

Abstract
An important limitation in the field of immunohistochemistry (IHC) is the inability to correlate stain intensity with specific analyte concentrations. Clinical immunohistochemical tests are not described in terms of analytic response curves, namely, the analyte concentrations in a tissue sample at which an immunohistochemical stain (1) is first visible, (2) increases in proportion to the analyte concentration, and (3) ultimately approaches a maximum color intensity. Using a new immunostaining tool (IHControls), we measured the analytic response curves of the major clinical immunohistochemical tests for human epidermal growth factor receptor type II (HER-2), estrogen receptor (ER), and progesterone receptor (PR). The IHControls comprise the analytes HER-2, ER, and PR at approximately log concentration intervals across the range of biological expression, from 100 to 1,000,000 molecules per test microbead. We stained IHControls of various concentrations using instruments, reagents, and protocols from three major IHC vendors. Stain intensity at each analyte concentration was measured, thereby generating an analytic response curve. We learned that for HER-2 and PR, there is significant variability in test results between clinical kits for samples with analyte concentrations of approximately 104 molecules/microbead. We propose that the characterization of immunostains is an important step toward standardization.
Keywords: analyte, analytic response curve, HER-2, IHControl, immunohistochemistry, peptide
Introduction
Of all the disciplines in laboratory medicine, quantitative measurement of analytes in histopathological specimens has presented some of the most challenging hurdles. Some of these unique hurdles include the requirement for tissue fixation, dehydration, embedding, and microtomy before analysis. In addition, the field is hampered by the unavailability of calibrators and standards as well as the lack of objective units of measure that are traceable to the number of analyte molecules. Against this backdrop, there are increasing demands for standardization, accuracy, and day-to-day reproducibility, especially for companion diagnostics. Methods and guidelines to address these clinical needs are well established in clinical laboratory medicine but have been difficult to apply to clinical IHC. Technical limitations frustrate the application of practical solutions that have proven effective in other clinical laboratory disciplines. For example, IHC is unusual among laboratory tests because the test output is expressed in terms of color: stain intensity, or percentage of cells that achieve a detectable stain intensity, or presence/absence of a stain. In all of these measures, there is no traceability to the actual analyte concentration. This would be analogous to expressing a serum glucose concentration as “dark pink” or “3+” based on the appearance of the colorimetric glucose oxidase reaction. Our inability to routinely correlate stain intensity to analyte concentration is a serious limitation in the field, frustrating the goals of standardization and reproducibility.
All quantitative laboratory assays are characterized by analytic response curves. Such curves correlate signal intensity with analyte concentration. At low analyte concentrations, no signal is detected. As the analyte concentration is increased, the signal becomes detectable and then increases in proportion to the analyte concentration. This is conventionally termed the linear range of measurement, or “analytic measurement range.” With even higher analyte concentrations, the increases in signal intensity eventually become smaller and ultimately reach a maximum, or “plateau.”
The inability to generate analytic response curves for commonly used clinical immunohistochemical tests complicates the standardization of test results. Previously published studies demonstrated that, like other immunoassays, human epidermal growth factor receptor type II (HER-2) and estrogen receptor (ER) immunostains are no exception in having analytic response curves.1,2 However, there has never been a quantitative tool that can be broadly applied on a national level to characterize them. In this article, we describe the use of IHControls to quantitatively compare analytic sensitivities of the major clinical immunohistochemical tests used for HER-2, ER, and progesterone receptor (PR). The clinical tests we evaluated collectively comprise >95% of the tests used in the United States for breast cancer management.3
Materials and Methods
IHControls
The IHControls are similar to those previously described4 but with a few modifications. In this study, we developed additional breast cancer IHControls at multiple different analyte concentrations, ranging from approximately 102–106 copies per bead. The precise number is established by quantitative fluorescence microscopy (described below, next section). Briefly, IHControls are composed of two different microbeads: analyte-coated glass test microbeads (7–8 µm diameter) and color standard microbeads (4.5 µm diameter). The analyte-coated microbeads bear covalently linked peptide epitopes for HER-2, ER, and PR. Between all of the various IHControls products used in this study, peptide analytes for all of the major clinical HER-2, ER, and PR tests are represented. The various IHControls differ in the concentration of HER-2, ER, and PR analytes. The microbeads are suspended in a proprietary clear liquid that hardens after application to the glass microscope slide, thereby retaining the microbeads on the glass slide during baking, deparaffinization, antigen retrieval, and staining. Once dried, the droplet can be treated as one would treat a tissue sample. Each dried microliter droplet on the slide incorporates approximately 5000 analyte-coated (test) microbeads.
The IHControls microbead suspension also includes color standard microbeads, which are permanently colored dark brown regardless of the IHC staining procedure. The small size (4.5 µm diameter) of these microbeads distinguishes them from the test microbeads. The color standard microbeads serve as a color intensity reference for standardizing color intensity measurements by image analysis, independent of the camera and microscope optical settings.
The IHControls microbeads were manufactured at a series of different analyte (peptide) concentrations that differ by approximately 1 log, from 106/bead (termed “level 5,” the highest concentration) to 102 (“level 1,” the lowest concentration). Peptide conjugation reactions to the glass microbeads are always performed at the same total peptide concentration, thereby saturating the available glass cross-linking sites. The peptides comprising HER-2, ER, and PR epitopes are 18–30 amino acids long. Each peptide incorporates end-capped amino acids, that is, N-terminal acetylation and C-terminal amidation. Moreover, each peptide has a single (terminal) chemically available (epsilon) amine group for cross-linking to aminosilane on the glass microbead. Each peptide is also synthesized with an additional lysine located near the carboxy or amino termini, distant from the antibody epitope, conjugated with fluorescein. The epsilon amine group of any other internal lysines that derive from the native protein sequence were blocked with an ivDde [1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)-3-methylbutyl] cleavable protecting group. This ivDde group blocks the epsilon amine, preventing it from binding to aminosilane during the cross-linking step to glass microbeads.
All peptide conjugations to glass microbeads were performed with an excess amount of peptide, thereby saturating the available aminosilane cross-linking sites on the microbead. For level 5 (highest analyte concentration) microbeads, only one peptide was conjugated, saturating the microbead with a single type of peptide. For other (lower analyte concentration) microbeads (levels 1–4), we performed the conjugation at defined molar ratios of a mixture of fluorescein-conjugated peptides, keeping the total peptide concentration constant. The molar concentration of each peptide in solution was calculated by spectrophotometry (492 nm), based on the molar extinction coefficient of fluorescein (74,000). An irrelevant peptide, constructed with the same design constraints as described above, was used to dilute out the relevant peptides so as to obtain any desired ratio. This maintained a constant (saturating) peptide concentration during the cross-linking reactions. For level 1–4 IHControls, the microbeads bear all of the HER-2, ER, and PR analytes at equivalent concentrations. The concentration of any individual peptide analyte is estimated by multiplying the molar ratio of that peptide in the mix of peptides used for conjugation to the glass microbeads by the total amount of peptide attached to the bead (as measured by quantitative fluorescence microscopy).
The IHControls also include a second type of glass microbead that is not immunoreactive with the antibody in question. The antigenically irrelevant microbeads serve as an unstained, internal negative control (illustrated in Fig. 2). For level 5 (highest analyte concentration) IHControls, the unstained, internal negative control comprises the microbeads coated with peptides to a different primary antibody. For example, the microbead coated with the SP3-immunoreactive peptide is unstained when immunostaining with the HercepTest antibody. For level 1–4 IHControls, the unstained, internal negative control bead bears an irrelevant peptide that is not immunoreactive with any of the HER-2, ER, or PR antibodies.
Figure 2.
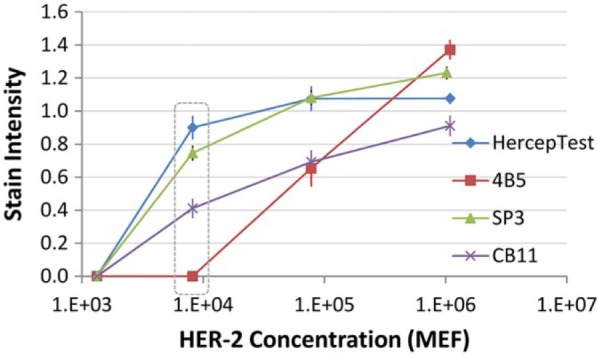
Stain intensity curve as a function of HER-2 concentration. The x-axis reflects analyte concentration (in scientific notation) on a logarithmic scale. Analyte concentration is expressed in MEF, molecules of equivalent fluorochrome, as described in the “Materials and Methods” section. The y-axis, stain intensity, is measured by image analysis. The stain intensity number is a ratio of the stain intensity of the stained test microbeads to the stain intensity of the color standard bead. Scores ≥1.0 represent strong stains. The data points in the dotted rectangle are depicted in Fig. 3. Each data point is the mean ± SD of triplicate slides, that is, three independent staining replicates. The stain intensity value for each slide was derived by sampling three different areas of the same IHControls spot. Abbreviations: HER-2, human epidermal growth factor receptor type II; SD, standard deviation.
Quantitative Fluorescence Microscopy
Analyte concentration on the glass microbeads is calculated using a calibration curve that correlates fluorescence intensity with molecular concentration. This calibration curve is generated using commercial fluorescein calibrator microbeads (cat. no ECFP-F1-5K, Spherotech FITC calibration particle kit; Lake Forest, IL). Fluorescence intensity is quantified after photomicroscopy using a cooled-CCD Spot Imaging camera, Model 2.3.0 (Diagnostic Instruments Inc., Sterling Heights, MI). To measure fluorescence intensity, 1 µl of a bead suspension is mixed with 2 µl of a fluorescence quenching inhibitor (SlowFade Gold; Life Technologies/Thermo Fisher Scientific, Waltham, MA), deposited on a slide, and coverslipped. Photomicroscopy was performed using a 40× objective magnification with an 8-bit depth data capture (gray scale).
The number of fluorescein molecules per calibrator bead is expressed in units of “molecular equivalents of fluorochrome” (MEF). This unit of measure is used in the field of flow cytometry. Soluble fluorochrome, such as fluorescein, can be directly correlated with molar concentration by virtue of fluorescein’s molar extinction coefficient. However, these parameters change slightly for bead-bound fluorescein. The extinction coefficient and fluorescent yield of immobilized (insoluble) fluorescein is not exactly the same as for soluble fluorescein. MEF is a similar measure to the absolute number of molecules except it accounts for this difference.
A 2-point calibration is performed using calibrator microbeads from Spherotech. Each calibration data point is measured in triplicate. Fluorescence intensity of analyte-coated (test) microbeads, coated with a fluorescein-conjugated analyte peptide, is then measured by quantitative fluorescence microscopy. The fluorescence intensity measurement of the test microbeads is then interpolated on the calibration curve, thereby identifying the MEF of HER-2, ER, or PR on each batch of test microbeads. Fluorescence quantification was performed using the thresholding function in ImageJ (provided as a free download at imagej.nih.gov).
Photomicroscopy
Images were acquired with (1) a Nikon Eclipse E400 microscope fitted with a Spot Imaging Solutions RT cooled-CCD color camera, Model 2.3.0 (Diagnostic Instruments Inc.), or (2) a Zeiss Axioskop microscope fitted with a Spot Imaging Solutions Insight Gigabit CCD camera (Diagnostic Instruments Inc.). For any single experiment, the same camera was used for photomicroscopy. For brightfield photomicroscopy of IHControls, the microscope optics are first set for Köhler illumination. Once Köhler illumination was established, the condenser aperture is then opened wide because the microbeads have more than sufficient contrast. With this adjustment, unstained test microbeads are faintly visible alongside stained microbeads. The camera software was set at a gamma of 1.0, using manual (fixed) photographic exposure times. Before photography, the camera was white-balanced and a flat-field correction was performed. Whole slide imaging was not used. Each slide’s color intensity was measured by averaging three images per spot (slide). Each data point in the “Results” section represents the mean ± standard deviation (SD) of triplicate slides.
Image Analysis
To promote consistency, we kept the photomicroscopy settings constant within each experiment. This includes both the optical settings, such as condenser and illumination apertures, as well as camera settings, such as exposure time. For the quantification of IHControls stain intensity, we developed a custom algorithm embedded in MATLAB, as previously described.4 The algorithm measures image intensity of the test microbeads’ rims relative to an internal color intensity standard bead. Because of its smaller size, the color standard bead is easily distinguished from a test bead. Consequently, IHControls stain intensity is expressed as a ratio. A score of 1.0 means that the test microbeads, stained for HER-2, ER, or PR, are equally intense in color as the color standard microbeads. Scores ≥1 represent strong stains.
Immunohistochemistry Staining
IHC staining was performed using three automated immunostainers, in three separate sites. For stains using HER-2, ER, or PR antibodies supplied by Dako Corp./Agilent, a Dako Autostainer (Dako Corp., Carpinteria, CA) was used. These stains included HercepTest, PR 636, PR 1294, and ER 1D5/2-123. The HER-2 and ER/PR PharmDx kits are sold with prediluted solutions and reagents. Slides were initially baked at approximately 57C for 40 min, deparaffinized in xylene, and then hydrated in decreasing grades of ethanol. Antigen retrieval was performed using Dako’s antigen retrieval solutions provided with the HER-2 or ER/PR kits. For HER-2, antigen retrieval was performed in a 97–98C water bath for 40 min, as per the manufacturer’s instructions. For ER/PR antigen retrieval, the slides were processed for 25 min in a Biocare Medical Decloaking Chamber pressure cooker (Biocare Medical Inc., Concord, CA). For all subsequent steps, the manufacturer’s reagents, buffers, and instructions were followed. PR antibody 636 was purchased separately from Dako and coupled with the ER/PR PharmDx kit detection system.
For immunostains using antibodies supplied by Leica Corp. (Leica Biosystems Inc., Buffalo Grove, IL), we performed the testing on a Bond III instrument. Slides were baked at 60–62C for 20–30 min. Depa-raffinization and antigen retrieval were performed on the instrument using the manufacturer’s reagents and protocols. We used the Leica ER 6F11, PR 16, and HER-2 CB11 antibodies with the kit detection reagents.
For immunostains using antibodies supplied by Ventana Medical/Roche (Ventana Medical Systems, Tucson, AZ), we performed the testing on a Benchmark XT. These samples were not baked, as per the usual protocol for that clinical IHC laboratory. Deparaffinization and antigen retrieval were performed on the instrument, using the manufacturer’s solutions and protocols. We used Ventana ER SP1, PR 1E2, and HER-2 4B5 antibodies coupled with the kit detection reagents.
At the end of each immunostaining protocol, the slides were removed from the instruments, dehydrated through increasing grades of ethanol, immersed in xylene, and coverslipped using Permount (ThermoFisher Corp., Waltham, MA).
We also included the HER-2 SP3 rabbit monoclonal antibody in these evaluations. The SP3 antibody was supplied as a hybridoma cell culture supernatant from Abcam (Cambridge, MA). The manufacturer does not supply the antibody concentration. Before use, the antibody preparation was diluted 1:100 in Tris-buffered saline with 0.05% Tween-20, pH 7.4 (TBST), as suggested by the manufacturer. The SP3 stain was performed on a Dako Autostainer because of the instrument’s open architecture. There is no company-specified detection kit for the SP3 antibody, so we paired it with the Ventana/Roche detection kit. The slides were incubated with the primary antibody at room temperature for 30 min. Subsequent steps were performed with the Ventana detection kit diluted 1:3 in TBST, for the following incubations: 30 min (detection reagent), 10 min (DAB), 10 min (copper enhancer), 10 min (hematoxylin), and 10 min (bluing reagent). Ventana detection reagents were diluted 3-fold for use on the Dako Autostainer so as to approximately match the on-slide concentration on the Ventana Benchmark XT. (Reagents on the Benchmark XT are dispensed into a residual buffer volume already on the slide, thereby diluting the reagent approximately 3-fold on the slide.)
Statistical Analysis
Each data point represents the mean ± SD from triplicate slides. Each slide bears a spot of IHControls containing approximately 5000 analyte-coated microbeads. To quantify a single IHControls spot, we sampled three different areas. This is analogous to sampling three fields of a patient’s breast carcinoma for the assessment of HER-2 or ER/PR. From these three fields, we calculated the mean stain intensity per spot (slide). Each data point in the graphs represents the mean ± SD of three separate IHControls spots, each on a separate slide. The data illustrated in this article are representative from multiple similar experiments.
Results
Generation of Test Samples Across the Analytic Range
Measuring analytic response curves requires a series of test samples at regularly spaced, well-characterized analyte concentrations. For the analytes HER-2, ER, and PR, those concentrations are ideally expressed as the number of molecules per cell. Until now, samples with known analyte concentrations, at regularly spaced concentration intervals, were not readily available (see the “Discussion” section). To overcome this problem, we created test samples comprising cell-sized glass microbeads coated with HER-2, ER, or PR peptide analytes (Fig. 1). We use these microbeads as a model solid phase substrate in lieu of cells, accepting one important approximation—test microbeads (8 µm diameter) and cells have slightly different surface areas. Therefore, 100,000 analyte molecules on a microbead will be packed at a slightly different density than in a cell. Nonetheless, it is reasonable to compare different clinical immunostains’ analytic response curves provided that the comparison data are all derived using the same solid phase object (microbeads or cells). The analyte response curves measured using cells will parallel those measured with microbeads, except that they will be shifted right or left to account for the slightly different spatial density of the analyte.
Figure 1.

Schematic illustration of a peptide analyte (e.g., HER-2, ER, or PR). Each sphere represents a single amino acid in a peptide. The letters represent the single-letter abbreviations for amino acids. This particular sequence is for illustrative purposes only and does not correspond to an actual epitope. The peptide is covalently linked to a glass microbead (left). At one end, a fluorescein reporter moiety (illustrated as a small yellow glowing sphere) is attached to an epsilon amine group of lysine. The fluorescein facilitates calculation of the peptide concentration per bead, as the bead’s fluorescence intensity is proportional to the peptide concentration. Abbreviations: HER-2, human epidermal growth factor receptor type II; ER, estrogen receptor; PR, progesterone receptor.
We measured the analyte concentration of IHControls test microbeads by synthesizing the analytes with a fluorescein reporter molecule (Fig. 1). This fluorescein served both to quantify the molar concentration of the peptide in solution (before glass microbead conjugation) and to quantify the total amount of peptide bound to glass microbeads. Both of these measurements depend on the molar extinction coefficient of fluorescein (see the “Materials and Methods” section). Consequently, fluorescence intensity of the IHControls test microbeads correlates with molecular concentration. As there is one fluorescein per peptide analyte, the number of fluorescein moieties equals the number of peptide analytes. We generated a fluorescence intensity—molecular concentration calibration curve using commercial fluorescent microbeads that are calibrated in units of fluorescein molecules per microbead (see the “Materials and Methods” section). The unit of measure for these commercial calibrator microbeads is “molecules of equivalent fluorochrome” (“MEF,” see the “Materials and Methods” and “Discussion” sections). The IHControls microbeads are suspended within a proprietary clear liquid that hardens on the application to a glass slide, thus retaining the microbeads on the slide during baking, deparaffinization, antigen retrieval, and immunostaining. The HER-2, ER, and PR peptide analytes coating the microbeads are composed of 18–30 amino acids that incorporate the linear epitope where the primary antibody binds.4–6
Analytic Response Curves for Clinical HER-2 IHC Tests
Figure 2 illustrates the stain intensity curve of four clinical HER-2 tests across a 3-log range of HER-2 concentration. The HercepTest, 4B5, and CB11 primary antibodies all bind to the same carboxy-terminal HER-2 peptide. The SP3 antibody binds to a separate HER-2 peptide. To fit the broad analyte concentration range being tested, the graph is log-linear. A sample with an even lower concentration than shown in the figure, at the 140 molecules per bead, was also tested but yielded no measurable stain. For each HER-2 test, the stain intensity increases from zero to a maximum over the span of a 1–2 log concentration difference in HER-2.
Figure 2 illustrates that the HercepTest and SP3 tests have similar analytic response curves. Stain intensity is zero at 103 molecules (MEF) per bead and approaches a maximum at approximately 105 molecules (MEF) per bead. Beyond 105 molecules (MEF) per bead, increasing analyte concentrations produce progressively smaller increases in stain intensity until a response plateau is reached. Analyte concentrations on a response plateau yield the same maximal signal regardless of analyte concentration.
The analytic response curves of the other two HER-2 antibodies, 4B5 and CB11, are shifted to the right relative to the HercepTest and SP3 stains. They achieve their maximal stain intensities at approximately 106 molecules (MEF) per bead. In fact, their response curves are shifted sufficiently far to the right so as to not demonstrate a response plateau over the tested range of analyte molecules per bead. The strong staining on this level of HER-2 expression (106 molecules per bead) parallels observations on cells; namely, tumor cells that express approximately 106 molecules per cell also stain strongly positive (3+).7,8
The exact concentrations of each of the different concentration levels of IHControls are shown in Table 1. The analytes on the IHControls microbeads are categorized by levels 1 (lowest concentration) to 5 (highest concentration). Each level is approximately 1 log different (in analyte concentration) than the next. The left-hand column of Table 1 describes the analytes on the IHControls microbeads. The analytes are defined by both the name of the protein (i.e., HER-2, ER, PR) and the specific peptides comprising antibody epitopes. For this reason, analytes are designated by both the name of the protein and the names of antibodies to which they bind. The right-hand column describes the analyte concentration, in molecules (MEF) per microbead.
Table 1.
Analyte Concentrations (in MEF) For Each Antibody
| Analytea | Concentration (MEF) |
|---|---|
| Level 5 IHControl | |
| HER-2 | |
| For HercepTest, 4B5, CB11b | 1,086,658 |
| For SP3 | 1,023,265 |
| ER | |
| For 1D5 | 934,651 |
| For 6F11, 2-123 | 703,460 |
| For SP1 | 904,259 |
| PR | |
| For 636 | 800,503 |
| For 1294, 1E2 | 1,379,113 |
| For 16 | 1,089,808 |
| Level 4 IHControl (All)c | 77,913 |
| Level 3 IHControl (All) | 8187 |
| Level 2 IHControl (All) | 1331 |
| Level 1 IHControl (All) | 140 |
Abbreviations: MEF, molecular equivalents of fluorochrome; HER-2, human epidermal growth factor receptor type II; ER, estrogen receptor; PR, progesterone receptor.
The analytes are defined not only by the molecule to which they bind but also by their antibody epitopes. For this reason, specific antibodies are listed in the “Analyte” column.
The HercepTest, 4B5, and CB11 antibodies all bind to the same HER-2 peptide, on the same microbead.
The term All refers to all of the HER-2, ER, and PR analytes together on the same microbead.
The different analytical sensitivities illustrated in Fig. 2 mean that samples at the 104 molecules (MEF) concentration range can produce completely different test results depending on which the kit is used. A sample expressing 104 molecules (MEF) HER-2 is unstained by the 4B5 stain, moderately stained by the CB11 stain, and strongly stained by HercepTest and SP3. This concentration level is highlighted by the dotted area in Fig. 2 and illustrated in Fig. 3. To orient the reader to the objects in Fig. 3, there are (1) HER-2-coated test microbeads; (2) permanently stained color standard microbeads, as an optical (color) reference; and (3) negative control microbeads with an irrelevant analyte. Frames A (SP3) and B (HercepTest) show strongly stained microbeads, frame C (CB11) shows moderate staining of HER-2-coated microbeads, whereas frame D (4B5) is unstained. Figure 3 illustrates that a single HER-2 concentration, at 8187 molecules per microbead, produces dramatically different test results depending on which clinical HER-2 stain is used.
Figure 3.

Photomicroscopic images of representative areas of IHControls that express 8187 molecules (MEF) of HER-2 per bead. These images relate to data points within the dotted rectangle of Fig. 2. Each of the stains illustrated in Fig. 2 is included: HercepTest (A), SP3 (B), CB11 (C), and 4B5 (D). In frame D (4B5 antibody), the test microbeads are unstained. The smaller 4.5-mm-diameter brown microbeads are color standard microbeads that are always colored brown, regardless of the immunostain. These microbeads serve as an internal stain intensity reference, for image analysis quantification of test bead stain intensity. Scale bar, 10 µm. Abbreviations: MEF, molecular equivalents of fluorochrome; HER-2, human epidermal growth factor receptor type II.
It is important to point out that the HercepTest, 4B5, and CB11 antibodies bind to the very same peptide antigen, on the same test microbead. All three antibodies are immunoreactive with the same carboxy-terminal protein sequence of HER-2. Consequently, the differences in the analytic response curves cannot be due to variability in the IHControl as it is constant across all three stains. Differences in the analytic response curves also cannot be due to preanalytic variables such as length of tissue fixation, cold ischemic time, dehydration, or embedding in paraffin. None of those preanalytic variables apply in this context. The differences in the analytic response curves among the three HER-2 stains are a direct measure of analytic sensitivity, incorporating aspects of both antigen retrieval and the immunostain (see the “Discussion” section).
Analytic Response Curves for Clinical ER and PR IHC Tests
Figure 4 illustrates the stain intensity curve of three clinical ER tests across a broad 4-log range of ER concentration. All three ER tests demonstrate a similar analytic range, each approaching a maximum stain intensity in the 104 molecules per microbead range. The data point for the 6F11 ER stain at the highest analyte concentration (106 molecules per bead) is not shown. Background staining of the matrix confounded accurate image quantification using our MATLAB-based algorithm. The background staining was present across multiple experiments but only at the highest analyte concentration and only with the 6F11 stain.
Figure 4.
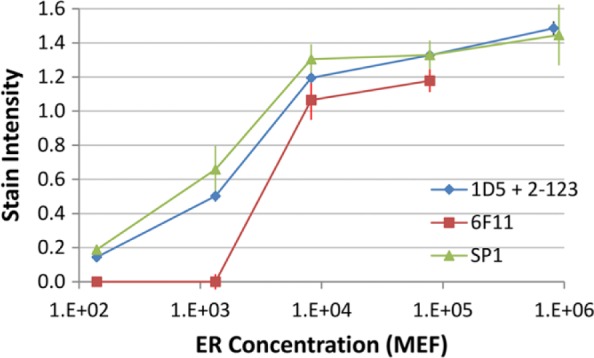
Stain intensity curve as a function of ER concentration per microbead. Data are the means ± SDs of triplicate slides. Each data point is the mean ± SD of three replicate slides. Abbreviations: ER, estrogen receptor; MEF, molecular equivalents of fluorochrome; SD, standard deviation.
Figure 5 illustrates the analytic response curve of four clinical PR tests across a similar, broad 4-log range of PR concentration. These curves, like those associated with HER-2, demonstrate substantial variability for samples in the 104 molecules (MEF) per microbead concentration range. The IHControls at this concentration are almost imperceptibly reactive with the 1E2 antibody but yield a dark brown stain with the 1294 or 636 stains. It is important to note that the 1294 and 1E2 antibodies bind to adjacent epitopes on the same exact peptide, on the same bead. As the analyte-coated microbead is constant for these two immunostains, the different stain intensity must be due to another factor. The difference in stain intensity most likely relates to analytic variables, such as reagent concentrations, primary antibody affinity, time and temperature of incubations, buffers, staining protocol, and so forth (see the “Discussion” section).
Figure 5.
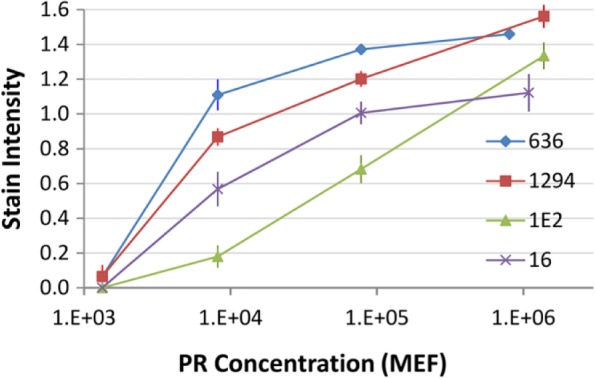
Stain intensity curve as a function of PR concentration per microbead. Each data point is the mean ± SD of three replicate slides. Abbreviations: PR, progesterone receptor; MEF, molecular equivalents of fluorochrome; SD, standard deviation.
Discussion
The most important new finding in this study is the use of IHControls for the quantitative characterization of immunohistochemical analytic response curves. For the first time, we have characterized widely used clinical tests for HER-2, ER, and PR, correlating stain intensity with analyte concentration. These immunostains are among the prototypical IHC companion diagnostic tests, requiring the highest level of performance. They are among the most rigorously validated immunohistochemical tests. Their Food & Drug Administration (FDA) Class II classification reflects this; most of these immunostains went through a 510(k) submission. Most of these kits are produced under current Good Manufacturing Practices (cGMP) conditions. If one were to expect a high level of standardization and reproducibility for any set of immunohistochemical stains, it would be these. Nonetheless, we find log differences in analytical sensitivity that produces discrepancies in the 104 molecules per microbead of analyte concentration range. These findings emphasize the importance of analytical tools for characterizing immunostain performance. Generating analytic response curves is a requisite step toward standardization among clinical tests.
Generating an analytic response curve is a well-established protocol in clinical laboratory medicine but new to the field of IHC. The reason for this is largely because of technical limitations. Calibrated test samples, traceable to an objective standard of measure, were not previously available in diagnostic IHC. We overcame that limitation with the IHControls, which represent a set of samples with a broad spectrum of analyte concentrations. The IHControls comprise microbead-bound analytes, a concept previously described by Shi et al.9 The method we describe in this report is a step toward creating traceability of test results to an objective unit of measure. Quantitative assessment of analytic sensitivity is an important step in transitioning IHC from a “stain” into the world of quantitative clinical diagnostic assays.
In this article, we introduce a new standard of measure that is traceable to molecules of equivalent fluorochrome, or MEF, also known as molecules of equivalent soluble fluorochrome, or MESF. The measure derives from the field of flow cytometry where commercially available microbeads with defined MEF values are used in verifying the linearity of fluorescence measurements on flow cytometers. MEF units are determined by comparing the fluorescence intensity of the fluorochrome-conjugated microbeads with the soluble fluorochrome. Equal numbers of conjugated and soluble fluorochrome molecules are not necessarily of equivalent fluorescent brightness because the conjugation of the fluorochrome to the microbead can affect the extinction coefficient and fluorescence quenching. Despite this limitation, traceability to MEF units has two important advantages: (1) The unit of measure is traceable to a physical constant, the molar extinction coefficient of the fluorescein molecule and (2) we do not require separate protein standards for each cellular protein (HER-2, ER, PR). As long as we can attach a fluorescein moiety to the peptide analyte, any analyte can be quantified.
Although the MEF scale does not exactly represent the number of analyte molecules in cells, our data suggest that they are similar. For example, the SKOV-3 cell line (in one study) was characterized as expressing a mean concentration of 657,088 HER-2 molecules per cell.7 The SK-BR-3 cell line was characterized as expressing 1,400,000–2,390,000 HER-2 molecules per cell.8 In those studies, strong (3+) immunostaining was observed for both cell lines.8,10 Our data parallel these published reports. Figure 2 illustrates that these analyte concentrations, in units of molecules (MEF) per bead, result in strong staining (Fig. 2).
The analytic response curve data indicate that all clinical HER-2, ER, and PR kits produce a strong stain in the 105–106 molecules (MEF) per microbead range. Patient samples expressing HER-2, ER, or PR in this concentration range will generate similar results regardless of the vendor selected by the laboratory. However, there are significant sensitivity differences in the range of approximately 104 molecules (MEF) per microbead. At this concentration range, some clinical HER-2 and PR kits produced positive test results, whereas others were negative. If patient samples were to express a comparable concentration per cell, then these differences can lead to different classifications and treatments.
It is important to note that preanalytic variables, such as fixation, cold ischemia time, dehydration, embedding in paraffin, and microtomy, are not relevant to this study. Although preanalytic variables are important for patient testing, they are distinct from the factors that affect the analytic range of an assay. Instead, the quantification of stain intensity with the IHControls solely reflects analytic and postanalytic variables. Many analytic factors can affect stain intensity, including primary antibody affinity. For example, an 8-fold difference in ER monoclonal antibody affinity was described between two widely used clones.11 Other factors include the amplification factor of the detection kit, concentration of reagents, buffers and wash reagents, instrument protocol, time and temperatures of incubations, reagent volume of dispense (dilution on-slide), and conditions for antigen retrieval.
This study may help explain numerous published studies describing discrepant HER-2, ER, or PR stain results in serial sections of the exact same formalin-fixed paraffin-embedded samples.12–18 The patient samples in those studies have analyte concentrations that are unknown but likely randomly distributed from low to high. The studies involved staining patient samples using two or more clinical immunohistochemical HER-2, ER, or PR tests. The fact that the studies used serial sections of the same samples controlled for preanalytic variables. Consequently, discrepancies in test results between two different clinical stains are most likely due to analytic differences, that is, antigen retrieval or immunostaining. Our findings suggest that these discrepant samples may have expressed HER-2, ER, or PR analytes at a concentration that was above the threshold of detection for one clinical stain but below the threshold of another. Moreover, our data suggest that the discrepant samples may likely fall into a concentration range (per cell) that would be comparable with the 104 molecules (MEF) per microbead range.
Theoretically, any set of test samples with well-defined analyte concentrations will suffice in generating analytic response curves. An obvious candidate set of samples for this role is transformed cell lines. Feasibility has already been demonstrated. The analytic response curve for an HER-2 stain was published in 2005.1 The analytic response curve for an ER stain was published in 2011.2 In those published descriptions, HER-2 and ER protein concentrations (per cell) were quantified from cell lysates of a series of transformed cell lines. Cell lines have also been widely used as samples in proficiency testing and as HER-2 kit controls. Therefore, it is surprising that cell lines have not been used to characterize the analytic response curves of important clinical immunohistochemical tests that are used for patient management.
A call for the development of a cell line HER-2 measurement standard in 200319 led to a HER-2 genomic DNA standard this year (2016)20,21 but, at least so far, no HER-2 protein standard. Other descriptions regarding the use of cell lines in IHC testing, sometimes in the context of proficiency testing, often do not characterize cell lines in terms of the number of HER-2 molecules per cell.22–24 The impediments appear to relate to the difficulties in reproducibly obtaining well-defined, homogeneous cells at regularly spaced protein concentration intervals. From a commercial manufacturer’s perspective, the production of cell line calibrators is complicated by variability in the level of analyte expression. Protein expression in cell cultures can vary with culture confluence and passage number.25 Moreover, there is no objective national protein standard for HER-2, ER, and PR. In measuring analytes from cell lysates, a manufacturer must establish their own calibration curve without traceability to a national protein standard. Technical challenges in HER-2 quantification were recently summarized.26
These difficulties do not apply to IHControls, which rely on chemical rather than biological methods of manufacture. Calibration of the IHControls is based on an already-established fluorescent bead standard that derives from the field of flow cytometry. In the absence of a national protein standard, the calibration curve is traceable to a physical constant (the molar extinction coefficient of fluorescein). The number of molecules (MEF) per microbead does not directly equate with the number of molecules per cell, but our data suggest that the scale is similar. Moreover, it is adaptable to any protein in question that bears a fluorescein as all analytes would trace to the same fluorescein molecular standard.
Our test data should not be interpreted as a definitive characterization of the various clinical HER-2, ER, and PR tests in all clinical IHC laboratories. To best reflect each manufacturer’s test performance, we performed our testing on the instrument platforms manufactured by the respective companies. Nonetheless, our conditions may not exactly replicate those of other clinical IHC laboratories. Variability in reagent dispense volumes, antigen retrieval and deparaffinization conditions, and even differences among multiple different instrument models from a single vendor can affect stain sensitivity, rendering our data different from other clinical IHC laboratories. These data are not yet sufficiently definitive to indicate one vendor’s assay to be more sensitive to another’s across a broad consensus of customers. That will soon follow as we conduct a large multicenter survey.
Looking to the future, we believe that this work will have several important ramifications. First, it enables manufacturers to compare test kits from lot to lot during manufacture. In this context, measuring the analytic response curve could be a useful quality control check of a finished product. A second ramification relates to the use of reproducible immunohistochemical tests for clinical trials. The ability to compare the sensitivity of different manufacturers’ test kits provides the basis for ultimately establishing a required standard for test sensitivity. Standardizing staining leads to reproducibility in patient diagnosis and treatment stratification. Finally, a third ramification is how it can affect clinical IHC laboratories. IHControls can provide important feedback to clinical laboratory staff when used as a positive on-slide control. Lower than expected stain intensity, relative to an already-established baseline, may indicate the presence of a staining problem.
The evaluation of on-slide stain performance may be a useful quality check for promoting inter-laboratory standardization and detecting problems that might otherwise be missed.
Acknowledgments
We are grateful to Dr. Ron Zeheb for his ongoing advice during the course of this project and review of this manuscript; Mr. Jason Badrinarain and Ms. Annette Barry for their technical assistance in operating the Bond III and Benchmark XT immunostainers; Ms. Drorit Bogen for administrative support; Dr. Farzad Noubary for advice regarding statistical analysis; and to the National Cancer Institute, National Institutes of Health, for funding this work.
Footnotes
Competing Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Three of the authors (SRS, KV, and SAB) have a patent (ownership) interest in the IHControls technology. The other two authors (AKS and AB) declare they have no competing interests.
Author Contributions: SRS contributed to the conception of the technology, manufacture of the IHControls, and execution of experiments. KV contributed to the execution of the experiments and image quantification. AKS contributed to the image quantification. AB contributed to the experimental design and development of quality systems for manufacture. SAB contributed to the conception of the technology, experimental design, data review, and drafting of the manuscript. All authors have read and approved the manuscript.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (NIH), National Cancer Institute (grant R44CA183203 to SAB) and the National Center for Advancing Translational Sciences (also NIH; grant UL1TR001064).
Literature Cited
- 1. McCabe A, Dolled-Filhart M, Camp R, Rimm D. Automated quantitative analysis (AQUA) of in situ protein expression, antibody concentration, and prognosis. J Natl Cancer Inst. 2005;97:1808–15. [DOI] [PubMed] [Google Scholar]
- 2. Welsh A, Moeder C, Kumar S, Gershkovich P, Alarid E, Harigopal M, Haffty B, Rimm D. Standardization of estrogen receptor measurement in breast cancer suggests false-negative results are a function of threshold intensity rather than percentage of positive cells. J Clin Oncol. 2011;29:2978–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. CAP Immunohistochemistry Committee. HER-2 A: immunohistochemistry tissue microarray participant survey. CAP Proficiency Test Survey Results, College of American Pathologists, Northfield, IL; 2016. [Google Scholar]
- 4. Sompuram S, Vani K, Tracey B, Kamstock D, Bogen S. Standardizing immunohistochemistry: a new reference control for detecting staining problems. J Histochem Cytochem. 2015;63:681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sompuram S, Kodela V, Ramanathan H, Wescott C, Radcliffe G, Bogen S. Synthetic peptides identified from phage-displayed combinatorial libraries as immunodiagnostic assay surrogate quality-control targets. Clin Chem. 2002;48:410–20. [PubMed] [Google Scholar]
- 6. Sompuram S, Kodela V, Zhang K, Ramanathan H, Radcliffe G, Falb P, Bogen S. A novel quality control slide for quantitative immunohistochemistry testing. J Histochem Cytochem. 2002;50:1425–34. [DOI] [PubMed] [Google Scholar]
- 7. DeFazio-Eli L, Strommen K, Dao-Pick T, Parry G, Goodman L, Winslow J. Quantitative assays for the measurement of HER1-HER2 heterodimerization and phosphorylation in cell lines and breast tumors: applications for diagnostics and targeted drug mechanism of action. Breast Cancer Res. 2011;13:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rasmussen O, Jorgensen R. Controls. In: Taylor CR, Rudbeck L, editors. Immunohistochemical staining methods. Glostrup, Denmark: Dako Corporation; 2014. p.165–8. [Google Scholar]
- 9. Shi S-R, Liu C, Perez J, Taylor C. Protein-embedding technique: a potential approach to standardization of immunohistochemistry for formalin-fixed, paraffin-embedded tissue sections. J Histochem Cytochem. 2005;53:1167–70. [DOI] [PubMed] [Google Scholar]
- 10. Rhodes A, Jasani B, Anderson E, Dodson AR, Balaton AJ. Evaluation of HER-2/neu immunohistochemical assay sensitivity and scoring on formalin-fixed and paraffin-processed cell lines and breast tumors: a comparative study involving results from laboratories in 21 countries. Am J Clin Pathol. 2002;118:408–17. [DOI] [PubMed] [Google Scholar]
- 11. Huang Z, Zhu W, Szekeres G, Xia H. Development of new rabbit monoclonal antibody to estrogen receptor: immunohistochemical assessment on formalin-fixed, paraffin-embedded tissue sections. Appl Immunohistochem Mol Morphol. 2005;13:91–5. [DOI] [PubMed] [Google Scholar]
- 12. Bogina G, Zamboni G, Sapino A, Bortesi L, Marconi M, Lunardi G, Coati F, Massocco A, Molinaro L, Pegoraro C, Venturini M. Comparison of anti–estrogen receptor antibodies SP1, 6F11, and 1D5 in breast cancer. Amer J Clin Pathol. 2012;138:697–702. [DOI] [PubMed] [Google Scholar]
- 13. Cheang M, Treaba D, Speers C, Olivotto I, Bajdik C, Chia S, Goldstein L, Gelmon K, Huntsman D, Gilks C, Nielsen T, Gown A. Immunohistochemical detection using the new rabbit monoclonal antibody SP1 of estrogen receptor in breast cancer is superior to mouse monoclonal antibody 1D5 in predicting survival. J Clin Oncol. 2006;24:5637–44. [DOI] [PubMed] [Google Scholar]
- 14. Dekker TJ, Borg ST, Hooijer GKJ, Meijer SL, Wesseling J, Boers JE, Schuuring E, Bart J, van Gorp J, Mesker WE, Kroep JR, Smit VTBM, van de Vijver MJ. Determining sensitivity and specificity of HER2 testing in breast cancer using a tissue micro-array approach. Breast Cancer Res. 2012;14:R93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gouvea A, Milanezi F, Olson S, Leitao D, Schmitt F, Gobbi H. Selecting antibodies to detect HER2 overexpression by immunohistochemistry in invasive mammary carcinomas. Appl Immunohistochem Mol Morphol. 2006;14:103–8. [DOI] [PubMed] [Google Scholar]
- 16. Rhodes A, Sarson J, Assam EE, Dean SJ, Cribb EC, Parker A. The reliability of rabbit monoclonal antibodies in the immunohistochemical assessment of estrogen receptors, progesterone receptors, and HER2 in human breast carcinomas. Amer J Clin Pathol. 2010;134:621–32. [DOI] [PubMed] [Google Scholar]
- 17. Ricardo S, Milanezi F, Carvalho S, Leitao D, Schmitt F. HER2 evaluation using the novel rabbit monoclonal antibody SP3 and CISH in tissue microarrays of invasive breast carcinomas. J Clin Pathol. 2007;60:1001–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rossi S, Laurino L, Furlanetto A, Chinellato S, Orvieto E, Canal F, Facchetti F, DeiTos A. Rabbit monoclonal antibodies: A comparative study between a novel category of immunoreagents and the corresponding mouse monocloncal antibodies. Amer J Clin Pathol. 2005;124:295–302. [DOI] [PubMed] [Google Scholar]
- 19. Hammond M, Barker P, Taube S, Gutman S. Standard reference material for Her2 testing: report of a National Institute of Standards and Technology-sponsored consensus workshop. Appl Immunohistochem Mol Morphol. 2003;11:103–6. [PubMed] [Google Scholar]
- 20. He H-J, Almeida J, Lund S, Steffen C, Choquette S, Cole K. Development of NIST standard reference material 2373: genomic DNA standards for HER2 measurements. Biomol Detect Quantif. 2016;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lih C, Si H, Das B, Harrington R, Harper K, Sims D, McGregor P, Camalier C, Kayserian A, Williams P, He H, Almeida J, Lund S, Choquette S, Cole K. Certified DNA reference materials to compare HER2 gene amplification measurements using next-generation sequencing methods. J Mol Diagn. 2016;18:753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rhodes A. Developing a cell line standard for HER2/neu. Cancer Biomark. 2005;1:229–32. [DOI] [PubMed] [Google Scholar]
- 23. Rhodes A, Jasani B, Couturier J, McKinley M, Morgan J, Dodson A, Navabi H, Miller K, Balaton A. A formalin-fixed, paraffin-processed cell line standard for quality control of immunohistochemical assay of HER-2/neu expression in breast cancer. Am J Clin Pathol. 2002;117:81–9. [DOI] [PubMed] [Google Scholar]
- 24. Xiao Y, Gao X, Maragh S, Telford W, Tona A. Cell lines as candidate reference materials for quality control of ERBB2 amplification and expression assays in breast cancer. Clin Chem. 2009;55:1307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Riera J, Simpson J, Tamayo R, Battifora H. Use of cultured cells as a control for quantitative immunocytochemical analysis of estrogen receptor in breast cancer. Am J Clin Pathol. 1999;111:329–35. [DOI] [PubMed] [Google Scholar]
- 26. Nuciforo P, Radosevic-Robin N, Ng T, Scaltriti M. Quantification of HER family receptors in breast cancer. Breast Cancer Res. 2015;17:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]