.
Estimate based on the value for BT-urea, which has the same chemical linker, as determined by multi-frequency EPR at 80 K. [31]
The exchange interaction, -2JS1·S2, contributes to the state mixing required for CE DNP in the same way as the dipolar interaction, d(3S1zS2z-S1·S2). The non-secular terms S1xS2x and S1yS2y introduce the off-diagonal term Do = -(d+2J). [18a] Therefore it seems advantageous for CE DNP to maximize the interaction between the two radical moieties. The good performance of AMUPol is in part contributed to its relatively strong dipolar interaction. [26] The strength of the interaction, however, should not be raised indefinitely. The simplified matching condition, ω0e1 – ω0e2 = ±ω01H, which is generally used in discussions of the CE in the literature, assumes Do ≪ ω01H. When Do and ω01H are of similar size, as is the case for TEMTriPols, it should be amended to ω01H ≈ ((ω0e1 – ω0e2)2 + Do2)1/2. [35] [36] [37] Hence, |Do| ≤ ω01H, or otherwise the degeneracy required for CE DNP cannot be accomplished.
It is now clear why TEMTriPol-0 (|J| = 441 MHz) and ProxTriPol (|J| = 820 MHz) are inefficient for CE DNP at all field strengths. Their exchange interaction is too strong to fulfill the CE matching condition, even at 800 MHz. Also, TEMTriPol-1 clearly owes its superior performance at 800 MHz at least in part to a relatively strong exchange interaction (|J| = 73 MHz). The field dependence of the enhancements observed for TEMTriPol-1, TEMTriPol-PPT and the TEMPOL-trityl mixture, however, suggests that the theoretical description given in the paragraph above is incomplete. At 211 MHz ε(mixture) > ε(TEMTriPol-PPT) > ε(TEMTriPol-1), while |J|(mixture) < |J|(TEMTriPol-PPT) < |J|(TEMTriPol-1) < 211/2 MHz (assuming d is negligible), see Table 1. On the other hand, at 600 MHz ε(TEMTriPol-1) > ε(TEMTriPol-PPT) > ε(mixture) and also at 800 MHz ε(TEMTriPol-1) > ε(TEMTriPol-PPT). The data thus suggest that there is an optimum |Do|/ω01H with Do considerably smaller than ω01H. It is possible that we approach this optimum at 600 and 800 MHz with TEMTriPol-1.
The origin of this optimum is currently not fully understood. Numerical simulations of static CE DNP on bis-nitroxide biradicals performed by Hu et al. [38] (Figure S6) show that the CE enhancement is maximum for d ≈ 26 MHz when ω0e1 = 140 GHz and for d ≈ 46 MHz when ω0e1 = 250 GHz. These optimal d-values were obtained with a T1e of 2 μs. As T1e becomes longer, dopt becomes smaller. Possibly a stronger electron-electron interaction reduces the steady-state polarization difference between the two electrons and thereby the CE enhancement.
The average strength of the exchange interaction determined for TEMTriPol-2 is similar to that of TEMTriPol-1, as is the field dependence of the enhancement, i.e. an optimal enhancement at 600 MHz (Table 1). The enhancement factor is, however, much lower. For TEMTriPol-4 the average exchange interaction approaches that of TEMTriPol-PPT. Again, the field dependence is similar, i.e. a moderate decrease in enhancement going from 211 to 600 to 800 MHz, but the enhancement itself is lower. The poor performance of TEMTriPol-2 and -4 might be related to the increased length and flexibility of the chemical linker (Scheme 1). The solution X-band EPR and frozen solution EPR at 140 GHz suggest a broad distribution in |J|-values for both TEMTriPol-2 and -4 (Reference [30c] and Figures S1 and S2), which means that at any field only a small fraction of the biradicals has the optimal interaction strength.
The asymmetry of CE DNP field profiles is associated with different electronic relaxation rates for electrons 1 and 2. If the directly excited electron is more easily saturated, this alters the steady state populations of the magnetic sublevels such that the final nuclear polarization increases. This effect was observed experimentally in CE DNP experiments on mixtures of two different types of radicals [28],[39] and is reproduced in simulations in which the relaxation is included via the stochastic Liouville equation. [28] [18b] EPR experiments show that the electronic relaxation rates T1e and Tm both are shorter for a nitroxide than for trityl in frozen solution around 100 K. [40] [41] [42] [29] The asymmetry of the field profiles of the TEMTriPol biradicals varies: |εmax|/|εmin| = 5.8, 5.4, 3.3, and 4.3 for TEMTriPol-1, -2, -4, and -PPT, respectively at 600 MHz (see SI). For TEMTriPol-1 the asymmetry increases to 8.1 at 800 MHz. Clearly the asymmetry is influenced by other factors as well.
To conclude, these de novo TEMTriPol biradicals show that CE DNP can efficiently enhance the sensitivity of MAS NMR experiments at high magnetic fields (i.e., ≥ 600 and 800 MHz). The 1H NMR signal enhancement of 65 we observe with TEMTriPol-1 at 800 MHz is the largest enhancement achieved by any DNP mechanism at 18.8 T to date. As resolution, and also sensitivity, in NMR increase with field, CE DNP with TEMTriPol-1 opens up the possibility to investigate yet larger and more complex chemical structures. The observed influence of the electron-electron interaction on the field dependence of the enhancement suggests that polarizing agents should be tailored appropriately for the field at which the CE DNP experiment is performed.
We are currently working to obtain more in-depth and quantitative understanding of the effects of strong electron-electron interaction and a difference in electronic relaxation rates between the radical moieties by means of numerical simulation of CE DNP under MAS.[43] This will be the topic of a forthcoming publication.
Supplementary Material
Acknowledgments
This research was supported by the NIBIB-NIH (EB-002804, EB-002026, and EB-016096) and the National Natural Science Foundation of China (81201126). GM gratefully acknowledges the Rubicon Fellowship from the Netherlands Organization for Scientific Research (NWO). VKM is grateful to NSERC and the Government of Canada for a Banting Postdoctoral Fellowship. Work at the 800 DNP spectrometer at UU was supported by NWO grants to MB (VICI: 700.10.443 and NWO Groot: 700.26.121) Kent Thurber is acknowledged for helpful discussion. Joe Walish is acknowledged for help with the DNP experiments at 211 MHz and 140 GHz EPR. Johan van der Zwan is acknowledged for technical support during the recording of the 800 MHz data.
Footnotes
The enhancement at 400 MHz reported by Sauvée et al. was on 0.25 M U-13C,15N-proline. Enhancements on proline tend to be slightly lower than those on urea. Sauvée et al. report an enhancement of 128 at 600 MHz on 0.25 M U-13C,15N-proline.
Supporting information for this article is given via a link at the end of the document.
References
- 1.Bajaj VS, Mak-Jurkauskas ML, Belenky M, Herzfeld J, Griffin RG. Proc Am Acad Sci. 2009;106:9244–9249. doi: 10.1073/pnas.0900908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lesage A, Lelli M, Gajan D, Caporini MA, Vitzthum V, Miéville P, Alauzun J, Roussey A, Thieuleux C, Mehdi A, Bodenhausen G, Copéret C, Emsley L. J Am Chem Soc. 2010;132:15459–15461. doi: 10.1021/ja104771z. [DOI] [PubMed] [Google Scholar]
- 3.Bayro MJ, Debelouchina GT, Eddy MT, Birkett NR, MacPhee CE, Rosay M, Maas WE, Dobson CM, Griffin RG. J Am Chem Soc. 2011;133:13967–13974. doi: 10.1021/ja203756x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lelli M, Gajan D, Lesage A, Caporini MA, Vitzthum V, Mieville P, Heroguel F, Rascon F, Roussey A, Thieuleux C, Boualleg M, Veyre L, Bodenhausen G, Coperet C, Emsley L. J Am Chem Soc. 2011;133:2104–2107. doi: 10.1021/ja110791d. [DOI] [PubMed] [Google Scholar]
- 5.Renault M, Pawsey S, Bos MP, Koers EJ, Nand D, Tommassen-van Boxtel R, Rosay M, Tommassen J, Maas WE, Baldus M. Angew Chem Int Edit. 2012;51:2998–3001. doi: 10.1002/anie.201105984. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi H, Lee D, Dubois L, Bardet M, Hediger S, De Paepe G. Angew Chem Int Ed Engl. 2012;51:11766–11769. doi: 10.1002/anie.201206102. [DOI] [PubMed] [Google Scholar]
- 7.Rossini AJ, Zagdoun A, Lelli M, Canivet J, Aguado S, Ouari O, Tordo P, Rosay M, Maas WE, Coperet C, Farrusseng D, Emsley L, Lesage A. Angew Chem Int Ed Engl. 2012;51:123–127. doi: 10.1002/anie.201106030. [DOI] [PubMed] [Google Scholar]
- 8.Lee D, Takahashi H, Thankamony AS, Dacquin JP, Bardet M, Lafon O, Paepe GD. J Am Chem Soc. 2012;134:18491–18494. doi: 10.1021/ja307755t. [DOI] [PubMed] [Google Scholar]
- 9.Fricke P, Demers JP, Becker S, Lange A. Chemphyschem. 2014;15:57–60. doi: 10.1002/cphc.201300994. [DOI] [PubMed] [Google Scholar]
- 10.a Koers EJ, van der Cruijsen EA, Rosay M, Weingarth M, Prokofyev A, Sauvee C, Ouari O, van der Zwan J, Pongs O, Tordo P, Maas WE, Baldus M. J Biomol NMR. 2014;60:157–168. doi: 10.1007/s10858-014-9865-8. [DOI] [PubMed] [Google Scholar]; b Kaplan M, Cukkemane A, van Zundert GCP, Narasimhan S, Daniels M, Mance D, Waksman G, Bonvin AMJJ, Fronzes R, Folkers GE, Baldus M. Nature Methods. 2015 doi: 10.1038/nmeth.3406. In press. [DOI] [PubMed] [Google Scholar]
- 11.a Gerfen GJ, Becerra LR, Hall DA, Griffin RG, Temkin RJ, Singel DJ. J Chem Phys. 1995;102:9494–9497. [Google Scholar]; b Hall DA, Maus DC, Gerfen GJ, Inati SJ, Becerra LR, Dahlquist FW, Griffin RG. Science. 1997;276:930–932. doi: 10.1126/science.276.5314.930. [DOI] [PubMed] [Google Scholar]
- 12.Kessenikh AV, Lushchikov VI, Manenkov AA, Taran YV. Sov Phys Solid State. 1963;5:321–329. [Google Scholar]
- 13.Kessenikh AV, Manenkov AA, Pyatnitskii GI. Sov Phys Solid State. 1964;6:641–643. [Google Scholar]
- 14.Hwang CF, Hill DA. Phys Rev Lett. 1967;18:110–112. [Google Scholar]
- 15.Hwang CF, Hill DA. Phys Rev Lett. 1967;19:1011–1014. [Google Scholar]
- 16.Wollan DS. Phys Rev B. 1976;13:3671–3685. [Google Scholar]
- 17.Wollan DS. Phys Rev B. 1976;13:3686–3696. [Google Scholar]
- 18.a Hu KN, Debelouchina GT, Smith AA, Griffin RG. J Chem Phys. 2011;134:125105. doi: 10.1063/1.3564920. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hovav Y, Feintuch A, Vega S. J Magn Reson. 2012;214:29–41. doi: 10.1016/j.jmr.2011.09.047. [DOI] [PubMed] [Google Scholar]
- 19.Hu K-N, Yu H-h, Swager TM, Griffin RG. J Am Chem Soc. 2004;126:10844–10845. doi: 10.1021/ja039749a. [DOI] [PubMed] [Google Scholar]
- 20.Song C, Hu KN, Joo CG, Swager TM, Griffin RG. J Am Chem Soc. 2006;128:11385–11390. doi: 10.1021/ja061284b. [DOI] [PubMed] [Google Scholar]
- 21.Matsuki Y, Maly T, Ouari O, Karoui H, Moigne FL, Rizzato E, Lyubenova S, Herzfeld J, Prisner T, Tordo P, Griffin RG. Angew Chem Int Ed. 2009;48:4996–5000. doi: 10.1002/anie.200805940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zagdoun A, Casano G, Ouari O, Lapadula G, Rossini AJ, Lelli M, Baffert M, Gajan D, Veyre L, Maas WE, Rosay M, Weber RT, Thieuleux C, Coperet C, Lesage A, Tordo P, Emsley L. J Am Chem Soc. 2012;134:2284–2291. doi: 10.1021/ja210177v. [DOI] [PubMed] [Google Scholar]
- 23.Dane EL, Corzilius B, Rizzato E, Stocker P, Maly T, Smith AA, Griffin RG, Ouari O, Tordo P, Swager TM. J Org Chem. 2012;77:1789–1797. doi: 10.1021/jo202349j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiesewetter MK, Corzilius B, Smith AA, Griffin RG, Swager TM. J Am Chem Soc. 2012;134:4537–4540. doi: 10.1021/ja212054e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao J, Akhmetzyanov D, Ouari O, Denysenkov V, Corzilius B, Plackmeyer J, Tordo P, Prisner TF, Glaubitz C. J Am Chem Soc. 2013;135:19275–19281. doi: 10.1021/ja409840y. [DOI] [PubMed] [Google Scholar]
- 26.Sauvée C, Rosay M, Casano G, Aussenac F, Weber RT, Ouari O, Tordo P. Angew Chem Int Ed. 2013;52:10858–10861. doi: 10.1002/anie.201304657. [DOI] [PubMed] [Google Scholar]
- 27.Maly T, Debelouchina GT, Bajaj VS, Hu K-N, Joo C-G, Mak–Jurkauskas ML, Sirigiri JR, Wel PCAvd, Herzfeld J, Temkin RJ, Griffin RG. J Chem Phys. 2008;128:052211. doi: 10.1063/1.2833582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu KN, Bajaj VS, Rosay M, Griffin RG. J Chem Phys. 2007;126:044512. doi: 10.1063/1.2429658. [DOI] [PubMed] [Google Scholar]
- 29.Lumata L, Kovacs Z, Sherry AD, Malloy C, Hill S, van Tol J, Yu L, Song L, Merritt ME. Phys Chem Chem Phys. 2013;15:9800–9807. doi: 10.1039/c3cp50186h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a Liu Y, Villamena FA, Rockenbauer A, Zweier JL. Chem Comm. 2010;46:628–623. doi: 10.1039/b919279d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu Y, Villamena FA, Song Y, Sun J, Rockenbauer A, Zweier JL. J Org Chem. 2010;775:7796–7802. doi: 10.1021/jo1016844. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liu Y, Villamena FA, Rockenbauer A, Song Y, Zweier JL. J Am Chem Soc. 2013;135:2350–2356. doi: 10.1021/ja311571v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu K-N, Song C, Yu H-h, Swager TM, Griffin RG. J Chem Phys. 2008;128:052302. doi: 10.1063/1.2816783. [DOI] [PubMed] [Google Scholar]
- 32.Engström M, Owenius R, Vahtras O. Chem Phys Lett. 2001;338:407–413. [Google Scholar]
- 33.Bowman MK, Mailer C, Halpern HJ. J Magn Reson. 2005;172:254–267. doi: 10.1016/j.jmr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Trukhan SN, Yudanov VF, Tormyshev VM, Rogozhnikova OY, Trukhin DV, Bowman MK, Krzyaniak MD, Chen H, Martyanov ON. J Magn Reson. 2013;233:29–36. doi: 10.1016/j.jmr.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanhouten J, Wenckebach WT, Poulis NJ. Physica B & C. 1977;92:201–209. [Google Scholar]
- 36.Stoll S, Epel B, Vega S, Goldfarb D. J Chem Phys. 2007;127 doi: 10.1063/1.2794329. [DOI] [PubMed] [Google Scholar]
- 37.Can TV, Ni QZ, Griffin RG. Journal of Magnetic Resonance. 2015;253:23–35. doi: 10.1016/j.jmr.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu KN. PhD Thesis. MIT; 2006. [Google Scholar]
- 39.Michaelis VK, Smith AA, Corzilius B, Haze O, Swager TM, Griffin RG. J Am Chem Soc. 2013;135:2935–2938. doi: 10.1021/ja312265x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakagawa K, Candelaria MB, Chik WWC, Eaton SS, Eaton GR. J Magn Reson. 1992;98:81–91. [Google Scholar]
- 41.Zecevic A, Eaton GR, Eaton SS, Lindgren M. Mol Phys. 1998;95:1255–1263. [Google Scholar]
- 42.Fielding AJ, Carl PJ, Eaton GR, Eaton SS. Appl Magn Reson. 2005;28:231–238. [Google Scholar]
- 43.a Thurber KR, Tycko R. J Chem Phys. 2012;137:084508. doi: 10.1063/1.4747449. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mentink-Vigier F, Akbey Ü, Hovav Y, Vega S, Oschkinat H, Feintuch A. J Magn Reson. 2012;224:13–21. doi: 10.1016/j.jmr.2012.08.013. [DOI] [PubMed] [Google Scholar]; c Thurber KR, Tycko R. Isr J Chem. 2014;54:39–46. [Google Scholar]; d Thurber KR, Tycko R. J Chem Phys. 2014;140 doi: 10.1063/1.4874341. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mentink-Vigier F, Akbey Ü, Oschkinat H, Vega S, Feintuch A. Journal of Magnetic Resonance. 2015 doi: 10.1016/j.jmr.2015.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.