

Abstract
Background
Ethanol (EtOH) consumption leads to an increase of proinflammatory signaling via activation of Toll‐like receptors (TLRs) such as TLR3 and TLR4 that leads to kinase activation (ERK1/2, p38, TBK1), transcription factor activation (NF κB, IRF3), and increased transcription of proinflammatory cytokines such as TNF‐α, IL‐1β, and IL‐6. This immune signaling cascade is thought to play a role in neurodegeneration and alcohol use disorders. While microglia are considered to be the primary macrophage in brain, it is unclear what if any role neurons play in EtOH‐induced proinflammatory signaling.
Methods
Microglia‐like BV2 and retinoic acid‐differentiated neuron‐like SH‐SY5Y were treated with TLR3 agonist Poly(I:C), TLR4 agonist lipopolysaccharide (LPS), or EtOH for 10 or 30 minutes to examine proinflammatory immune signaling kinase and transcription factor activation using Western blot, and for 24 hours to examine induction of proinflammatory gene mRNA using RT‐PCR.
Results
In BV2, both LPS and Poly(I:C) increased p‐ERK1/2, p‐p38, and p‐NF κB by 30 minutes, whereas EtOH decreased p‐ERK1/2 and increased p‐IRF3. LPS, Poly(I:C), and EtOH all increased TNF‐α and IL‐1β mRNA, and EtOH further increased TLR2, 7, 8, and MD‐2 mRNA in BV2. In SH‐SY5Y, LPS had no effect on kinase or proinflammatory gene expression. However, Poly(I:C) increased p‐p38 and p‐IRF3, and increased expression of TNF‐α, IL‐1β, and IL‐6, while EtOH increased p‐p38, p‐IRF3, p‐TBK1, and p‐NF κB while decreasing p‐ERK1/2 and increasing expression of TLR3, 7, 8, and RAGE mRNA. HMGB1, a TLR agonist, was induced by LPS in BV2 and by EtOH in both cell types. EtOH was more potent at inducing proinflammatory gene mRNA in SH‐SY5Y compared with BV2.
Conclusions
These results support a novel and unique mechanism of EtOH, TLR3, and TLR4 signaling in neuron‐like SH‐SY5Y and microglia‐like BV2 that likely contributes to the complexity of brain neuroimmune signaling.
Keywords: Ethanol, Innate Immune, Microglia, Neuron, Toll‐Like Receptor
Ethanol (EtOH) consumption causes increased proinflammatory signaling in brain that is linked with neurodegeneration (Collins et al., 1996; Crews et al., 2004, 2006; Qin et al., 2008; Reynolds et al., 2015), alcoholism (He and Crews, 2008; Vetreno et al., 2013), fetal alcohol syndrome disorder (Drew and Kane, 2014), and drinking behavior (Agrawal et al., 2011; Blednov et al., 2005, 2012). EtOH treatment in mice increases transcription of proinflammatory cytokines such as TNF‐α, IL‐1β, and IL‐6 in brain (Alfonso‐Loeches et al., 2010; Qin and Crews, 2012a; Qin et al., 2008). EtOH is thought to promote this proinflammation in brain through activation of Toll‐like receptors (TLRs), a family of 11 receptors (13 in mice) that react to viral and bacterial components (pathogen‐associated molecular patterns [PAMPs]). In particular, TLR4 and TLR3 have been shown to be involved in proinflammatory innate immune signaling by EtOH. EtOH treatment in mice increases both mRNA expression and number of immunoreactive cells for TLR3 and TLR4 in brain, as well as increased TLR3 and TLR4 immunoreactive cells in postmortem human alcoholic brain (Crews et al., 2013). Knockout of TLR4 in mice prevents EtOH‐induced increases in proinflammatory cytokines TNF‐α, IL‐1β, and IL‐6 (Alfonso‐Loeches et al., 2010), and knockout of TLR3 in mice decreases EtOH consumption (Jang et al., 2016). We have previously found that EtOH exposure potentiates both TLR4 agonist‐induced (Qin et al., 2008) and TLR3 agonist‐induced (Qin and Crews, 2012a) expression of proinflammatory innate immune signaling molecules (e.g., TNF‐α, IL‐1β), implying that EtOH activates both TLR3 and TLR4 pathways. However, it is not known to what extent EtOH mimics activation of either TLR3 or TLR4.
Both TLR3 and TLR4 respond to different agonists, yet ultimately both promote proinflammatory signaling through kinase activation. TLR3 is traditionally activated by double‐stranded RNA, with polyinosinic:polycytidylic acid [Poly(I:C)] being the most commonly used mimetic, and initiates signaling through a Myd88‐independent pathway (Narayanan and Park, 2015). The canonical agonist for TLR4 is lipopolysaccharide (LPS), and unlike TLR3, TLR4 uses coadaptor proteins (CD14 and MD‐2) as well as signaling through both Myd88‐dependent and Myd88‐independent pathways (Narayanan and Park, 2015). However, activation of both these receptors leads to activation of downstream signaling kinases (e.g., MAPKs like ERK1/2 and p38; TBK1), leading to activation of kinases such as NFκB and IRF3 which result in increased transcription of various proinflammatory cytokines, such as IL‐1β, IL‐6 and TNF‐α, as well as additional TLR expression (Narayanan and Park, 2015). These cytokines then signal through their respective receptors to promote a proinflammatory state (Newton and Dixit, 2012). EtOH has been shown to affect various components of this immune signaling pathway in brain, with EtOH increasing expression of Myd88 and CD14 (Alfonso‐Loeches et al., 2010) in whole mouse brain. Further, EtOH increases activation of p38, ERK1/2, NFκB, and IRF3 in cultured primary microglia (Fernandez‐Lizarbe et al., 2009).
As the brain is a sterile environment, lacking viral and bacterial products (PAMPS) that are present in the periphery, discovery of innate immune signaling in brain has led to increased interest in endogenous TLR agonists. Damage‐associated molecular patterns (DAMPs), have been shown to be released from cells following stress, inflammation, or cell death, and lead to activation of TLRs (Chen and Nunez, 2010). One such DAMP, HMGB1, functions as an agonist at TLR2, TLR4, and TLR9 (Park et al., 2004; Yang et al., 2013). HMGB1 also binds to receptor for advanced glycation end products (RAGE) (Kokkola et al., 2005), a receptor which leads to increased activation of NFκB (Han et al., 2011). EtOH up‐regulates both HMGB1 (Crews et al., 2013; Lippai et al., 2013) and RAGE (Vetreno et al., 2013) in brain, suggesting a role for these molecules in EtOH‐induced proinflammatory signaling. While DAMPs are commonly thought to be passively released with cell death, HMGB1 is also released actively by immune‐competent cells (Andersson and Tracey, 2011). Interestingly, EtOH‐induced release of HMGB1 in hippocampal brain slices occurs without any evidence of cell death, and may occur via a histone deacetylase‐dependent manner (Zou and Crews, 2014).
The aforementioned TLR signaling pathway has been established in macrophages, such as the canonical macrophage in brain, microglia (Rivest, 2009). Microglia indeed play an integral role in EtOH‐induced innate immune signaling (Fernandez‐Lizarbe et al., 2009, 2013) and neuronal toxicity (Boyadjieva and Sarkar, 2010). While another cell type, astrocytes, has been implicated alcohol use disorders and displays proinflammatory innate immune signaling following EtOH treatment (Adermark and Bowers, 2016; Alfonso‐Loeches et al., 2010), neurons have largely been overlooked in having a possible role in the innate immune system. However, TLRs have recently been reported to be on neurons as well. In particular, primary mouse cortical neurons were found to express TLR3 and TLR4 using both single cell PCR and immunocytochemistry (Tang et al., 2007), and expression TLR4 on primary mouse cortical neurons using Western blot was also reported (Lok et al., 2015). Previously, we have also shown colocalization of TLR3 and TLR4 on rat neurons using immunohistochemistry (Vetreno and Crews, 2012). Furthermore, there has been evidence that TLR3 agonist Poly(I:C) increases p38 kinase activation and TLR3 expression in neuronal cell line SH‐SY5Y (Nessa et al., 2006). In addition, Poly(I:C) increases mRNA expression of TNF‐α and IL‐6 in neuronal cell line NT2‐N (Prehaud et al., 2005). However, despite evidence for neuronal TLR4, previous reports suggest that TLR4 agonist LPS does not affect TNF‐α, IL‐6, or IL‐1β release in SH‐SY5Y (Klegeris and McGeer, 2001) nor does it increase cytokine release in neuronal cell line NT2‐N (Prehaud et al., 2005). In addition, in vivo studies using flow cytometry suggest that TLR4 is predominantly located on microglia (Schwarz et al., 2013), suggesting that neurons lack significant TLR signaling. Despite some conflicting evidence, these data suggest that some level of proinflammatory innate immune activity may exist in neurons, but it is unclear whether neurons demonstrate a macrophage‐like signaling pathway activation (e.g., activation of kinases such as MAPKs that lead to NFκB activation). It is also unknown whether EtOH affects innate immune signaling in neurons.
In order to gain insight into the respective involvement of microglia and neurons in EtOH‐induced proinflammatory innate immune signaling, we utilized a mouse microglial cell line (BV2) and a retinoic acid‐differentiated human neuroblastoma cell line (SH‐SY5Y). The BV2 cell line is well established as a model for microglia, with studies finding that the LPS response in primary glia is highly similar in BV2 (Henn et al., 2009). Neuron‐like SH‐SY5Y express synaptic proteins and mature neuronal markers after differentiation with retinoic acid (Cheung et al., 2009), and are commonly used to model dopamine neurons in Parkinson's disease, a condition known to involve neuroinflammation (Korecka et al., 2013). While it is unknown exactly how EtOH affects proinflammatory gene expression in brain (e.g., increases in TNF‐α, IL‐1β, IL‐6), it is possible that it has direct effect on transcription through factors such as NFκB, or through release of TLR agonist HMGB1. Therefore, we examined both kinases (ERK1/2, p38, TBK1) and transcription factors (IRF3, NFκB) as well as HMGB1 release in conjunction with mRNA expression of various proinflammatory cytokines, TLRs, and TLR‐associated proteins, and compared the EtOH findings to both TLR3 and TLR4 stimulation in both microglia‐like BV2 and neuron‐like SH‐SY5Y.
Given the aforementioned data, we hypothesized that (i) neuron‐like SH‐SY5Y express various components of the innate immune signaling pathway, including TLRs, TLR‐associated proteins, and cytokines; (ii) neuron‐like SH‐SY5Y display activation of kinases and induction of proinflammatory gene expression in response to TLR3 stimulation, but not TLR4; (iii) EtOH causes activation of kinases and up‐regulation of proinflammatory gene expression in neuron‐like SH‐SY5Y in a manner unique to that to microglia‐like BV2; and (iv) EtOH activates kinases and induces of proinflammatory genes unique to either TLR3 or TLR4 stimulation. Our results indicate that EtOH has a unique and broader pattern of proinflammatory gene induction compared with either TLR3/TLR4 stimulation alone in both cell types and that EtOH and Poly(I:C) both activate unique proinflammatory innate immune signaling kinase and gene induction in neuron‐like SH‐SY5Y.
Materials and Methods
Cell Culture and Treatment
BV2 were acquired from ICLC (#ATL03001; Genoa, Italy). BV2 were cultured using Dulbecco's modified Eagle's serum (DMEM; Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Life Technologies), 1× GlutaMAX (Life Technologies), and 1× antibiotic–antimycotic (Life Technologies). Sixteen hours prior to treatments, media was changed to 2% FBS.
SH‐SY5Y were acquired from ATCC (#CRL‐2266; Manassas, VA). SH‐SY5Y were cultured using DMEM/F‐12+ GlutaMAX (Life Technologies), 10% FBS, and 1× antibiotic–antimycotic. Prior to treatments, SH‐SY5Y were differentiated using 10 μM retinoic acid (RA; #R2625; Sigma‐Aldrich, St. Louis, MO) for 4 days in Neurobasal media (Life Technologies) containing 2% B27 supplement (Life Technologies), 0.5 mM GlutaMAX, and 1× antibiotic–antimycotic. Media was refreshed 16 hours prior to treatments.
Both cell types were maintained in a humidified 5% CO2 incubator at 37°C. Passage number at the time of treatment did not exceed 10. Cells were treated at ~90% confluency with either 50 μg/ml Poly(I:C) (#27‐4732‐01; Amersham/GE Healthcare, Pittsburgh, PA), 100 ng/ml LPS (#L2630; Sigma‐Aldrich), or varying concentrations of EtOH. Concentrations were chosen due to prior literature on Poly(I:C) (Nessa et al., 2006) and LPS (Shin et al., 2014) treatment in these cells. Cells treated with EtOH were placed in a separate EtOH‐saturated chamber to prevent evaporation of EtOH from the media. Cell death following treatments was measured using trypan blue exclusion assay.
Western Blot
Cells were washed with ice‐cold PBS and lysed on ice for 15 minutes using cold lysis buffer (20 mM Tris, 0.25 M sucrose, 2 mM EDTA, 10 mM EGTA, 1% Triton X‐100) and 1 tablet of Complete Ultra protease inhibitor cocktail tablets (Roche, Mannheim, Germany) and PhosSTOP phosphatase inhibitor cocktail tablet (Roche)/10 ml. Samples were centrifuged at 16,000×g for 15 minutes, and protein‐containing supernatant was transferred to a separate tube. Total protein concentration was assessed using a BCA protein assay kit (Pierce, Rockford, IL). Ten micrograms of protein was mixed with 1× loading buffer (Pierce), boiled for 5 minutes, and ran on Mini‐PROTEAN TGX Stain‐Free gels (Bio‐Rad, Hercules, CA). Following transfer to a Trans‐Blot Turbo nitrocellulose membrane (Bio‐Rad), the blot was blocked with Odyssey Blocking Buffer (Li‐Cor, Lincoln, NE), and incubated with primary antibodies neuron‐specific enolase (NSE; #ab53025, 1:500; Abcam, Cambridge, UK), β‐tubulin‐III (Tubb3; #ab18207, 1:1000; Abcam), Cd11b (#NB110‐89474, 1:500; Novus Biologicals, Littleton, CO), HMGB1 (#ab18256, 1:400; Abcam), TLR4 (#sc‐293072, 1:500; Santa Cruz, Dallas, TX), RAGE (#ab3611, 1:500; Abcam), phospho(p)‐ERK1/2 (#4370, 1:500; Cell Signaling), p‐p38 MAPK (#4511, 1:500; Cell Signaling), p‐TBK1 (#5483, 1:500; Cell Signaling), pIRF3 (#4947, 1:500; Cell Signaling), p‐NFκB‐p65 (#3033, 1:500; Cell Signaling), and β‐actin (#sc‐47778, 1:500; Santa Cruz) overnight at 4°C, followed by IRDye 700DX anti‐rabbit (#611‐730‐127; Rockland, Limerick, PA) or IRDye 800DX anti‐mouse (#610‐731‐124; Rockland) secondary antibodies for 2 hours at room temperature. Protein bands were visualized using an Odyssey fluorescent scanner, with a protein ladder (Odyssey; Li‐Cor) used as a size reference.
Flow Cytometry
Cells were detached and stained with violet LD (#L34955; Invitrogen, Carlsbad, CA) as a live–dead discrimination marker. Cells were then stained with Cd11b‐PE/Cy7 (1:500; #25‐0112‐82; eBioscience, San Diego, CA), washed with PBS and then permeabilized using Fix/Perm buffer (BD Biosciences) and stained with primary antibody Tubb3 (1:1,000, #ab18207; Abcam) followed by secondary antibody D649 (1:800, #406406; Biolegend, San Diego, CA). Isotype controls (rabbit IgG, #171870; Abcam; rat IgG, #25‐4031‐82; eBioscience) were used to account for background staining. Cells were fixed using 4% paraformaldehyde and analyzed using a CyAN cytometer. FlowJo (Treestar, OR) was used to analyze flow cytometry data.
Real‐Time PCR
Total RNA was extracted from cell lysates using TRIzol (Invitrogen). RNA concentration was determined using a NanoDrop (Thermo Scientific, Waltham, MA) and was reverse‐transcribed to cDNA. The SYBR Green PCR Master Mix (Life Technologies) was used for real‐time (RT)‐PCR analysis. The relative differences in expression between groups were expressed using cycle time (Ct) values normalized with β‐actin, and relative differences between control and treatment groups were calculated and expressed as relative increases setting control as 100%. For genes that were not detectable within 40Ct in control samples, controls were arbitrarily set at 35Ct to calculate a fold change compared with treated samples, as utilized in previous studies (Tuomela et al., 2013). This method allows an estimated fold change while reducing possible bias (McCall et al., 2014). Primers used are listed in Table 1.
Table 1.
Primers Used in RT‐PCR Analysis
| Gene | Species | Forward (5′‐3′) | Reverse (5′‐3′) |
|---|---|---|---|
| β‐actin | Human | GAT GCA GAA GGA GAT CAC TGC | ATA CTC CTG CTT GCT GAT CCA |
| TLR2 | Human | GGCTTCTCTGTCTTGTGACC | GGGCTTGAACCAGGAAGACG |
| TLR3 | Human | TTG CCT TGT ATC TAC TTT TGG GG | TCA ACA CTG TTA TGT TTG TGG GT |
| TLR4 | Human | CTC TGG GGA GGC ACA TCT TC | CCC AGG TGA GCT GTA GCA TT |
| TLR7 | Human | GATAACAATGTCACAGCCGTCC | GTTCCTGGAGTTTGTTGATGTTC |
| TLR8 | Human | ATG TTC CTT CAG TCG TCA ATG C | TTG CTG CAC TCT GCA ATA ACT |
| CD14 | Human | AGA GGC AGC CGA AGA GTT CAC | GCG CTC CAT GGT CGA TAA GT |
| MD‐2 | Human | GAA GCT CAG AAG CAG TAT TGG GTC | GGT TGG TGT AGG ATG ACA AAC TCC |
| Myd88 | Human | CCGCGCTGGCGGAGGAGATGGAC | GCAGATGAAGGCATCGAAACGCTC |
| RAGE | Human | CTA CCG AGT CCG TGT CTA CCA | CAT CCA AGT GCC AGC TAA GAG |
| HMGB1 | Human | GGA GAT CCT AAG AAG CCG AGA | CAT GGT CTT CCA CCT CTC TGA |
| TNF‐α | Human | CCC AGG CAG TCA GAT CAT CTT CT | ATG AGG TAC AGG CCC TCT GAT |
| IL‐1β | Human | ATG ATG GCT TAT TAC AGT GGC AA | GTCGGAGATTCGTAGCTGGA |
| IL‐6 | Human | ACT CAC CTC TTC AGA ACG AAT TG | CCA TCT TTG GAA GGT TCA GGT TG |
| β‐actin | Mouse | GTA TGA CTC CAC TCA CGG CAA A | GGT CTC GCT CCT GGA AGA TG |
| TLR2 | Mouse | GCA AAC GCT GTT CTG CTC AG | AGG CGT CTC CCT CTA TTG TAT T |
| TLR3 | Mouse | GTG AGA TAC AAC GTA GCT GAC TG | TCC TGC ATC CAA GAT AGC AAG T |
| TLR4 | Mouse | ATG GCA TGG CTT ACA CCA CC | GAG GCC AAT TTT GTC TCC ACA |
| TLR7 | Mouse | ATG TGG ACA CGG AAG AGA CAA | GGT AAG GGT AAG ATT GGT GGT G |
| TLR8 | Mouse | GAA AAC ATG CCC CCT CAG TCA | CGTCACAAGGATAGCTTCTGGAA |
| CD14 | Mouse | GCC AAA TTG GTC GAA CAA GC | CCA TGG TCG GTA GAT TCT GAA AGT |
| MD‐2 | Mouse | CGC TGC TTT CTC CCA TAT TGA | CCT CAG TCT TAT GCA GGG TTC A |
| Myd88 | Mouse | TCA TGT TCT CCA TAC CCT TGG T | AAA CTG CGA GTG GGG TCA G |
| RAGE | Mouse | GAA GGC TCT GTG GGT GAG TC | CCG CTT CCT CTG ACT GAT TC |
| HMGB1 | Mouse | CGC GGA GGA AAA TCA ACT AA | TCA TAA CGA GCC TTG TCA GC |
| TNF‐α | Mouse | GAC CCT CAC ACT CAG ATC ATC TTC T | CCT CCA CTT GGT GGT TTG CT |
| IL‐1β | Mouse | CTG GTG TGT GAC GTT CCC ATT A | CCG ACA GCA CGA GGC TTT |
| IL‐6 | Mouse | ACA AGT CGG AGG CTT AAT TAC ACA T | TTG CCA TTG CAC AAC TCT TTT C |
Enzyme‐Linked Immunosorbent Assays
Cells were lysed using ice‐cold lysis buffer (20 mM Tris, 0.25 M sucrose, 2 mM EDTA, 10 mM EGTA, 1% Triton X‐100), 1 tablet of Complete Ultra protease inhibitor cocktail (Roche), and 1 table of PhosSTOP phosphatase inhibitor (Roche)/10 ml. Lysate was spun at 21,000×g for 15 minutes and protein‐containing supernatant was collected. Media was collected and spun down at 500×g to eliminate cell debris. Protein concentration was determined using a BCA kit (Thermo Scientific). Cell lysates and/or media were analyzed using HMGB1 (IBL, Hamburg, Germany) and IFN‐β (R&D, Minneapolis, MN) enzyme‐linked immunosorbent assays (ELISAs) as per manufacturers' instructions.
Statistical Analysis
Data are expressed as mean ± standard error of the mean (SEM). t‐Tests were used to compare basal expression of mRNA and protein between BV2 and SH‐SY5Y (Fig. 2). In order to examine the treatment effect in each respective cell type, 1‐way ANOVAs were utilized for comparisons between LPS, EtOH, and Poly(I:C) followed by Dunnett's test to account for multiple comparison (Figs 3–8). Excluding Fig. 2, which directly compares basal levels of immune signaling molecules, direct statistical comparisons are not made between cell types. A p‐value <0.05 was considered statistically significant. Data were analyzed and figures designed using Prism software (GraphPad, La Jolla, CA).
Results
BV2 Microglia and SH‐SY5Y Neurons Display Typical Microglial and Neuronal Markers, Respectively
To clearly define the cell type of BV2 and SH‐SY5Y, expression of microglial and neuronal protein markers was assessed using Western blot and flow cytometry. RA‐differentiated SH‐SY5Y expressed neuronal markers NSE and Tubb3, but not microglial marker Cd11b (Fig. 1 A). Accordingly, BV2 expressed microglial marker Cd11b but not neuronal markers NSE or Tubb3, whereas SH‐SY5Y expressed NSE and Tubb3 but not Cd11b (Fig. 1 A). Flow cytometry analysis confirmed that essentially all BV2 (>99%) express Cd11b, whereas RA‐differentiated SH‐SY5Y predominantly express neuronal marker Tubb3 (Fig. 1 B). These data confirm that the culture preparations are representative of microglial and neuronal cells.
Figure 1.
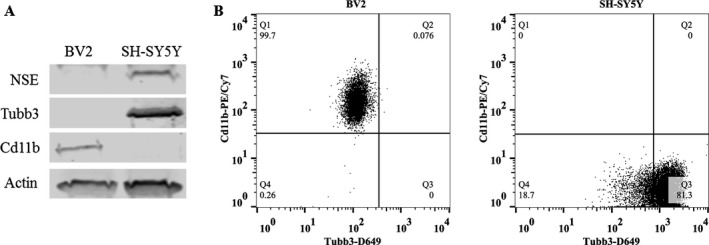
BV2 express microglial markers and SH‐SY5Y express neuronal markers, indicating they are microglial‐like and neuronal‐like cells, respectively. (A) Western blot analysis of basal expression of microglial and neuronal cell type markers in cell lysates of BV2 and SH‐SY5Y. SH‐SY5Y were differentiated into a neuron‐like phenotype using retinoic acid (RA) for 4 days. Microglia‐like BV2 express microglial marker Cd11b. Neuron‐like SH‐SY5Y express neuronal markers neuron‐specific enolase (NSE) and β‐III tubulin (Tubb3). β‐actin was used as a loading control. (B) Flow cytometry analysis of basal expression of microglial and neuronal markers in BV2 and SH‐SY5Y. The majority of BV2 cells express microglial marker Cd11b, whereas the majority of SH‐ SY5Y express neuronal marker Tubb3. n = 3 per group. Gates were established using unstained and isotype controls for each antibody.
Microglia‐Like BV2 and Neuron‐Like SH‐SY5Y Have Unique Basal Expression of Innate Immune Signaling Molecules
The expression of proinflammatory signaling molecules in BV2 and SH‐SY5Y was examined using both RT‐PCR and Western blot. As expected, microglia‐like BV2 were found to express a wide variety of proinflammatory genes (Table 2). Neuron‐like SH‐SY5Y also express most proinflammatory genes examined, although in lower relative amounts compared with BV2 for most genes assessed. Both BV2 and SH‐SY5Y were found to express protein and mRNA for TLR4, RAGE, and HMGB1 (Fig. 2). Consistent with their expected cellular phenotypes, BV2 express more TLR4 protein and mRNA as well as many fold greater levels of mRNA for multiple TLRs, cytokines, and other proinflammatory genes. Neuron‐like SH‐SY5Y had less than 1% of the BV2 level of TLR4 expression (Fig. 2 A). Western blot analysis also found a detectable but relatively lower expression of TLR4 in SH‐SY5Y compared with BV2 (Fig. 2 B). SH‐SY5Y had greater relative expression of TLR3 than the other TLRs; however, this was only 20% of the level in BV2s (Table 2). Further, SH‐SY5Y had no detectable levels of TLR4 adaptor protein MD‐2, or proinflammatory cytokines IL‐1β and IL‐6 (Table 2, using a 40 cycle cutoff). In contrast, SH‐SY5Y express more RAGE and HMGB1 compared with BV2. Interestingly, Myd88, a critical adapter signaling transducer for TLRs and cytokine receptor signaling, has similar levels in SH‐SY5Y and BV2. These data indicate that SH‐SY5Y and BV2 both express TLR and other proinflammatory genes, although at different basal levels.
Table 2.
Summary Basal mRNA Expression of Proinflammatory Genes in Microglia‐Like BV2 and Neuron‐Like SH‐SY5Y
| ddCt | ||
|---|---|---|
| BV2 | SH‐SY5Y | |
| RAGE | 1.03 | 21.4600 |
| HMGB1 | 1.00 | 4.6000 |
| Myd88 | 1.00 | 0.9500 |
| TLR3 | 1.03 | 0.1800 |
| TLR8 | 1.01 | 0.1600 |
| CD14 | 1.01 | 0.0360 |
| TLR2 | 1.01 | 0.0290 |
| TLR7 | 1.01 | 0.0035 |
| TLR4 | 1.01 | 0.0031 |
| TNF‐α | 1.00 | 0.0017 |
| MD‐2 | 1.01 | n.d. |
| IL‐1β | 1.06 | n.d. |
| IL‐6 | 1.02 | n.d. |
Average basal mRNA levels of various immune signaling molecules in BV2 and SH‐SY5Y (n = 3 per group) were analyzed using RT‐PCR and expressed as ddCt, setting BV2 as 1.
Figure 2.
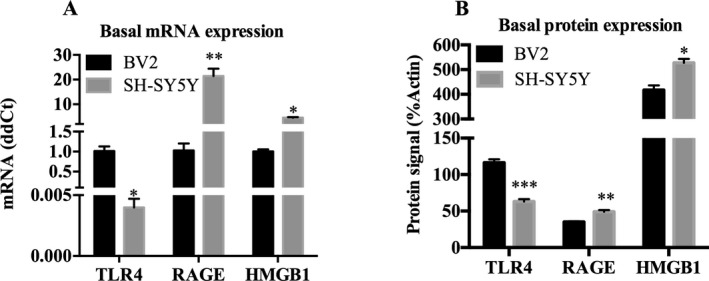
Microglia‐like BV2 and neuron‐like SH‐SY5Y express mRNA and protein of various proinflammatory molecules. (A) Comparison of basal mRNA expression of TLR4, RAGE, and HMGB1 in BV2 and SH‐SY5Y. Data are expressed in ddCt normalized to BV2 expression level. SH‐SY5Y express less TLR4 but greater RAGE and HMGB1 mRNA. (B) Basal protein levels of TLR4, RAGE, and HMGB1 were examined using Western blot in BV2 and SH‐SY5Y. Data are expressed in %β‐actin units. SH‐SY5Y express less TLR4 but greater RAGE and HMGB1 protein. n = 3 per group; *p < 0.05, **p < 0.01, ***p < 0.001 versus BV2.
LPS, Poly(I:C), and EtOH Differentially Activate Kinases/Transcription Factors that Modulate TLR Pathways in Microglia‐Like BV2
Activation of TLR3 and TLR4 involves key kinases the lead to activation of NFκB and other transcription factors that mediate induction of proinflammatory genes. To better understand the role of EtOH in relation to TLR4 and TLR3 activation, we treated microglia‐like BV2 TLR4 agonist LPS, TLR3 agonist Poly(I:C), or EtOH for 10 or 30 minutes. Activated kinases/transcription factors were measured by examining phosphorylated forms of these proteins in cell lysates using Western blot. As expected, both LPS and Poly(I:C) activated kinases related to TLR signaling. Following 10 minutes of LPS treatment, phospho(p)‐ERK1/2 was increased by 29% (±5.9%, p < 0.01) and p‐p38 by 98% (±17%, p < 0.001). p‐NFκB, a transcription factor downstream of TLR activation that is involved in transcription of a variety of proinflammatory molecules, was increased by 42% (±20, p < 0.05) (Fig. 3 A). After 30 minutes, LPS caused an even more robust effect, by increasing p‐ERK1/2 by 68% (±5.6%, p < 0.0001), p‐p38 by 568% (±61%, p < 0.0001), and p‐NFκB by 80% (±18%, p < 0.001) relative to controls (Fig. 3 A). Thus, LPS stimulation displayed a canonical activation of kinases in BV2.
Figure 3.
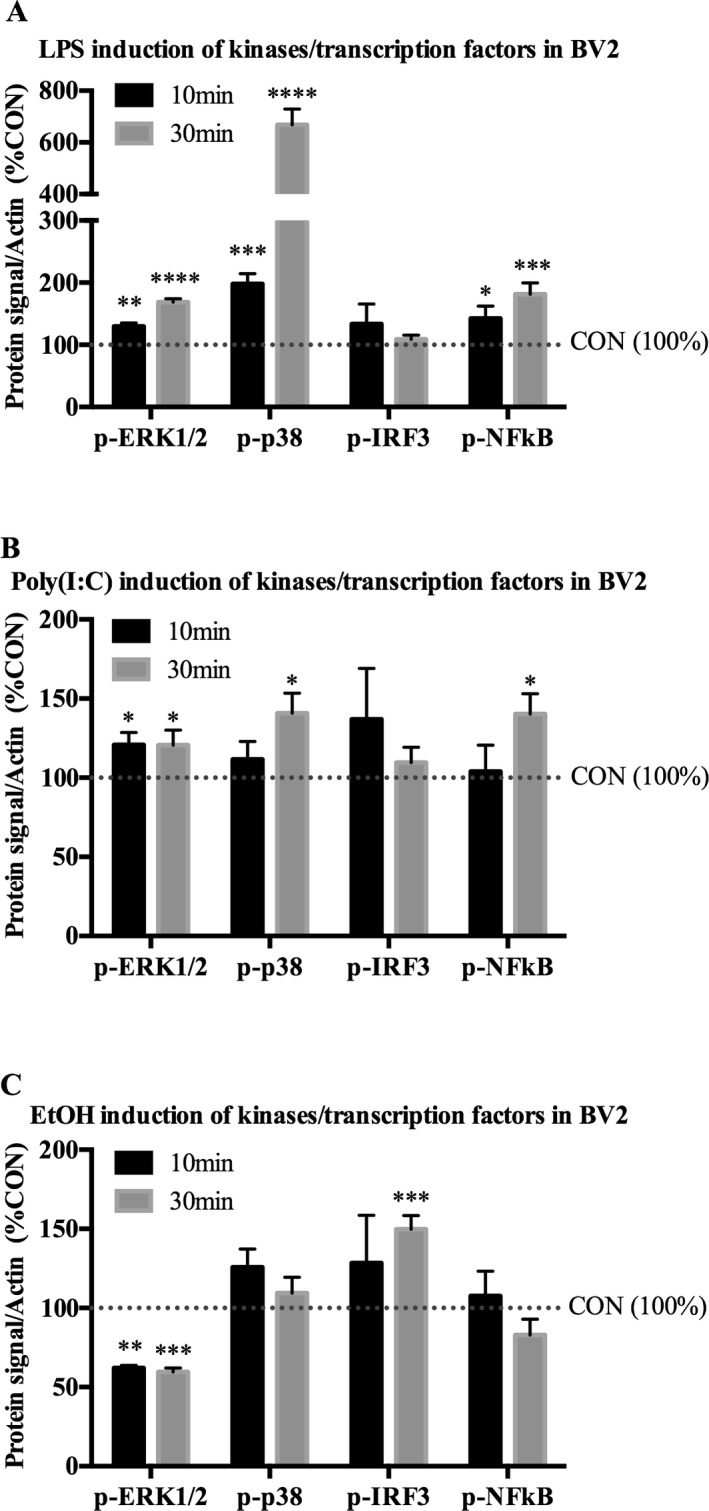
LPS, Poly(I:C), and EtOH activate proinflammatory‐related kinases/transcription factors in microglia‐like BV2. Microglia‐like BV2 were treated with indicated drug for 10 or 30 minutes. Cell lysates were collected for Western blot analysis. (A) Ten minutes of LPS (100 ng/ml) increased p‐ERK1/2 by 29%, p‐p38 by 98%, and p‐NF κB‐p65 by 42%. Thirty minutes of LPS increased p‐ERK1/2 by 68%, p‐p38 by 568%, and p‐NF κB by 80%. (B) Ten minutes of Poly(I:C) (50 μg/ml) increased p‐ERK1/2 by 21%. Thirty minutes of Poly(I:C) increased p‐ERK1/2 by 21%, p‐p38 by 41%, and p‐NF κB by 40%. (C) Ten minutes of EtOH (150 mM) decreased p‐ERK1/2 by 38%. Thirty minutes of EtOH decreased p‐ERK1/2 by 40% and increased p‐IRF3 by 50%. Data are represented as %control (%CON) following β‐actin normalization. n = 3 per group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus CON.
Treatment of BV2 with Poly(I:C) had a slightly less robust effect compared with LPS, with only p‐ERK1/2 increased by 21% (±7.8%, p < 0.05) by 10 minutes of treatment. However, by 30 minutes Poly(I:C) increased p‐ERK1/2 by 21% (±9.4, p < 0.05), p‐p38 by 41% (±13, p < 0.05), and p‐NFκB by 40% (±13%, p < 0.05) (Fig. 3 B), indicating activation of kinases by Poly(I:C) signaling in BV2.
Unlike either Poly(I:C) or LPS, EtOH treatment at 10 and 30 minutes significantly decreased p‐ERK1/2 by 38% (±1.6%, p < 0.01) and 40% (±2.5%, p < 0.001), respectively, whereas p‐IRF3 was increased by 50% (±8.7%, p < 0.001) after 30 minutes (Fig. 3 C). Thus, LPS, Poly(I:C), and EtOH activate proinflammatory signaling kinases/transcription factors in BV2. However, EtOH displays a unique pattern of activation compared with either TLR3 or TLR4 signaling pathways.
LPS, Poly(I:C), and EtOH Increase Proinflammatory Gene Expression in Microglia‐Like BV2
Studies of canonical macrophage TLR signaling find that activation of transcription factors such as NFκB and IRF3 leads to further proinflammatory signaling (Narayanan and Park, 2015). Thus, we next assessed subsequent induction of proinflammatory genes following 24‐hour treatment of TLR3 agonist Poly(I:C), TLR4 agonist LPS, or EtOH in BV2. Cell death was assayed using trypan blue exclusion and no significant effects were found in any treatment (data not shown). LPS induced a typical microglia‐like response in BV2, with significant increases in mRNA expression of TNF‐α (814 ± 28%, p < 0.001), IL‐1β (2,775 ± 370%, p < 0.0001), IL‐6 (209 ± 24%, p < 0.05) (Fig. 4 A), and TLR4 coreceptor CD14 (242 ± 21%, p < 0.01) (Table 3). LPS treatment also increased HMGB1 in media of BV2 by 37% (±8.5%, p < 0.01) relative to controls (Fig. 4 B). TLR3 agonist Poly(I:C) treatment increased mRNA expression of TNF‐α (217 ± 29%, p < 0.05) and IL‐1β (366 ± 33%, p < 0.01) (Fig. 4 C), whereas Poly(I:C) had no significant effect on HMGB1 release in media (Fig. 4 D). Additionally, TLR8 was down‐regulated by both Poly(I:C) (49 ± 15%, p < 0.05) and LPS (21 ± 2.7%, p < 0.01), whereas LPS up‐regulated TLR2 (588 ± 69%, p < 0.01) and CD14 (242 ± 21%, p < 0.01) as depicted in Table 3. Raw Ct values are depicted in Supplementary Table S1. These data are consistent with BV2 displaying a characteristic macrophage‐like proinflammatory signaling induction following TLR4 and TLR3 agonist treatment.
Figure 4.
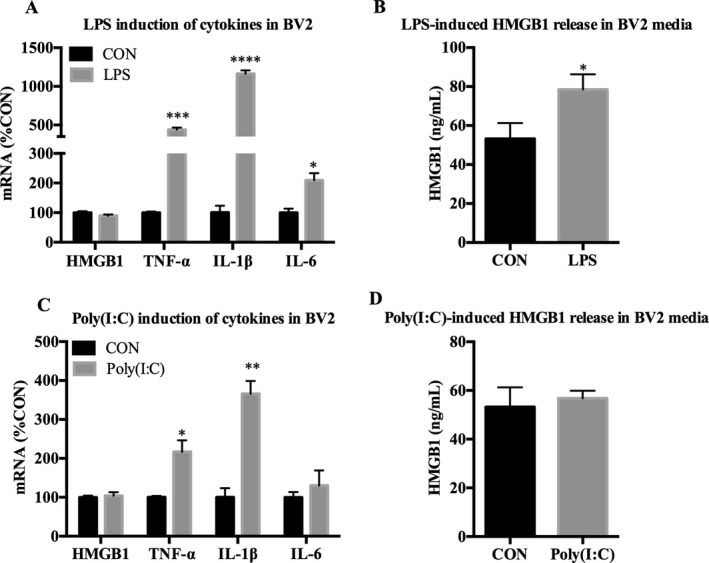
LPS and Poly(I:C) up‐regulates cytokine expression in microglia‐like BV2. BV2 were treated with either LPS (100 ng/ml) or Poly(I:C) (50 μg/ml) for 24 hours followed by mRNA analysis in cell lysates and analysis of HMGB1 release in media using ELISA. (A) LPS increased TNF‐α (814%), IL‐1β (2,775%), and IL‐6 (209%) mRNA. (B) LPS increased HMGB1 release in media by 37%. (C) Poly(I:C) increased TNF‐α (217%) and IL‐1β (366%) mRNA. (D) Poly(I:C) did not induce HMGB1 release in media. Data for mRNA are expressed as %control (%CON), with CON set at 100%. n = 3 per group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus CON.
Table 3.
Summary of Poly(I:C), LPS, and EtOH Induction of Proinflammatory Gene mRNA in BV2 and SH‐SY5Y
| CON | Poly(I:C) | LPS | EtOH | |
|---|---|---|---|---|
| BV2 | ||||
| TLR2 | 100 ± 10 | 323 ± 70 | 588 ± 69 ** | 170 ± 15 * |
| TLR3 | 100 ± 16 | 158 ± 13 | 133 ± 9.3 | 120 ± 9.1 |
| TLR4 | 100 ± 9.3 | 100 ± 19 | 58 ± 2.2 | 90 ± 8.6 |
| TLR7 | 100 ± 6.0 | 121 ± 24 | 75 ± 3.5 | 158 ± 8.4 ** |
| TLR8 | 100 ± 11 | 49 ± 15 * | 21 ± 2.7 ** | 194 ± 9.5 ** |
| CD14 | 100 ± 7.5 | 154 ± 13 | 242 ± 21 ** | 104 ± 3.7 |
| MD‐2 | 100 ± 11 | 118 ± 31 | 98 ± 13 | 156 ± 4.3 ** |
| Myd88 | 100 ± 1.7 | 113 ± 6.2 | 96 ± 8.5 | 89 ± 7.2 |
| RAGE | 100 ± 16 | 134 ± 7.7 | 98 ± 4.8 | 104 ± 4.4 |
| HMGB1 | 100 ± 4.9 | 104 ± 9.2 | 90 ± 4.3 | 67 ± 9.7 |
| TNF‐α | 100 ± 3.6 | 217 ± 29 * | 440 ± 27 *** | 204 ± 15 ** |
| IL‐1β | 100 ± 9.3 | 366 ± 33 ** | 1,163 ± 45 **** | 304 ± 29 ** |
| IL‐6 | 100 ± 14 | 130 ± 39 | 210 ± 24 * | 10 ± 2.4 ** |
| SH‐SY5Y | ||||
| TLR2 | 100 ± 19 | 230 ± 31 * | 121 ± 30 | 177 ± 23 |
| TLR3 | 100 ± 46 | 920 ± 260 * | 98 ± 35 | 398 ± 80 * |
| TLR4 | 100 ± 23 | 158 ± 12 | 67 ± 18 | 147 ± 14 |
| TLR7 | 100 ± 46 | 234 ± 59 | 153 ± 79 | 575 ± 100 * |
| TLR8 | 100 ± 33 | 103 ± 22 | 121 ± 60 | 320 ± 17 ** |
| CD14 | 100 ± 18 | 146 ± 17 | 61 ± 21 | 127 ± 11 |
| MD‐2 | n.d. | n.d. | n.d. | n.d. |
| Myd88 | 100 ± 13 | 530 ± 30 *** | 79 ± 4.7 | 128 ± 5.9 |
| RAGE | 100 ± 6.6 | 117 ± 8.9 | 61 ± 20 | 170 ± 15 * |
| HMGB1 | 100 ± 4.2 | 125 ± 5.2 * | 84 ± 11 | 105 ± 2.4 |
| TNF‐α | 100 ± 9.5 | 10,426 ± 2,130 ** | 65 ± 17 | 67 ± 13 |
| IL‐1β | n.d. | 2,860 ± 135 | n.d. | n.d. |
| IL‐6 | n.d. | 591 ± 100 | n.d. | n.d. |
Microglia‐like BV2 and neuron‐like SH‐SY5Y were treated with TLR3 agonist Poly(I:C) (50 μg/ml), TLR4 agonist LPS (100 ng/ml), or EtOH (150 mM) for 24 hours. Cell lysates were collected and analyzed for mRNA expression using RT‐PCR. Data for mRNA are expressed as %control (%CON), with CON set at 100%. Data for HMGB1, TNF‐α, IL‐1β, and IL‐6 for LPS and Poly(I:C) treatment in BV2 are also represented in Fig. 4 A,C. Data for HMGB1, TNF‐α, IL‐1β, IL‐6, TLR2, 3, 4, 7, 8, Myd88, Cd14, MD‐2, and RAGE for EtOH treatment in BV2 are also represented in Fig. 5 A,B. Data for HMGB1, TNF‐α, IL‐1β, and IL‐6 for LPS and Poly(I:C) treatment in SH‐SY5Y are also represented in Fig. 7 A,C. Data for HMGB1, TNF‐α, IL‐1β, IL‐6, TLR2, 3, 4, 7, 8, Myd88, Cd14, MD‐2, and RAGE for EtOH treatment in SH‐SY5Y are also represented in Fig. 8 A,B. n.d. = not detectable; n = 3 per group.
Bolded values represent data significantly different versus control. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus CON.
BV2 were next treated with EtOH for 24 hours to determine whether EtOH causes induction of proinflammatory genes in microglia‐like cells. EtOH increased transcription of both cytokines and TLRs. Significant increases were found for TNF‐α (204 ± 15%, p < 0.01), IL‐1β (304 ± 29%, p < 0.01), TLR2 (193 ± 28%, p < 0.01), TLR7 (158 ± 8.4%, p < 0.01), TLR8 (194 ± 9.6%, p < 0.01), and the TLR4 accessory protein MD‐2 (155 ± 4.4%, p < 0.01) mRNA (Fig. 5 A,B), as well as HMGB1 release in media (Fig. 5 C). A concentration curve indicated that EtOH induces transcription of proinflammatory genes at a concentration of over 50 mM (Fig. 5 D). These data indicate that EtOH treatment of microglia‐like BV2 activates proinflammatory gene induction.
Figure 5.
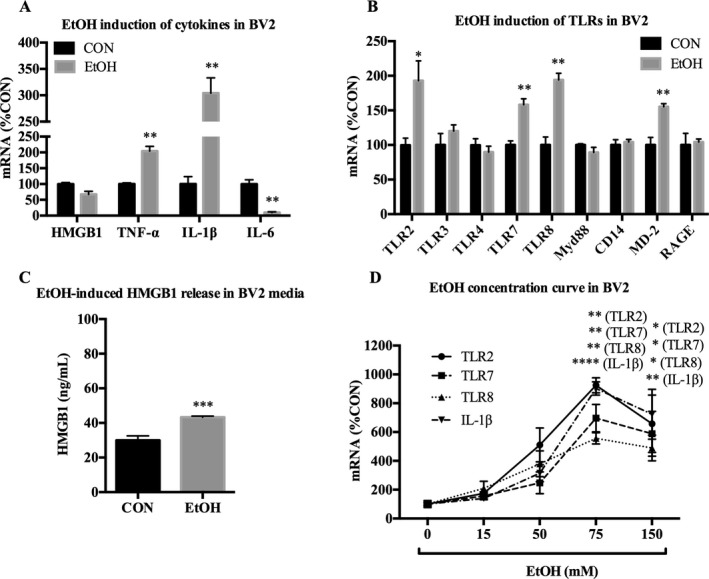
EtOH up‐regulates proinflammatory genes in microglia‐like BV2. BV2 were treated with EtOH (150 mM) for 24 hours and analyzed for mRNA expression of cytokines, TLRs and TLR‐associated signals, and HMGB1 release in media. (A) EtOH increased TNF‐α (204%) and IL‐1β (304%), and decreased IL‐6 (10%) mRNA. (B) EtOH increased TLR2 (193%), TLR7 (158%), TLR8 (194%), and MD‐2 (155%) mRNA. (C) EtOH increased HMGB1 release in media by 44%. (D) Concentration curve of 24hr of EtOH treatment on mRNA expression in BV2. EtOH increased TLR2, TLR7, TLR8, and IL‐1β at 75 and 150 mM. Data for mRNA are expressed as %control (%CON), with CON set at 100%. n = 3 per group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus CON.
Poly(I:C) and EtOH, But Not LPS, Activate TLR Signaling Kinases/Transcription Factors Pathway in Neuron‐Like SH‐SY5Y
To determine whether TLR3 or TLR4 pathways are activated in neuron‐like SH‐SY5Y, the cells were treated for 10 or 30 minutes with either TLR4 agonist LPS, TLR3 agonist Poly(I:C), or EtOH followed by Western blot analysis of phosphorylated (activated) kinases. While LPS had no measurable effect on SH‐SY5Y at either time point (Fig. 6 A), Poly(I:C) increased p‐p38 by 88% (±24, p < 0.01) and p‐IRF3 by 38% (±6.6%, p < 0.05) at 30 minutes (Fig. 6 B). A 10‐minute treatment of EtOH decreased p‐ERK1/2 by 55% (±4.7%, p < 0.001), and increased p‐p38 by 49% (±17%, p < 0.05), p‐IRF3 by 41% (±19%, p < 0.05), p‐TBK1 by 74% (±17%, p < 0.001), and p‐NFκB by 56% (±17%, p < 0.01) (Fig. 6 C). By 30 minutes of treatment, EtOH decreased p‐ERK1/2 by 49% (±6.7, p < 0.01), and caused a broad range of effects, increasing p‐p38 by 222% (±24, p < 0.0001), p‐IRF3 by 125% (±14%, p < 0.0001), p‐TBK1 by 94% (±11%, p < 0.0001), and p‐NFκB by 72% (±13%, p < 0.001) (Fig. 6 C). These data indicate that Poly(I:C) but not LPS activates kinases in SH‐SY5Y. Importantly, EtOH induces a broad range of activation of proinflammatory signaling kinases/transcription factors in SH‐SY5Y, with several effects unique to EtOH.
Figure 6.
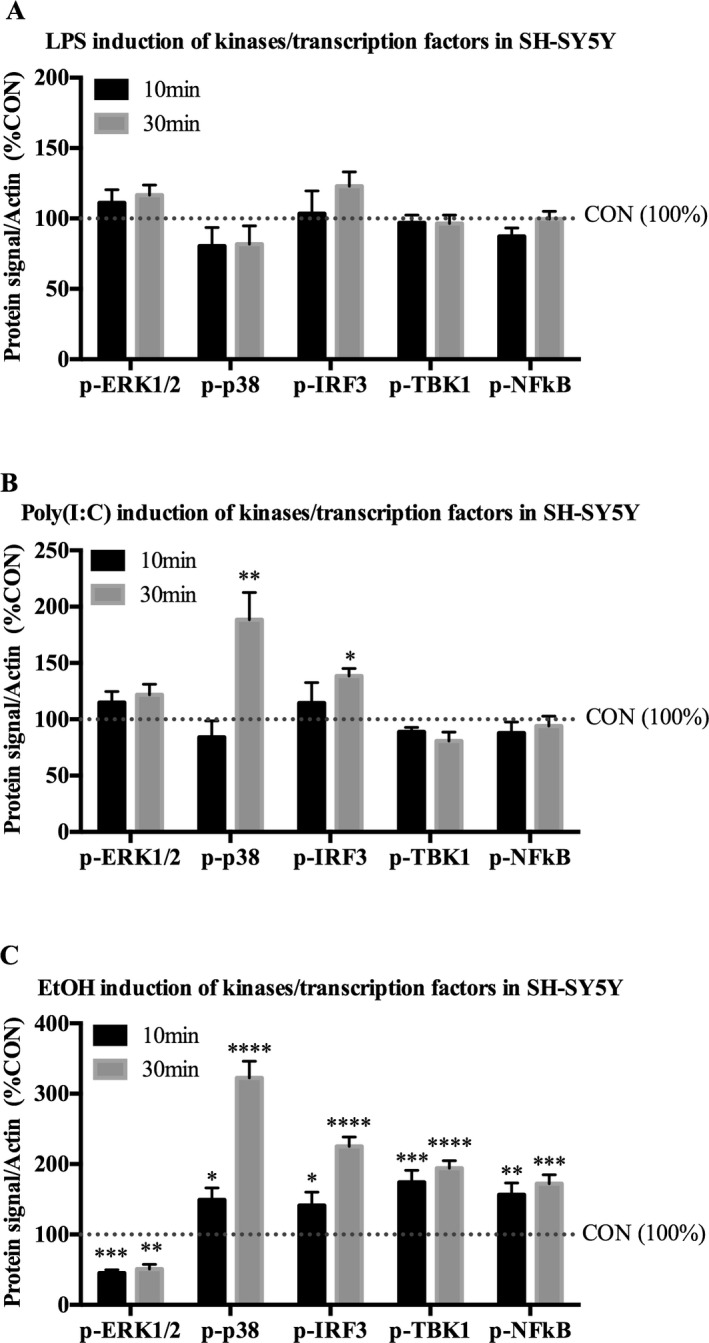
Poly(I:C) and EtOH, but not LPS, activate proinflammatory‐related kinases/transcription factors in neuron‐like SH‐SY5Y. SH‐SY5Y were treated with indicated drug for 10 or 30 minutes. Cell lysates were collected for Western blot analysis. (A) LPS (100 ng/ml) had no detectable effect on any of the kinases/transcription factors examined. (B) Poly(I:C) (50 μg/ml) increased p‐p38 by 88% and p‐IRF3 by 38% by 30 minutes. (C) Ten minutes of EtOH (150 mM) decreased p‐ERK1/2 by 55%, and increased p‐p38 by 49%, p‐IRF3 by 41%, p‐TBK1 by 74%, and p‐NF κB by 56%. Thirty minutes of EtOH decreased p‐ERK1/2 by 49%, and increased p‐p38 by 222%, p‐IRF3 by 125%, p‐TBK1 by 94%, and p‐NF κB by 73%. Data are represented as %CON following β‐actin normalization. n = 3 per group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus CON.
Poly(I:C) and EtOH, But Not LPS, Increase Proinflammatory Gene Expression in Neuron‐Like SH‐SY5Y
Subsequent proinflammatory gene expression was then examined in neuron‐like SH‐SY5Y. Cell death was assayed using trypan blue exclusion, and no significant effects were found in any treatment (data not shown). In accordance with the lack of kinase activation, SH‐SY5Y did not have any detectable changes in proinflammatory cytokine expression or HMGB1 release in response to LPS (Fig. 7 A,B). However, Poly(I:C) increased mRNA expression of TNF‐α (10,426 ± 2,132%, p < 0.01) and HMGB1 (125 ± 5.2%, p < 0.05), and further increased levels of IL‐1β and IL‐6 from nondetectable (>40 Ct) to detectable ranges, with estimated fold changes of 2,860 (± 135%) and 591 (± 100%), respectively (Fig. 7 C). Poly(I:C) also increased expression of its own receptor, TLR3 (920 ± 260%, p < 0.05) as well as TLR2 (230 ± 31%, p < 0.05) and TLR adaptor protein Myd88 (530 ± 30%, p < 0.001), as shown in Table 3. Poly(I:C) had no effect on HMGB1 release (Fig. 7 D), and no change in IFN‐β protein in cell lysates was observed (data not shown). Raw Ct values are depicted in Table S1. These data indicate that neuron‐like SH‐SY5Y appear to lack a detectable LPS response. However, SH‐SY5Y are highly responsive to TLR3 stimulation. Poly(I:C) induced strong up‐regulation of cytokines in SH‐SY5Y, as well as TLR receptors and HMGB1.
Figure 7.
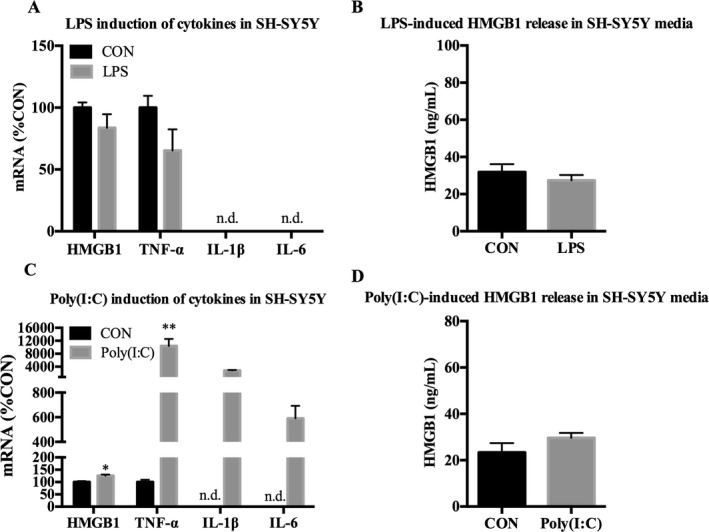
Poly(I:C), but not LPS, up‐regulates cytokine expression in neuron‐like SH‐SY5Y. SH‐SY5Y were treated with either LPS (100 ng/ml) or Poly(I:C) (50 μg/ml) for 24 hours followed by mRNA analysis in cell lysates and analysis of HMGB1 release in media using ELISA. (A) LPS had no detectable effect on expression of HMGB1, TNF‐α, IL‐1β, or IL‐6 mRNA. (B) LPS had no effect on HMGB1 release in media. (C) Poly(I:C) increased expression of HMGB1 (125%), TNF‐α (10,426%), IL‐1β (2,860%), and IL‐6 (591%) mRNA. (D) Poly(I:C) had no effect on HMGB1 release in media. Data for mRNA are expressed as %control (%CON), with CON set at 100%. n = 3 per group; n.d. = not detectable; *p < 0.05, **p < 0.01 versus CON.
SH‐SY5Y were next treated with EtOH to examine whether EtOH causes induction of proinflammatory genes in neuron‐like cells. While no significant changes were seen in mRNA of the cytokines we assessed (Fig. 8 A), EtOH did increase TLR3 (398 ± 80%, p < 0.05), TLR7 (574 ± 100%, p < 0.05), TLR8 (319 ± 17%, p < 0.01), and RAGE (170 ± 15%, p < 0.05) (Fig. 8 B). EtOH also caused HMGB1 release in media (Fig. 8 C). Concentrations as low as 15 mM EtOH caused increases in TLR3, TLR7, and RAGE mRNA (Fig. 8 D). In summary, EtOH causes induction of TLR3, TLR7, TLR8, and RAGE in SH‐SY5Y even at low concentrations, and in a unique manner, different from either TLR3 or TLR4 stimulation alone.
Figure 8.
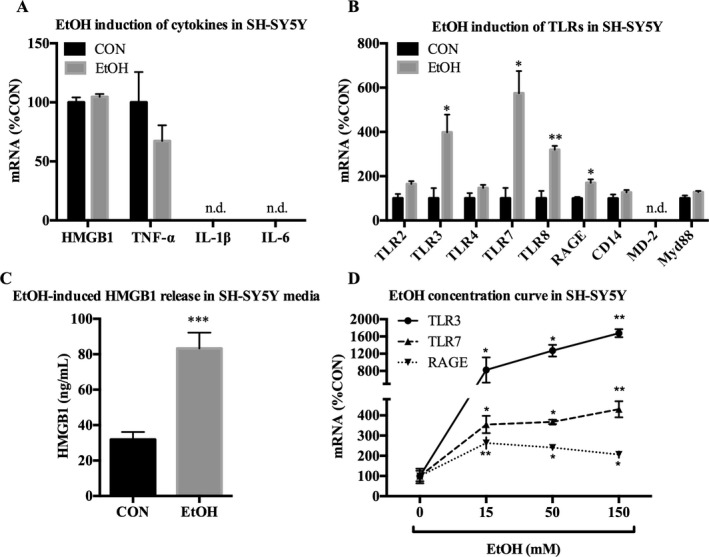
EtOH up‐regulates proinflammatory genes in neuron‐like SH‐SY5Y. SH‐SY5Y were treated with EtOH (150 mM) for 24 hours and analyzed for mRNA expression of cytokines, TLRs and TLR‐associated signals, and HMGB1 release in media. (A) EtOH had no detectable effect on HMGB1, TNF‐α, IL‐1β, or IL‐6 mRNA. (B) EtOH increased expression of TLR3 (398%), TLR7 (574%), TLR8 (319%), and RAGE (170%) mRNA. (C) EtOH increased release of HMGB1 in the media by 161%. (D) Concentration curve of 24 hours of EtOH treatment on mRNA expression in SH‐SY5Y. EtOH increased expression of TLR3, TLR7, and RAGE mRNA by 15 mM. Data for mRNA are expressed as %control (%CON), with CON set at 100%. n = 3 per group; n.d. = not detectable; *p < 0.05, **p < 0.01, ***p < 0.001 versus CON.
Discussion
This study compares TLR4, TLR3, and EtOH‐induced kinase activation and proinflammatory gene induction in BV2 microglia‐like cells and differentiated SH‐SY5Y neuron‐like cells (Fig. 9). As expected, BV2 express greater levels of most proinflammatory signaling molecules compared with SH‐SY5Y. BV2 responded to all treatments as indicated by activation of proinflammatory signaling kinases and induction of proinflammatory signaling gene mRNA (Fig. 9 A–C, red labels). Although all 3 treatments induced a BV2 response, TLR3 and TLR4 activation increased canonical kinases, for example, p‐ERK1/2, p‐p38, and p‐NFκB, whereas EtOH uniquely increased p‐IRF3 in BV2, a different canonical proinflammatory signaling pathway not changed by LPS or Poly(I:C) treatment. Further, EtOH decreased p‐ERK1/2 in BV2, whereas both LPS and Poly(I:C) increased p‐ERK1/2, further distinguishing the signaling of EtOH from TLR3 and TLR4. Proinflammatory cytokines TNF‐α and IL‐1β were increased by all 3 treatments in BV2, but EtOH further increased TLR2, TLR7, TLR8, and MD‐2 (Fig. 9 A–C, red labels). In neuron‐like SH‐SY5Y, we found TLR4 protein and mRNA, but no detectable LPS responses (Fig. 9 A). However, Poly(I:C) and EtOH both increased p‐p38 and p‐IRF3, with EtOH also increasing p‐NFκB and decreasing p‐ERK1/2 in SH‐SY5Y (Fig. 9 B,C, blue labels). Interestingly, Poly(I:C) increased proinflammatory cytokine mRNA, for example, TNF‐α, IL‐1β, and IL‐6 in SH‐SY5Y, some from nondetectable levels, without increasing p‐NFκB, the key proinflammatory transcription factor in monocytes, whereas EtOH increased p‐NFκB, but did not induce TNF‐α, IL‐1β, or IL‐6 in SH‐SY5Y. Both Poly(I:C) and EtOH increased multiple TLR mRNA in SH‐SY5Y, but only EtOH increased RAGE expression, which uniquely had greater basal expression in SH‐SY5Y than in BV2. HMGB1, a TLR and RAGE agonist, was induced and released by LPS in BV2, but not in SH‐SY5Y, whereas EtOH released HMGB1 from both cell types (Fig. 9 A,C). EtOH induced TLR mRNA at lower concentrations in SH‐SY5Y (15 mM, Fig. 8 D) compared with BV2 (75 mM, Fig. 5 D). This suggests greater neuronal sensitivity to EtOH induction of TLR mRNA and unique cellular differences in EtOH induction of TLR and other proinflammatory genes that contribute to the complexity in brain.
Figure 9.
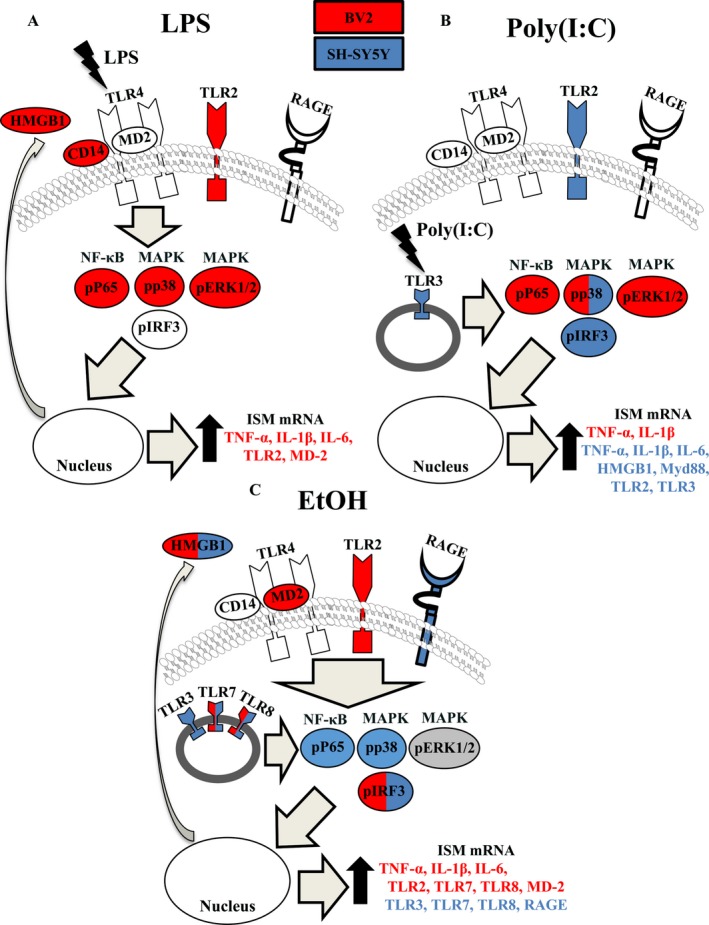
EtOH instigates broader proinflammatory changes than TLR4‐LPS or TLR3‐Poly(I:C) alone in either microglia‐like BV2 or neuron‐like SH‐SY5Y. Red = increased in BV2; blue = increased in SH‐SY5Y; gray = decreased in both cell types; white = unchanged in both cell types.
EtOH is known to modulate innate immune responses in brain (Coller and Hutchinson, 2012; Crews and Vetreno, 2016; Mayfield et al., 2013; Montesinos et al., 2016) and drinking behavior (Agrawal et al., 2011; Blednov et al., 2005, 2012). Microglia and astrocytes have been found to play critical roles in the proinflammatory responses to EtOH, particularly through TLR4 activation (Alfonso‐Loeches et al., 2010; Blanco et al., 2005; Fernandez‐Lizarbe et al., 2009, 2013). However, neuronal proinflammatory signaling in response to EtOH has not previously been described. This study utilized microglial and neuronal cell lines to determine commonalities and differences in proinflammatory signaling to EtOH. As expected, microglia‐like BV2 have canonical activation to EtOH. EtOH treatment increased activation of IRF3 and increased transcription of NFκB‐dependent genes (e.g.,TNF‐α, IL‐1β, TLR2) consistent with previous reports (Alfonso‐Loeches et al., 2010; Crews et al., 2013; Fernandez‐Lizarbe et al., 2013). However, we now report that EtOH causes proinflammatory signaling in neuronal cells that is distinct from microglia. In neuron‐like SH‐SY5Y, EtOH activated a broader range of transcription factors and proinflammatory modulating kinases (NFκB, p38, TBK1) than in microglia‐like BV2. EtOH also increased transcription of immune receptors RAGE, TLR3, TLR7, and TLR8 in neuron‐like SH‐SY5Y. Although microglia‐like BV2 basally express more TLRs and proinflammatory genes than neuron‐like SH‐SY5Y, SH‐SY5Y had a lower concentration threshold of response to EtOH compared with BV2 (15 mM vs. 75 mM), indicating a higher sensitivity of neurons to EtOH‐induced induction of proinflammatory signaling. We further found that EtOH has a unique pattern of activation of kinases/proinflammatory genes compared with either TLR3 or TLR4 stimulation alone, again pointing toward a broader effect of EtOH on immune signaling. Our data support a role for EtOH‐induced proinflammatory innate immune signaling in neurons unique from that of microglia as well as TLR3 or TLR4 activation alone.
Previously, we found that EtOH treatment of mice for 10 days (5 g/kg/d) induces brain TLR3 and TLR4 expression, as well as increased induction of proinflammatory cytokines following systemic treatment with LPS (Qin et al., 2008) and Poly(I:C) (Qin and Crews, 2012a), implying that EtOH sensitizes TLR responses through up‐regulation of these pathways to instigate proinflammatory signaling. Our findings here expand upon these data by suggesting that EtOH has a broad range of proinflammatory activation rather than through TLR3 or TLR4 alone. In BV2, EtOH had a less robust effect on kinases/transcription factors compared to treatment with TLR3 agonist Poly(I:C) or TLR4 agonist LPS. In SH‐SY5Y, however EtOH actually had a broader range of effects, with EtOH activating several kinases/transcription factors (TBK1 and NFκB) that were not activated by Poly(I:C). We also found that ERK1/2 was decreased by EtOH in both cell types, whereas p38 was increased in EtOH‐treated SH‐SY5Y. A similar study showed that EtOH (100 mM, 10 minutes) inhibited p‐ERK1/2 in cultured human cortical neurons (Kalluri and Ticku, 2003), and p‐ERK1/2 is decreased in cortex in brains of adolescent‐intermittent EtOH‐treated rats (Liu and Crews, 2015). Poly(I:C) increased activation of IRF3, a transcription factor that controls interferon production (Hiscott, 2007), in SH‐SY5Y, and EtOH increased IRF3 in both BV2 and SH‐SY5Y. Consistent with our studies, EtOH (50 mM, 1 hour) increases activated IRF3 in cultured primary mouse microglia (Fernandez‐Lizarbe et al., 2009). However, we observed no Poly(I:C) or EtOH‐induced changes in IFN‐β in SH‐SY5Y cell lysates, suggesting that either different IFNs are being targeted or that a noncanonical role for IRF3 exists in neurons. It is important to note that while we observed that 30 minutes of EtOH increased p‐NFκB and p‐p38 in SH‐SY5Y, no change was seen in BV2 at this time point, although other studies in our laboratory find an increase in p‐NFκB at 6 hours in BV2 (data not shown). This may suggest greater importance of other transcription factors (e.g., IRF3) early after EtOH exposure in BV2. Given these results, we surmise that EtOH causes a unique activation of kinases/transcription factors in SH‐SY5Y and BV2, with a broader range of activation in neuron‐like SH‐SY5Y. While it is not clear why these neuron‐like and microglia‐like cells show different responses to EtOH, it may be related to differential kinase activation in neurons versus microglia. Immune induction by EtOH is complex and includes TLR clustering in lipid rafts, release of endogenous TLR agonists, and oxidative stress activation of NFκB (Fernandez‐Lizarbe et al., 2013; Qin and Crews, 2012b; Zou and Crews, 2014). Although complex, our findings suggest EtOH uniquely activates proinflammatory signaling pathways in both microglia‐like BV2 and neuron‐like SH‐SY5Y.
Microglia‐like BV2 and neuron‐like SH‐SY5Y demonstrated unique responses to TLR3 and TLR4 agonists. Microglial responses were canonical with both TLR3 agonist Poly(I:C) and TLR4 agonist LPS increasing activated p‐NFκB, p‐p38, and p‐ERK1/2, consistent with findings in primary microglia cultures (Town et al., 2006). However, neuron‐like SH‐SY5Y displayed a response only to TLR3 stimulation. TLR3 agonist Poly(I:C) activated p38 and IRF3 in SH‐SY5Y, consistent with previous studies in SH‐SY5Y (Nessa et al., 2006). We also found that Poly(I:C) also greatly up‐regulated TNF‐α, IL‐1β, and IL‐6 mRNA in SH‐SY5Y, whereas LPS had no effect, consistent with previous studies (Klegeris and McGeer, 2001; Prehaud et al., 2005). Importantly, Poly(I:C) can bind other receptors (RIG‐I, MDA5) in some cells, but studies in human neuronal cells indicate that extracellular Poly(I:C) specifically utilizes TLR3 (Peltier et al., 2010). Although we detected TLR4 mRNA and protein, we were unable to detect MD‐2 expression in SH‐SY5Y. MD‐2 is a critical accessory protein that is necessary for both LPS‐ and HMGB1‐induced TLR4 signaling (Nagai et al., 2002; Yang et al., 2015). This absence of MD‐2 might explain the lack of detectable TLR4 response in SH‐SY5Y. Thus, our findings indicate LPS and Poly(I:C) activate canonical immune signaling kinases and induce immune signaling mRNA in microglia‐like BV2, but only Poly(I:C) and not LPS activates proinflammatory signaling in neuron‐like SH‐SY5Y.
EtOH did not increase TLR4 expression in either cell type at the concentration and time point (24 hours) utilized in this study, although our previous in vivo study found TLR4 mRNA up‐regulated in mouse brain following 10 daily binges of 5 g/kg EtOH (Crews et al., 2013). Primary rodent microglia, however, have maximally up‐regulated TLR4 protein at 30 minutes that decreases to nonsignificantly elevated levels by 24 hours (Fernandez‐Lizarbe et al., 2013), indicating a time point specificity of EtOH‐induced TLR4 expression in cultured microglia. Despite the lack of TLR4 induction, we found that TLR3 mRNA was increased by EtOH and Poly(I:C) in SH‐SY5Y, suggesting possible EtOH activation of TLR3 signaling in neurons. Fewer studies have examined possible EtOH proinflammatory signaling through TLR3, although a recent study showed that TLR3 KO mice consume more EtOH (Jang et al., 2016). More research is needed to elucidate the possible neuronal role of TLR3 in EtOH‐induced innate immune signaling. Furthermore, the up‐regulation of both TLR7 and TLR8 by EtOH in both BV2 and SH‐SY5Y indicates a commonalty shared between the 2 cell types, indicating more research is needed to understand the complexity of EtOH proinflammatory signaling.
We also found that EtOH increased HMGB1 release in both BV2 and SH‐SY5Y, consistent with EtOH‐induced HMGB1 in hippocampal brain slices, brains of binge EtOH‐treated mice, and postmortem human alcoholic brain (Crews et al., 2013). Similar to our data, HMGB1 is also released from EtOH‐treated (50 mM, 24 hours) nondifferentiated neuroblastoma SH‐SY5Y (Wang et al., 2015). As HMGB1 activates multiple TLRs (Park et al., 2004; Yang et al., 2013), RAGE (Kokkola et al., 2005), and can enhance signaling of cytokines such as IL‐1β (Sha et al., 2008), the EtOH‐induced HMGB1 release we observe may play a role in proinflammatory signaling in both microglia and neurons. We also found that RAGE, a receptor that binds HMGB1 and enhances NFκB signaling (Han et al., 2011), was up‐regulated in EtOH‐treated SH‐SY5Y but not in BV2, consistent with an increase in neuronal RAGE in adolescent‐intermittent EtOH‐treated rats (Vetreno et al., 2013). Overall, we report here that EtOH up‐regulates proinflammatory genes and releases HMGB1 in both cell types, as well as specific TLR3 and RAGE mRNA up‐regulation in neuron‐like SH‐SY5Y but not in microglia‐like BV2.
It is important to note that SH‐SY5Y is a neuroblastoma cell line derived from human tissue and may be different than rodent neurons. However, primary rodent cortical neuronal cultures express and release HMGB1 similar to our findings in human SH‐SY5Y (Perez‐Carrion and Cena, 2013). We also found that TLR2, 3, 4, 7 and 8 mRNA are expressed in neuron‐like SH‐SY5Y, consistent with previous studies in mouse (Lehmann et al., 2012; Lok et al., 2015; Tang et al., 2007). However, as BV2 and SH‐SY5Y are an in vitro model consisting of immortalized and cancer‐derived cells, respectively, their responses may differ from a microglial or neuronal phenotype in vivo. Nonetheless, reports suggest a negligible difference between gene induction by LPS in primary microglia and BV2 (Henn et al., 2009), and SH‐SY5Y are a well‐accepted model for studying dopamine neurons in Parkinson's disease (Korecka et al., 2013). Furthermore, examination of any single cell type outside of its intact environment may alter results, making it worthwhile for our future studies to examine both interaction between these cell types as well using in vivo studies.
Although not utilized in this study, astrocytes are also involved in alcohol use disorders and display proinflammatory immune signaling following EtOH treatment (Adermark and Bowers, 2016; Alfonso‐Loeches et al., 2010). Astrocytes would then make an interesting future point of comparison of innate immune signaling versus the canonical macrophage signaling in microglia.
In summary, this study supports that neurons may contribute to proinflammatory signaling, and suggests a novel role of neurons in EtOH‐induced innate immune signaling. Microglia‐like BV2 displayed activated kinases following LPS, Poly(I:C), and EtOH treatment, as well as induction of cytokines like TNF‐α and IL‐1β. Although neuron‐like SH‐SY5Y had no detectable response to LPS, Poly(I:C) and EtOH increased activated kinases and proinflammatory genes. EtOH displayed a different pattern of activated kinases/transcription factors compared with Poly(I:C) or LPS treatment in either cell type. EtOH also increased release of HMGB1 in both cell types and increased expression of RAGE in SH‐SY5Y, indicating a possible role of HMGB1/RAGE signaling in this model. These findings are consistent with EtOH activating proinflammatory signaling in both neuron‐like SH‐SY5Y and microglia‐like BV2. Further work is needed to examine EtOH‐induced immune signaling in both neurons and microglia.
Supporting information
Table S1. Summary of mRNA Ct values in BV2 and SH‐SY5Y.
Sources of Support
This work was supported in part by the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism (AA019767, AA11605, AA007573, and AA021040), and the Bowles Center for Alcohol Studies. Flow cytometry data were collected in collaboration with the UNC Flow Cytometry Core Facility, which is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. The authors report no conflicts of interest.
References
- Adermark L, Bowers MS (2016) Disentangling the role of astrocytes in alcohol use disorder. Alcohol Clin Exp Res 40:1802–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal RG, Hewetson A, George CM, Syapin PJ, Bergeson SE (2011) Minocycline reduces ethanol drinking. Brain Behav Immun 25(Suppl 1):S165–S169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso‐Loeches S, Pascual‐Lucas M, Blanco AM, Sanchez‐Vera I, Guerri C (2010) Pivotal role of TLR4 receptors in alcohol‐induced neuroinflammation and brain damage. J Neurosci 30:8285–8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson U, Tracey KJ (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29:139–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco AM, Valles SL, Pascual M, Guerri C (2005) Involvement of TLR4/type I IL‐1 receptor signaling in the induction of inflammatory mediators and cell death induced by ethanol in cultured astrocytes. J Immunol 175:6893–6899. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Bergeson SE, Walker D, Ferreira VM, Kuziel WA, Harris RA (2005) Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behav Brain Res 165:110–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF, Harris RA (2012) Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addict Biol 17:108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyadjieva NI, Sarkar DK (2010) Role of microglia in ethanol's apoptotic action on hypothalamic neuronal cells in primary cultures. Alcohol Clin Exp Res 34:1835–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GY, Nunez G (2010) Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10:826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung YT, Lau WK, Yu MS, Lai CS, Yeung SC, So KF, Chang RC (2009) Effects of all‐trans‐retinoic acid on human SH‐SY5Y neuroblastoma as in vitro model in neurotoxicity research. Neurotoxicology 30:127–135. [DOI] [PubMed] [Google Scholar]
- Coller JK, Hutchinson MR (2012) Implications of central immune signaling caused by drugs of abuse: mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacol Ther 134:219–245. [DOI] [PubMed] [Google Scholar]
- Collins MA, Corso TD, Neafsey EJ (1996) Neuronal degeneration in rat cerebrocortical and olfactory regions during subchronic “binge” intoxication with ethanol: possible explanation for olfactory deficits in alcoholics. Alcohol Clin Exp Res 20:284–292. [DOI] [PubMed] [Google Scholar]
- Crews F, Nixon K, Kim D, Joseph J, Shukitt‐Hale B, Qin L, Zou J (2006) BHT blocks NF‐kappaB activation and ethanol‐induced brain damage. Alcohol Clin Exp Res 30:1938–1949. [DOI] [PubMed] [Google Scholar]
- Crews FT, Collins MA, Dlugos C, Littleton J, Wilkins L, Neafsey EJ, Pentney R, Snell LD, Tabakoff B, Zou J, Noronha A (2004) Alcohol‐induced neurodegeneration: when, where and why? Alcohol Clin Exp Res 28:350–364. [DOI] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP, Zou J (2013) High mobility group box 1/Toll‐like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biol Psychiatry 73:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Vetreno RP (2016) Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology 233:1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PD, Kane CJ (2014) Fetal alcohol spectrum disorders and neuroimmune changes. Int Rev Neurobiol 118:41–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Lizarbe S, Montesinos J, Guerri C (2013) Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J Neurochem 126:261–273. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Lizarbe S, Pascual M, Guerri C (2009) Critical role of TLR4 response in the activation of microglia induced by ethanol. J Immunol 183:4733–4744. [DOI] [PubMed] [Google Scholar]
- Han SH, Kim YH, Mook‐Jung I (2011) RAGE: the beneficial and deleterious effects by diverse mechanisms of actions. Mol Cells 31:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Crews FT (2008) Increased MCP‐1 and microglia in various regions of the human alcoholic brain. Exp Neurol 210:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M (2009) The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex 26:83–94. [DOI] [PubMed] [Google Scholar]
- Hiscott J (2007) Triggering the innate antiviral response through IRF‐3 activation. J Biol Chem 282:15325–15329. [DOI] [PubMed] [Google Scholar]
- Jang Y, Lee MH, Park JH, Han SY, Kim DK (2016) TLR3 deficiency increases voluntary alcohol consumption. NeuroReport 27:356–360. [DOI] [PubMed] [Google Scholar]
- Kalluri HS, Ticku MK (2003) Regulation of ERK phosphorylation by ethanol in fetal cortical neurons. Neurochem Res 28:765–769. [DOI] [PubMed] [Google Scholar]
- Klegeris A, McGeer PL (2001) Inflammatory cytokine levels are influenced by interactions between THP‐1 monocytic, U‐373 MG astrocytic, and SH‐SY5Y neuronal cell lines of human origin. Neurosci Lett 313:41–44. [DOI] [PubMed] [Google Scholar]
- Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, Nawroth P, Andersson U, Harris RA, Harris HE (2005) RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol 61:1–9. [DOI] [PubMed] [Google Scholar]
- Korecka JA, van Kesteren RE, Blaas E, Spitzer SO, Kamstra JH, Smit AB, Swaab DF, Verhaagen J, Bossers K (2013) Phenotypic characterization of retinoic acid differentiated SH‐SY5Y cells by transcriptional profiling. PLoS ONE 8:e63862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, Habbel P, Kalin R, Franzoni E, Rybak A, Nguyen D, Veh R, Ninnemann O, Peters O, Nitsch R, Heppner FL, Golenbock D, Schott E, Ploegh HL, Wulczyn FG, Lehnardt S (2012) An unconventional role for miRNA: let‐7 activates Toll‐like receptor 7 and causes neurodegeneration. Nat Neurosci 15:827–835. [DOI] [PubMed] [Google Scholar]
- Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt‐Jones EA, Szabo G (2013) Alcohol‐induced IL‐1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J Leukoc Biol 94:171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Crews FT (2015) Adolescent intermittent ethanol exposure enhances ethanol activation of the nucleus accumbens while blunting the prefrontal cortex responses in adult rat. Neuroscience 293:92–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok KZ, Basta M, Manzanero S, Arumugam TV (2015) Intravenous immunoglobulin (IVIg) dampens neuronal toll‐like receptor‐mediated responses in ischemia. J Neuroinflammation 12:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J, Ferguson L, Harris RA (2013) Neuroimmune signaling: a key component of alcohol abuse. Curr Opin Neurobiol 23:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, McMurray HR, Land H, Almudevar A (2014) On non‐detects in qPCR data. Bioinformatics 30:2310–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos J, Alfonso‐Loeches S, Guerri C (2016) Impact of the innate immune response in the actions of ethanol on the central nervous system. Alcohol Clin Exp Res 40:2260–2270. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K (2002) Essential role of MD‐2 in LPS responsiveness and TLR4 distribution. Nat Immunol 3:667–672. [DOI] [PubMed] [Google Scholar]
- Narayanan KB, Park HH (2015) Toll/interleukin‐1 receptor (TIR) domain‐mediated cellular signaling pathways. Apoptosis 20:196–209. [DOI] [PubMed] [Google Scholar]
- Nessa BN, Tanaka T, Kamino K, Sadik G, Ansar AB, Kimura RYO, Tanii H, Okochi M, Morihara T, Tagami S, Kudo T, Takeda M (2006) Toll‐like receptor 3 mediated hyperphosphorylation of tau in human SH‐SY5Y neuroblastoma cells. Psychiatry Clin Neurosci 60:S27–S33. [Google Scholar]
- Newton K, Dixit VM (2012) Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4:a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E (2004) Involvement of toll‐like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279:7370–7377. [DOI] [PubMed] [Google Scholar]
- Peltier DC, Simms A, Farmer JR, Miller DJ (2010) Human neuronal cells possess functional cytoplasmic and TLR‐mediated innate immune pathways influenced by phosphatidylinositol‐3 kinase signaling. J Immunol 184:7010–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Carrion MD, Cena V (2013) Knocking down HMGB1 using dendrimer‐delivered siRNA unveils its key role in NMDA‐induced autophagy in rat cortical neurons. Pharm Res 30:2584–2595. [DOI] [PubMed] [Google Scholar]
- Prehaud C, Megret F, Lafage M, Lafon M (2005) Virus infection switches TLR‐3‐positive human neurons to become strong producers of beta interferon. J Virol 79:12893–12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Crews FT (2012a) Chronic ethanol increases systemic TLR3 agonist‐induced neuroinflammation and neurodegeneration. J Neuroinflammation 9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Crews FT (2012b) NADPH oxidase and reactive oxygen species contribute to alcohol‐induced microglial activation and neurodegeneration. J Neuroinflammation 9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT (2008) Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AR, Berry JN, Sharrett‐Field L, Prendergast MA (2015) Ethanol withdrawal is required to produce persisting N‐methyl‐D‐aspartate receptor‐dependent hippocampal cytotoxicity during chronic intermittent ethanol exposure. Alcohol 49:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9:429–439. [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Smith SH, Bilbo SD (2013) FACS analysis of neuronal‐glial interactions in the nucleus accumbens following morphine administration. Psychopharmacology 230:525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha Y, Zmijewski J, Xu Z, Abraham E (2008) HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol 180:2531–2537. [DOI] [PubMed] [Google Scholar]
- Shin JH, Lee HK, Lee HB, Jin Y, Lee JK (2014) Ethyl pyruvate inhibits HMGB1 phosphorylation and secretion in activated microglia and in the postischemic brain. Neurosci Lett 558:159–163. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP (2007) Pivotal role for neuronal Toll‐like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A 104:13798–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA (2006) Microglia recognize double‐stranded RNA via TLR3. J Immunol 176:3804–3812. [DOI] [PubMed] [Google Scholar]
- Tuomela S, Autio R, Buerki‐Thurnherr T, Arslan O, Kunzmann A, Andersson‐Willman B, Wick P, Mathur S, Scheynius A, Krug HF, Fadeel B, Lahesmaa R (2013) Gene expression profiling of immune‐competent human cells exposed to engineered zinc oxide or titanium dioxide nanoparticles. PLoS One 8:e68415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT (2012) Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll‐like receptors in the adult prefrontal cortex. Neuroscience 226:475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Qin L, Crews FT (2013) Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiol Dis 59:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chu G, Yang Z, Sun Y, Zhou H, Li M, Shi J, Tian B, Zhang C, Meng X (2015) Ethanol directly induced HMGB1 release through NOX2/NLRP1 inflammasome in neuronal cells. Toxicology 334:104–110. [DOI] [PubMed] [Google Scholar]
- Yang H, Antoine DJ, Andersson U, Tracey KJ (2013) The many faces of HMGB1: molecular structure‐functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol 93:865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ju Z, Ragab AA, Lundback P, Long W, Valdes‐Ferrer SI, He M, Pribis JP, Li J, Lu B, Gero D, Szabo C, Antoine DJ, Harris HE, Golenbock DT, Meng J, Roth J, Chavan SS, Andersson U, Billiar TR, Tracey KJ, Al‐Abed Y (2015) MD‐2 is required for disulfide HMGB1‐dependent TLR4 signaling. J Exp Med 212:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JY, Crews FT (2014) Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS One 9:e87915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of mRNA Ct values in BV2 and SH‐SY5Y.