

Abstract
Induced pluripotent stem (iPS) cells are generated from mouse and human somatic cells by forced expression of defined transcription factors using different methods. Here, we produced iPS cells from mouse amniotic fluid cells, using a non-viral-based transposon system. All obtained iPS cell lines exhibited characteristics of pluripotent cells, including the ability to differentiate toward derivatives of all three germ layers in vitro and in vivo. This strategy opens up the possibility of using cells from diseased fetuses to develop new therapies for birth defects.
Keywords: Developmental Biology, Issue 120, Amniotic fluid cells, induced pluripotent stem cells, PiggyBac transposon, regenerative medicine, reprogramming, mouse.
Introduction
Prenatal diagnosis is an important clinical tool to evaluate genetic diseases (i.e. chromosomal aberrations, monogenetic or polygenetic/multifactorial diseases) and congenital malformations (i.e. congenital diaphragmatic hernia, cystic lung lesions, exomphalos, gastroschisis). Amniotic fluid (AF) cells are simple to obtain from routinely scheduled procedures during the second trimester of pregnancy (i.e. amniocentesis and amnioreduction) or caesarean sections1,2. The availability of AF cells from prenatal or neonatal patients provides the possibility of using this source for regenerative medicine, and several researchers investigated the possibility to treat different tissue damages or diseases using a stem cell population isolated from AF3,4,5,6,7,8,9,10,11,12. The possibility of easily obtaining AF cells from diseased patients, in a time window in which the disease is often stationary, opens the way to the idea of using this cell source for reprogramming purposes. Indeed, induced pluripotent stem (iPS) cells derived from AF cells could be differentiated in the cells of interest for in vitro drug testing or for tissue engineering approaches, in order to prepare an adequate patient-specific therapy before childbirth. Many studies have already demonstrated the ability of AF cells to be reprogrammed and differentiated into a wide range of cell types13,14,15,16,17,18,19,20,21,22,23,24,25,26,27.
Since the discovery by Takahashi and Yamanaka28 of reprogrammed somatic cells through the forced expression of four transcription factors (Oct4, Sox2, cMyc and Klf4), progress has been made in the field of reprogramming. Considering the different methods, we can distinguish between viral and non-viral approaches. The first expects the usage of viral vectors (retroviruses and lentiviruses), which have high efficiency but usually incomplete silencing of the retroviral transgene, with both the consequence of a partially reprogrammed cell line and the risk of insertional mutagenesis29,30,31. The non-viral method uses different strategies: i.e. plasmids, vectors, mRNA, protein, transposons. The derivation of iPS cells free of transgenic sequences aims to circumvent the potentially harmful effects of leaky transgene expression and insertional mutagenesis. Among all the above mentioned non-viral strategies, the PiggyBac (PB) transposon/transposase system requires only the inverted terminal repeats flanking a transgene and transient expression of the transposase enzyme to catalyse insertion or excision events32. The advantage in using transposons over other methods for iPS cell generation is the possibility of obtaining vector-free iPS cells with a non-viral vector approach that shows the same efficiency of retroviral vectors. This is possible by trace-less excision of the integrated transposon encoding for the reprogramming factors following a new transient expression of the transposase in the iPS cells33. Given that PB is efficient in different cell types34,35,36,37, is more suitable for a clinical approach with respect to viral vectors, and allows for the production of xeno-free iPS cells contrary to current viral production protocols that use xenobiotic conditions, this system is used to obtain iPS cells from murine AF.
Here we propose a detailed protocol following already published work to show the production of pluripotent iPS clones from mouse AF cells (iPS-AF cells)38.
Protocol
All procedures were in accordance with Italian law. Murine AF samples were harvested from pregnant mice at 13.5 days post coitum (d.p.c.) from C57BL/6-Tg(UBC-GFP)30Scha/J mice called GFP.
1. Transposon Production
NOTE: Transposon expression vectors were generated using standard cloning procedures. The plasmid DNA for mouse AF cells transfection was prepared using commercial kits.
Mix 10 ng of plasmid DNA with 50 µl of DH5α bacteria in a 1.5 ml microcentrifuge tube. Incubate the microcentrifuge tube on ice for 20 min.
Heat shock at 42 °C for 40 sec.
Place microcentrifuge tube on ice for 2 min.
Add 250 µl of pre-warmed (37 °C) lysogeny broth (LB) broth. Shake (300 rpm) at 37 °C for 30 min.
Spread 30 µl of each transformation onto LB plates with 0.1 mg/ml ampicillin. Allow plates to dry and incubate inverted at 37 °C overnight.
The next day, in the afternoon, pick 3 colonies, using sterile 200 µl tips, from each transformation and transfer onto 0.1 mg/ml LB amp.
Shake (300 rpm) bacteria overnight at 37 °C.
For plasmid purification use commercial kits. Stock plasmids at -20 °C.
2. Mouse Embryonic Fibroblast (MEF) Culture
- Coating of the 6-well Plates with 0.1% Gelatin
- Coat plates under the hood adding 1 ml of sterile 0.1% gelatin to each of the six wells. Let the gelatin polymerize on the incubator (37 °C) for 1 hr.
- Discard the excess, and dry the plates for 1 hr.
- Use parafilm to seal and store plates at room temperature.
- Inactivation of MEF with Mitomycin C
- Thaw a vial of 4 x 106 MEF cells using a 37 °C bath.
- Seed MEF in the hood on a Petri dish (150 mm) with MEF medium.
- Cultivate cells at 37 °C and 5% CO2.
- (CAUTION) Add mitomycin C (5 mg/mL) when cells are confluent.
- Place cells into an incubator (37 °C and 5% CO2) for 3 h.
- Remove medium with mitomycin C. Wash cells three times with 1x phosphate buffer saline (PBS).
- Add 2 ml of the commercial trypsin and place cells into the 37 °C incubator for 5 min. After, add 18 ml of blocking medium to stop the trypsin reaction.
- Count the cells using a burker chamber according to manufacturer's protocol.
- Centrifuge cells at 145 x g for 5 min. Discard the supernatant.
- Seed 60,000 cells/cm2 of inactivated MEF on 0.1% gelatin coated plates.
- Cultivate cells for 24 h at 37 °C and 5% CO2, before seeding AF and/or iPS-AF cells.
3. Mouse AF Cell Isolation
Set up timed matings and check vaginal plugs after 24 h of co-housing.
Observe until 13.5 d.p.c and sacrifice pregnant dam through cervical dislocation.
Clean the abdominal wall of the animal using 70% ethanol.
Using scissors, perform a midline laparotomy incising the length of the abdominal wall to gain access into the abdominal cavity.
Expose the uterus using forceps. Remove the uterus using scissors and transfer into a 100 mm Petri dish filled with sterile 1x PBS placed on ice.
Use a stereomicroscope to collect the AF at 10X magnification.
Hold the uterus with forceps and remove the uterine wall with scissors. Be careful to avoid any damage to the fetal membranes and consequent amniotic fluid leakage.
Collect fetuses into a 100 mm Petri dish filled with sterile 1x PBS and place on ice.
Grasp the placenta of one fetus with a fine point forceps and place in a clean 100 mm Petri dish.
Remove the yolk sac using fine point forceps.
Disrupt the amnion using fine point forceps and collect 30 µl of AF for each fetus using an insulin syringe into a 15 ml conical vial.
After AF collection, wash each fetus with sterile 1x PBS supplemented with 1% fetal bovine serum (FBS) and collect into the 15 ml conical tube containing the AF.
Centrifuge AF at 145 x g for 5 min. Remove the supernatant under the hood and seed the pelleted AF cells.
4. Mouse AF Cell Culture
- Seeding of Mouse AF Cells
- One day prior to the seeding, prepare a 0.1% gelatin-coated 6-well plate with mitomycin C-treated MEF.
- After AF collection, seed the cells obtained from an amniocentesis (around 1 x 107 cells obtained from 6 fetuses) onto the mitotically inactivated MEF using AF culture medium.
- Cultivate cells at 37 °C and 5% CO2, changing the medium every other day.
- After 7 days of culture, transfect AF cells following the procedure described below.
5. Transfection, iPS-AF Cell Generation and Culture
NOTE: Mouse AF cells were transfected with the PB-tetO2-IRES-OKMS transposon plasmid, in conjunction with a transposase expression plasmid (mPBase) and with the reverse tetracycline transactivator (PB-CAG-rtTA) transposon plasmid. All plasmids were provided by Professor Andras Nagy33.
Mix 1 µg of PB-tetO2-IRES-OKMS with 0.5 µg of mPBase and 0.5 µg of PB-CAG-rtTA (Total DNA: 2 µg) in a 1.5 ml microcentrifuge tube.
Dilute the DNA to 100 µL with serum-free medium (Dulbecco's Modified Eagle Medium - DMEM).
Add 8 µL of the transfection reagent (e.g., FUGENE) (at the ratio of plasmid DNA:transfection agent of 2 µg (DNA) to 8 µL transfection agent) into the 1.5 mL microcentrifuge tube containing DNA.
Incubate transfection reagent/DNA mixture for 15 min at room temperature.
Add dropwise 100 µL of the transfection reagent/DNA mixture per well to a 6-well plate with mouse AF cells and distribute by gentle swirling, with a final volume of 2 mL/well. Leave for 24 h.
The day after, induce the expression of the Yamanaka's factors (Oct4, Klf4, cMyc, Sox2) by feeding the cells with fresh iPS-AF medium supplemented with 1.5 µg/mL of doxycycline (2 mL of media for each well).
Feed cells every day, without passage, with fresh dox-containing medium (maintain cells in media supplemented with doxycycline until point 5.13).
Observe cells within 24 h for mCherry expression and daily for colony formation (colonies typically appear between 20 and 30 days after transfection).
One day before colony picking, prepare a 96-well tissue culture plate with mitomycin C-treated MEF.
Pick single colonies in 50 µL of volume using a 200 µL pipette, and transfer them sequentially into a 96-well plate in individual wells.
The day after, dissociate every colony into single cells by adding 50 µl of the commercial trypsin and incubate for 5 min at 37 °C.
Pipette up and down to disaggregate the colony.
Transfer 50 µL of the trypsin containing the dissociated colony into a new well with inactivated MEF on a 0.1% gelatin coated 96 well-plate filled with 100 µL of fresh iPS-AF medium supplemented with doxycycline.
When cells reach 60-70% confluence, split into a well of a 24-well plate.
When cells reach 60-70% confluence, split 1:2 to two wells, one with doxycycline and the other without doxycycline, to verify if colonies appear also in the condition without doxycycline.
Continue to maintain iPS-AF clones, doxycycline independent, on inactivated MEF in the iPS-AF medium.
Cultivate cells at 37 °C and 5% CO2, changing medium daily.
6. Alkaline Phosphatase Staining
Perform alkaline phosphatase staining following the manufacturer's protocol whenever there is the appearance of doxycycline independent iPS-AF cells.
7. Immunofluorescence
Whenever there is the appearance of doxycycline independent iPS-AF cells, seed 150,000 cells/cm2 on a well of a 24-well plate with inactivated MEF, and grow cells for 48 hr at 37 °C and 5% CO2.
(CAUTION) Fix the cells by adding 300 µl per well of 4% paraformaldehyde (PFA) for 15 min at room temperature.
Add 300 µl of 0.1% NP-40 to permeabilize cells.
Rinse cells with 300 µl of 0.1% PBS-Tween 20.
Add 300 µl of the blocking buffer (10% horse serum in 1x PBS) for 1 hr at room temperature.
Use the following primary antibodies: Oct4 (1:80), Sox2 (1:80), Klf4 (1:80), Nanog (1:100) and SSEA1 (1:80). Dilute SSEA1 with 1% bovine serum albumin (BSA), 10% normal goat serum, 0.3 M glycine in 0.1% PBS-Tween-20. Dilute all other antibodies with 10% horse serum in 1x PBS.
Incubate antibodies overnight at 4 °C.
Rinse with 300 µl of 0.1% PBS-Tween-20.
Use the secondary antibodies diluted in 10% horse serum in 1x PBS: Alexa594-conjugated goat anti-mouse IgG (1:200), Alexa594-conjugated chicken anti-goat IgG (1:200), Alexa594-conjugated chicken anti-rabbit IgG (1:200), Alexa568-conjugated goat anti-mouse IgM (1:200). Incubate for 2 hr at room temperature.
Rinse with 1x PBS.
Stain nuclei with Hoechst solution (1:10,000) in 1x PBS.
8. In Vitro Cell Differentiation
To form hanging drop embryoid bodies (EBs), cultivate single drops (2,000 cells/20 µl) onto the lid of Petri dishes, using the differentiation medium.
Incubate the dishes at 37 °C and 5% CO2 for 4 days.
Transfer EBs into 100 mm bacterial-grade dishes in the suspension culture for 3 days in differentiation medium.
Plate EBs in 24 well-plates coated with basement membrane matrix (diluted 1:10 in DMEM) for another 14 days.
For immunofluorescence analyses, use primary antibodies: T (1:100), αfp (1:200) and Tubb3 (1:500)37.
9. In Vivo Teratoma Formation
Inject 100 µl of 1x PBS containing 1 x 106 iPS-AF cells into the muscle of the hindlimb of Rag2-/-γc-/- mice.
Sacrifice mice through cervical dislocation 6 weeks after cell injection.
Remove the tumor masses, distinguished by their size, from the hindlimb of the mice for the following analyses.
Cut 7-10 µm thick transverse sections of the isopentane-frozen tumor using a cryostat.
- For Haematoxylin and Eosin (HE) Stain:
- Stain with HE to evaluate the tumor tissue composition, following the manufacturer's protocol.
- For immunoperoxidase Stain:
- Fix teratoma sections with 4% PFA for 15 min at room temperature.
- Permeabilize with 0.1% Triton X-100.
Incubate with the following antibodies: Tubb3 (1:100), αfp (1:50) and αSMA (1:100) diluted in 1% BSA in 1x PBS. Incubate for 1 hr at 37 °C (for Tubb3) or overnight at 4 °C (for the other antibodies).
Block peroxidase using 100% methanol for 20 min.
Incubate for 45 min at 37 °C with secondary antibody anti-mouse HRP-conjugated (1:150).
Rinse with 1x PBS.
Incubate with peroxidase (HRP) substrate (See Materials Table) for 5 min.
Rinse with tap water for 5 min.
Stain with Haematoxylin QS for 9 sec.
Rinse with tap water.
10. RNA Extraction from Cells
Use a commercial RNA extraction kit according to the manufacturer's recommended protocol.
11. RNA Extraction from Teratoma
Homogenize the teratoma using a pestle and mortar and liquid nitrogen to avoid thawing the sample.
Weigh 25 mg of tissue.
Add 1 mL of commercial reagent for RNA extraction and 200 µL of chloroform.
Incubate at room temperature for 5 min.
Centrifuge at 13,000 x g and 4 °C for 15 min.
Transfer the cleared supernatant to a new tube.
To purify the RNA use a commercial RNA extraction kit according to the manufacturer's protocol.
12. Reverse Transcription-polymerase Chain Reaction Analysis
Synthesize the first-strand cDNA using a reverse transcriptase for reverse transcription (RT)-polymerase chain reaction (PCR) and oligo (dT) according to manufacturer's protocol, to obtain 50 ng/µL of cDNA. Dilute cDNA with RNase-free water at a concentration of 10 ng/µL.
Perform RT-PCR using the DNA polymerase according to manufacturer's protocol, utilizing 1 ng/µL of cDNA.
Representative Results
To evaluate the capacity of reprogramming, mouse AF cells were collected from fetuses of GFP mice. Cells were transfected with the circular transposon plasmid PB-tetO2-IRES-OKMS, which expresses the Yamanaka factors (Oct4, Sox2, cMyc and Klf4) linked to the mCherry fluorescent protein in a doxycycline-inducible manner, and reverse tetracycline transactivator (PB-CAG-rtTA) plasmids together with the transposase expression plasmid (mPBase). Mouse AF cells were transfected with Oct4, Klf4, cMyc and Sox2 after 1 week of in vitro expansion over a MEF feeder layer. For the expression of the exogenous factors, doxycycline was added the day after the transfection at a concentration of 1.5 µg/mL, as previously described39. The expression of the mCherry was visible after 24 h from the doxycycline addition, indicating transfection. The first colonies with an ES-like morphology appeared 20 days after doxycycline induction and were picked 30 days post-transfection. These colonies were seeded on inactivated fibroblast feeder layers. After picking, surviving clones were maintained in medium supplemented with doxycycline for some passages until found to be doxycycline independent in replicated wells33.
Mouse iPS-AF clones were evaluated for different parameters: mCherry expression, alkaline phosphatase staining, protein expression of the pluripotency markers Oct4, Sox2, Klf4, Nanog and SSEA1, expression of exogenous and endogenous pluripotency genes (Oct4, Klf4, cMyc and Sox2), in vitro (EBs) and in vivo (teratoma assay) differentiation. They fulfilled all the criteria evaluated to validate their pluripotency, as summarized in the Figure 1.
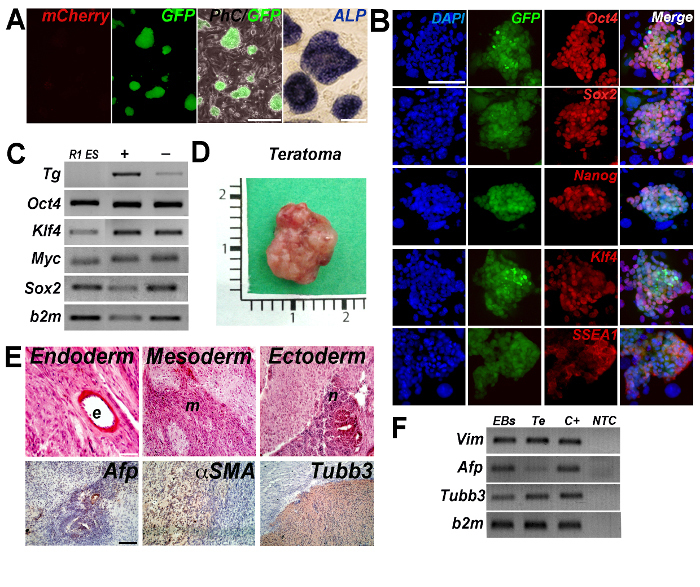 Figure 1: Reprogramming of mouse AF GFP+ cells. (A) Doxycycline independent colonies of iPS-AF GFP+ cells were negative for the expression of mCherry and positive for the alkaline phosphatase (ALP) staining. Scale bars = 250 µm and 100 µm. (B) Representative images of stable doxycycline-independent iPS-AF GFP+ cell expressing Oct4, Sox2, Nanog, Klf4 and SSEA1. Scale bar = 100 µm. (C) RT-PCR analysis for the expression of exogenous (vector-based primers, Tg) and endogenous (Oct4, Klf4, cMyc and Sox2) genes. Beta-2-microglobulin (b2m) was considered as a housekeeping gene. Reprogrammed cell lines were grown in the presence (+) or absence (-) (96 hr) of doxycycline. R1 ES cells were used as a control. (PCR panel has been modified from Bertin et al.38). (D) Gross appearance of teratoma obtained 6 weeks after the injection of iPS-AF GFP+ cells into the hind limbs of Rag2-/-γc-/- mouse. (E) Teratoma histology and immunostaining confirmed cell differentiation into all three germ layers (ectoderm, mesoderm and endoderm). αfetoprotein (Afp, endoderm), αsmooth muscle actin (αSMA, mesoderm), and β3 tubulin (Tubb3, ectoderm) were detected in tumor masses. Scale bar = 100 μm. (F) RT-PCR analysis of EBs and teratoma (Te). C+, positive control; NTC, non-template control. Vimentin (Vim, mesoderm), αfetoprotein (Afp, endoderm) and β3 tubulin (Tubb3, ectoderm). (PCR panel has been modified from reference38). Please click here to view a larger version of this figure.
Figure 1: Reprogramming of mouse AF GFP+ cells. (A) Doxycycline independent colonies of iPS-AF GFP+ cells were negative for the expression of mCherry and positive for the alkaline phosphatase (ALP) staining. Scale bars = 250 µm and 100 µm. (B) Representative images of stable doxycycline-independent iPS-AF GFP+ cell expressing Oct4, Sox2, Nanog, Klf4 and SSEA1. Scale bar = 100 µm. (C) RT-PCR analysis for the expression of exogenous (vector-based primers, Tg) and endogenous (Oct4, Klf4, cMyc and Sox2) genes. Beta-2-microglobulin (b2m) was considered as a housekeeping gene. Reprogrammed cell lines were grown in the presence (+) or absence (-) (96 hr) of doxycycline. R1 ES cells were used as a control. (PCR panel has been modified from Bertin et al.38). (D) Gross appearance of teratoma obtained 6 weeks after the injection of iPS-AF GFP+ cells into the hind limbs of Rag2-/-γc-/- mouse. (E) Teratoma histology and immunostaining confirmed cell differentiation into all three germ layers (ectoderm, mesoderm and endoderm). αfetoprotein (Afp, endoderm), αsmooth muscle actin (αSMA, mesoderm), and β3 tubulin (Tubb3, ectoderm) were detected in tumor masses. Scale bar = 100 μm. (F) RT-PCR analysis of EBs and teratoma (Te). C+, positive control; NTC, non-template control. Vimentin (Vim, mesoderm), αfetoprotein (Afp, endoderm) and β3 tubulin (Tubb3, ectoderm). (PCR panel has been modified from reference38). Please click here to view a larger version of this figure.
Discussion
The method chosen to obtain the induction of pluripotency is relevant for cell clinical safety with respect to long term transplantation. Nowadays, there are several methods suitable for the reprogramming. Among the non-integrative methods, the Sendai viral (SeV) vector is an RNA virus that can produce large amounts of protein without integrating into the nucleus of the infected cells40 and could be a strategy to obtain iPS cells. SeV vectors could be an attractive candidate for the generation of translational-grade iPS cells, but it presents some shortcomings. The viral replicase is sensitive to the nature of the transgenic sequences. Since SeV vectors constitutively replicate, they can be difficult to eliminate from the host cells, even if different strategies are used to improve this aspect41,42. It requires special handling, such as a Level 2 biological safety cabinet. There is only one commercial vendor available. There is a current lack of clinical-grade SeV for reprogramming and there are high costs of iPS cell production.
Due to these aspects, the transposon system can be considered a better approach with respect to SeV vectors, and here we establish that this system is a suitable method for reprogramming mouse AF cells with various advantages. It allows the production of xenofree iPS cells contrary to current viral production protocols that use xenobiotic conditions. It has no apparent species barrier and has a transgene cargo capacity bigger than viruses. It allows simple and safe delivery of plasmid transposons through the use of standard cell transfection method, which can be easily performed in every laboratory equipped with a Level 1 biological safety cabinet. Transposase re-expression permits the removal of PB elements with consequent generation of footprint-free mouse and human iPS cells, as demonstrated in various cell lines 32,34,35. Different studies showed that the transposon systems display a lower frequency in targeting near cancer related genes and less bias in targeting active genes compared to viral-based vectors43,44,45,46,47. Reprogramming based on transposons depends on the chosen delivery method, and this is a limitation due to the resistance or toxicity related to DNA transfection methods (lipofection, electroporation or nucleofection). Here we used a non-liposomal formulation designed to transfect DNA which worked well with mouse AF cells. If other cells are employed, tests will be necessary to use the best delivery method in terms of high efficiency and low toxicity. Summarily, the PB transposon method is considered to be safe and low cost but with a lower efficiency compared with SeV vectors.
Regarding the procedure to obtain mouse AF cells, it is critical to have pregnant female mice available. As such, it is necessary to set up an adequate number of matings (at least six females with two females per male).
More experiments could be performed specifically on human AF cells to produce iPS cells from healthy and diseased fetuses. Considering the wide range of congenital diseases that can be detected through the prenatal diagnosis, cells obtained from the AF during gestation represent the best source to derive iPS cells for both studying the disease48 and also for planning a regenerative medicine approach in case of tissue defects such as cardiac or skeletal muscle49,50.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by CARIPARO Foundation Grant number 13/04 and Fondazione Istituto di Ricerca Pediatrica Città della Speranza Grant number 10/02. Martina Piccoli, Chiara Franzin and Michela Pozzobon are funded by Fondazione Istituto di Ricerca Pediatrica Città della Speranza. Enrica Bertin is funded by CARIPARO Foundation Grant number 13/04. Paolo De Coppi is funded by Great Ormond Street Hospital Children's Charity.
References
- You Q, et al. Isolation of human mesenchymal stem cells from third-trimester amniotic fluid. Int J Gynaecol Obstet. 2008;103(2):149–152. doi: 10.1016/j.ijgo.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Prusa AR, Hengstschlager M. Amniotic fluid cells and human stem cell research: a new connection. Med Sci Monit. 2002;8(11):RA253–RA257. [PubMed] [Google Scholar]
- Ditadi A, et al. Human and murine amniotic fluid c-Kit+ Lin- cells display hematopoietic activity. Blood. 2009;113(17):3953–3960. doi: 10.1182/blood-2008-10-182105. [DOI] [PubMed] [Google Scholar]
- Piccoli M, et al. Amniotic fluid stem cells restore the muscle cell niche in a HSA-Cre, Smn(F7/F7) mouse model. Stem Cells. 2012;30(8):1675–1684. doi: 10.1002/stem.1134. [DOI] [PubMed] [Google Scholar]
- Zani A, et al. Amniotic fluid stem cells improve survival and enhance repair of damaged intestine in necrotising enterocolitis via a COX-2 dependent mechanism. Gut. 2014;63(2):300–309. doi: 10.1136/gutjnl-2012-303735. [DOI] [PubMed] [Google Scholar]
- Rota C, et al. Human amniotic fluid stem cell preconditioning improves their regenerative potential. Stem Cells Dev. 2012;21(11):1911–1923. doi: 10.1089/scd.2011.0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabella T, et al. Pro-angiogenic soluble factors from Amniotic Fluid Stem Cells mediate the recruitment of endothelial progenitors in a model of ischemic fasciocutaneous flap. Stem Cells Dev. 2012;21(12):2179–2188. doi: 10.1089/scd.2011.0639. [DOI] [PubMed] [Google Scholar]
- Mirabella T, Cili M, Carlone S, Cancedda R, Gentili C. Amniotic liquid derived stem cells as reservoir of secreted angiogenic factors capable of stimulating neo-arteriogenesis in an ischemic model. Biomaterials. 2011;32(15):3689–3699. doi: 10.1016/j.biomaterials.2011.01.071. [DOI] [PubMed] [Google Scholar]
- Yeh YC, et al. Cardiac repair with injectable cell sheet fragments of human amniotic fluid stem cells in an immune-suppressed rat model. Biomaterials. 2010;31(25):6444–6453. doi: 10.1016/j.biomaterials.2010.04.069. [DOI] [PubMed] [Google Scholar]
- Kim JA, et al. MYOD mediates skeletal myogenic differentiation of human amniotic fluid stem cells and regeneration of muscle injury. Stem Cell Res Ther. 2013;4(6):147. doi: 10.1186/scrt358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadasz S, et al. Second and third trimester amniotic fluid mesenchymal stem cells can repopulate a de-cellularized lung scaffold and express lung markers. J Pediatr Surg. 2014;49(11):1554–1563. doi: 10.1016/j.jpedsurg.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Turner CG, et al. Preclinical regulatory validation of an engineered diaphragmatic tendon made with amniotic mesenchymal stem cells. J Pediatr Surg. 2011;46(1):57–61. doi: 10.1016/j.jpedsurg.2010.09.063. [DOI] [PubMed] [Google Scholar]
- Galende E, et al. Amniotic Fluid Cells Are More Efficiently Reprogrammed to Pluripotency Than Adult Cells. Cloning Stem Cells. 2009;12(2):117–125. doi: 10.1089/cell.2009.0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, et al. Pluripotency can be rapidly and efficiently induced in human amniotic fluid-derived cells. Hum Mol Genet. 2009;18(22):4340–4349. doi: 10.1093/hmg/ddp386. [DOI] [PubMed] [Google Scholar]
- Ye L, et al. Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc Natl Acad Sci U S A. 2009;106(24):9826–9830. doi: 10.1073/pnas.0904689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum K, et al. The LARGE Principle of Cellular Reprogramming: Lost, Acquired and Retained Gene Expression in Foreskin and Amniotic Fluid-Derived Human iPS Cells. PLoS ONE. 2010;5(10):137–138. doi: 10.1371/journal.pone.0013703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, et al. Generation of induced pluripotent stem cells using site-specific integration with phage integrase. Proc Natl Acad Sci U S A. 2010;107(45):19467–19472. doi: 10.1073/pnas.1012677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HE, et al. Selection of alkaline phosphatase-positive induced pluripotent stem cells from human amniotic fluid-derived cells by feeder-free system. Exp Cell Res. 2011;317(13):1895–1903. doi: 10.1016/j.yexcr.2011.05.017. [DOI] [PubMed] [Google Scholar]
- Lu HE, et al. Modeling Neurogenesis Impairment in Down Syndrome with Induced Pluripotent Stem Cells from Trisomy 21 Amniotic Fluid Cells. Exp Cell Res. 2012;319(4):498–505. doi: 10.1016/j.yexcr.2012.09.017. [DOI] [PubMed] [Google Scholar]
- Kobari L, et al. Human induced pluripotent stem cells can reach complete terminal maturation: in vivo and in vitro evidence in the erythropoietic differentiation model. Haematologica. 2012;97(12):1795–1803. doi: 10.3324/haematol.2011.055566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Fan Y, Sun X, Yu Y. Generation of Induced Pluripotent Stem Cells from Human Amniotic Fluid Cells by Reprogramming with Two Factors in Feeder-free Conditions. J Reprod Dev. 2012;59(1):72–77. doi: 10.1262/jrd.2012-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Luo Y, Chen X, Li Q, Sun X. Generation of Human β-thalassemia Induced Pluripotent Stem Cells from Amniotic Fluid Cells Using a Single Excisable Lentiviral Stem Cell Cassette. J Reprod Dev. 2012;58(4):404–409. doi: 10.1262/jrd.2011-046. [DOI] [PubMed] [Google Scholar]
- Liu T, et al. High efficiency of reprogramming CD34+ cells derived from human amniotic fluid into induced pluripotent stem cells with Oct4. Stem Cells Dev. 2012;21(12):2322–2332. doi: 10.1089/scd.2011.0715. [DOI] [PubMed] [Google Scholar]
- Jiang G, et al. Human transgene-free amniotic-fluid-derived induced pluripotent stem cells for autologous cell therapy. Stem Cells Dev. 2014;23(21):2613–2625. doi: 10.1089/scd.2014.0110. [DOI] [PubMed] [Google Scholar]
- Drozd AM, et al. Generation of human iPSC from cells of fibroblastic and epithelial origin by means of the oriP/EBNA-1 episomal reprogramming system. Stem Cell Res Ther. 2015;6(122) doi: 10.1186/s13287-015-0112-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschidou D, et al. Human mid-trimester amniotic fluid stem cells cultured under embryonic stem cell conditions with Valproic acid acquire pluripotent characteristics. Stem Cells Dev. 2012;22(3):444–458. doi: 10.1089/scd.2012.0267. [DOI] [PubMed] [Google Scholar]
- Moschidou D, et al. Valproic Acid Confers Functional Pluripotency to Human Amniotic Fluid Stem Cells in a Transgene-free Approach. Mol Ther. 2012;20(10):1953–1967. doi: 10.1038/mt.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454(7200):49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan R, et al. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136(2):364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight S, Collins M, Takeuchi Y. Insertional mutagenesis by retroviral vectors: current concepts and methods of analysis. Curr Gene Ther. 2013;13(3):211–227. doi: 10.2174/1566523211313030006. [DOI] [PubMed] [Google Scholar]
- Fraser MJ, Ciszczon T, Elick T, Bauser C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol Biol. 1996;5(2):141–151. doi: 10.1111/j.1365-2583.1996.tb00048.x. [DOI] [PubMed] [Google Scholar]
- Woltjen K, Kim SI, Nagy A. The PiggyBac Transposon as a Platform Technology for Somatic Cell Reprogramming Studies in Mouse. Methods Mol Biol. 2015;1357:1–22. doi: 10.1007/7651_2015_274. [DOI] [PubMed] [Google Scholar]
- Wu SC, et al. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A. 2006;103(41):15008–15013. doi: 10.1073/pnas.0606979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, et al. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122(3):473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Wang W, et al. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105(27):9290–9295. doi: 10.1073/pnas.0801017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadiñanos J, Bradley A. Generation of an inducible and optimized PiggyBac transposon system. Nucleic Acids Res. 2007;35(12):e87. doi: 10.1093/nar/gkm446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin E, et al. Reprogramming of mouse amniotic fluid cells using a PiggyBac transposon system. Stem Cell Research. 2015;15(3):510–513. doi: 10.1016/j.scr.2015.09.009. [DOI] [PubMed] [Google Scholar]
- Woltjen K, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458(7239):766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaki N, et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85(8):348–362. doi: 10.2183/pjab.85.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishishita N, et al. Generation of virus-free induced pluripotent stem cell clones on a synthetic matrix via a single cell subcloning in the naive state. PLoS One. 2012;7(9):e38389. doi: 10.1371/journal.pone.0038389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M, et al. Generation of induced pluripotent stem cells from human nasal epithelial cells using a Sendai virus vector. PLoS One. 2012;7(8):e42855. doi: 10.1371/journal.pone.0042855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant SR, et al. High-resolution genome-wide mapping of transposon integration in mammals. Mol Cell Biol. 2005;25(6):2085–2094. doi: 10.1128/MCB.25.6.2085-2094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MH, Coates CJ, George AL. PiggyBac transposon-mediated gene transfer in human cells. Mol Ther. 2007;15(1):139–145. doi: 10.1038/sj.mt.6300028. [DOI] [PubMed] [Google Scholar]
- Galvan DL, et al. Genome-wide mapping of PiggyBac transposon integrations in primary human T cells. J Immunother. 2009;32(8):837–844. doi: 10.1097/CJI.0b013e3181b2914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, et al. Gene transfer efficiency and genome-wide integration profiling of Sleeping Beauty, Tol2, and piggyBac transposons in human primary T cells. Mol Ther. 2010;18(10):1803–1813. doi: 10.1038/mt.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir YJ, et al. Genome-wide target profiling of PiggyBac and Tol2 in HEK 293: pros and cons for gene discovery and gene therapy. BMC Biotechnol. 2011;11(28) doi: 10.1186/1472-6750-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363(15):1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- Filareto A, et al. An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun. 2013;4(1549) doi: 10.1038/ncomms2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabi R, et al. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 2012;10(5):610–619. doi: 10.1016/j.stem.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]