

Abstract
Atrial fibrillation (AF) is an all-too-common and often challenging reality of clinical care. AF leads to significant morbidity and mortality; however, currently available treatments for AF have modest efficacy and high recurrence rates. In recent years, genetic therapy approaches have been explored in preclinical models of AF, and offer potential as a treatment modality with targeted delivery, tissue specificity, and therapy tailored to address mechanisms underlying the arrhythmia. However, many challenges remain before gene therapy can advance to a clinically relevant AF treatment. In this review, we will summarize the available published data on gene therapy and discuss the challenges, opportunities, and limitations of this approach.
Keywords: action potentials, antagomirs, genetic loci, genetic predisposition to disease, gene therapy, microRNAs
Atrial fibrillation (AF) is the most common sustained arrhythmia, affecting millions of patients worldwide with increasing prevalence (1). This arrhythmia carries significant morbidity and is well known for its stroke risk; however, the risks of congestive heart failure (CHF), dementia, and death are also significantly increased with AF (1,2). Although several epidemiological factors, such as obesity, diabetes, and CHF influence AF incidence, there is also a genetic predisposition to AF. For instance, having a first-degree relative with AF increases the chance of AF incidence (3). In recent years, genome-wide association studies (GWAS) have identified at least 14 distinct genetic loci associated with the arrhythmia (4), in addition to other loci identified through familial linkage studies. Although it is clear that genetics alone does not explain AF incidence, efforts have focused on genetics as an avenue to understand the molecular underpinnings of AF, as well as to hopefully identify novel treatment paradigms to combat this arrhythmia.
Treatment of AF often starts with the decision of rate versus rhythm control. There has been no proven mortality benefit demonstrated for rhythm control despite several randomized trials; hence, rhythm control is commonly pursued for specific patient populations and patients symptomatic from AF (5). Medical therapy is the most common preliminary strategy for rhythm control, yet antiarrhythmic drug use is limited by modest efficacy and significant possible toxicities, including proarrhythmic effects. Hence, use of these medications is limited to a subset of patient populations and not applied to the majority of AF patients. Nonpharmacological therapy in the form of catheter ablation is quite effective in certain patient populations, such as young patients with few comorbidities and those with paroxysmal AF, yet this represents a minority of patients with AF (6). When ablation is applied to patients with persistent AF, for example, efficacy rates are lower (7). Therefore, developing new therapies that could be applied more broadly would certainly be beneficial and is a focus of intense research interest. In this review, we will briefly describe some of the mechanisms and remodeling that occur in AF, and how these mechanisms may be targeted through a genetic approach to decrease the burden of AF.
MECHANISMS OF AF
Initiation and maintenance of AF requires both a driver for the arrhythmia and the appropriate substrate to maintain the rhythm. Two principal driving mechanisms are felt to be responsible for AF: focal ectopic firing and re-entry. Both of these mechanisms are supported by the particular expression of ion channels and the tissue architecture of the pulmonary veins, which underlie the reason that pulmonary vein isolation has become the cornerstone of ablation therapy for AF.
Focal ectopic activity often arises from the pulmonary veins due, in large part, to decreased coupling to the surrounding atrial substrate, a relatively depolarized resting membrane potential, and short action potential duration (APD) (8). Similar to the relative electrical insulation surrounding the human sinus node (9), decreased coupling reduces the electrotonic load on pulmonary vein cells, which increases the safety factor of conduction from the pulmonary veins to the surrounding atrial myocardium and supports automaticity (10,11). A higher resting membrane potential in pulmonary vein cells is likely due to decreased inward-rectifier potassium current (IK1) expression (12). Elevation of the resting membrane potential decreases the net inward sodium current (INa) due to partial inactivation of sodium channels, which will slow conduction, and may contribute to the slow conduction in the proximal pulmonary vein that has been seen experimentally (10,13). However this loss of depolarizing current through partial INa inactivation may not affect propagation from the pulmonary veins because decreased coupling has a larger effect on the safety factor of conduction, allowing propagation to occur (10). The APD is shorter in pulmonary vein cells compared with atrial cells, due to higher expression of channels underlying the outward repolarization currents, IKr and IKs, which reduces the effective refractory period, facilitates rapid firing, and contributes to heterogeneous repolarization, thereby supporting re-entry (12,13). In addition, altered intracellular ionized calcium (Ca2+) homeostasis can lead to afterdepolarizations, triggering focal ectopic activity as well (14). Finally, the pulmonary vein tissue architecture supports re-entry due to anisotropic, heterogeneous conduction at the junction of the pulmonary veins and the atrium (13,15).
Electrical and structural remodeling of the atrial substrate, autonomic modulation, and Ca2+-handling abnormalities all frequently occur in response to cardiac pathology (14). Electrical remodeling is typically characterized by a decrease in the L-type Ca2+ current (ICaL) and an increase in the inward-rectifier current, IK1, both of which act to shorten the atrial APD. However, this may vary, depending on comorbid conditions. For instance, in the setting of CHF with AF, an APD change is less pronounced, whereas remodeling of Ca2+-handling proteins is more apparent and unique from that seen in AF or CHF alone (16–18). In addition, alterations in the level of the constitutively-active acetylcholine-induced potassium current (IKAchC) are frequently present, which can increase the heterogeneity of the APD in the atrial substrate, favoring the maintenance of AF (19). Structural remodeling, characterized by left atrial enlargement and atrial fibrosis, also contributes to heterogeneous conduction and slower conduction velocity, facilitating the maintenance of AF. Any one of these abnormalities could potentially be the target of gene therapy to reduce the likelihood of AF.
GENE THERAPY FOR AF
The use of targeted genetic alterations to customize treatment for AF is an intriguing approach, particularly in an era of increasing calls for the personalization of medical therapy, yet it has been challenging for these efforts to progress beyond investigational tools. The advantages of gene therapy for AF include tissue specificity with less off-target effects and hopefully increased therapeutic specificity as well. For instance, one could envision tailored genetic therapy for an individual patient on the basis of certain characteristics of their disease, yet many challenges exist. Conceptually, the heterogeneity of the AF substrate may decrease the efficacy of a single genetic alteration. For clinical practice, the inherent safety concern of using gene therapy to modify the myocardium is of paramount importance. Another concern is how the genetic material would be delivered: viral vectors, plasmids, or nanoparticles are all possible. Although viral vectors are likely the most practical, legitimate concerns about their safety exist. The establishment of lasting gene expression is also a significant hurdle that must be addressed. While many challenges remain, effective and lasting therapy for AF has proven elusive and therefore innovative approaches are necessary to increase therapeutic options.
MYOCARDIAL GENE DELIVERY
Gene delivery can be accomplished with either viral or nonviral vectors, with varying degrees of gene incorporation and expression. A nonviral vector consists of a DNA plasmid containing the gene of interest, with or without other coating agents to improve the uptake of DNA into cells (20). Viral vectors have the advantage of incorporation of genetic material into the genome of the target tissue. Both adenoviral vectors and adeno-associated viral (AAV) vectors have been used in preclinical models (20–22). Although the adenovirus is able to deliver larger gene sizes, AAV vectors may produce longer (if not permanent) gene expression and have a more favorable safety profile (20,23).
Delivery of the vector to the myocardium can take multiple approaches (Figure 1). Infusion of the vector into a coronary artery is possible, and has been used in preclinical studies for rate control to deliver vectors to the atrioventricular node (24–26). For rhythm control in paroxysmal AF, however, suppression of ectopic foci in the pulmonary veins would be an ideal target, yet the pulmonary veins do not fit a common coronary distribution. In preclinical models, one typical method is to inject the vector into the myocardium, and then electroporate the tissue to improve DNA uptake (27,28). One could envision a similar delivery system via a catheter at locations that are currently treated with ablation lesions. Epicardial gene “painting” has also been validated in preclinical models with transmural gene transfer using an adenoviral vector, a weak protease to improve tissue penetration, and a polymerization agent to allow the vector to adhere to the myocardium (21). Using this technique, nearly 100% of cells examined transmurally had evidence of gene transfer. This technique in its current form could be applied during cardiac surgery, but future possibilities could include a pericardial injection or coated-balloon delivery via an endocardial route to the pulmonary veins, similar to drug-coated balloons currently used in peripheral vascular disease (29).
FIGURE 1. Possible Methods of Gene Delivery to the Left Atrium.
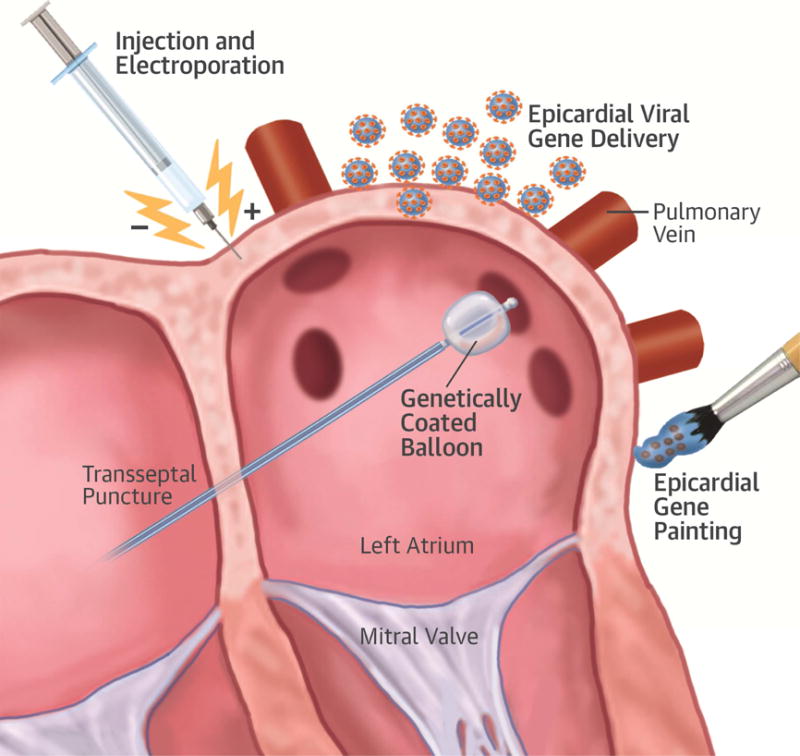
Preclinical models have demonstrated the feasibility of epicardial injection of a plasmid carrying a gene of interest coupled with electroporation, epicardial viral gene delivery, and epicardial gene painting. Future delivery methods could include balloons coated with either a plasmid or a viral delivery vector delivered to the pulmonary veins with a similar approach to that used currently for pulmonary vein isolation. LIPV = left inferior pulmonary vein; LSPV = left superior pulmonary vein; RIPV = right inferior pulmonary vein; RSPV =right superior pulmonary vein.
TARGETS FOR GENE THERAPY
A variety of targets may be appropriate candidates for gene therapy of AF, and have been the subject of 2 recent reviews by Lugenbiel et al. (30) and Donahue (20). The targets are derived from our current molecular understanding of AF, and can be largely divided by their contributions to re-entry: shortened action potential (ion channels, autonomic modulation) or delayed conduction (gap junctions, structural remodeling). Parsing the changes that are responsible for AF, as opposed to those that occur secondary to the arrhythmia, may have a significant impact on the therapeutic strategy. In the future, further targets on the basis of ever-expanding population-based genetic studies will undoubtedly present themselves as well. A review of preclinical studies that have used gene therapy to modify atrial electrophysiology is provided later and summarized in Table 1.
TABLE 1.
Summary of Preclinical Studies Modulating Atrial Electrophysiology Via Gene Therapy
| Target Class | Study | Model | Genetic Material Delivered | Vector | Delivery Approach | Goal | Outcome |
|---|---|---|---|---|---|---|---|
| Ion channel | Kikuchi et al., 2005 | Pig | KCNH2: DN mutant (G628S) | Adenovirus | Epicardial painting | ↓ IKr | ↑ APD, AERP |
| Perlstein et al., 2005 | Pig | KCNE2: sensitizing mutant (Q9E) | Plasmid | Injection | ↓ IKr | ↑ APD | |
| Amit et al., 2010 | Pig: RA tachypacing 7–21 days | KCNH2: DN mutant (G628S) | Adenovirus | Epicardial painting day 0 | ↓ IKr | ↑ APD → ↓ AF | |
| Soucek et al., 2012 | Pig: RA tachypacing 14 days | KCNH2: DN mutant (G627S) | Adenovirus | Injection + electroporation day 0 | ↓ IKr | ↑ APD, AERP → ↓ AF | |
|
| |||||||
| Autonomic innervation | Donahue et al., 2000 | Pig: acute AF | WT Gαi2 | Adenovirus | Intracoronary infusion to AVN | Rate control via ↑ Gαi2 | Slower AVN conduction, ↑ AVN ERP, ↓ heart rate |
| Bauer et al., 2004 | Pig: RA tachypacing, tCMP | Gαi2: GOF mutant (Q205L) | Adenovirus | Intracoronary infusion to AVN on day 21 | Rate control via ↑ Gαi2 | ↓ heart rate, ↑ LVEF w/constitutively active Gαi2 | |
| Aistrup et al., 2011 | Dog: acute vagal stim, carbachol | Gαi and Gαo C-terminal peptides | Plasmid | Injection + electroporation posterior LA | ↓ vagal effect and carbachol on ERP | Attenuated vagal effect on AERP, ↓ carbachol induced AF | |
| Lugenbiel et al., 2012 | Pig: RA tachypacing | Gαs siRNA | Adenovirus | Intracoronary infusion to AVN on day 0 | Rate control via ↓ Gα(s) | ↓ heart rate, preservation of LVEF | |
|
| |||||||
| Gap Junctions | Bikou et al., 2011 | Pig: RA tachypacing | Cx43 | Adenovirus | Injection + electroporation | ↑ Cx43 | ↑ Cx43 → ↓ AF |
| Igarashi et al., 2012 | Pig: RA tachypacing | Cx40 or Cx43 | Adenovirus | Epicardial painting | ↑ Cx43 or ↑ Cx40 | Homogenized conduction, ↓ AF | |
|
| |||||||
| Substrate modifier | Trappe et al., 2013 | Pig: RA tachypacing | Caspase 3 siRNA | Adenovirus | Injection + electroporation to RA and LA | ↓ caspase-3 → ↓ cardiomyocyte apoptosis | ↓ AF incidence, delayed onset of AF, preserved atrial conduction time |
| Zhang et al., 2015 | Dog: RA tachypacing | MiR-206 and anti-miR-206 | Lentivirus | Injection (left superior fat pad) | Identify miR-206 effect on autonomic remodeling, AERP | MiR 206 ↑ autonomic nerve sprouting, ↓ AERP | |
| Kunamalla et al., 2016 | Dog: RV pacing 3 weeks, tCMP | Type II TGF-β receptor: DN mutant | Plasmid | Injection + electroporation posterior LA | ↓ TGF-β signaling | ↓ LA fibrosis, homogenized conduction, ↓ restitution slope, ↓ AF duration | |
AERP = atrial effective refractory period; AF = atrial fibrillation; APD = action potential duration; AVN = atrioventricular node; Cx = connexin; DN = dominant negative; ERP = effective refractory period; GOF = gain-of-function; LA = left atrium; LVEF = left ventricular ejection fraction; miR = microRNA; RA = right atrium; RV = right ventricle; siRNA = small interfering RNA; tCMP = tachycardia-induced cardiomyopathy; TGF-β = transforming growth factor beta.
ION CHANNELS
Pharmacological manipulation of ion-channel function is the basis for currently available antiarrhythmic drugs (31–34). In particular, prolongation of the atrial action potential with consequent increased refractoriness may reduce or prevent AF. Amit et al. (35) reported a gene-therapy-based approach using this tactic in a porcine model of AF. Epicardial delivery by atrial painting of an adenovirus containing a dominant-negative variant of the alpha subunit of KCNH2 (the channel responsible for the IKr current) resulted in significant APD prolongation, with reduction in AF burden and inducibility. Differential levels of KCNH2 between the treated and untreated groups were observed at 7 days post-treatment, but not 21 days post-treatment. A similar approach was taken by Soucek et al. (36), using a different method of delivery to supply the atrium with a dominant-negative version of KCNH2. Further proof of concept that the atrial action potential can be modified by gene therapy was provided by Perlstein et al. (37). Transfection of a clarithromycin-responsive subunit mutation of the IKr regulatory subunit, hMiRP-1, allowed prolongation of the APD by administration of clarithromycin 2 weeks later. However, the efficacy of this approach in reducing or preventing atrial arrhythmia was not studied.
AUTONOMIC INNERVATION
The left atrium is richly innervated by the parasympathetic nervous system, activation of which via left cervical vagal stimulation causes shortening of the atrial effective refractory period (AERP) and increased vulnerability to AF, whereas local pharmacological blockade is protective (38). In contrast, vagal nerve stimulation has also been shown to increase atrial refractoriness, possibly through up-regulation of atrial connexin expression, and to reduce the inducibility of AF in both canine and lupine models (39,40). From a gene therapy perspective, Aistrup et al. (41) endeavored to attenuate vagal signaling in the left atrium by inhibiting the primary effector molecules of the system, Gαi and Gαo. Introduction of plasmids containing the complementary DNA for the Gαi and Gαo C-terminus peptides to the left atrium prolonged the action potential and resulted in diminished AF inducibility during vagal stimulation (41).
GAP JUNCTIONS
Reduced expression or abnormal localization of connexins 40 and 43 are associated with impaired electrical conduction in the atrium and an increased risk of developing AF (42,43). Although thought to be a secondary phenomenon, investigators have postulated that restoration of connexin biology to the undiseased state might be beneficial in the management of AF. Accordingly, gene transfer of both of these connexins using an epicardial painting approach significantly improved expression and localization of the proteins, and was associated with improved conduction and a reduction in arrhythmia burden in a porcine atrial-burst pacing model of AF (22). A separate study of connexin-43 alone in the same type of model resulted in similar findings, with a marked reduction in the development of persistent AF in treated animals compared with untreated controls (44).
SUBSTRATE MODIFIERS: APOPTOSIS
AF has been associated with local and systemic inflammation, one facet of which is cardiomyocyte apoptosis (45). In a canine model of AF, apoptosis associated with the arrhythmia was also associated with increased activity of calpain, an intracellular calcium-activated protease, and caspase-3, an apoptotic enzyme (46). With this basis for target selection, genetic knockdown of caspase-3 with an adenovirus-mediated silencing RNA in a porcine model of AF resulted in prolonged atrial conduction and delayed onset of AF with atrial burst pacing (28).
MicroRNAs are short, noncoding RNAs that are increasingly recognized to play a role in the pathogenesis of AF. Sequencing of microRNAs from the ganglionic plexus of dogs that underwent tachypacing revealed differential expression of distinct miRNAs compared with controls. The most significantly dysregulated was miRNA 206 (miR-206), which targets superoxide dismutase 1 (SOD1), a well-known mediator of apoptosis. Gene therapy of a canine model with a lentivirus containing miR-206 resulted in truncation of the atrial APD beyond what was seen with atrial tachypacing alone. Accordingly, treatment with a lentivirus containing the antagonist anti-miR-206 resulted in AERP prolongation and reduced inducibility of AF (47).
SUBSTRATE MODIFIERS: FIBROSIS
Atrial fibrosis is also a well-known factor in the pathogenesis of AF, and may particularly explain the increasing prevalence of the arrhythmia with age. A central feature of age-related fibrosis is up-regulation of transforming growth factor beta (TGF-β) (48). Kunamalla et al. (27) attempted gene-therapy-based modulation of atrial fibrosis by delivering a dominant-negative type II TGF-β receptor to the posterior left atrium in a canine model of AF. The therapy resulted in decreased fibrosis and a reduction in pacing-induced AF in the treated versus control animals (27). Interestingly, this approach also flattened the restitution slope in the treated animals, rendering the tissue more resistant to AF. TGF-β is known to affect the expression of ion channels and the magnitude of currents, such as INa. The finding of a change in restitution slope in this study suggests that targeting TGF-β not only affected structural remodeling, but electrical remodeling as well. In future studies, carefully selecting targets, such as TGF-β, that affect several aspects of remodeling may prove to be an effective strategy to combat AF. A summary of the targets discussed in this review, and other potential targets, is shown in the Central Illustration.
CENTRAL ILLUSTRATION. Possible Targets for Gene Therapy in AF.
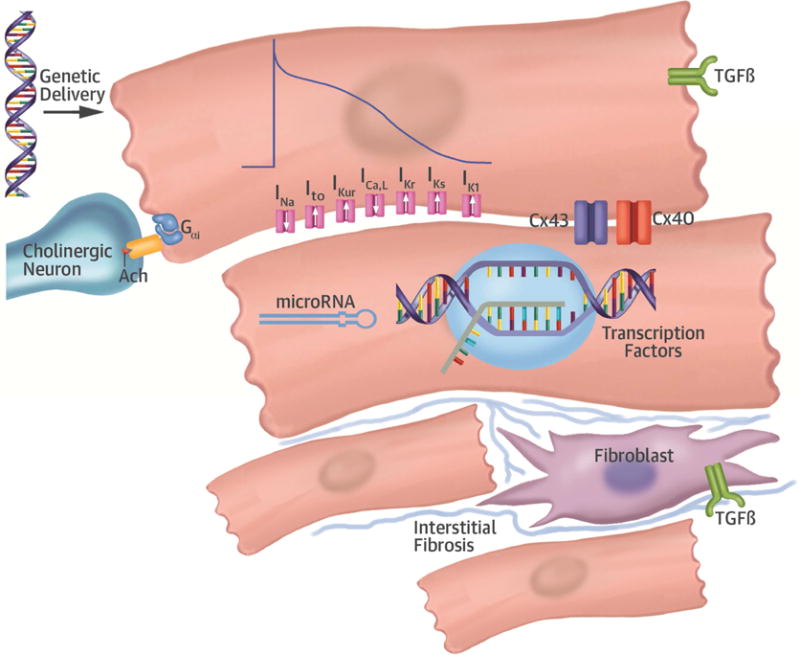
Gene therapy delivered to the atrium could target several different pathways involved in atrial remodeling underlying AF. Gene therapy targeting fibroblast proliferation and/or interstitial fibrosis, differentially expressed ion channels involved in electrical remodeling, gap junctions underlying cardiomyocyte coupling, or autonomic modulation have all been attempted in preclinical models. Future targets of gene therapy may include transcription factors, such as PITX2, affecting several pathways involved in AF susceptibility or microRNAs to modulate translation of AF targets. AChR = acetylcholine receptor; AF = atrial fibrillation; Cx = connexin; PITX2 = paired like homeodomain-2 transcription factor; TGF-β = transforming growth factor beta.
INSIGHT FROM GENETIC STUDIES
As detailed earlier, the targets for gene therapy that have been attempted thus far involve specific genetic targets aimed at addressing the electrical or structural remodeling that occurs in AF. In large GWAS studies of AF patients, the number of loci corresponding to clear electrophysiological phenotypes (ion channels, gap junctions) is relatively few. In contrast, many of the loci that have been identified in GWAS correspond to transcription factors or loci with no currently known function in cardiac physiology, yet their identification through GWAS of thousands of patients with AF strongly suggests a role in the disease. One such locus is near the paired like homeodomain-2 transcription factor (PITX2) gene, encoding a transcription factor implicated in left atrial development and ion-channel expression, which has the strongest statistical association with AF in GWAS (49). Similar to the idea of targeting TGF-β to manipulate several aspects of remodeling, targeting a transcription factor, such as PITX2, that is upstream to several pathways involved in left atrial development with a gene-therapy approach may have far-reaching effects on the atrial substrate beyond that of a single-gene approach. Such a strategy may be very useful in AF, where there is such diversity in remodeling. Intriguingly, a recent study of mice with heterozygous knockdown of PITX2 were shown to have improved sensitivity to flecainide, suggesting the possibility of using gene therapy to improve the response to antiarrhythmic therapy as well (50).
GENE THERAPY AS AN INVESTIGATIVE TOOL
Rather than using gene therapy as a treatment, an alternative use of gene therapy could be to induce AF in a preclinical model. Inducing AF in animal models is challenging due to the relatively young age of animals used, the size of certain species, and the time required for remodeling to occur. Mouse models have been invaluable in studies of basic mechanisms of AF; however, translating findings in mice to humans is inherently limited for several reasons. First, the resting heart rate of a mouse is approximately an order of magnitude faster than that of a human; therefore, the currents underlying the mouse action potential are fundamentally different, and the APD is much shorter at baseline. This difference influences the susceptibility to AF, as well as the relative impact of specific electrical remodeling (e.g., down-regulation of ICaL or increase of IK1) in mouse versus human. Second, susceptibility to re-entry is influenced by the size of the tissue, and the diminutive size of the mouse atrium influences the probability of initiating a re-entrant circuit, affecting the balance of triggered activity and re-entry in AF induced in a mouse compared with human AF. Third, the mechanisms of and propensity for triggered beats in mice are potentially different than in humans, due to differences in intracellular calcium handling. These inherent differences between mice and humans suggest the need for robust AF models in larger species.
Generating AF models in larger animal species is challenging due to their size, age, and expense. In addition, initiating AF can be difficult and time-consuming, often taking weeks of rapid atrial pacing. However genetic alteration of large animal species may offer an alternative method of creating a reproducible model of AF, or perhaps facilitate AF induction and maintenance after rapid atrial pacing. For instance, inducing atrial arrhythmias and AF in a mouse is facilitated by chamber-specific knockout of PITX2 or inactivation of microRNAs regulated by PITX2 (51,52). Applying a similar approach in large animal models may produce a reliable model that could be used to further study mechanisms of AF that are more translatable to human AF, or the efficacy of novel pharmacotherapy and ablation techniques.
The field of gene therapy has many significant challenges that must be overcome before its use will become a clinical reality. Nevertheless, the management of AF remains in urgent need of new treatment paradigms, and gene therapy offers some unique advantages that may be useful in the treatment of AF. In the near term, gene therapy may provide unique opportunities to increase our understanding of the disease, as well as possibly expanding the ability to study AF in a preclinical model. Over a longer time horizon, advances in this field may expand our treatment options for AF, but there is still a long way to go until these approaches will be broadly implemented in clinical practice.
CONDENSED.
Atrial fibrillation (AF) is the most common sustained arrhythmia, associated with increased rates of stroke, congestive heart failure, dementia, and death. Available pharmacological and invasive management strategies continue to have limited efficacy, and a different treatment paradigm is needed. Gene therapy may offer an individualized and targeted approach to AF management with potentially less off target effects than pharmacotherapy, although many challenges exist before this can become a clinical reality. Preclinical studies on this approach to AF and the challenges that remain are reviewed.
Acknowledgments
Funding: Dr. Ellinor is supported by grants from the National Institutes of Health (1RO1HL092577, R01HL128914, K24HL105780). He is also supported by an Established Investigator Award from the American Heart Association (13EIA14220013) and by the Fondation Leducq (14CVD01). Dr. Hucker was supported by Award Number T32HL007208 from the National Heart, Lung, And Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. Dr. Hanley was supported by the Brian McGovern travelling fellowship awarded by the Irish Cardiac Society.
ABBREVIATIONS AND ACRONYMS
- AAV
adeno-associated virus
- AF
atrial fibrillation
- APD
action potential duration
- CHF
congestive heart failure
- GWAS
genome-wide association study
- PITX2
paired like homeodomain-2 transcription factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: Dr. Ellinor is the principal investigator on a grant from Bayer HealthCare to the Broad Institute focused on the genetics and therapeutics of atrial fibrillation. The other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
References
- 1.Piccini JP, Hammill BG, Sinner MF, et al. Incidence and prevalence of atrial fibrillation and associated mortality among Medicare beneficiaries, 1993–2007. Circ Cardiovasc Qual Outcomes. 2012;5:85–93. doi: 10.1161/CIRCOUTCOMES.111.962688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunch TJ, Weiss JP, Crandall BG, et al. Atrial fibrillation is independently associated with senile, vascular, and Alzheimer’s dementia. Heart Rhythm. 2010;7:433–7. doi: 10.1016/j.hrthm.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Lubitz SA, Yin X, Fontes JD, et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA. 2010;304:2263–9. doi: 10.1001/jama.2010.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tucker NR, Clauss S, Ellinor PT. Common variation in atrial fibrillation: navigating the path from genetic association to mechanism. Cardiovasc Res. 2016;109:493–501. doi: 10.1093/cvr/cvv283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.January CT, Wann LS, Alpert JS, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society [Published correction appears in J Am Coll Cardiol 2014;64:2305–7] J Am Coll Cardiol. 2014;64:e1–76. doi: 10.1016/j.jacc.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 6.Calkins H, Kuck KH, Cappato R, et al. 2012 HRS/EHRA/ECAS Expert Consensus Statement on Catheter and Surgical Ablation of Atrial Fibrillation: recommendations for patient selection, procedural techniques, patient management and follow-up, definitions, endpoints, and research trial design. Europace. 2012;14:528–606. doi: 10.1093/europace/eus027. [DOI] [PubMed] [Google Scholar]
- 7.Bhargava M, Di Biase L, Mohanty P, et al. Impact of type of atrial fibrillation and repeat catheter ablation on long-term freedom from atrial fibrillation: results from a multicenter study. Heart Rhythm. 2009;6:1403–12. doi: 10.1016/j.hrthm.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Nattel S, Dobrev D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol. 2016;13:575–90. doi: 10.1038/nrcardio.2016.118. [DOI] [PubMed] [Google Scholar]
- 9.Fedorov VV, Glukhov AV, Chang R, et al. Optical mapping of the isolated coronary-perfused human sinus node. J Am Coll Cardiol. 2010;56:1386–94. doi: 10.1016/j.jacc.2010.03.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–41. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 11.Morley GE, Danik SB, Bernstein S, et al. Reduced intercellular coupling leads to paradoxical propagation across the Purkinje-ventricular junction and aberrant myocardial activation. Proc Natl Acad Sci U S A. 2005;102:4126–9. doi: 10.1073/pnas.0500881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehrlich JR, Cha TJ, Zhang L, et al. Cellular electrophysiology of canine pulmonary vein cardiomyocytes: action potential and ionic current properties. J Physiol. 2003;551:801–13. doi: 10.1113/jphysiol.2003.046417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arora R, Verheule S, Scott L, et al. Arrhythmogenic substrate of the pulmonary veins assessed by high-resolution optical mapping. Circulation. 2003;107:1816–21. doi: 10.1161/01.CIR.0000058461.86339.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol. 2014;63:2335–45. doi: 10.1016/j.jacc.2014.02.555. [DOI] [PubMed] [Google Scholar]
- 15.Hocini M, Ho SY, Kawara T, et al. Electrical conduction in canine pulmonary veins: electrophysiology and anatomic correlation. Circulation. 2002;105:2442–8. doi: 10.1161/01.cir.0000016062.80020.11. [DOI] [PubMed] [Google Scholar]
- 16.Schotten U, Verheule S, Kirchhof P, Goette A. Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol Rev. 2011;91:265–325. doi: 10.1152/physrev.00031.2009. [DOI] [PubMed] [Google Scholar]
- 17.Cha TJ, Ehrlich JR, Zhang L, Nattel S. Atrial ionic remodeling induced by atrial tachycardia in the presence of congestive heart failure. Circulation. 2004;110:1520–6. doi: 10.1161/01.CIR.0000142052.03565.87. [DOI] [PubMed] [Google Scholar]
- 18.Lugenbiel P, Wenz F, Govorov K, Schweizer PA, Katus HA, Thomas D. Atrial fibrillation complicated by heart failure induces distinct remodeling of calcium cycling proteins. PLoS One. 2015;10:e0116395. doi: 10.1371/journal.pone.0116395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makary S, Voigt N, Maguy A, et al. Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ Res. 2011;109:1031–43. doi: 10.1161/CIRCRESAHA.111.253120. [DOI] [PubMed] [Google Scholar]
- 20.Donahue JK. Biological therapies for atrial fibrillation: ready for prime time? J Cardiovasc Pharmacol. 2016;67:19–25. doi: 10.1097/FJC.0000000000000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kikuchi K, McDonald AD, Sasano T, Donahue JK. Targeted modification of atrial electrophysiology by homogeneous transmural atrial gene transfer. Circulation. 2005;111:264–70. doi: 10.1161/01.CIR.0000153338.47507.83. [DOI] [PubMed] [Google Scholar]
- 22.Igarashi T, Finet JE, Takeuchi A, et al. Connexin gene transfer preserves conduction velocity and prevents atrial fibrillation. Circulation. 2012;125:216–25. doi: 10.1161/CIRCULATIONAHA.111.053272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jessup M, Greenberg B, Mancini D, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–13. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bauer A, McDonald AD, Nasir K, et al. Inhibitory G protein overexpression provides physiologically relevant heart rate control in persistent atrial fibrillation. Circulation. 2004;110:3115–20. doi: 10.1161/01.CIR.0000147185.31974.BE. [DOI] [PubMed] [Google Scholar]
- 25.Donahue JK, Heldman AW, Fraser H, et al. Focal modification of electrical conduction in the heart by viral gene transfer. Nat Med. 2000;6:1395–8. doi: 10.1038/82214. [DOI] [PubMed] [Google Scholar]
- 26.Lugenbiel P, Thomas D, Kelemen K, et al. Genetic suppression of Gαs protein provides rate control in atrial fibrillation. Basic Res Cardiol. 2012;107:265. doi: 10.1007/s00395-012-0265-5. [DOI] [PubMed] [Google Scholar]
- 27.Kunamalla A, Ng J, Parini V, et al. Constitutive expression of a dominant negative TGF-β type II receptor in the posterior left atrium leads to beneficial remodeling of atrial fibrillation substrate. Circ Res. 2016;119:69–82. doi: 10.1161/CIRCRESAHA.115.307878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trappe K, Thomas D, Bikou O, et al. Suppression of persistent atrial fibrillation by genetic knockdown of caspase 3: a pre-clinical pilot study. Eur Heart J. 2013;34:147–57. doi: 10.1093/eurheartj/ehr269. [DOI] [PubMed] [Google Scholar]
- 29.Rosenfield K, Jaff MR, White CJ, et al. LEVANT 2 Investigators Trial of a paclitaxel-coated balloon for femoropopliteal artery disease. N Engl J Med. 2015;373:145–53. doi: 10.1056/NEJMoa1406235. [DOI] [PubMed] [Google Scholar]
- 30.Lugenbiel P, Schweizer PA, Katus HA, Thomas D. Antiarrhythmic gene therapy – will biologics replace catheters, drugs and devices? Eur J Pharmacol. 2016;791:264–273. doi: 10.1016/j.ejphar.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Brugada P, Smeets JL, Brugada J, Farré J. Mechanism of action of sotalol in supraventricular arrhythmias. Cardiovasc Drugs Ther. 1990;4(Suppl 3):619–23. doi: 10.1007/BF00357040. [DOI] [PubMed] [Google Scholar]
- 32.Kodama I, Kamiya K, Toyama J. Cellular electropharmacology of amiodarone. Cardiovasc Res. 1997;35:13–29. doi: 10.1016/s0008-6363(97)00114-4. [DOI] [PubMed] [Google Scholar]
- 33.Murray KT. Ibutilide. Circulation. 1998;97:493–7. doi: 10.1161/01.cir.97.5.493. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z, Pagé P, Nattel S. Mechanism of flecainide’s antiarrhythmic action in experimental atrial fibrillation. Circ Res. 1992;71:271–87. doi: 10.1161/01.res.71.2.271. [DOI] [PubMed] [Google Scholar]
- 35.Amit G, Kikuchi K, Greener ID, Yang L, Novack V, Donahue JK. Selective molecular potassium channel blockade prevents atrial fibrillation. Circulation. 2010;121:2263–70. doi: 10.1161/CIRCULATIONAHA.109.911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soucek R, Thomas D, Kelemen K, et al. Genetic suppression of atrial fibrillation using a dominant-negative ether-a-go-go-related gene mutant. Heart Rhythm. 2012;9:265–72. doi: 10.1016/j.hrthm.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 37.Perlstein I, Burton DY, Ryan K, et al. Posttranslational control of a cardiac ion channel transgene in vivo: clarithromycin-hMiRP1-Q9E interactions. Hum Gene Ther. 2005;16:906–10. doi: 10.1089/hum.2005.16.906. [DOI] [PubMed] [Google Scholar]
- 38.Arora R, Ulphani JS, Villuendas R, et al. Neural substrate for atrial fibrillation: implications for targeted parasympathetic blockade in the posterior left atrium. Am J Physiol Heart Circ Physiol. 2008;294:H134–44. doi: 10.1152/ajpheart.00732.2007. [DOI] [PubMed] [Google Scholar]
- 39.Chen M, Zhou X, Liu Q, et al. Left-sided noninvasive vagus nerve stimulation suppresses atrial fibrillation by upregulating atrial gap junctions in canines. J Cardiovasc Pharmacol. 2015;66:593–9. doi: 10.1097/FJC.0000000000000309. [DOI] [PubMed] [Google Scholar]
- 40.Yuan Y, Jiang Z, He Y, et al. Continuous vagal nerve stimulation affects atrial neural remodeling and reduces atrial fibrillation inducibility in rabbits. Cardiovasc Pathol. 24:395–8. doi: 10.1016/j.carpath.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 41.Aistrup GL, Cokic I, Ng J, et al. Targeted nonviral gene-based inhibition of Gαi/o-mediated vagal signaling in the posterior left atrium decreases vagal-induced atrial fibrillation. Heart Rhythm. 2011;8:1722–9. doi: 10.1016/j.hrthm.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tuomi JM, Tyml K, Jones DL. Atrial tachycardia/fibrillation in the connexin 43 G60S mutant (Oculodentodigital dysplasia) mouse. Am J Physiol Heart Circ Physiol. 2011;300:H1402–11. doi: 10.1152/ajpheart.01094.2010. [DOI] [PubMed] [Google Scholar]
- 43.Chaldoupi SM, Loh P, Hauer RNW, de Bakker JMT, van Rijen HVM. The role of connexin40 in atrial fibrillation. Cardiovasc Res. 2009;84:15–23. doi: 10.1093/cvr/cvp203. [DOI] [PubMed] [Google Scholar]
- 44.Bikou O, Thomas D, Trappe K, et al. Connexin 43 gene therapy prevents persistent atrial fibrillation in a porcine model. Cardiovasc Res. 2011;92:218–25. doi: 10.1093/cvr/cvr209. [DOI] [PubMed] [Google Scholar]
- 45.Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. 2015;12:230–43. doi: 10.1038/nrcardio.2015.2. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Gong ZH, Sheng L, et al. Anti-apoptotic effects of a calpain inhibitor on cardiomyocytes in a canine rapid atrial fibrillation model. Cardiovasc Drugs Ther. 2009;23:361–8. doi: 10.1007/s10557-009-6199-y. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, Zheng S, Geng Y, et al. MicroRNA profiling of atrial fibrillation in canines: miR-206 modulates intrinsic cardiac autonomic nerve remodeling by regulating SOD1. PLoS One. 2015;10:e0122674. doi: 10.1371/journal.pone.0122674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies L, Jin J, Shen W, et al. Mkk4 is a negative regulator of the transforming growth factor beta 1 signaling associated with atrial remodeling and arrhythmogenesis with age. J Am Heart Assoc. 2014;3:e000340. doi: 10.1161/JAHA.113.000340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tucker NR, Ellinor PT. Emerging directions in the genetics of atrial fibrillation. Circ Res. 2014;114:1469–82. doi: 10.1161/CIRCRESAHA.114.302225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Syeda F, Holmes AP, Yu TY, et al. PITX2 modulates atrial membrane potential and the antiarrhythmic effects of sodium-channel blockers. J Am Coll Cardiol. 2016;68:1881–1894. doi: 10.1016/j.jacc.2016.07.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chinchilla A, Daimi H, Lozano-Velasco E, et al. PITX2 insufficiency leads to atrial electrical and structural remodeling linked to arrhythmogenesis. Circ Cardiovasc Genet. 2011;4:269–79. doi: 10.1161/CIRCGENETICS.110.958116. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Bai Y, Li N, et al. Pitx2-microRNA pathway that delimits sinoatrial node development and inhibits predisposition to atrial fibrillation. Proc Natl Acad Sci U S A. 2014;111:9181–6. doi: 10.1073/pnas.1405411111. [DOI] [PMC free article] [PubMed] [Google Scholar]