

Abstract
Heparanase, a heparan sulfate (HS)–specific endoglycosidase, plays an important role in inflammation and mediates acute pulmonary and renal injuries during sepsis. To explore its role in septic intestinal injury, a non-anticoagulant heparanase inhibitor, N-desulfated/re-N-acetylated heparin (NAH), was administrated to a mouse sepsis model induced by cecal ligation and puncture (CLP). Immunohistochemical staining revealed massive shedding of HS from the intestinal mucosal surfaces after CLP, and effective inhibition of heparanase by NAH was confirmed by markedly reduced HS shedding. Following CLP, intestinal expression of heparanase was increased, whereas pretreatment with NAH reduced the sepsis-induced upregulation of heparanase expression. Meanwhile, CLP led to shedding of syndecan-1 and upregulated expression of proteases such as matrix metalloprotease-9 and urokinase-type plasminogen activator in the intestine, whereas NAH markedly suppressed syndecan-1 shedding and protease upregulation following CLP. In addition, pretreatment with NAH attenuated intestinal injury, inhibited neutrophil infiltration and suppressed the production of inflammatory cytokines (tumor necrosis factor–α, interleukin-1β, and interleukin-6) in the intestine during sepsis, and it also significantly reduced the elevation of inflammatory cytokines in the serum 24 hr after CLP. Our findings demonstrate that the activation of intestinal heparanase contributes to intestinal injury during early sepsis by facilitating the destruction of mucosal epithelial glycocalyx and promoting inflammatory responses.
Keywords: heparan sulfate, heparanase, intestinal injury, mucosal epithelial glycocalyx, sepsis, syndecan-1
Introduction
Heparan sulfate (HS) is a ubiquitous polysaccharide present both on cell surfaces and in extracellular matrix (ECM). By covalently binding to and regulating numerous ECM proteins, growth factors, morphogens, cytokines, chemokines, and coagulation factors, HS plays critical roles in a variety of biological functions, including cell signaling, motility, cytoskeleton organization, and tissue barrier integrity.1,2 In addition, the deregulation of HS chains is associated with several pathological processes such as tumor formation, metastasis, and angiogenesis.3,4 Heparanase is currently the only known mammalian endoglycosidase capable of degrading HS glycosaminoglycan.5 Early studies have demonstrated that heparanase promotes cancer cell metastasis by not only breaking down extracellular barriers but also regulating bioavailability and activity of growth factors.6 In addition to its well-characterized functions in cancer, heparanase is recently implicated in the pathogenesis of several inflammatory disorders.7 Heparanase was found overexpressed and activated in the colonic epithelium of patients and experimental animals with ulcerative colitis and Crohn’s disease.8,9 Moreover, the inhibition of heparanase activity prevented endotoxemia-associated loss of pulmonary endothelial glycocalyx, and thus attenuated sepsis-induced inflammatory lung injury in mice.10
Heparin and synthetic heparin-mimicking species exhibit strong inhibitory activities toward heparanase.11 Heparin and heparin derivatives have been proven clinically and experimentally to be efficacious in treating inflammatory disorders,12–14 and their anti-inflammatory effect was believed attributed to the inhibition of heparanase activity.9 Our group previously reported that unfractionated heparin (UFH) reduced heparanase activation and attenuated intestinal injury in septic mice, and we postulated that UFH may exert the protective action by inhibiting heparanase activity.15 However, heparin is well known for its anticoagulant properties.16 An earlier study demonstrated that treatment with UFH reduced coagulation and alleviated inflammation in endotoxemic mice,17 implying that the anticoagulant activity of UFH may also contribute to its anti-inflammatory effect in sepsis. Hence, whether the enzymatic activity of heparanase is a vital player in inflammatory response and tissue injury during sepsis remains to be elucidated.
In humans, the plasma level and activity of heparanase are elevated in septic shock.18 Several animal studies indicate that heparanase plays critical roles in sepsis-associated acute pulmonary injury10 and renal failure,19 yet its role in septic intestinal injury is unclear. In this study, we investigated the role of heparanase in septic intestinal injury using a mouse sepsis model of cecal ligation and puncture (CLP). To exclude the confounding anticoagulant activity of UFH, a non-anticoagulant N-desulfated/re-N-acetylated heparin (NAH) was used to specifically inhibit the enzymatic activity of heparanase.
Materials and Methods
Sepsis Model and Treatment
The animal experiment protocol was reviewed and approved by the Animal Ethics Committee in the First Affiliated Hospital of Hainan Medical College. Male C57BL/6 mice of 6 to 8 weeks were obtained from the Experimental Animal Centre of China Medical University. The mice were randomly assigned to three groups: sham, CLP, and CLP+NAH. CLP was conducted as previously described.15 Briefly, the mice were anesthetized with 10% chloral hydrate by intraperitoneal injection. The cecum was exteriorized, ligated at a distal position to the ileocecal valve, and perforated by a single through-and-through puncture with a 23G needle. Subsequently, the cecum was gently squeezed to extrude a small amount of feces and then relocated into the abdominal cavity. The sham-operated mice were subjected to the same procedures except for ligation and perforation. The mice in the CLP+NAH group were subcutaneously injected with 150 µg NAH (diluted in 200 µl sterile saline; ZZStandard, Shanghai, People’s Republic of China) 2 hr before CLP, and the mice in the other two groups received an equal volume of saline via subcutaneous injection. The mice were sacrificed 4 or 24 hr after CLP for the following examinations.
Histopathological Examination
The intestines were fixed in 4% paraformaldehyde, embedded in paraffin, and processed into 5-µm thick sections. Routine hematoxylin and eosin staining was performed, and the sections were examined under a DP microscope (Olympus, Tokyo, Japan) at ×200 magnification.
Immunohistochemistry
The intestines were fixed, paraffin embedded, and cut into 5-µm sections. The sections were dewaxed in xylene and re-hydrated in graded ethanol, followed by antigen retrieval in a citrate buffer at 40C for 10 min. After the inactivation of endogenous peroxidase by 3% H2O2, the sections were blocked with goat serum, and incubated with a primary antibody against HS (1:100; Boster, Wuhan, People’s Republic of China), syndecan-1 (1:100; Bioss, Beijing, People’s Republic of China), or heparanase (1:100; Bioss) overnight at 4C. Thereafter, the sections were incubated sequentially with a biotin-conjugated secondary antibody and horseradish peroxidase (HRP)–labeled streptavidin (Beyotime, Haimen, People’s Republic of China) at room temperature for 30 min each. The immune complexes were visualized by brief incubation with 3,3′-diaminobenzidine, and the cell nuclei were counterstained with hematoxylin. The sections were dehydrated, mounted, and observed by microscopy at ×400 magnification.
Real-Time Polymerase Chain Reaction
Total RNA was extracted from the intestinal tissues with an RNAsimple Total RNA Extraction Kit (Tiangen, Beijing, People’s Republic of China) following the manufacturer’s instructions. RNA concentration and purity was determined by NanoDrop 2000 (ThermoScientific, Wilmington, DE). cDNA was prepared from 1 µg total RNA by reverse transcription using Super M-MLV reverse transcriptase (BioTeke, Beijing, People’s Republic of China) and oligo(dT)15. The mRNA levels of heparanase, tumor necrosis factor–α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6) were measured by real-time polymerase chain reaction (PCR) in an ExicyclerTM 96 Real-Time Quantitative Thermal Block (Bioneer, Daejeon, Korea) using SYBR Green Master Mix (Solarbio, Beijing, People’s Republic of China); the primers are listed in Table 1. β-actin was used as the internal reference, and the relative mRNA level was calculated using the 2−ΔΔCT method.
Table 1.
Real-Time PCR Primers.
| Gene Name | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| Heparanase | AAGCGTGAGTCCCTCGTTC | GGCTCAGACCTGCAAATATC |
| TNF-α | TTCTACTGAACTTCGGGGTGAT | CACTTGGTGGTTTGCTACGA |
| IL-1β | TTTGAAGTTGACGGACCCC | ATCTCCACAGCCACAATGAGTG |
| IL-6 | ACTTCCATCCAGTTGCCTTCTT | TCATTTCCACGATTTCCCAGA |
| β-actin | CTGTGCCCATCTACGAGGGCTAT | TTTGATGTCACGCACGATTTCC |
Abbreviations: TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; IL-6, interleukin-6.
Enzyme-Linked Immunosorbent Assay
The serum heparanase level was measured with a Heparanase Assay Kit (USCN, Wuhan, People’s Republic of China). The levels of TNF-α, IL-1β, and IL-6 in the serum were determined using the corresponding commercial enzyme-linked immunosorbent assay (ELISA) kits (Boster) according to the manufacturer’s instructions.
Then 10% (m/v) intestinal tissue homogenates were prepared with saline, and the level of myeloperoxidase (MPO) in the intestinal tissue homogenate was measured with a MPO Assay Kit (JianchengBio, Nanjing, People’s Republic of China) following the manufacturer’s protocol.
Western Blotting
Total proteins were extracted from the intestinal tissues using RIPA lysis buffer (Beyotime). An equal amount of proteins from each sample were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The separated proteins were transferred onto a polyvinylidene fluoride membrane. After blocking with 5% skimmed milk, the membrane was incubated with a primary antibody against matrix metalloprotease-9 (MMP-9; abcam, Cambridge, UK), urokinase-type plasminogen activator (uPA; Santa Cruz, Dallas, TX) or β-actin (Santa Cruz) overnight at 4C. After washing, the membrane was incubated with HRP-conjugated secondary antibody (Beyotime) at 37C for 45 min. Thereafter, the immune complexes were detected using Enhanced ECL Detection System (Beyotime), and the signals were exposed to the x-ray films. The films were then scanned and analyzed by Gel-Pro-Analyzer software to determine the densitometric values of the target bands, using β-actin as the internal control.
Statistical Analysis
The data are presented as the means ±standard deviations (SDs). Differences between two groups were analyzed using unpaired t-test. A p value <0.05 was considered statistically significant.
Results
NAH Prevented Intestinal HS Shedding Following CLP
Cleavage of HS chain is an indicator of heparanase activity. Here, we detected intestinal heparanase activity by assessing the level of HS in the intestinal tissues by immunohistochemistry. As shown in Figure 1, HS, which was abundantly present at the surfaces of intestinal villi and crypts in the sham-operated mice, was shed off to a considerable extent in 4 hr following CLP, and most villi lost HS 24 hr after CLP. By contrast, pretreatment with NAH significant preserved HS proteoglycans at the intestinal surfaces at both 4 and 24 hr post-CLP, suggesting efficient inhibition of heparanase activity by NAH in the intestine.
Figure 1.

NAH protected intestinal HS from shedding during CLP-induced sepsis. The mice received CLP operation with or without pretreatment with NAH. The levels of HS in the intestinal tissues at 4 and 24 hr after CLP were detected by immunohistochemistry. This figure shows the representative image from each group (n=5; scale = 50 µm). Abbreviations: NAH, N-desulfated/re-N-acetylated heparin; HS, heparan sulfate; CLP, cecal ligation and puncture.
Pretreatment With NAH Inhibited the Upregulation of Heparanase Expression in the Intestine of CLP-Treated Mice
Our group previously reported that intestinal expression and serum level of heparanase were elevated in mice with CLP-induced sepsis.15 In the present study, pretreatment with NAH markedly decreased upregulation of heparanase transcription in the intestine 4 and 24 hr after CLP (Fig. 2A). Moreover, a significant amount of heparanase protein was released to the serum 24 hr after CLP, and NAH almost abolished the elevation of serum heparanase during CLP-induced sepsis (Fig. 2B). Furthermore, immunohistochemical staining revealed that the expression of heparanase in the intestinal epithelium increased with time following CLP, and NAH pretreatment minimized CLP-induced upregulation of heparanase (Fig. 2C). Taken together, NAH inhibited the upregulation of heparanase expression in the intestinal epithelium and reduced elevation of serum heparanase in CLP-induced sepsis.
Figure 2.

NAH suppressed the upregulation of heparanase expression following CLP. (A) The level of heparanase mRNA in the intestinal tissues was determined by real-time PCR. (B) The level of heparanase protein in the serum was measured by ELISA. (C) Intestinal heparanase protein was detected by immunohistochemistry (scale = 50 µm). The figure shows the representative image from each group, and the data are presented as the means ± SD (n=5). Abbreviations: NAH, N-desulfated/re-N-acetylated heparin; CLP, cecal ligation and puncture; PCR, polymerase chain reaction; ELISA, enzyme-linked immunosorbent assay. *p<0.01, **p<0.001.
NAH Inhibited CLP-Induced Shedding of Syndecan-1 and Upregulation of Proteases in the Intestine
Syndecan-1 is a member of HS proteoglycan family and is involved in maintaining the homeostasis of intestinal epithelial barrier.20 As shown in Figure 3A, syndecan-1 was distributed over the surfaces of intestinal villi and crypts in the healthy intestine, with a great abundance in the crypts. Following CLP, syndecan-1 at the villous surface was shed off, whereas NAH reduced the shedding of villous surface syndecan-1 following CLP operation.
Figure 3.

NAH inhibited shedding of syndecan-1 and upregulation of proteases in the intestine of CLP-treated mice. (A) The levels of syndecan-1 in the intestine at 4 hr and 24 hr after CLP were detected by immunohistochemistry (scale = 50 µm). The expressions of (B) MMP-9 and (C) uPA in the intestinal tissues were assayed by Western blotting. This figure presents the representative images, and the data are presented as the means ± SD (n = 5). Abbreviations: NAH, N-desulfated/re-N-acetylated heparin; CLP, cecal ligation and puncture; MMP-9, matrix metalloprotease-9; uPA, urokinase-type plasminogen activator. *p<0.01, **p<0.001.
To explore the mechanism underlying the association between heparanase upregulation and syndecan-1 shedding during sepsis, we examined the intestinal expression levels of several proteases that were implicated in heparanase-stimulated syndecan-1 shedding.21 CLP led to the marked upregulation of MMP-9 and uPA in the intestinal tissues 4 hr and 24 hr following the operation, whereas NAH pretreatment markedly suppressed CLP-induced upregulation of MMP-9 and uPA in the intestine (Fig. 3B and C). These data suggest that MMP-9 and uPA may mediate the shedding of syndecan-1 on heparanase activation in the intestine during CLP-induced sepsis.
Inhibition of Heparanase Attenuated Intestinal Injury and Inflammation Following CLP
Acute intestinal injury is a hallmark of early sepsis. As shown in Figure 4A, the erosion of upper villous surfaces was noticed 4 hr after CLP, and the widespread destruction of intestinal villi was observed 24 hr after CLP. Pretreatment with NAH maximally preserved the intestinal structure 4 hr after CLP, and the histopathological changes in the intestine were markedly attenuated by NAH 24 hr after CLP. Next, we assessed neutrophil infiltration into the intestinal tissues during CLP-induced sepsis. The level of MPO, a marker of neutrophil, in the intestinal tissues was significantly increased 24 hr after CLP, and NAH eliminated sepsis-induced elevation of intestinal MPO level (Fig. 4B), implying that NAH pretreatment inhibited neutrophil infiltration into the intestinal tissues during early sepsis. We further assessed the production of inflammatory cytokines by the intestinal tissues during CLP-induced sepsis by real-time PCR. The mRNA levels of TNF-α, IL-1β, and IL-6 in the intestinal tissues were increased with time following CLP operation, whereas CLP-induced upregulation of these inflammatory cytokines was significantly reduced by NAH pretreatment (Fig. 4C–E). These results demonstrated that the inhibition of heparanase activity by NAH during early sepsis suppressed intestinal inflammation and attenuated septic intestinal injury in mice.
Figure 4.

NAH reduced sepsis-induced intestinal injury and inflammation. (A) Histopathological examination of intestine by H&E staining (scale = 100 µm). (B) MPO activity in the intestinal tissues was determined using a commercial MPO assay kit. (C–E) The mRNA levels of TNF-α, IL-1β, and IL-6 in the intestinal tissues were measured by real-time PCR. This figure shows the representative image from each group, and the data are presented as the means ± SD (n=5). Abbreviations: NAH, N-desulfated/re-N-acetylated heparin; CLP, cecal ligation and puncture; MPO, myeloperoxidase; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; IL-6, interleukin-6; PCR, polymerase chain reaction; H&E, hematoxylin and eosin. *p<0.01, **p<0.001.
Inhibition of Heparanase Reduced Serum Levels of Inflammatory Cytokines During Early Sepsis
To evaluate the systemic inflammatory status, we measured the serum levels of TNF-α, IL-1β, and IL-6 in CLP-treated mice with or without pretreatment with NAH. The ELISA results indicated that the serum levels of TNF-α, IL-1β, and IL-6 started increasing 4 hr after CLP and rose dramatically at 24 hr (Fig. 5). Pretreatment with NAH, however, significantly inhibited the elevation of inflammatory cytokines in the serum 24 hr after CLP, suggesting that the inhibition of heparanase activity by NAH may attenuate systemic inflammation in CLP-treated mice.
Figure 5.
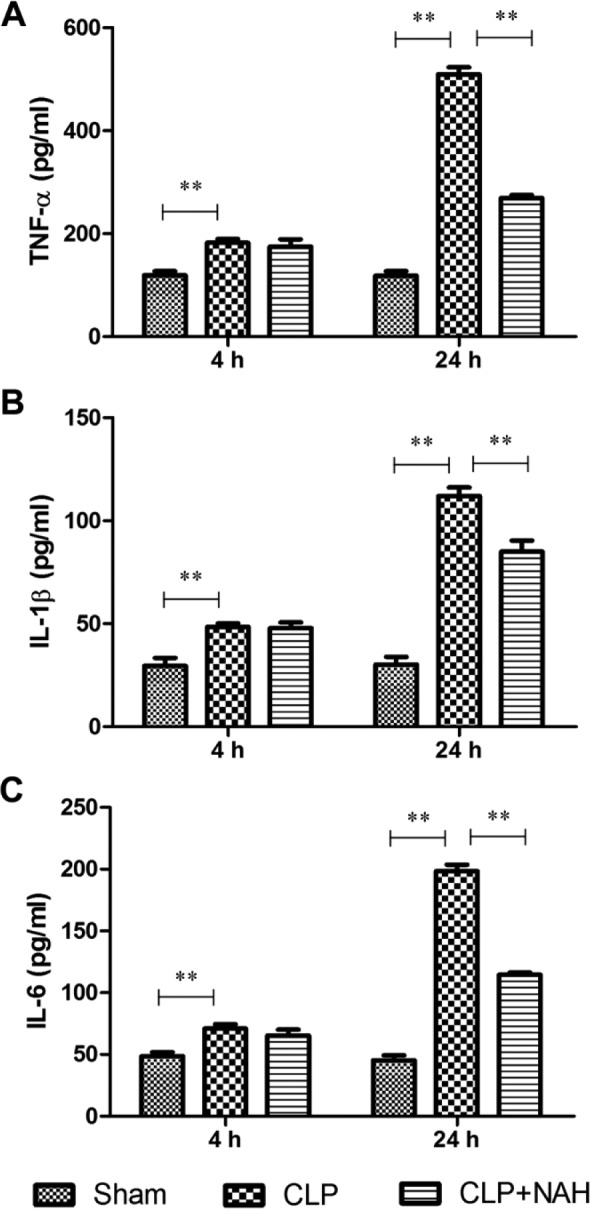
NAH inhibited elevation of serum inflammatory cytokines following CLP. The levels of (A) TNF-α, (B) IL-1β, and (C) IL-6 in the serum of CLP-treated mice with or without NAH pretreatment were determined by ELISA. The data are presented as the means ± SD (n=5). Abbreviations: NAH, N-desulfated/re-N-acetylated heparin; CLP, cecal ligation and puncture; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; IL-6, interleukin-6; ELISA, enzyme-linked immunosorbent assay. **p<0.001.
Discussion
The small intestine plays a central role in the pathophysiology of sepsis, and it is referred to as the “motor” of systemic inflammatory response syndrome.22 Our group previously reported that UFH that presented inhibitory activities toward both heparanase and coagulation attenuated intestinal injury and systemic inflammation in mouse models of sepsis.15,17 In the present study, we treated the mice with a non-anticoagulant heparanase inhibitor, NAH, before the induction of sepsis, to elucidate the role of heparanase in sepsis. Our results demonstrated that the inhibition of heparanase activity by NAH, as evidenced by the prevention of HS shedding, reduced sepsis-induced upregulation of heparanase expression in the intestinal epithelial barrier, inhibited shedding of syndecan-1, attenuated intestinal injury, and suppressed intestinal and systemic inflammation during CLP-induced sepsis in mice.
Mucosal epithelial glycocalyx consists of transmembrane syndecans and bound HS. This structure maintains physiological permeability, physical barrier, and homeostasis of innate immunity.23 Here we showed that the inhibition of heparanase activity by NAH prevented the destruction of intestinal mucosal glycocalyx, inhibited neutrophil infiltration, protected mucosal integrity, and suppressed inflammatory responses in the mouse model of sepsis. These data suggest that restricted activity of heparanase is crucial for maintaining intestinal mucosal homeostasis. In addition, Schmidt et al. demonstrated that the heparanase-mediated degradation of pulmonary endothelial glycocalyx contributes to neutrophil adhesion and inflammation in sepsis-associated acute lung injury.10 Taken together, deregulated heparanase during early sepsis destroys epithelial and endothelial glycocalyx in multiple organs, resulting in the aggravation of local and systemic inflammation, multiple organ injury/failure, and high mortality.
Syndecans are transmembrane HS proteoglycans that can be cleaved by sheddases such as heparanase and matrix metalloproteinases to release the HS-containing ectodomain. Syndecan-1, primarily known as the epithelial syndecan, has been recently implicated in the homeostasis of intestinal epithelial barrier.20 Moreover, activated shedding of syndecan-1 and elevated levels of heparanase are found to be associated with the clinicopathological features and severity of Crohn’s disease.24 In the present study, CLP led to the activation of heparanase and shedding of syndecan-1 from the intestinal mucosal epithelium, and NAH markedly reduced syndecan-1 shedding following CLP operation. These data suggest an association between heparanase activation and syndecan-1 shedding during early sepsis. Heparanase has been demonstrated to stimulate syndecan-1 shedding in tumors.25,26 Furthermore, it was shown that blocking the proteolytic activity of MMP-9 or uPA inhibited heparanase-induced syndecan-1 shedding in myeloma cells,21 proposing that MMP-9 and uPA play a role in mediating syndecan-1 shedding on heparanase activation. In our study, the inhibition of heparanase activation by NAH significantly suppressed CLP-induced upregulation of MMP-9 and uPA, implying that MMP-9 and uPA may be involved in mediating snydecan-1 shedding in response to heparanase activation. In addition, prominent shedding of syndecans is commonly observed in inflammation, and the shed syndecans are believed to be inflammatory mediators.27,28 In this study, syndecan-1 shedding was coincident with intestinal inflammation and injury following CLP, implying that shed syndecan-1 may contribute to intestinal inflammation and injury during sepsis.
Circulating HS fragments are highly potent endogenous molecules that can trigger inflammatory responses through pro-inflammatory pathways such as Toll-like receptor-dependent pathways.29 Hence, by liberating membrane- and ECM-bound HS, heparanase contributes to excessive inflammatory response in sepsis. In our study, the CLP-induced activation of heparanase led to the production of TNF-α, IL-1β, and IL-6 in the intestinal tissues and elevation of these inflammatory cytokines in the serum. Meanwhile, the expression of heparanase in the intestine was also upregulated with time following CLP operation, whereas the inhibition of heparanase activity by NAH reduced the upregulation of intestinal heparanase expression in CLP-treated mice. Moreover, we observed a correlation between the severity of inflammation and the expression level of heparanase during early sepsis. It was demonstrated that heparanase was upregulated during chronic inflammation in a TNF-α-dependent mechanism.8 Thus, during early sepsis, TNF-α induces upregulation of heparanase expression, and activated heparanase produces HS fragments that trigger the production of TNF-α and other inflammatory cytokines. Consequently, the TNF-α-heparanase circuit promotes regional inflammation and contributes to systemic inflammatory response syndrome.
In conclusion, our study demonstrates that heparanase contributes to sepsis-associated acute intestinal injury by destroying mucosal epithelial glycocalyx barrier and generating HS fragments that exacerbate inflammatory responses during early sepsis.
Footnotes
Competing Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: XM and GJ conceived the study, reviewed the data, and amended the manuscript; SC designed the experiments and drafted the manuscript; SC, ZH, SL, and XY performed the animal work, and conducted the hematoxylin and eosin and IHC assays; YH and CL carried out polymerase chain reaction, Western blotting, and enzyme-linked immunosorbent assay; and SC and YH analyzed and interpreted all the data.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant from the Key Medical Research Foundation of Department of Health, Hainan Province (2012-ZD-10).
Literature Cited
- 1. Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–7. [DOI] [PubMed] [Google Scholar]
- 2. Kramer KL, Yost HJ. Heparan sulfate core proteins in cell-cell signaling. Annu Rev Genet. 2003;37:461–84. [DOI] [PubMed] [Google Scholar]
- 3. Iozzo RV, Sanderson RD. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J Cell Mol Med. 2011;15:1013–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2:521–8. [DOI] [PubMed] [Google Scholar]
- 5. Vreys V, David G. Mammalian heparanase: what is the message? J Cell Mol Med. 2007;11:427–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal I, Spector L, Pecker I. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med. 1999;5:793–802. [DOI] [PubMed] [Google Scholar]
- 7. Goldberg R, Meirovitz A, Hirshoren N, Bulvik R, Binder A, Rubinstein AM, Elkin M. Versatile role of heparanase in inflammation. Matrix Biol. 2013;32:234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lerner I, Hermano E, Zcharia E, Rodkin D, Bulvik R, Doviner V, Rubinstein AM, Ishai-Michaeli R, Atzmon R, Sherman Y, Meirovitz A, Peretz T, Vlodavsky I, Elkin M. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J Clin Invest. 2011;121:1709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Waterman M, Ben-Izhak O, Eliakim R, Groisman G, Vlodavsky I, Ilan N. Heparanase upregulation by colonic epithelium in inflammatory bowel disease. Mod Pathol. 2007;20:8–14. [DOI] [PubMed] [Google Scholar]
- 10. Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, Smith LP, Cheng SS, Overdier KH, Thompson KR, Geraci MW, Douglas IS, Pearse DB, Tuder RM. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18:1217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bar-Ner M, Eldor A, Wasserman L, Matzner Y, Cohen IR, Fuks Z, Vlodavsky I. Inhibition of heparanase-mediated degradation of extracellular matrix heparan sulfate by non-anticoagulant heparin species. Blood. 1987;70:551–7. [PubMed] [Google Scholar]
- 12. Dotan I, Hallak A, Arber N, Santo M, Alexandrowitz A, Knaani Y, Hershkoviz R, Brazowski E, Halpern Z. Low-dose low-molecular weight heparin (enoxaparin) is effective as adjuvant treatment in active ulcerative colitis: an open trial. Dig Dis Sci. 2001;46:2239–44. [DOI] [PubMed] [Google Scholar]
- 13. Folwaczny C, Wiebecke B, Loeschke K. Unfractioned heparin in the therapy of patients with highly active inflammatory bowel disease. Am J Gastroenterol. 1999;94:1551–5. [DOI] [PubMed] [Google Scholar]
- 14. Young E. The anti-inflammatory effects of heparin and related compounds. Thromb Res. 2008;122:743–52. [DOI] [PubMed] [Google Scholar]
- 15. Chen S, Zhang X, Sun Y, Hu Z, Lu S, Ma X. Unfractionated heparin attenuates intestinal injury in mouse model of sepsis by inhibiting heparanase. Int J Clin Exp Pathol. 2015;8:4903–12. [PMC free article] [PubMed] [Google Scholar]
- 16. Hirsh J, Anand SS, Halperin JL, Fuster V, American Heart A. Guide to anticoagulant therapy: heparin: a statement for healthcare professionals from the American Heart Association. Circulation. 2001;103:2994–3018. [DOI] [PubMed] [Google Scholar]
- 17. Ding R, Zhao D, Guo R, Zhang Z, Ma X. Treatment with unfractionated heparin attenuates coagulation and inflammation in endotoxemic mice. Thromb Res. 2011;128:e160–5. [DOI] [PubMed] [Google Scholar]
- 18. Martin L, De Santis R, Koczera P, Simons N, Haase H, Heinbockel L, Brandenburg K, Marx G, Schuerholz T. The synthetic antimicrobial peptide 19-2.5 interacts with heparanase and heparan sulfate in murine and human sepsis. PLoS ONE. 2015;10:e0143583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lygizos MI, Yang Y, Altmann CJ, Okamura K, Hernando AA, Perez MJ, Smith LP, Koyanagi DE, Gandjeva A, Bhargava R, Tuder RM, Faubel S, Schmidt EP. Heparanase mediates renal dysfunction during early sepsis in mice. Physiol Rep. 2013;1:e00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qing Q, Zhang S, Chen Y, Li R, Mao H, Chen Q. High glucose-induced intestinal epithelial barrier damage is aggravated by syndecan-1 destruction and heparanase overexpression. J Cell Mol Med. 2015;19:1366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Purushothaman A, Chen L, Yang Y, Sanderson RD. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clark JA, Coopersmith CM. Intestinal crosstalk: a new paradigm for understanding the gut as the “motor” of critical illness. Shock. 2007;28:384–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wells JM, Loonen LM, Karczewski JM. The role of innate signaling in the homeostasis of tolerance and immunity in the intestine. Int J Med Microbiol. 2010;300:41–8. [DOI] [PubMed] [Google Scholar]
- 24. Zhang S, Qing Q, Wang Q, Xu J, Zhi F, Park PW, Zhang Y, Chen Y. Syndecan-1 and heparanase: potential markers for activity evaluation and differential diagnosis of Crohn’s disease. Inflamm Bowel Dis. 2013;19:1025–33. [DOI] [PubMed] [Google Scholar]
- 25. Mahtouk K, Hose D, Raynaud P, Hundemer M, Jourdan M, Jourdan E, Pantesco V, Baudard M, De Vos J, Larroque M, Moehler T, Rossi JF, Reme T, Goldschmidt H, Klein B. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood. 2007;109:4914–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang Y, Macleod V, Miao HQ, Theus A, Zhan F, Shaughnessy JD, Jr, Sawyer J, Li JP, Zcharia E, Vlodavsky I, Sanderson RD. Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–33. [DOI] [PubMed] [Google Scholar]
- 27. Gotte M. Syndecans in inflammation. FASEB J. 2003;17:575–91. [DOI] [PubMed] [Google Scholar]
- 28. Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006;20:9–22. [DOI] [PubMed] [Google Scholar]
- 29. Johnson GB, Brunn GJ, Platt JL. Cutting edge: an endogenous pathway to systemic inflammatory response syndrome (SIRS)-like reactions through Toll-like receptor 4. J Immunol. 2004;172:20–4. [DOI] [PubMed] [Google Scholar]