

Abstract
Objective
A non-apoptotic role for Fas signaling has been implicated in the regulation of inflammation and innate immunity. These studies were performed to elucidate the role of Fas signaling on macrophages in the development of arthritis.
Methods
K/BxN serum transfer arthritis was induced in a mouse line with Fas conditionally deleted in the myeloid lineage (Fasf/f, LyzMcre). The arthritis was assessed clinically and histologically. IL-1β, CXCL5, IL-10, IL-6 and gp96 expression was determined by ELISA. Bone marrow derived macrophages were activated by IL-1β and gp96. Cells were analyzed for phenotype and apoptosis by flow cytometry.
Results
The onset of arthritis in Fasf/f, LyzMcre mice was comparable to that observed in control mice, however, resolution was accelerated during the chronic phase. The attenuated arthritis was associated with reduced articular expression of the endogenous TLR2 ligand gp96 and the neutrophil chemokine CXCL5, and the enhanced expression of IL-10. Activation with IL-1β or gp96 induced increased IL-10 in Fas deficient, compared with control, macrophages. IL-10 suppressed IL-1β plus gp96 induced IL-6 and CXCL5. IL-1β-mediated activation of ERK, which regulates IL-10 expression, was increased in Fas-deficient macrophages.
Conclusions
Together, our observations suggest that impaired Fas signaling results in the enhanced expression of anti-inflammatory IL-10 and reduced gp96, which are associated with accelerated resolution of inflammation in the chronic phase of arthritis. These observations suggest that strategies to reduce endogenous TLR ligands and increase IL-10 may be beneficial in patients with RA.
INTRODUCTION
Myeloid cells, including macrophages and neutrophils, are critical to the pathogenesis of rheumatoid arthritis (RA) through the release of proinflammatory cytokines, chemokines and other mediators such a prostaglandins and matrix metalloproteinases (1-3). Monocytes, macrophages and granulocytes express the death receptor Fas (4). RA synovial tissue (5) and RA synovial fluid macrophages also express both Fas and FasL (6). We previously demonstrated that RA synovial fluid macrophages were resistant to FasL mediated apoptosis, due to the increased expression of the anti-apoptotic molecule FLIP (6). Further, RA peripheral blood monocytes and synovial fluid macrophages are resistant to FasL-mediated apoptosis induced by activated CD4+CD25-responder T cells (7).
Mounting evidence supports a non-apoptotic role for Fas which is dependent upon cell type and context (8). Fas signaling has been implicated in T cell proliferation and activation, liver regeneration after partial hepatectomy and nerve regeneration after a crush injury (8). Previously, we demonstrated reduced collagen-induced arthritis in DBA/J mice that expressed a mutant Fas receptor (lpr) (9). Interruption of Fas-FasL interactions on human macrophages resulted in reduced TLR4 and IL-1 receptor signaling, mediated at least in part by damping of MyD88 signaling due to the increased availability of FADD, which was no longer recruited to the Fas receptor (9). In order to specifically examine the role of Fas on myeloid cells in vivo, we recently generated a myeloid linage Fas deficient line by crossing Fasf/f and LysM cre mice (10). These mice revealed no phenotypic abnormalities at 2-4 months of age (young), however, when they reached 6-8 months of age (old) they developed a systemic lupus erythematosus-like disease associated with leukocytosis, splenomegaly, antinuclear antibodies and glomerulonephritis.
In order to determine the role of myeloid specific Fas in the effector phase of arthritis, we employed young Fasf/f, LyzMcre mice to examine the immune complex-mediated K/BxN serum transfer model of arthritis. These young Fasf/f, LyzMcre mice demonstrated no alterations in the numbers of monocytes, macrophages or neutrophils. There was no alteration in the initial induction of arthritis, however, once the clinical arthritis peaked, there was a more rapid amelioration of disease in Fasf/f, LyzMcre mice This improvement was associated with reduced inflammation and neutrophil infiltration. There was no intrinsic decrease in the ability of neutrophils or monocytes to migrate to an inflammatory site and no increase of neutrophil apoptosis. While there was no difference in proinflammatory IL-1β or IL-6 within the involved joints, there was a significant increase in IL-10 and a significant reduction of the neutrophil chemokine CXCL5 and the endogenous TLR2 ligand gp96. There was no significant difference in the ability of IL-1β to induce IL-6 or CXCL5 in Fasf/f, LyzMcre or control macrophages, however, IL-1β and gp96-induced IL-10 was significantly increased with Fasf/f, LyzMcre macrophages. Further, IL10 suppressed the synergistic induction of IL-6 and CXCL5 in macrophages in response to IL-1β and gp96. These observations demonstrate that intact macrophage Fas signaling promotes ongoing inflammation by lessening the expression of IL-10 and enhancing the expression of the endogenous TLR2 ligand gp96.
MATERIALS AND METHODS
Generation and characterization of mouse line with Fas deficient in myeloid lineage
The genetically modified mouse line with myeloid linage specific deletion of floxed Fas(Fasf/f) was generated as described (10), by crossing Fasf/f mice with those expressing Cre driven by the myeloid linage-specific LysM promoter (LysMcre). Fas and LysMcre were genotyped by PCR employing genomic DNA extracted from tail biopsies. Mice genotyped with Fasf/f and LysMcre/cre (or cre/+) have Fas deleted in myeloid cells (Fasf/f, LysMcre). Littermates or gender and age matched mice with Fasf/f, LysM+ or Fas+, LysMcre served as controls. All animal procedures were approved by the Office of Research Safety and the Institutional Animal Care and Use Committee of Northwestern University.
Immunophenotypingand blood count
Fas cell surface expression was determined by multi-color fluorochrome-conjugated antibody cocktails, employing antibodies to Fas or isotype matched control IgGs. Cell types were determined by immunophenotyping employing antibodies to CD11b, F4/80, CD115, CD3, CD19, CD11c and Gr-1 (ebioscience or BD Pharmingen). Cell membrane integrity was assessed by exclusion of DAPI. Apoptosis was assessed by detecting the expression of Annexin V (BD Pharmingen). Data was acquired by BD LSR II flow cytometer (BD FACSDIVA software) and analyzed by Flowjo (TreeStar, Inc.). The complete blood counts and differentials were determined by Hemavet 950 from Drew Scientific Inc.
K/BxN serum transfer arthritis and analysis
K/BxN mice were generated and anti-GPI serum collected at 8-9 weeks of age as described (11). Arthritis was induced in 7-16 week old of Fasf/f, LyzMcre mice and age/gender matched controls employing 100 μl anti-GPI serum administrated intraperitoneally on day 0. The development of arthritis was assessed by measuring hind ankle swelling (width) using a caliper and grading the clinical index of all 4 paws/ankles on a scale of 0-3 (maximum =12) according to published methods (12, 13). Arthritis was evaluated between day 0 to 11 post induction.
Ankles were harvested at day 11 for histological analysis, cytokine/chemokine quantification, and immunophenotyping by flow cytometry. For histology, ankles were fixed in 10% neutral buffered formalin and then incubated in EDTA decalcification buffer for 2 weeks, embedded in paraffin, and then 4 μm sections were stained with hematoxylin and eosin (H&E). H&E ankle sections were evaluated by a blinded observer for inflammation (0-5), bone erosion (0-5), pannus formation (0-5) and median synovial lining thickness, neutrophil infiltration, and cartilage destruction, as previously described (13-15).
For quantification of cytokines and chemokines, ankles were homogenized in PBS containing 1x of a proteinase inhibitor cocktail and supernatants collected by centrifugation as described (12). Levels of IL-1β, IL-10, IL-6 and CXCL5 were determined by ELISA (DuoSets, R&D, Minneapolis, MN) following manufacturer’s instructions. Gp96 concentrations were determined by a mouse gp96 ELISA, as previously described (12, 16). The concentration of each protein was adjusted to mg of total ankle homogenate protein determined by BCA protein assay reagents (Thermo scientific, Rockford IL).
To further analyze the cells infiltrating the joints, tibias and paws were dissected, carefully removing muscle without opening the bone, employing an established protocol (17). The intact tibias and paws, with the ankles and knees opened, were incubated in 1 mg/ml collagenase in PBS buffer for 1 hour. The cells eluted from the knee and ankle joints were immunophenotyped by flow cytometry.
Macrophage differentiation and activation
Bone marrow-derived macrophages were generated from whole bone marrow cells isolated from the femurs and tibias of control and Fasf/f, LysMcre mice followed by in vitro differentiation in RPMI 1640 media, supplemented with 10% (v/v) FBS, 20 ng/ml of mouse GM-CSF (R&D) and 0.1 % (v/v) of 2-mercaptoethanol (GIBCO/invitrogen), as described (18). At day 7-9, the in vitro differentiated macrophages were collected by gentle pipetting, washed with PBS and were allowed to rest for 1 hour prior to activation.
Recombinant human IL-1β was purchased from R&D. Recombinant N-terminal domain of gp96 was prepared and purified with extensive endotoxin removal procedures, as previously described (16, 19). The endotoxin level in the recombinant gp96 used was below the level of detection by the limulus amebocyte lysate assay (16). Macrophages were seeded at 1×105/200 μl/well in 96 well cell culture plates. Cell activation was performed in RPMI 1640 medium supplemented with 10% FBS and 1μg/ml of polymyxin B. Macrophages were incubated with IL-1β or gp96 for 4 or 20 hours and the supernatants were harvested and examined for IL-10, IL-6 and CXCL5 by ELISA. The concentration of each cytokine was adjusted for cell number employing the MTT assay (O.D. 490 nm) at the time of terminating the activation.
Immunoblotting
IL-1β mediated macrophage Akt, GSK3, ERK, and p38 signaling was assessed by immunoblot analysis. Bone marrow-derived macrophages were incubated with IL-1β (20 ng/ml) for 15 minutes to 20 hours. Cells were lysed in buffer supplemented with 1x protease inhibitor and 1x phosphatase inhibitor cocktails (Sigma). 10-20 μg of whole cell protein extracts were resolved by 10% SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membranes (immobilon-P, Millipore Co. Bedford, MA) as described (20). Individual blots were probed with phospho-specific antibodies (Cell Signaling Technology)to Akt, GSK3, ERK, or p38. After stripping (Thermo scientific. Rockford, IL), the same blot was re-probed with antibodies to each total protein and GAPDH (Cell Signaling Technology). The immunoblots were developed employing enhanced ECL Prime Western Blotting detection reagent (GE Healthcare, Buckinghamshire, UK) and image acquisition system (Ultralum). Densitometry analysis of immunoblots was performed by ImageJ software.
Statistical analysis
The two-sided t-test was employed to analyze differences between two groups. For samples that failed the normality test, Mann-Whitney rank test was performed. Correlations were determined by Pearson linear correlation or Spearman’s non-parametric correlation if the data analyzed had a non-Gaussian distribution. Significance levels were set at 0.05.
RESULTS
Characterization of a genetically modified mouse line with Fas deficient in the myeloid lineage
In order to characterize the in vivo role of Fas on myeloid cells, we generated a genetically modified mouse line with Fas deleted in the myeloid lineage (Fasf/f, LyzMcre), which has been recently characterized (10). The Fasf/f, LyzMcre mice developed normally and demonstrated no phenotypic abnormalities prior to 6-8 months of age (10). The mice employed in this study were examined between 2 and 4 months of age. These young Fasf/f, LyzMcre mice expressed Fas on B and T lymphocytes isolated from peripheral blood (data not shown and (10)) and peritoneal cavity (Figure 1A). In contrast, Fas was deleted in peritoneal macrophages (Figure 1A) and the expression of Fas on the surface of peritoneal macrophages in Fasf/f, LyzMcre was significantly (p < 0.001) reduced compared with littermate controls (data not shown). Fas on neutrophils and monocytes in peripheral blood was also reduced compared to the controls (data not shown, and (10)).
Figure 1. Reduced anti-GPI serum induced arthritis in mice with Fas deficiency in the myeloid lineage.
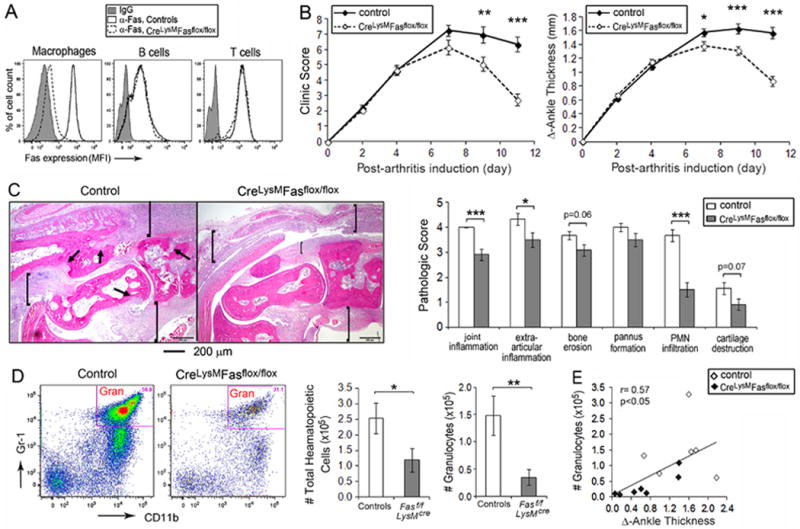
(A) Fas membrane expression on cells isolated from the resident peritoneal cavities of Fasf/f, LyzMcre and the controls were examined by Flow cytometry. Representative imaging for Fas expression level is indicated by the mean fluorescent intensity (MFI) on the surface of peritoneal macrophages, B cells and T cells. (B) The course of the arthritis was determined by the clinical activity score (0-3 for each limb, maximum of 12, right panel) and the change of ankle swelling (∆ ankle thickness in mm for hind ankles, left panel), determined between day s 2 and11 post arthritis induction. Data w ascombined from 5 individual experiments, with a total of 19-36 mice at each time point for each group. (C) Representative H&E histology imaging from the ankles of control and Fasf/f, LyzMcre mice (left panel) and the comparison of histological scoring (mean ± SE) between ankles from 9 controls and 12 Fasf/f, LyzMcre mice harvested on day 11 post -arthritis induction (right panel). The arrows identify bone erosion and pannus formation and the brackets articular and extra-articular inflammation. (D) Flow cytometry analysis of cells isolated from the joints harvested on day 11 post-arthritis induction, and the total number cells isolated from the joints (mean ± SE) for the control (n=6) and Fasf/f, LyzMcre (n=7) mice. Hematopoietic cells were identified by CD45 positivity. Granulocytes were identified by CD45+, CD11c-, CD11b+ and Gr1+ staining. Gran, granulocyte. (E) The correlation between the number of granulocytes and ankle swelling (∆ ankle thickness) was analyzed by data obtained from the same ankles indicated in panel (C). * represents p< 0.05, ** p < 0.01 and *** p < 0.001 between the indicated groups.
Deletion of Fas in myeloid cells results in more rapid resolution of K/BxN serum transfer-induced arthritis
The effect of Fas deletion in myeloid cells on the effector phase of immune complex-mediated arthritis was examined employing the K/BxN anti-glucose-6-phosphate isomerase (GPI) serum transfer model (11, 21, 22). Fasf/f, LyzMcre mice developed arthritis that was initially comparable to that observed in the controls (Figure 1B). However, after the peak of clinical disease at day 7, there was a significantly greater improvement in Fasf/f, LyzMcre mice compared with controls when evaluated by clinical score or ankle thickness (Figure 1B). Consistent with this observation, histological examination of ankles on day 11 demonstrated a significant reduction of joint (p < 0.001) and extra-articular (p < 0.05) inflammation, as well as a reduction of neutrophils (p < 0.001) (Figure 1C). A decrease in the number of neutrophils (p < 0.01) was also identified by flow cytometry employing cells lavaged from the joints of the Fasf/f, LyzMcre mice, compared with the controls (Figure 1D), and the number of neutrophils in the lavage fluids strongly correlated (r = 0.81, p < 0.01) with the clinical exam just prior to sacrifice (Figure 1E). There was a trend toward reduction of bone erosion and cartilage destruction that did not reach statistical significance (Figure 1C).
Effect of Fas deletion on monocyte and neutrophil migration
A previous study demonstrated that deletion of FasL in myeloid cells resulted in reduced thioglycollate-induced recruitment of macrophages and neutrophils, which was mediated by Fas-FasL interactions (23). Therefore, studies were performed to determine if there was an intrinsic defect of recruitment of myeloid cells to an inflammatory site in Fasf/f, LyzMcre mice. In the absence of stimulation there was no difference of total neutrophils or monocytes in the circulation between Fasf/f, LyzMcre and control mice (Figure 2A). Following the intraperitoneal injection of thioglycollate there was no difference in the recruitment of macrophages at 72 hours or neutrophils at 6 or 16 hours (Figure 2B) between Fasf/f, LyzMcre and control mice. The decrease in neutrophils in the joints of the Fasf/f, LyzMcre mice might be explained by increased apoptotic cell death. However, there was no increase of annexin V apoptotic neutrophils following the intraperitoneal injection of thioglycollate at 6 or 16 hours in the Fasf/f, LyzMcre mice (Figure 2C,D). These observations suggest there was no intrinsic difference in recruitment of Fas deficient neutrophils or monocytes to an inflammatory stimulus, and no increase of apoptosis at an inflammatory site by Fas deficient neutrophils.
Figure 2. Neutrophil migration and survival are not affected by Fas deletion.
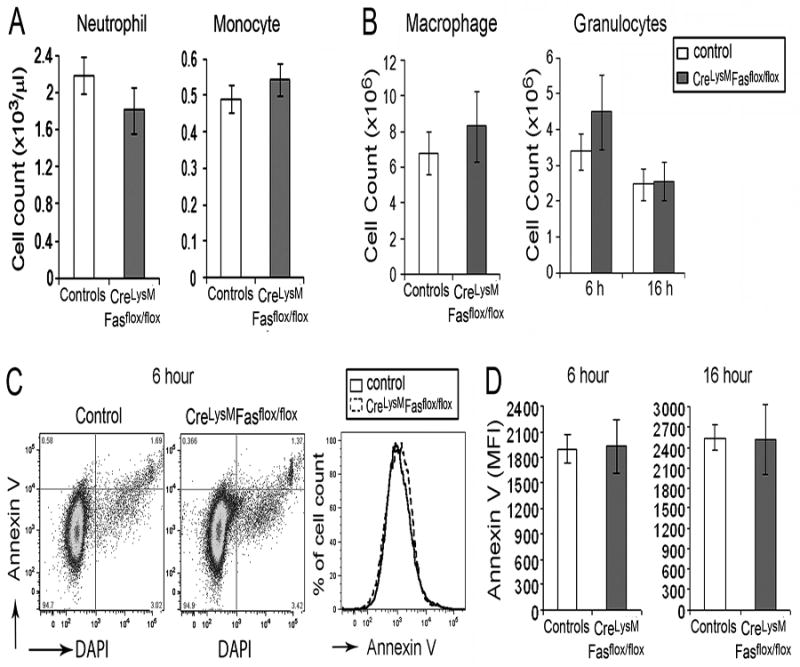
(A) The number of neutrophils and monocytes in peripheral blood, determined by complete blood count profiles (n= 37 control and 40 Fasf/f, LyzMcre mice ). (B) Number of thioglycollate induced macrophages at 72 hours and granulocytes at 6 and 16 hours analyzed by flow cytometry (n=10 in each group). (C) Representative Annexin V and DAPI flow gating for granulocytes harvested at 6 hours. (D) The MFI of Annexin V expression at6 hours (n=6) and 16 hours (n=7) on thioglycollate-induced peritoneal granulocytes.
Increased IL-10 and decreased CXCL5and gp96 in ankles from Fasf/f, LyzMcre mice
IL-1β is essential for the development of anti-GPI serum transfer-induced arthritis (24). Therefore, ankles were examined on day 11 to determine if altered expression of cytokines might contribute to the reduced arthritis in the myeloid cell-specific Fas deficient mice. Contrary to expectations, both IL1β and IL-6 (Figure 3A) were comparably expressed in the ankles of Fasf/f, LyzMcre and control mice on day 11. In contrast, the neutrophil chemokine CXCL5 was significantly (p < 0.001) reduced in the joints of the Fasf/f, LyzMcre mice, and the concentration of CXCL5 correlated (r = 0.77, p < 0.001) with ankle swelling (Figure 3B). Further, IL-10 was significantly (p < 0.001) increased in the ankles of the Fasf/f, LyzMcre mice (Figure 3C), and the concentration in the joints was inversely correlated (r = -0.59, p < 0.01) with ankle thickness. We recently demonstrated that the endogenous TLR2 ligand gp96 was strongly expressed in anti-GPI serum transfer-induced arthritis at the peak of clinical disease and that neutralization promoted disease amelioration (12). The expression of gp96 was significantly (p < 0.001) reduced in the ankles of the Fasf/f, LyzMcre mice and the concentration of gp96 strongly correlated (r = 0.86, p < 0.001) with joint swelling (Figure 3D). These observations suggest that the increased expression of IL-10 and reduction of CXCL5 and gp96 may contribute to the enhanced resolution of arthritis noticed in the Fasf/f, LyzMcre mice.
Figure 3. Increased IL10 and reduced CXCL5 and gp96 in mice with Fas deficiency in the myeloid lineage.
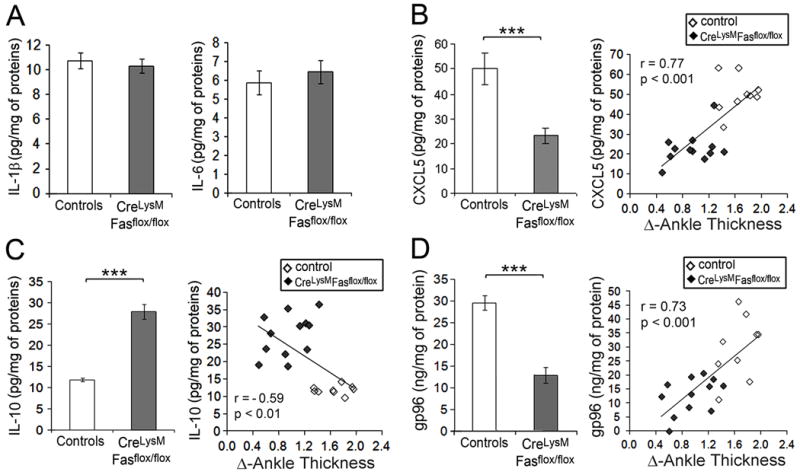
The concentration of cytokinesIL -1β, IL-6, IL-10, chemokine CXCL5 and gp96 in ankles on day11 wasdetermined by ELISA (A-D). Data present mean ± SE of 9 control and 12 Fasf/f, LyzMcre mice The correlation between the clinical arthritis (∆ of ankle thickness) and CXCL5, gp96 and IL-10 is presented on the right of each panel (B-D). The correlation coefficient (r) and the p value are indicated. *** represents p< 0.001 between the indicated groups.
Altered activation of Fas deficient macrophages by IL -1β and the TLR2 ligand gp96
Since IL-1β is essential for disease induction in the K/BxN serum transfer model (24), while gp96 promotes disease progression (12), the responses of bone marrow-derived macrophages to IL-1β and very low endotoxin gp96 were examined. No difference in the induction of either IL-6 or CXCL5 by IL-1β or gp96 inFasf/f, LyzMcre macrophages was observed, except for a reduction of IL-6 at 4 hours in response to gp96 (Figure 4A,B). In contrast, both IL-1β and gp96 induced significantly (p < 0.01) more IL-10 from Fas deficient, compared with control, macrophages (Figure 4C). These observations suggest that increased IL-1β-induced IL-10 by macrophages may contribute to the more rapid improvement noted in the Fasf/f, LyzMcre mice. Further, since gp96 was reduced in the ankles of the Fasf/f, LyzMcre mice, its diminished expression may also have contributed to the diminished disease activity later in the clinical course.
Figure 4. Increased induction of IL -10 by Fas deficient macrophages in response to IL-1β and gp96.
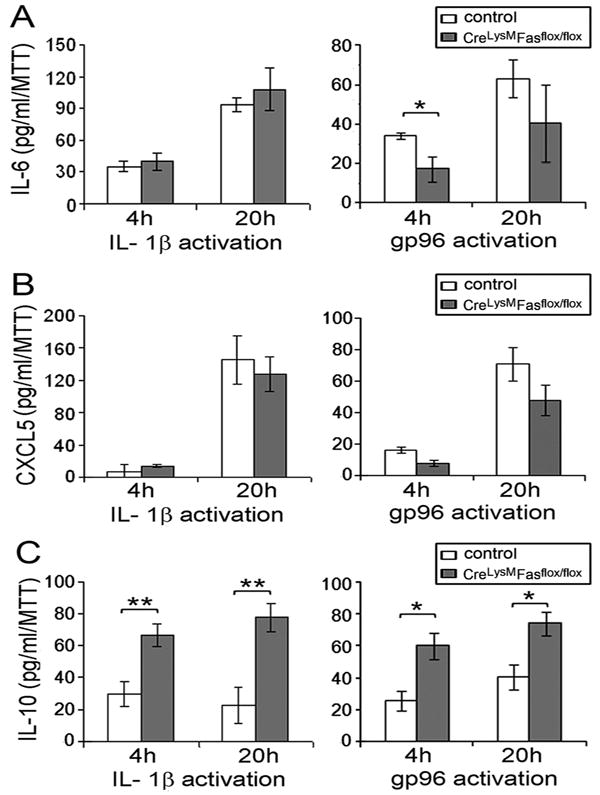
In vitro bone marrow-derived macrophages from controland Fasf/f, LyzMcre mice were activated by recombinant IL-1β (5 ng/ml) or gp96 (5 μg/ml) for 4 or 20 hours. The concentrations of IL-6 (A), CXCL5 (B) and IL-10 (C) in culture supernatants were determined by ELISA. Values were normalized for viable cell number by MTT. Data are mean ± SE of macrophages from 4 mice per group. * represents p < 0.05, and ** p < 0.01 between the indicated groups.
IL-10 suppresses macrophage activation by IL-1β and gp96
Both IL-1β and gp96 are each capable of promoting macrophage activation and are highly expressed in the ankles of wild type mice following the induction of serum transfer induced arthritis(12). Therefore the role of IL-10 in suppressing macrophage activation by these mediators was examined. The combination of IL-1β and gp96 synergistically induced the expression of IL-6 and CXCL5 (Figure 5A, B), but not IL-10 (data not shown). IL-10 significantly (p < 0.01-0.001) suppressed the expression of IL-6 and CXCL5 induced by IL-1β plus gp96 (Figure 5C,D). These observations suggest that the increased expression of IL-10 in the Fasf/f, LyzMcre mice may contribute to the reduced inflammation, in part due to suppression of proinflammatory cytokines and chemokines, such as CXCL5, downstream of IL-1β and gp96.
Figure 5. The synergistic activation of bone marrow-derived macrophages by IL-1β and gp96 is suppressed by IL-10.
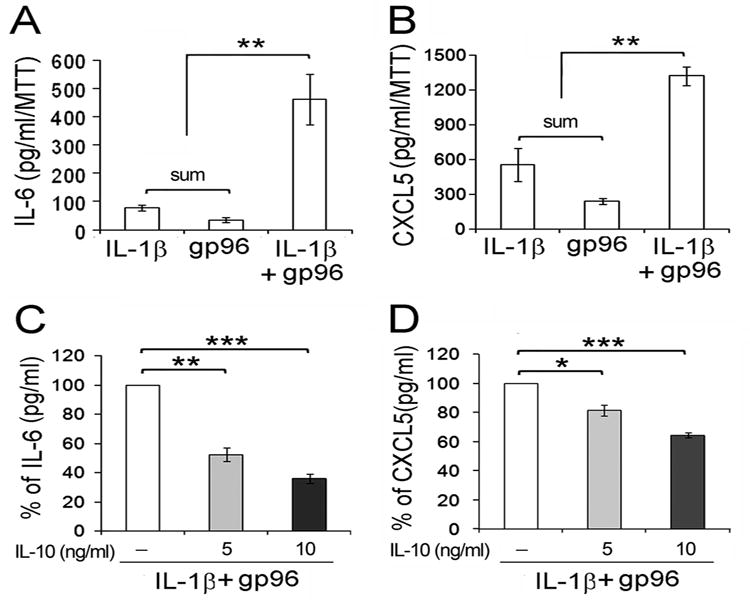
Bone marrow cellsfrom wild type B6 mice were in vitro differentiated to macrophages and incubated with recombinant IL -1β (5 ng/ml) or gp96 (10 μg/ml) alone or in combination, without or with pre -incubation with recombinant murine IL-10 at 5 or 10 ng/ml. Panels A and C represent the concen tration of IL-6 (panel A) and CXCL5 (panel B) induced without IL-10 pre-incubation. The values in panels A and B represent 100% for the data presented in panels C and D, following the addition of IL-10. The percent of activation (%) from cells pre-incubated with the indicated concentrations of IL-10 is presented in panels C and D. Data represent the mean ± SE of macrophagesfrom 4 mice per group. * represents p < 0.05, ** p< 0.01 and *** p < 0.001 between the indicated groups.
Mechanism for the increased induction of IL-10
To identify the potential mechanism for the increased IL-1β-mediated induction of IL-10 by Fas deficient macrophages, we examined pathways known to regulate the expression of IL-10 including PI3K/Akt, GSK3, ERK and p38. The activation of Akt, GSK3 and p38 were not different between the Fasf/f, LyzMcre mice and the controls (data not shown). In contrast, IL-1β-induced ERK activation was increased employing macrophages from the Fasf/f, LyzMcre mice compared with control mice (Figure 6). This observation suggests that intact Fas signaling modulates the IL-1β-induced expression of IL-10 by macrophages, mediated through ERK.
Figure 6. Increased IL-1β mediated ERK activation in Fas deficient macrophages.
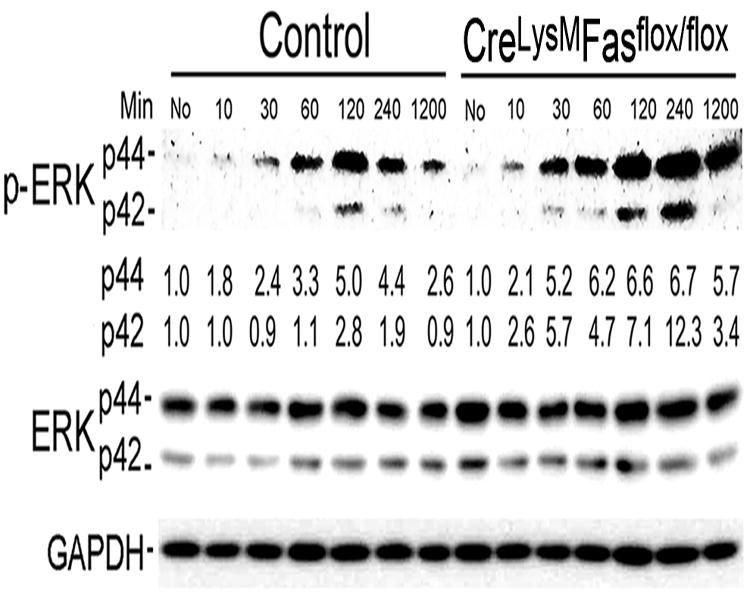
In-vitro differentiated macrophages were incubated without (no) or with IL-1β (20 ng/ml) for 15 to 1200 minutes (20 hours). Macrophage cell lysates were collected at indicated time points and analyzed by immunoblot analysis, employing antibodies to phospho-ERK of p44 and p42, total ERK of p44 and p42 and GAPDH. The numbers below each of the phosphorylated ERK proteins(p -44 and p42) represent the fold change compared to time 0, after adjusted loading with GAPDH. The relative expression of each protein is calculated by densitometry employing Image J. The gels presented in panels are representative of blots from 3 mice.
DISCUSSION
This study demonstrates that Fas expression on myeloid cells promotes anti-GPI serum transfer induced arthritis. The initial phase of the clinical course of the arthritis in Fasf/f, LyzMcre mice was comparable to that observed in control mice. However, the Fasf/f, LyzMcre mice demonstrated a more rapid amelioration during the chronic phase of the disease that was documented histologically by reduced inflammation and decreased neutrophils in the joints (Figure 1). At the time of sacrifice on day 11, the ankles from the Fasf/f, LyzMcre mice demonstrated significantly increased IL-10 and reduction of the neutrophil chemokine CXCL5 and of the endogenous TLR2 ligand gp96 (Figure 3). IL-1β is critical to the initiation of serum transfer induced arthritis (24), while the local expression of gp96 within the joint promotes progression of disease after peak clinical activity( 12). Activation of Fas deficient macrophages with IL-1β or gp96 resulted in significantly higher concentrations of IL-10 compared with control macrophages, and this was associated with increased phosphorylation of ERK. These observations suggest the absence of intact Fas signaling on macrophages results in reduced gp96 and increased IL-10 which promote resolution of the arthritis. This may be mediated, at least in part, by reduction of chemokines such as CXCL5.
A recent study demonstrated that myeloid cell FasL promotes central nervous system inflammation that results following injury (23). Mice deficient in FasL in myeloid cells demonstrated reduced spinal cord damage, reduced neutrophil infiltration and reduced neutrophil and monocyte recruitment to the peritoneum following injection with thioglycollate (23). In contrast, in the current study, although there was a reduction of neutrophils in the inflamed joints of Fasf/f, LyzMcre mice, there was no reduction of thioglycollate-induced recruitment of Fas deficient neutrophils. Neutrophil apoptosis is crucial to the resolution of inflammation (25) and reduction of neutrophil apoptosis exacerbates inflammatory arthritis (26). Fas signaling may promote neutrophil apoptosis, which is protected by the anti-apoptotic proteins Mcl-1 and Bcl-2 (27), while Mcl-1 is essential for neutrophil survival in the absence of a death signal( 28, 29). We observed no difference in spontaneous apoptosis by Fas deficient neutrophils. Therefore, although reduced neutrophils were observed in the ankles of Fasf/f, LyzMcre mice, there was no evidence for an intrinsic defect of neutrophil migration and no spontaneous increase of neutrophil cell death.
Neutrophils are critical to the pathogenesis of anti-GPI serum transfer induced arthritis (30). Neutrophil chemokines CXCL1, CXCL2, and CXCL5 and the receptor for these chemokines CXCR2 are increased in the joints of mice with serum transfer induced arthritis (31). Mice deficient in CXCR2 demonstrate attenuated serum transfer induced arthritis, with an onset similar to the control mice (31), consistent with the course observed in the Fasf/f, LyzMcre mice. We chose to examine CXCL5 because our prior study demonstrated that anti-CXCL5, but not anti-CXCL1, ameliorates IL-17 induced arthritis (32). Reduced CXCL5 was observed in the joints of the Fasf/f, LyzMcre mice, and at the time of tissue acquisition. The concentration of CXCL5 was highly correlated with joint swelling , which also correlated with the number of neutrophils in the joints. Since there was no intrinsic defect in the recruitment or survival of Fas deficient neutrophils, these observations suggest that reduced CXCL5 contributed to the amelioration during the chronic phase of the arthritis observed in the Fasf/f, LyzMcre mice.
Consistent with an earlier study in DBA/llpr/lpr mice with collagen induced arthritis, there was no reduction of proinflammatory cytokines IL-1β or IL -6 in the joints of the Fasf/f, LyzMcre mice. Therefore reduced IL-1β, which is necessary for the induction of the disease, does not appear to be the cause of the enhanced amelioration of arthritis in the Fasf/f, LyzMcre mice. Further, the induction of CXCL5 expression by IL-1β was not reduced in Fas deficient macrophages, suggesting that reduced IL-1β-induced CXCL5 was not responsible for the amelioration of arthritis. However, we observed that the endogenous TLR2 ligand gp96 was significantly reduced in the joints of the Fasf/f, LyzMcre mice on day11 (Figure 3C). Our recent studies demonstrated increased ankle gp96 during serum transfer induced arthritis, peaking at the time of maximal joint swelling and that neutralizing anti-gp96 antibody ameliorated the clinical course (12). Sine gp96 induced the expression of CXCL5 comparably in the Fas deficient and control macrophages, the reduction of gp96 in the Fasf/f, LyzMcre mice may have contributed to the reduced CXCL5 observed in the arthritic joints. Supporting this interpretation, in additional experiments employing 300 μl of K/BxN serum, a reduction of CXCL5, but not IL-10, was observed on day 4 in the ankles of the Fasf/f, LyzMcre, compared with the control, mice (data not shown).
The mechanism for the induction of gp96 in arthritis is unknown, although IL-2 and interferon γ are known to promote its expression (33, 34). However, it is highly expressed in RA synovial tissue and gp96 in RA synovial fluid is capable of activating macrophages through TLR2 (12, 16). These features have identified a pivotal role for gp96 in the persistent activation of macrophages, leading the self -perpetuating inflammatory process observed in RA (35). It is possible that other endogenous TLR ligands such as tenacin C or HMGB-1 may also contribute to the progression of arthritis(36-38). These observations suggest that a reduction of the endogenous TLR2 ligand gp96 may contribute to the amelioration of arthritis later in the clinical course in the Fasf/f, LyzMcre mice.
Previously, using human macrophages which express both Fas and FasL, we demonstrated that interrupting Fas signaling resulted in suppressed IL-1R1 and TLR4 signaling which was mediated by the interaction of FADD with MyD88, resulting in the reduced expression of IL-6 (9). Further, macrophages from lpr and gld mice demonstrated reduced TLR4-induced IL-6. Consistent with these observations, Fas deficient macrophages demonstrated reduced IL-6 at 4 hours in response to the endogenous TLR2 ligand gp96, as well as microbial TLR2 and TLR4 ligands (data not shown). However, in the current study, we observed no reduction in the induction of IL-6 or CXCL5 in response to IL-1β employing the Fas deficient murine macrophages. Nevertheless, with human macrophages interruption of Fas-FasL signaling employing an antagonistic anti-FasL antibody resulted in increased IL-1β-induced IL-6 expression and NF-κB activation (9). These differences may be due to the observation that we were not able to definitively detect FasL on the surface of murine macrophages (data not shown), consistent with the observations of others that the expression of FasL on the surface of unmanipulated macrophages is quite low (39). Therefore, the influence of Fas-FasL interactions between macrophages may be less dramatic in mice. Nonetheless, the DBA/llpr/lpr mice demonstrated no reduction of cellular or humeral immunity to collagen (9), consistent with the important role of myeloid expressed Fas observed in the current study.
IL-10 is important in the pathogenesis of rheumatoid and experimental arthritis. IL-10 is expressed in the joints of patients with RA (40). Employing ex vivo RA synovial tissue cultures, neutralization of IL-10 promoted, and the addition of exogenous IL-10 suppressed, the spontaneous expression of proinflammatory cytokines( 40), supporting the importance of IL-10 in controlling inflammation in RA. Further, the absence of IL-10 exacerbates (41, 42), while treatment with IL-10 suppresses, collagen induced arthritis (43). Additionally, IL-10 deficient mice experience increased K/BxN serum transfer induced arthritis (44), demonstrating that IL-10 suppresses, but does not prevent, arthritis in this model. IL-10 was significantly increased in the joints of the Fasf/f, LyzMcre mice compared to the controls, suggesting that increased IL-10 may contribute to the amelioration of the chronic phase of the arthritis in these mice. Other mechanisms, in addition to reduced gp96, are also possible. IL-1Ra is also increased in RA and, although only modestly effective therapeutically, is approved for the treatment of the disease. IL-1Ra deficient mice spontaneously develop arthritis (45) and develop more severe K/BxN serum transfer arthritis, while IL-1Ra transgenic mice are largely protected (44). Type I interferon β is also expressed by macrophages and other cell types and is capable of suppressing anti-GPI mediated arthritis (44), although it was not effective in RA (46). Together, these observations suggest that the enhanced expression of IL-10, and possibly other anti-inflammatory cytokines, contributed to the amelioration of arthritis observed in the Fasf/f, LyzMcre mice.
The mechanism for the increased expression of IL-10 in the joints of the Fasf/f, LyzMcre mice was examined. Incubation of Fas deficient macrophages with either IL-1β or gp96 resulted in the increased expression of IL-10. The activation of ERK and p38 are important for the induction of IL-10 by microbial TLR ligands and immune complexes, while the activation of Akt has a permissive effect by reducing the activity of GSK3 (47). In response to IL-1β, the expression of only the ERK pathway was differentially regulated between macrophages from Fasf/f, LyzMcre and control mice. There was no difference in the expression of markers of alternative macrophage differentiation including Fizz1, Arg1, Ym1 or IL-10 in GM-CSF bone marrow differentiated macrophages (data not shown), In addition, macrophages from old Fasf/f, LyzMcre mice demonstrated no increase in the constitutive activation of ERK, p38 or Akt (10). These observations suggest that increased ERK activation in response to IL-1β, and not a difference in the pattern of macrophage differentiation, may have contributed to the enhanced expression of IL-10 in the Fasf/f, LyzMcre mice.
Together these observations suggest that in the presence of intact Fas signaling on macrophages, the expression of IL-10, and possibly other suppressive cytokines, is restrained permitting the full expression of inflammation. Further, the induction of the endogenous TLR2 ligand gp96 is enhanced in the presence of intact macrophage Fas signaling, promoting disease progression and further joint destruction. These observations also support the rationale of therapeutic strategies in RA that reduce the expression of endogenous TLR ligands and promote the enhanced expression of IL-10.
References
- 1.Gierut A, Perlman H, Pope RM. Innate immunity and rheumatoid arthritis. Rheum Dis Clin North Am. 2010;36:271–96. doi: 10.1016/j.rdc.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365:2205–19. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 3.Li J, Hsu HC, Yang P, Wu Q, Li H, Edgington LE, et al. Treatment of arthritis by macrophage depletion and immunomodulation: testing an apoptosis-mediated therapy in a humanized death receptor mouse model. Arthritis Rheum. 2012;64:1098–109. doi: 10.1002/art.33423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perlman H, Pagliari LJ, Georganas C, Mano T, Walsh K, Pope RM. FLICE-inhibitory protein expression during macrophage differentiation confers resistance to fas-mediated apoptosis. J Exp Med. 1999;190:1679–88. doi: 10.1084/jem.190.11.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith MD, Weedon H, Papangelis V, Walker J, Roberts-Thomson PJ, Ahern MJ. Apoptosis in the rheumatoid arthritis synovial membrane: modulation by disease-modifying anti-rheumatic drug treatment. Rheumatology (Oxford) 2010;49:862–75. doi: 10.1093/rheumatology/kep467. [DOI] [PubMed] [Google Scholar]
- 6.Perlman H, Pagliari LJ, Liu H, Koch AE, Haines GK, 3rd, Pope RM. Rheumatoid arthritis synovial macrophages express the Fas-associated death domain-like interleukin-1beta-converting enzyme-inhibitory protein and are refractory to Fas-mediated apoptosis. Arthritis Rheum. 2001;44:21–30. doi: 10.1002/1529-0131(200101)44:1<21::AID-ANR4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 7.Jagger AL, Evans HG, Walter GJ, Gullick NJ, Menon B, Ballantine LE, et al. FAS/FAS-L dependent killing of activated human monocytes and macrophages by CD4+CD25-responder T cells, but not CD4+CD25+ regulatory T cells. J Autoimmun. 2012;38:29–38. doi: 10.1016/j.jaut.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 8.Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, et al. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–50. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 9.Ma Y, Liu H, Tu-Rapp H, Thiesen HJ, Ibrahim SM, Cole SM, et al. Fas ligation on macrophages enhances IL-1R1-Toll-like receptor 4 signaling and promotes chronic inflammation. Nat Immunol. 2004;5:380–7. doi: 10.1038/ni1054. [DOI] [PubMed] [Google Scholar]
- 10.Cuda CM, Agrawal H, Misharin AV, Haines GK, 3rd, Hutcheson J, Weber E, et al. Requirement of myeloid cell-specific Fas expression for prevention of systemic autoimmunity in mice. Arthritis Rheum. 2012;64:808–20. doi: 10.1002/art.34317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–22. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 12.Huang QQ, Koessler RE, Birkett R, Dorfleutner A, Perlman H, Kenneth Haines G, 3rd, et al. Gp96 prpetuates the persistent inflammation of rheumatoid arthritis. Arthritis Rheum. 2012;64:3638. doi: 10.1002/art.34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scatizzi JC, Hutcheson J, Bickel E, Woods JM, Klosowska K, Moore TL, et al. p21Cip1 is required for the development of monocytes and their response to serum transfer-induced arthritis. Am J Pathol. 2006;168:1531–41. doi: 10.2353/ajpath.2006.050555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scatizzi JC, Bickel E, Hutcheson J, Haines GK, 3rd, Perlman H. Bim deficiency leads to exacerbation and prolongation of joint inflammation in experimental arthritis. Arthritis Rheum. 2006;54:3182–93. doi: 10.1002/art.22133. [DOI] [PubMed] [Google Scholar]
- 15.Scatizzi JC, Hutcheson J, Bickel E, Haines GK, 3rd, Perlman H. Pro-apoptotic Bid is required for the resolution of the effector phase of inflammatory arthritis. Arthritis Res Ther. 2007;9:R49. doi: 10.1186/ar2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang QQ, Sobkoviak R, Jockheck-Clark AR, Shi B, Mandelin AM, 2nd, Tak PP, et al. Heat shock protein 96 is elevated in rheumatoid arthritisand activates macrophages primarily via TLR2 signaling. J Immunol. 2009;182:4965–73. doi: 10.4049/jimmunol.0801563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, Lu Y, Proulx ST, Guo R, Yao Z, Schwarz EM, et al. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res Ther. 2007;9:R118. doi: 10.1186/ar2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–44. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 19.Petsch D, Anspach FB. Endotoxin removal from protein solutions. J Biotechnol. 2000;76:97–119. doi: 10.1016/s0168-1656(99)00185-6. [DOI] [PubMed] [Google Scholar]
- 20.Huang Q, Ma Y, Adebayo A, Pope RM. Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 2007;56:2192–201. doi: 10.1002/art.22707. [DOI] [PubMed] [Google Scholar]
- 21.Scatizzi JC, Hutcheson J, Pope RM, Firestein GS, Koch AE, Mavers M, et al. Bim-Bcl-2 homology 3 mimetic therapy is effective at suppressing inflammatory arthritis through the activation of myeloid cell apoptosis. Arthritis Rheum. 2010;62:441–51. doi: 10.1002/art.27198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scatizzi JC, Mavers M, Hutcheson J, Young B, Shi B, Pope RM, et al. The CDK domain of p21 is a suppressor of IL-1beta-mediated inflammation in activated macrophages. Eur J Immunol. 2009;39:820–5. doi: 10.1002/eji.200838683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Letellier E, Kumar S, Sancho-Martinez I, Krauth S, Funke-Kaiser A, Laudenklos S, et al. CD95-ligand on peripheral myeloid cells activates Syk kinase to trigger their recruitment to the inflammatory site. Immunity. 2010;32:240–52. doi: 10.1016/j.immuni.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 24.Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, et al. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med. 2002;196:77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savill J. Apoptosis in resolution of inflammation. J Leukoc Biol. 1997;61:375–80. doi: 10.1002/jlb.61.4.375. [DOI] [PubMed] [Google Scholar]
- 26.Cox JH, Starr AE, Kappelhoff R, Yan R, Roberts CR, Overall CM. Matrix metalloproteinase 8 deficiency in mice exacerbates inflammatory arthritis through delayed neutrophil apoptosis and reduced caspase 11 expression. Arthritis Rheum. 2010;62:3645–55. doi: 10.1002/art.27757. [DOI] [PubMed] [Google Scholar]
- 27.Croker BA, O’Donnell JA, Nowell CJ, Metcalf D, Dewson G, Campbell KJ, et al. Fas-mediatedneutrophil apoptosis is accelerated by Bid, Bak, and Bax and inhibited by Bcl -2 and Mcl-1. Proc Natl Acad Sci U S A. 2011;108:13135–40. doi: 10.1073/pnas.1110358108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effect or function. Blood. 2009;113:2805–15. doi: 10.1182/blood-2008-05-159145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood. 2007;109:1620–6. doi: 10.1182/blood-2006-03-013771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167:1601–8. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 31.Jacobs JP, Ortiz-Lopez A, Campbell JJ, Gerard CJ, Mathis D, Benoist C. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis Rheum. 2010;62:1921–32. doi: 10.1002/art.27470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pickens SR, Chamberlain ND, Volin MV, Gonzalez M, Pope RM, Mandelin AM, 2nd, et al. Anti-CXCL5 therapy ameliorates IL-17-induced arthritis by decreasing joint vascularization. Angiogenesis. 2011;14:443–55. doi: 10.1007/s10456-011-9227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson SL, Shen T, Lou J, Xing L, Blachere NE, Srivastava PK, et al. The endoplasmic reticular heat shock protein gp96 is transcriptionally upregulated in interferon-treated cells. J Exp Med. 1994;180:1565–9. doi: 10.1084/jem.180.4.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen YG, Ashok BT, Liu X, Garikapaty VP, Mittelman A, Tiwari RK. Induction of heat shock protein gp96 by immune cytokines. Cell Stress Chaperones. 2003;8:242–8. doi: 10.1379/1466-1268(2003)008<0242:iohspg>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang QQ, Pope RM. The role of glycoprotein 96 in the persistent inflammation of rheumatoid arthritis. Arch Biochem Biophys. 2013;530:1–6. doi: 10.1016/j.abb.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–80. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 37.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 38.Page TH, Charles PJ, Piccinini AM, Nicolaidou V, Taylor PC, Midwood KS. Raised circulating tenascin-C in rheumatoid arthritis. Arthritis Res Ther. 2012;14:R260. doi: 10.1186/ar4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao PM, Tabas I. Free cholesterol loading of macrophages induces apoptosis involving the fas pathway. J Biol Chem. 2000;275:23807–13. doi: 10.1074/jbc.M002087200. [DOI] [PubMed] [Google Scholar]
- 40.Katsikis PD, Chu CQ, Brennan FM, Maini RN, Feldmann M. Immunoregulatory role of interleukin 10 in rheumatoid arthritis. J Exp Med. 1994;179:1517–27. doi: 10.1084/jem.179.5.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finnegan A, Kaplan CD, Cao Y, Eibel H, Glant TT, Zhang J. Collagen-induced arthritis is exacerbated in IL-10-deficient mice. Arthritis Res Ther. 2003;5:R18–24. doi: 10.1186/ar601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johansson AC, Hansson AS, Nandakumar KS, Backlund J, Holmdahl R. IL-10-deficient B10.Q mice develop more severe collagen-induced arthritis, but are protected from arthritis induced with anti-type II collagen antibodies. J Immunol. 2001;167:3505–12. doi: 10.4049/jimmunol.167.6.3505. [DOI] [PubMed] [Google Scholar]
- 43.Walmsley M, Katsikis PD, Abney E, Parry S, Williams RO, Maini RN, et al. Interleukin-10 inhibition of the progression of established collagen-induced arthritis. Arthritis Rheum. 1996;39:495–503. doi: 10.1002/art.1780390318. [DOI] [PubMed] [Google Scholar]
- 44.Corr M, Boyle DL, Ronacher LM, Lew BR, van Baarsen LG, Tak PP, et al. Interleukin 1 receptor antagonist mediates the beneficial effects of systemic interferon beta in mice: implications for rheumatoid arthritis. Ann Rheum Dis. 2011;70:858–63. doi: 10.1136/ard.2010.141077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191:313–20. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Holten J, Pavelka K, Vencovsky J, Stahl H, Rozman B, Genovese M, et al. A multicentre, randomised, double blind, placebo controlled phase II study of subcutaneous interferon beta-1a in the treatment of patients with active rheumatoid arthritis. Ann Rheum Dis. 2005;64:64–9. doi: 10.1136/ard.2003.020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–81. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]