

Abstract
HIV-1 envelope proteins engage cognate receptors on the target cell surface, which leads to viral-cell membrane fusion followed by the release of the viral capsid (CA) core into the cytoplasm. Subsequently, the viral Reverse Transcriptase (RT), as part of a namesake nucleoprotein complex termed the Reverse Transcription Complex (RTC), converts the viral single-stranded RNA genome into a double-stranded DNA copy (vDNA). This leads to the biogenesis of another nucleoprotein complex, termed the pre-integration complex (PIC), composed of the vDNA and associated virus proteins and host factors. The PIC-associated viral integrase (IN) orchestrates the integration of the vDNA into the host chromosomal DNA in a temporally and spatially regulated two-step process. First, the IN processes the 3' ends of the vDNA in the cytoplasm and, second, after the PIC traffics to the nucleus, it mediates integration of the processed vDNA into the chromosomal DNA. The PICs isolated from target cells acutely infected with HIV-1 are functional in vitro, as they are competent to integrate the associated vDNA into an exogenously added heterologous target DNA. Such PIC-based in vitro integration assays have significantly contributed to delineating the mechanistic details of retroviral integration and to discovering IN inhibitors. In this report, we elaborate upon an updated HIV-1 PIC assay that employs a nested real-time quantitative Polymerase Chain Reaction (qPCR)-based strategy for measuring the in vitro integration activity of isolated native PICs.
Keywords: Infection, Issue 120, Integration, HIV-1, Preintegration complex, Target DNA, nested PCR, in vitro
Introduction
HIV-1 replication in the target cell involves multiple steps, broadly grouped into two phases: early and late events. The early events begin when HIV-1 sequentially binds to the target cell surface receptor CD4 and one of the two co-receptors (CCR5 or CXCR4)1. Consequently, the virus-cell membranes fuse, and the viral capsid (CA) core is released into the cytoplasm2. The CA core contains the viral genomic single-stranded RNA (ssRNA) and several proteins, both viral and cellular in origin. The CA-associated viral proteins include Reverse Transcriptase (RT) and integrase (IN), two enzymes that mediate critical steps during early events in viral replication. The RT and IN function in the context of nucleoprotein complexes, namely the Reverse Transcription Complex (RTC) and the pre-integration complex (PIC), respectively3,4,5,6. The ends of the viral ssRNA genome harbor terminal repeat (R) elements adjacent to the unique sequences U5 (at the 5' end) and U3 (at the 3' end). The RTC converts the viral ssRNA genome into a double-stranded (ds) DNA copy (vDNA). This reverse transcription process also results in duplication of the U5 and U3 sequences, thus generating long terminal repeats (LTR) at both ends of the vDNA7,8. Subsequently, the PIC-associated IN catalyzes two sequential biochemical reactions that enable the integration of the vDNA into the host chromosome9,10,11. First, in the cytoplasm, IN multimers engage both LTRs12 and cleave a dinucleotide from each of the 3'-terminal ends. This 3'-end processing generates a hydroxyl group at each 3'-end (CAOH) of the vDNA. Next, PIC traffics to the nucleus, wherein IN uses the vDNA CAOH as a nucleophile to cut both strands of the chromosomal DNA in a staggered fashion. Simultaneously, IN coordinately splices both ends of the vDNA to the resulting phosphodiester bonds on opposite strands of the chromosomal DNA. Following this strand transfer step, the host cell machinery removes the two unpaired nucleotides at the 5' ends of the vDNA and repairs the ensuing single-stranded gaps at the junction of the integration site. The early events thus culminate with the establishment of an integrated DNA copy (provirus) of the HIV-1 genome. The provirus provides a suitable environment for efficient viral gene expression, which initiates later events, including the expression of the virus-encoded proteins; assembly of the immature virus; and budding, release, and maturation into infectious virions13.
Purified recombinant retroviral IN is competent to carry out, in vitro, both 3'-end processing and strand transfer activities on exogenously supplied LTR-like substrate DNA. Biochemical studies using such purified recombinant IN have revealed critical aspects of vDNA integration14,15,16,17. However, unlike in natural infection, this in vitro biochemical reaction does not support concerted integration of both ends of the substrate DNA into the target DNA. In contrast, retroviral PICs isolated from acutely infected cells concertedly integrate both ends of the endogenous vDNA into heterologous target DNA. This led to the widespread adoption and subsequent refinements of In Vitro Integration (IVI) assays using isolated native PICs.
In the first reported retroviral IVI assay, cytoplasmic extracts from cells infected with a recombinant Murine Leukemia Virus (MLV) harboring the E. coli supF gene in its LTR served as the source of IN activity, and the DNA of a mutant lambda phage defective in lytic growth was exogenously supplied as the target DNA. Successful integration of the recombinant MLV DNA copy into the phage DNA and the ensuing supF expression led to restoration of the plaque-forming ability of the recombinant phages. However, this experimental strategy is laborious and measures integration in an indirect manner. To address such caveats, a Southern blot-based indirect end-labeling assay, which quantifies the PIC-associated IVI activity as a measure of the endogenous vDNA integrated into exogenous linearized bacteriophage phiX174 DNA, was developed6,7,18. Though this method provides a direct measure of integration events, relatively high amounts of PIC preparation are required, which remains a technically challenging endeavor. To circumvent this, nested PCR-based assays requiring only modest amounts of PICs were developed4,5,19.
In this report, we describe an updated version of a nested PCR-based in vitro method devised to recapitulate the HIV-1 PIC-mediated integration activity occurring during natural infection. This method uses the cytoplasmic extracts of HIV-1-infected target cells as a source of endogenous PIC activity, which is measured with a real-time quantitative Polymerase Chain Reaction (qPCR). The procedures for isolating PICs and measuring their integration activities are adapted, with modifications, from protocols published by the Engelman laboratory20. In this method, the PIC-associated integration activity is initiated by providing an exogenous linear target DNA21, and the DNA products resulting from this integration reaction are purified and used as the starting material for subsequent PCR-based quantification. In the first-round conventional PCR, the viral-target DNA junctions are amplified by using appropriate primers. In the second-round qPCR, LTR-specific primers are used to specifically enrich the vDNA population from the first-round PCR products. Please see the protocol section for a detailed description of this method.
In vitro studies with retroviral PICs have led to significant advances in our understanding of the mechanism of retroviral DNA integration and in the development of IN inhibitors. Based on the recent increased focus on mapping HIV-1 integration sites, determining the role of viral and host factors in HIV-1 integration, and developing new antiviral drugs towards combating drug resistance, we envisage an increased interest in and widespread use of IVI assays, like the one described in this report. This, in turn, will lead to future refinements in the method, thereby diversifying the scope of its applications.
Protocol
1. Virus Production
NOTE: To generate high titers of infectious HIV-1, transfect the Human Embryonic Kidney (HEK) cell line 293T with an infectious HIV-1 molecular clone using an activated dendrimer-based transfection reagent. It has been observed that the calcium phosphate transfection method yields comparable results.
In a Biosafety Level 2 (BSL2) lab, seed 3 x 106 293T cells per 10 cm dish in 8 mL of Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS), penicillin, and streptomycin. Culture the cells in an incubator set at 37 °C and 5% CO2 O/N.
The next day, for transfecting cells in each 10 cm dish, dilute 8 µg of pNL4-3 plasmid DNA (see Materials Table) to a final volume of 300 µL of DMEM lacking serum and antibiotics. Add 80 µL of the transfection reagent, mix well by pipetting, and incubate at RT for 5 - 10 min for transfection complex formation.
Remove the medium from the dishes by aspiration and add 7 mL per dish of fresh DMEM containing serum and antibiotics.
Add 1 mL of DMEM containing serum and antibiotics to the transfection complex, mix by pipetting, and add this mixture to the cells. Gently swirl the dish to facilitate even distribution of the transfection complexes.
Immediately transfer the cells to the BSL3 lab, place in an incubator set at 37 °C and 5% CO2, and culture the cells for 48 h.
Collect the virus-containing tissue culture supernatant using a pipette and then transfer it to a centrifuge tube. Centrifuge at 300 x g for 5 min to pellet the cells and debris, and then filter the supernatant through a 0.45 µm pore-size syringe filter.
To eliminate any carryover plasmid DNA in this cell-free virus preparation, treat with DNase 1 (20 µg/mL) at 37 °C for 1 h.
To quantify the HIV-1 titer, use the p24-specific Enzyme-linked Immunosorbent Assay (ELISA), as it is a rapid and amenable method for routine use22. NOTE: It should be noted that the p24 ELISA measures the physical titer (number of virus particles). Alternatively, measuring the virus-associated RT activity (exogenous RT assay) or virus infectivity (TZM-bl indicator cell line-based infectivity assays) will provide exclusive readout of infectious virus particles.
2. Virus Infection of Sup-T1 Cells (BSL3 Lab)
- In a BSL2 lab, maintain Sup-T1 cells between 1.0 x 106 and 2.0 x 106 cells/mL in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% heat-inactivated FBS, penicillin, and streptomycin (split ratio of 1:2 every 2 - 3 d). Pool the Sup-T1 cells from the culture flasks, mix well by pipetting, and determine the viable cell count by using a hemocytometer and the trypan blue exclusion staining method.
- Prepare the required number of aliquots of 80 x 106 cells (per virus infection) in 50 mL conical centrifuge tubes.
- Centrifuge the cells in a swinging-bucket rotor at 500 x g and 25 °C for 5 min. Without removing the cell culture medium, transfer the tubes containing pelleted cells to the BSL3 lab for further processing. Carefully aspirate the cell culture medium from the tube without disturbing the cell pellet.
Add an appropriate amount of DNase I-treated virus inoculum to the cells in the tube and mix well by gentle pipetting. For 80 x 106 cells, use 30 mL of DNase I-treated virus (50 µg of p24-equivalent virus). NOTE: Isolation of functional PICs requires infecting the target cells with very high multiplicities of infection (MOI).
Distribute the mixture evenly between two 6-well tissue culture plates (i.e. 12 x 2.5-mL aliquots; use 2 plates per infection).
For spinoculation, place the culture plates in plate carriers and centrifuge using a swinging bucket rotor at 480 x g and 25 °C for 2 h.
Incubate the spinoculated cells at 37 °C and 5% CO2 for 5 h.
3. Harvesting and Lysis of Infected Cells (BSL3 lab)
Pool the cells corresponding to each infection from the culture plates into a 50 mL conical centrifuge tube (~30 mL in total).
Harvest the cells by centrifugation at 300 x g and 25 °C for 10 min. Carefully aspirate the supernatant from the tube (alternatively, use a pipette).
To remove any residual viral particles, wash each cell pellet by adding 2 mL of buffer K-/- (150 mM KCl, 20 mM HEPES, pH 7.6, 5 mM MgCl2) and centrifuging the sample at 300 x g and 25 °C for 10 min. Then, without disturbing the pellet, carefully remove the supernatant by aspiration or by using a pipette.
Repeat step 3.3.
Carefully remove any residual buffer by using a micropipette. Resuspend each cell pellet in 2 mL of ice-cold lysis buffer K+/+ (Buffer K-/- supplemented with 0.025% digitonin, 1 mM DTT, 20 µg/mL aprotinin), mix contents by gentle pipetting, and transfer to a 2 mL microcentrifuge tube.
4. Preparation of Cytoplasmic PICs (BSL3 lab)
Rock the samples on a rotary shaker at RT for 10 min.
Centrifuge the samples, with the rotor precooled to 4 °C, at 1,500 x g and 4 °C for 4 min.
Without disturbing the pellet, carefully transfer the supernatant to a fresh microcentrifuge tube and centrifuge at 16,000 x g and 4 °C for 1 min.
Carefully transfer the supernatant to a fresh microcentrifuge tube. Add RNase A (20 mg/mL) to a final concentration of 20 µg/mL (2 µL for 2 mL of PICs) and incubate the contents at RT for 30 min (no rocking).
Add sucrose (60% w/v in buffer K-/-) to a final concentration of 7% and mix well by gentle pipetting. Prepare aliquots (e.g., 200 µL) of the PICs in microcentrifuge tubes, flash-freeze in liquid nitrogen, and place in a -80 °C freezer for long-term storage.
5. In Vitro Integration (IVI) Assays Using HIV-1 PICs
- Prepare the target DNA used in the IVI assay by amplifying a 2.0 kb region from the phiX174 plasmid DNA with PCR using custom-made primers, phiX174-F and phiX174-R.
- Assemble the PCR mixture as described in Table 1. Perform PCR in a thermal cycler using the thermal cycling conditions recommended in Table 2. Gel-purify the PCR product using a gel-purification kit (following manufacturer-recommended protocol), and store the purified target DNA as aliquots at -80 °C until use.
Add 600 ng of target DNA to 200-µL aliquot of PICs, mix well, and incubate at 37 °C for 45 min. Note: IVI reactions omitting target DNA or including an integrase inhibitor (e.g., Raltegravir) are recommended control reactions.
Stop the IVI reaction by adding SDS, EDTA, and proteinase K to final concentrations of 0.5%, 8 mM, and 0.5 mg/mL, respectively. Mix the reaction contents thoroughly and incubate overnight at 56 °C.
The next day, purify the total DNA from the IVI reaction by phenol/chloroform extraction and ethanol precipitation. Carry out all centrifugation steps at 16,000 x g and RT.
Add an equal volume of phenol to the deproteinized sample, mix vigorously by vortexing, and centrifuge for 2 min. Carefully remove the upper aqueous phase and transfer it to a fresh tube.
Repeat step 5.5. Transfer the upper aqueous phase to a fresh tube.
Add an equal volume of 1:1 phenol:chloroform mixture, mix vigorously by vortexing, and centrifuge for 2 min. Transfer the upper aqueous phase to a fresh tube.
Add an equal volume of chloroform, mix vigorously by vortexing, and centrifuge for 2 min. Transfer the upper aqueous phase to a fresh tube.
To precipitate DNA, add glycogen to a final concentration of 0.1 µg/µL, 3 M of sodium acetate to a final concentration of 0.3 M, and 2.5 volumes of 100% ethanol (ice-cold). Vortex to mix, centrifuge briefly to collect the contents, and place the tube at -80 °C O/N.
The next day, centrifuge the sample for 30 min at 16,000 x g and 4 °C for 30 min. Carefully remove the supernatant without disturbing the pellet.
Add 500 µL of 80% ethanol (ice-cold) to the pellet and centrifuge at 16,000 x g and 4 °C for 10 min. Air-dry the DNA pellet at RT and thoroughly resuspend in 50 µL of nuclease-free water. Store the DNA samples at -20 °C.
6. PCR Assays for Quantifying PIC Activity
NOTE: The copy number of HIV-1 DNA integrated into the target DNA is quantified by a nested PCR strategy that employs 2 sets of primers and 2 sequential rounds of PCR.
- Perform the first-round conventional PCR using primers targeting the viral LTR and the phiX174 DNA (LTR and phiX174 primers, respectively) to amplify the junctions of the integrated HIV-1 DNA.
- Assemble the first-round PCR mixture as described in Table 3. Assemble a master mix of all components excluding the template DNA, dispense 45-µL aliquots into separate PCR tubes, and add 5 µL of template DNA or nuclease-free water (control).
- Perform PCR in a thermal cycler using the thermal cycling conditions recommended in Table 4.
- Perform the second-round qPCR using primers that flank a short region of the viral LTR (LTR-R and LTR-U primers) to selectively amplify the integrated vDNA sequences from the first-round PCR products. To determine the integrated vDNA copy numbers, generate a standard curve by using 10x serial dilutions of known copy numbers (e.g., 100 - 108) of the HIV-1 molecular clone.
- Assemble the second-round qPCR mixture as described in Table 5. Assemble a master mix of all components excluding the template DNA, carefully dispense 25 µL aliquots into the appropriate number of wells in a 96-well PCR reaction plate, and then add 5 µL of the unpurified first-round PCR products or nuclease-free water (control). Perform all qPCR reactions in triplicate.
- Load the PCR plate into the real-time PCR instrument and perform qPCR using the cycling conditions recommended in Table 6.
- Analyze the data using a suitable software.
Representative Results
Isolation of HIV-1 PICs
A schematic of the protocol used for the isolation of HIV-1 PICs from acutely infected Sup-T1 cells is depicted in Figure 1. This protocol is derived from the methods described by Engelman et al.19,20. HIV-1 PICs are nucleoprotein complexes that are assembled in infected cells and are comprised of the reverse-transcribed vDNA, viral proteins, and cellular factors19,20. The method described in this report uses the cytoplasmic extracts of HIV-1-infected cells as the source of PIC-associated integration activity.
In vitro integration activity of HIV-1 PICs
A schematic of the method used to measure the IVI activity of HIV-1 PICs is shown in Figure 2. This method utilizes a nested qPCR-based strategy for measuring PIC-associated IVI activity, and the procedures are adapted, with modifications, from protocols published by the Engelman laboratory19,20,21. In this method, the PIC-associated integration activity is initiated by providing a heterologous linear target DNA21, and the DNA products resulting from this integration reaction are purified and used as the starting material for subsequent PCR-based quantification. In the first-round conventional PCR, the viral-target DNA junctions are amplified using appropriate primers. In the second-round qPCR, LTR-specific primers are used to specifically enrich the vDNA population from the first-round PCR products. Please see the protocol section for a detailed description of this method.
Representative results from an IVI assay performed using HIV-1 PICs are shown in Figure 3. Sup-T1 cells (80 x 106 cells/infection) were spinoculated with 30 mL of DNase 1-treated HIV-1 (~50 µg of p24 equivalent) in the absence or presence of 1 µM HIV-1 IN inhibitor Raltegravir at 480 x g and 25 °C for 2 h. Cells were subsequently cultured for 5 h at 37 °C and 5% CO2. PICs were isolated as described in the protocol section. The IVI reactions were carried out, each using 200 µL of PICs and 600 ng of the target DNA (a 2 kb DNA fragment amplified from phiX174 DNA by PCR), at 37 °C for 45 min. The reactions were deproteinized by proteinase K treatment in the presence of SDS and EDTA at 56 °C O/N. The reaction products were extracted twice with phenol, once with a phenol:chloroform (1:1) mixture, and once with chloroform. Subsequently, the DNA was ethanol-precipitated in the presence of salt and the co-precipitant glycogen, and the DNA pellet was washed once with 80% ethanol. The purified DNA was resuspended in 50 µL of nuclease-free water. A nested qPCR strategy was used to determine the number of vDNA integrated into the phiX174 DNA. In the first-round conventional PCR, primers complementary to sequences on the phiX174 target DNA and viral LTR region were used to amplify the integrated viral-target DNA junctions from 5 µL of the integration reaction DNA products. In the second-round qPCR, LTR-spanning primers were used to exclusively amplify the HIV-specific DNA sequences from 5 µL of the first-round amplicon products and from 10-fold serial dilutions of known copy numbers (108-100) of the HIV-1 molecular clone (Standards).
All qPCR reactions were performed in triplicate and the data were analyzed using commercial software. The values derived from the standards were plotted to generate a standard curve, from which the integrated vDNA copy numbers were interpolated. Error bars represent standard deviations. As shown in the Figure 3, the HIV-1 PICs robustly integrate the associated vDNA into the target DNA (Lane 5). Importantly, the IVI efficiency of PICs was greatly reduced in reactions supplemented with the IN inhibitor Raltegravir (Lane 6). This strongly attributes the integration activity measured in this assay to HIV-1 IN. Further, the control reactions containing cytoplasmic lysates from uninfected cells (Lane 1), PICs alone (Lane 2), target DNA alone (Lane 3), and buffer alone (Lane 4) did not show any integration activity. In addition, the control that excluded Taq polymerase in the first round PCR (Lane 7) did not amplify integration products. Notably, the data from the PICs-alone control (Lane 2) indicated that the nested qPCR strategy specifically measures the integrated vDNA but not the PIC-associated vDNA. Collectively, these data demonstrate the measurement of PIC-associated integration activity.
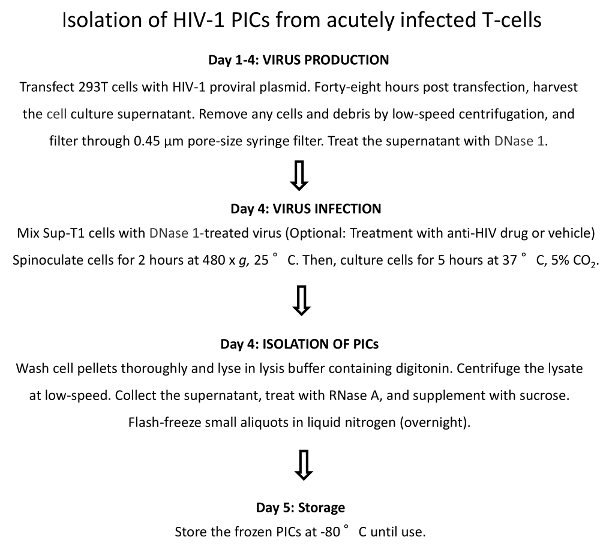 Figure 1: Schematic Representation of the Protocol for the Preparation of HIV-1 PICs.
Please click here to view a larger version of this figure.
Figure 1: Schematic Representation of the Protocol for the Preparation of HIV-1 PICs.
Please click here to view a larger version of this figure.
Cytoplasmic extracts containing PICs are isolated from HIV-1-infected Sup-T1 cells. These PICs are used as the source of integration activity in the in vitro integration assay.
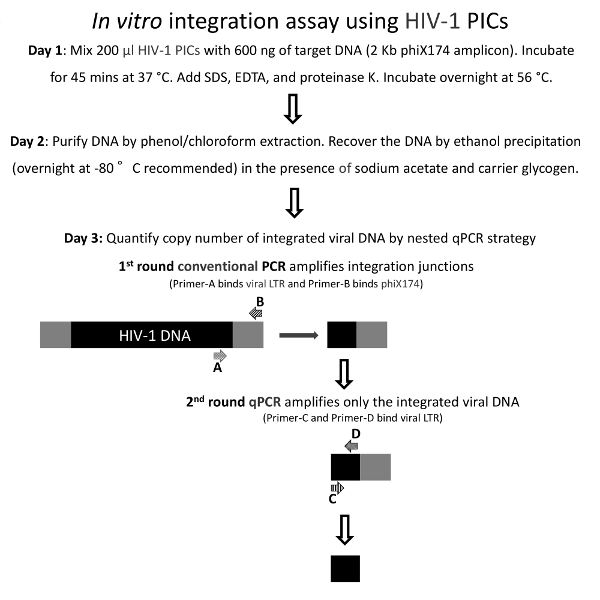 Figure 2: Schematic Representation of the in Vitro Integration Assay of HIV-1 PICs.
Please click here to view a larger version of this figure.
Figure 2: Schematic Representation of the in Vitro Integration Assay of HIV-1 PICs.
Please click here to view a larger version of this figure.
Integration assays were carried out in vitro using HIV-1 PICs and the heterologous target phiX174 DNA. The integrated HIV-1 DNA was measured by a nested PCR strategy, wherein the first-round conventional PCR amplifies the integration junctions from which vDNA-specific sequences are preferentially amplified in a second-round qPCR.
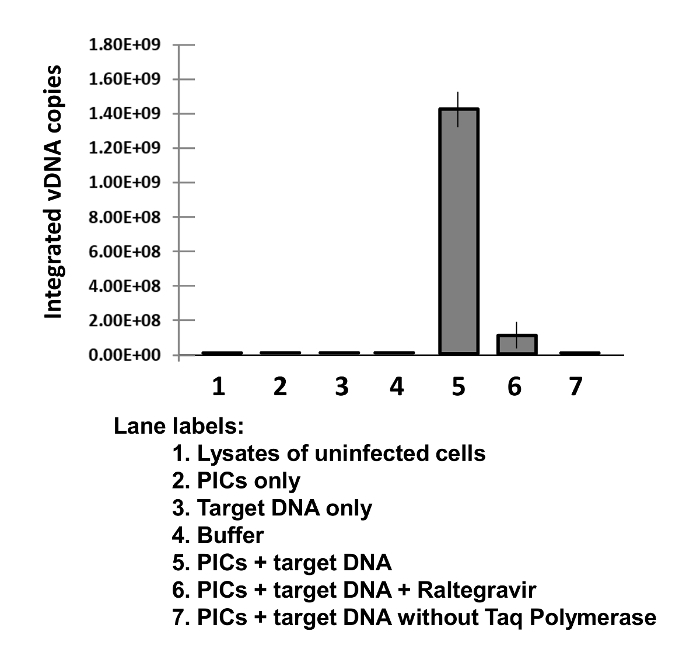 Figure 3: HIV-1 PIC-associated vDNA Integration Activity.
Please click here to view a larger version of this figure.
Figure 3: HIV-1 PIC-associated vDNA Integration Activity.
Please click here to view a larger version of this figure.
Representative results of the HIV-1 PIC-associated integration activity assay. The integrated vDNA copy numbers were determined by plotting values derived from a standard curve generated using 10x serial dilutions of known copy numbers of an HIV-1 molecular clone. All reactions were performed in triplicate and the data were analyzed using commercial software. Error bars represent standard deviations.
| Component | Stock | Final volume for 50 μL reaction |
| phiX174 plasmid DNA | 10 ng/µL | 5.0 µL |
| GoTaq reaction buffer | 5x | 10.0 µL |
| dNTP nucleotide mix | 10 mM each | 1.0 µL |
| phiX174-F primer | 10 μM | 2.5 µL |
| phiX174-R primer | 10 μM | 2.5 µL |
| GoTaq DNA Polymerase | (5 U/µL) | 0.25 µL |
| Nuclease-free distilled water | - | 28.75 µL |
Table 1.
| 95 °C = 2 min |
| Cycle 34 repetitions of: 95 °C = 30 s 53 °C = 30 s 72 °C = 2 min |
| 72 °C = 10 min |
| 4 °C = Hold |
Table 2.
| Component | Stock | Final volume for 50 μL reaction |
| IVI reaction DNA product | - | 5.0 µL |
| GoTaq reaction buffer | 5x | 10.0 µL |
| dNTP nucleotide mix | 10 mM each | 1.0 µL |
| LTR primer | 10 μM | 2.5 µL |
| phiX174 primer | 10 μM | 2.5 µL |
| GoTaq DNA Polymerase | (5 U/µL) | 0.25 µL |
| Nuclease-free distilled water | - | 28.75 µL |
Table 3.
| 95 °C = 5 min |
| Cycle 23 repetitions of: 94 °C = 30 s 55 °C = 30 s 72 °C = 4 min |
| 72 °C = 10 min |
| 4 °C = Hold |
Table 4.
| Component | Stock | Final volume for 30 μL reaction |
| 1st round PCR products | - | 5.0 µL |
| LTR-R primer | 10 μM | 0.9 µL |
| LTR-U primer | 10 μM | 0.9 µL |
| Taqman Probe | 10 μM | 0.3 µL |
| iQ Supermix | 2x | 15.0 µL |
| Nuclease-free distilled water | - | 7.9 µL |
Table 5.
| 95 °C = 3 min |
| Cycle 39 repetitions of: 94 °C = 15 s 58 °C = 30 s 72 °C = 30 s Plate Read |
Table 6.
Discussion
Biochemical analyses of retroviral PICs have provided critical insights into the mechanism of retroviral DNA integration. Measurement of the integration activity of retroviral PICs can be achieved by plaque formation assay, Southern blot analysis, and nested qPCR. The experimental strategy of plaque assay is laborious and utilizes an indirect method to measure integration6,7,18. Southern blot-based assays measure integration directly but require relatively high amounts of PICs6,7,18.
This report details a nested qPCR strategy for measuring HIV-1 PIC-associated integration activity in vitro. The major advantage and significance of this method is the requirement of relatively low amounts of PIC preparation per reaction4,5,19. The most critical steps of this method are the generation of a high-titer virus for infection and the handling of PIC preparation. It should be noted here that the first round of PCR involves exponential amplification of the integration junctions. Consequently, the copy numbers of integrated vDNA determined in the second round qPCR are semi-quantitative and, hence, have the limitation of not giving a measure of PIC-associated vDNA copies. However, the PIC-associated vDNA can be determined by qPCR of the DNA purified from PICs with LTR-specific primers. The ratio of the integrated vDNA copy numbers to the corresponding PIC-associated vDNA can provide information about the integration efficiency of an in vitro integration reaction. This method can be modified by incorporating other heterologous target DNA of choice and other primers to detect integration at both orientations. In summary, the method described here measures the integration activity of HIV-1 PICs quantitatively and can be adopted for research developing an understanding of the biochemical details of retroviral integration.
Disclosures
The authors declare no competing financial and nonfinancial interests.
Acknowledgments
This work is partly supported by grants DA024558, DA30896, DA033892, and DA021471 from the NIDA/NIH to CD. We also acknowledge the RCMI grant G12MD007586, the Vanderbilt CTSA grant UL1RR024975, the Meharry Translational Research Center (MeTRC) CTSA grant U54 RR026140 from the NCRR/NIH, the U54 grant MD007593 from the NIMHD/NIH, and the Tennessee CFAR grant P30 AI110527.
References
- Sattentau QJ, Weiss RA. The CD4 antigen: physiological ligand and HIV receptor. Cell. 1988;52(5):631–633. doi: 10.1016/0092-8674(88)90397-2. [DOI] [PubMed] [Google Scholar]
- Wilen CB, Tilton JC, Doms RW. Molecular mechanisms of HIV entry. Adv Exp Med Biol. 2012;726:223–242. doi: 10.1007/978-1-4614-0980-9_10. [DOI] [PubMed] [Google Scholar]
- Bowerman B, Brown PO, Bishop JM, Varmus HE. A nucleoprotein complex mediates the integration of retroviral DNA. Genes Dev. 1989;3(4):469–478. doi: 10.1101/gad.3.4.469. [DOI] [PubMed] [Google Scholar]
- Bukrinsky MI, Sharova N, Dempsey MP, et al. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc Natl Acad Sci. 1992;9(14):6580–6584. doi: 10.1073/pnas.89.14.6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MS, Smith GJ, 3rd, Kafri T, Molteni V, Siegel JS, Bushman FD. Integration complexes derived from HIV vectors for rapid assays in vitro. Nature biotechnol. 1999;17(6):578–582. doi: 10.1038/9886. [DOI] [PubMed] [Google Scholar]
- Farnet CM, Haseltine WA. Integration of human immunodeficiency virus type 1 DNA in vitro. Proc Natl Acad Sci. 1990;87(11):4164–4168. doi: 10.1073/pnas.87.11.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Mizuuchi K. Retroviral DNA integration: structure of an integration intermediate. Cell. 1988;54(4):497–504. doi: 10.1016/0092-8674(88)90071-2. [DOI] [PubMed] [Google Scholar]
- Brown PO, Bowerman B, Varmus HE, Bishop JM. Correct integration of retroviral DNA in vitro. Cell. 1987;49(3):347–356. doi: 10.1016/0092-8674(87)90287-x. [DOI] [PubMed] [Google Scholar]
- Lewinski MK, Bushman FD. Retroviral DNA integration--mechanism and consequences. Adv Genet. 2005;55:147–181. doi: 10.1016/S0065-2660(05)55005-3. [DOI] [PubMed] [Google Scholar]
- Engelman A, Mizuuchi K, Craigie R. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell. 1991;67(6):1211–1221. doi: 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- Goff SP. Genetics of retroviral integration. Ann rev gen. 1992;26:527–544. doi: 10.1146/annurev.ge.26.120192.002523. [DOI] [PubMed] [Google Scholar]
- Panganiban AT, Temin HM. The terminal nucleotides of retrovirus DNA are required for integration but not virus production. Nature. 1983;306(5939):155–160. doi: 10.1038/306155a0. [DOI] [PubMed] [Google Scholar]
- Bieniasz PD. The cell biology of HIV-1 virion genesis. Cell host microbe. 2009;5(6):550–558. doi: 10.1016/j.chom.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman PA, Fyfe JA. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc Natl Acad Sci. 1990;87(13):5119–5123. doi: 10.1073/pnas.87.13.5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman M, Katz RA, Skalka AM, Leis J. The avian retroviral integration protein cleaves the terminal sequences of linear viral DNA at the in vivo sites of integration. J Virol. 1989;63(12):5319–5327. doi: 10.1128/jvi.63.12.5319-5327.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craigie R, Mizuuchi K, Bushman FD, Engelman A. A rapid in vitro assay for HIV DNA integration. Nucl acid res. 1991;19(10):2729–2734. doi: 10.1093/nar/19.10.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman FD, Craigie R. Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc Natl Acad Sci. 1991;88(4):1339–1343. doi: 10.1073/pnas.88.4.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauza CD. Two bases are deleted from the termini of HIV-1 linear DNA during integrative recombination. Virology. 1990;179(2):886–889. doi: 10.1016/0042-6822(90)90161-j. [DOI] [PubMed] [Google Scholar]
- Engelman A. Isolation and analysis of HIV-1 preintegration complexes. Meth Mol Biol. 2009;485:135–149. doi: 10.1007/978-1-59745-170-3_10. [DOI] [PubMed] [Google Scholar]
- Engelman A, Oztop I, Vandegraaff N, Raghavendra NK. Quantitative analysis of HIV-1 preintegration complexes. Methods. 2009;47(4):283–290. doi: 10.1016/j.ymeth.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addai AB, Pandhare J, Paromov V, Mantri CK, Pratap S, Dash C. Cocaine modulates HIV-1 integration in primary CD4+ T cells: implications in HIV-1 pathogenesis in drug-abusing patients. J Leuk Biol. 2015;97(4):779–790. doi: 10.1189/jlb.4A0714-356R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrly K, Chesebro B. p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods. 1997;12(4):288–293. doi: 10.1006/meth.1997.0481. [DOI] [PubMed] [Google Scholar]