

Abstract
The mammalian body is equipped with various layers of mechanisms that help to defend itself from pathogen invasions. Professional phagocytes of the immune system — such as neutrophils, dendritic cells, and macrophages — retain the innate ability to detect and clear such invading pathogens through phagocytosis1. Phagocytosis involves choreographed events of membrane reorganization and actin remodeling at the cell surface2,3. Phagocytes successfully internalize and eradicate foreign molecules only when all stages of phagocytosis are fulfilled. These steps include recognition and binding of the pathogen by pattern recognition receptors (PRRs) residing at the cell surface, formation of phagocytic cup through actin-enriched membranous protrusions (pseudopods) to surround the particulate, and scission of the phagosome followed by phagolysosome maturation that results in the killing of the pathogen3,4.
Imaging and quantification of various stages of phagocytosis is instrumental for elucidating the molecular mechanisms of this cellular process. The present manuscript reports methods to study the different phases of phagocytosis. We describe a microscope-based approach to visualize and quantify the binding, phagocytic cup formation, and the internalization of particulate by phagocytes. As phagocytosis occurs when innate receptors on phagocytic cells encounter ligands on a target particle bigger than 0.5 µm, the assays we present here comprise the use of pathogenic fungi Candida albicans and other particulates such as zymosan and IgG-coated beads.
Keywords: Microbiology, Issue 120, phagocytosis, lipids, microscopy, phagocytic cup, actin remodeling, zymosan, live imaging, Candida albicans
Introduction
Despite the continuous exposure to pathogens such as bacteria, viruses and fungi, our body is well equipped with immune mechanism that provide protection against infection. The innate immune system is the first line of defense against invading pathogens and relies mainly on phagocytic cells that recognize and internalize foreign targets.
Phagocytosis is an evolutionarily conserved cellular process that encompasses the engulfment of unwanted particulates greater than 0.5 µm. Phagocytic cells express a wide range of immune receptors (also known as pattern recognition receptors, PRRs) at the cell surface that enable them to recognize pathogen-associated molecular patterns (PAMPs) present on pathogens prior to engulfment3. Pathogen binding is followed by receptor clustering at the cell surface and triggers the formation of a phagocytic cup. This results in actin-driven membrane remodeling that protrudes around the target, eventually enveloping it and pinching-off to form a discrete phagosomal vacuole2,5. The phagosome then matures and acidifies by subsequent fusion with late endosomes and lysosomes that forms phagolysosome6.
Although phagocytosis is described as receptor-mediated and actin-driven event, this process also relies on spatial-temporal modification of lipids that compose the plasma membrane, such as phosphoinositides (PIs) and sphingolipids7,8. While actin polymerization is dictated by a local accumulation of phosphoinositol-4,5-biphosphate (PI(4,5)P2) at the base of the phagocytic cup, actin depolymerization depends on the conversion of (PI(4,5)P2 to phosphoinositol-3,4,5-biphosphate (PI(3,4,5)P3)3,9. Both modifications are essential as the former leads to successful extension of pseudopods around the target and the latter enables sinking of particles in the cytosol of the phagocyte10.
Cells that have the ability to phagocytose are either professional phagocytes, like macrophages/monocytes, granulocytes/neutrophils, and dendritic cells (DCs) or nonprofessional phagocytes, such as fibroblast and epithelial cells11. Phagocytosis performed by all phagocytes plays a central role in tissue maintenance and remodeling, while phagocytosis performed by professional phagocytes is responsible for the coordination of the innate and adaptive immune response against pathogens. Professional phagocytes do not only engulf and kill the pathogen, but also present antigens to the lymphoid cells of the adaptive immune system. This contributes to the release of pro-inflammatory cytokines and to the engagement of lymphoid cells, therefore leading to the successful blockade of infection12.
Conventional biochemical techniques have been instrumental in gaining knowledge regarding the molecular mechanism of different cellular processes during phagocytosis, such as post-translational modifications and various high-affinity associations between proteins. However, it is difficult to obtain information regarding the spatial and temporal dynamics of phagocytic events using the conventional biochemical methods. Live cell imaging not only allows us to monitor cellular events in a time sensitive manner but also enables us to gain information at a single cell level. Here we describe a method to investigate the different stages of phagocytosis, as well as to analyze the whole process spatiotemporally using confocal microscopy.
Protocol
1. Preparation of DC2.4 and RAW 264.7 Cell Lines
NOTE: The macrophage-like cell line RAW 264.7 and the dendritic cell line DC2.4 are both murine origin, and the following conditions were used to grow the cells.
Grow RAW 264.7 cells in DMEM (Dulbecco's Minimal Eagle's medium) supplemented with 10% inactivated Fetal Bovine Serum (FBS) at 37 °C in a humidified incubator with 5% CO2.
Grow DC2.4 cells in similar conditions with that of RAW 264.7 cells, except use RPMI (Roswell Park Memorial Institute) medium supplemented with L-Glutamine instead of DMEM.
2. Preparation of Fluorescent Conjugated Particulates: C. albicans, Zymosan, IgG-coated Beads
- Growing Candida albicans
- Inoculate C. albicans strain SC5314 from a frozen glycerol stock in 25 ml of YPD supplemented with Uri (2% Bacto-peptone, 1% yeast extract, 2% glucose and 80 µg/ml Uridine) overnight at 30 °C. NOTE: Avoid growing the Candida culture more than an OD600 of 12.
- Take 1 ml of the C. albicans and spin it down at 2,000 x g for 5 min at room temperature.
- Aspirate the supernatant and re-suspend the pellet with 1 ml of PBS.
- Collect the pellet by centrifugation at 2,000 x g for 5 min at room temperature.
- Repeat step 2.1.4.
- Calculate the multiplicity of infection (MOI) of C. albicans in relation to the number of phagocytes and proceed with the phagocytosis assay as described in section 3. An OD600 of 1 is equivalent to 2.5 x 107 Candida albicans per 1 ml. NOTE: Generation of Candida-BFP is described in reference13.
- Preparation of Zymosan and IgG-coated Beads NOTE: Zymosan is a β-glucan-containing fungal carbohydrate particulate preparation that derived from Saccharomyces cerevisiae. Zymosan is known to evoke inflammatory signals in macrophages and dendritic cells, and it has been used extensively in phagocytosis studies 13,14,15.
- Briefly vortex the tube that contains the particulate, and aliquot an amount sufficient for an experiment into a 1.5 ml tube. NOTE: Typically we use 1:10 ratio (for one cell, add 10 zymosan particles or IgG-coated beads).
- Add 1 ml of ice cold PBS, and collect the particles by centrifuging for 1 min at 10,000 x g.
- Aspirate the supernatant and resuspend the pellet in 1 ml ice-cold PBS.
- Repeat steps 2.2.2-2.2.3 NOTE: Proceed to step 2.2.5 or store the tube at 4 °C until use.
- Sonicate the particulates for 10 sec in ultrasonic bath sonicator (120 V, 50-60 kHz) in order to avoid any particle aggregation.
3. Phagocytosis
NOTE: Phagocytosis is a complex process that begins with binding of the particles on the cell surface of the phagocytes through interaction of PRRs with ligands on the surface of the particle. Binding is followed by the assembly of actin and its associated proteins at the site of contact, and the formation of a phagocytic cup. The subsequent actin disassembly occurs at the phagosome and results in the complete engulfment of the particulate. Below we describe the different stages of phagocytosis.
- Binding Assay NOTE: The purpose of this experiment is to monitor the first step of phagocytosis that involves the interaction between the ligands on the target particles and the receptors on the phagocytes.
- Seed about 5 x 104 cells in a chamber slide so that they are 60-70% confluent at the time of the experiment. Alternatively, plate cells on cover slips (in 24- or 12-well format) or glass-bottom 96-well plates. Incubate cells overnight at 37 °C humidified incubator with 5% CO2. NOTE: Cells can be counted using hemocytometer or a similar technique.
- Place the chamber slide (or plate) at 10 °C for 10 min.
- Gently remove the medium from each well using a pipette or aspirator. CAUTION: Perform this step carefully as each well of a chambered coverglass system is small; it is easy to perturb cells with the tip of a pipette or a strong airflow of an aspirator.
- Mix the fluorescently labeled particulates with a cell culture medium (prepared as described above). Add Candida-BFP (at an MOI of 10), fluorescently conjugated-zymosan or latex beads-rabbit IgG-FITC (10 particles per cell) to the cells. Use a final volume of 200 µl media for one chamber well to ensure all cells are covered.
- Spin down the chamber slide at 277 x g for 3 min at 10 °C. NOTE: This centrifugation step helps to increase the contact between the particulates and the cells, and it synchronizes the binding process.
- Immediately transfer the chamber slide to a 37 °C humidified incubator with 5% CO2 for 5 min.
- Remove the chamber slide from the incubator and aspirate the media.
- Wash the cells 3x with ice-cold PBS. NOTE: Check the presence of unbound particles with a light microscope and wash more times with PBS if necessary. It is essential to remove unbound particles from the well to minimize background noise during the analysis.
- Fix cells by adding 500 µl 4% paraformaldehyde (PFA) diluted in PBS by incubating for 30 min at room temperature. CAUTION: This step requires the use of 4% PFA which is highly toxic, corrosive, and potential carcinogen. Proper caution should be taken when using this liquid. Also, protect samples from light.
- Wash the cells by adding 500 µl of PBS into the chamber and aspirate the solution gently.
- Repeat 3.1.9 step twice. NOTE: At this step, refill each vial immediately after removing the previous PBS.
- Stop the fixation by incubating with 500 µl of 50 mM NH4Cl in PBS for 10 min.
- Wash cells twice with PBS as described in step 3.1.10.
- Permeabilize cells by adding 250 µl binding buffer (0.1% saponin, 0.2% BSA in PBS) for 30 min at room temperature.
- Stain F-actin by adding fluorescently conjugated phalloidin (diluted 1:500 in binding buffer). Incubate cells for 1 hr at room temperature.
- Wash cells three times with PBS as described in step 3.1.10.
- Capture images using a confocal microscope (63X objective, and 402 nm and 488 nm laser) and analyze images using ImageJ16. NOTE: A successful particle binding is characterized by the presence of a phagocytic cup at the site where the particulate contact the phagocytic cell (Figure 1). See step 3.2.5.
- Phagocytosis Uptake Assay NOTE: This experiment is designed to monitor the complete internalization of fluorescently labeled particulates by phagocytes using confocal microscopy (Figure 2).
- Seed about 5 x 104 cells in a chamber slide so that they are 60-70% confluent at the time of the experiment. Alternatively, plate cells on cover slips (in 24- or 12-well format) or glass-bottom 96-well plates. Incubate cells overnight at 37 °C humidified incubator with 5% CO2. NOTE: Cells can be counted using hemocytometer or a similar technique.
- Keep the chamber slides (or plate) at 10 °C for 10 min. NOTE: Cooling is necessary in order to block endocytosis as well as unwanted phagocytosis, and subsequently to have a synchronized phagocytosis.
- Perform steps from 3.1.3 to 3.1.5
- Transfer the chamber slide to a 37 °C humidified incubator with 5% CO2, and incubate for different time points. Fix cells after 30, 60 and 90 min post infection. Observe C. albicans begin to form hyphae at 60 min post infection; phagocytes begin to show cell death (pyroptosis) after 90 min8. NOTE: This is a recommendation, and optimization is essential depending on the cell type and the particulate used.
- Perform steps from 3.1.7 to 3.1.15. Analyze images using ImageJ16. NOTE: a successful uptake is characterized by a complete internalization of a particulate inside the cytoplasm of a phagocyte (Figure 2). Staining the phagocytes with cell surface marker such as phalloidin helps to delineate the contour of cells. It is also essential to perform z-stacks in order to determine whether the particulate is bound to cell surface or completely internalized.
- Live Imaging of Phagocytosis NOTE: In this protocol we describe the use of confocal microscopy to capture the different stages of phagocytosis using fluorescently conjugated zymosan. We use the dendritic cell line DC2.4 that stably expresses mCherry-tagged F-actin8, a biosensor that shows the distribution of filamentous actin in living cells. Since phagocytosis is an actin-driven process the live imaging in this cell not only enables us to capture the movement and dynamics of F-actin network, but also to visualize the different phagocytic events within the living cells (Figure 3, Movie 1).
- Seed about 5 x 104 cells in a chamber slide at a concentration so that they are 60-70% confluent at the time of the experiment. Incubate cells overnight at 37 °C humidified incubator with 5% CO2. NOTE: Cells can be counted using hemocytometer or a similar technique.
- Keep the chamber slides on ice for 10 min. NOTE: Cooling is necessary in order to block unwanted uptake, and to have a synchronized phagocytosis.
- Perform steps from 3.1.3 to 3.1.5
- Pre-warm the microscope stage to 37 °C and equilibrate the chamber with 5% CO2. Wait 30-45 min for the temperature and the CO2 to stabilize before starting the imaging. NOTE: Since the CO2 and temperature levels are critical for phagocytosis, it is important to turn on the microscope heater prior to the experiment to equilibrate the environmental chamber.
- Place the chamber slide containing the cells in the microscope incubator enclosure equilibrated at 37 °C and 5% CO2.
- Perform live imaging17,18. NOTE: The settings for the laser power (gain, pinhole, etc.) can vary depending on the type of the microscope and fluorescent probe used. Therefore, we recommend consulting the manual of the microscope for optimal condition for your live imaging 17,18.
- Use confocal laser scanning microscope (and the associated software) for live imaging. Use 63X 1.4NA oil objective with a pinhole of 1-1.1 Airy units.
- To start the imaging, used the bright field to find a representative region on the chamber slide. Next, select the field by clicking the Position option on the software. To detect GFP in Track 1 used the filter set for 488 nm laser with a beam splitter at 582 nm, to detect wavelengths between 488 and 582 nm.
- In Track 2 select a filter set optimal for mCherry (RFP) using a beam splitter at 578 nm and detecting wavelengths between 578 and 600 nm. Use 488 nm and 578 nm lasers with 50% laser power and a gain output of 600-700. Obtain images every 30 sec for a total of 90 min.
Representative Results
Microscope-based method to monitor the different stages of phagocytosis is presented. The different events during the phagocytosis of various fluorescent particulates by DC2.4 cells are shown. Using the techniques described here, we investigated the role of sphingolipids in the early stages of phagocytosis. For this purpose, DC2.4 dendritic cells genetically deficient in Sptlc2, the enzyme that catalyzes the first and rate-limiting step in the sphingolipid biosynthetic pathway, were used. As compared to wild type cells, Sptlc2-/- DC2.4 cells have significantly reduced level of sphingolipids including ceramide, sphingomyelin and glucosylceramide8. Sptlc2-/- DC2.4 cells are defective in binding, as well as in the uptake of C. albicans, zymosan and IgG latex beads8. In Figure 1, the binding of fluorescently conjugated-zymosan particles in DC2.4 cells is shown (see Section 3.1 for details). Sptlc2-/- DC2.4 cells showed significantly less binding of zymosan than the control DC2.4 cells (p = 0.0008; Figure 1A). The number of zymosan particles bound per cell was significantly higher for control cells than for Sptlc2-/- DC2.4 cells (p < 0.0001; Figure 1C). We next investigated the ability of Sptlc2-/- DC2.4 cells to phagocytose C. albicans. Sptlc2-/- DC2.4 cells showed significantly less phagocytosis of C. albicans (p < 0.0005) as compared to the control cells (Figure 2A, 2B). As expected, Sptlc2-deficient cells showed significantly higher number of non-phagocytic cells (p < 0.05; Figure 1C) and decreased number of C. albicans per cell (Figure 1C, D). These results underscore the role of an intact sphingolipid biosynthetic pathway in binding, as well as in the uptake of particulates. Figure 3 shows the time-lapse images demonstrating the early stages of phagocytosis (see Section 3.3 for detailed procedure). Movie 1 shows the live imaging of DC2.4 cells stably expressing F-actin, a biosensor that reveals the distribution of filamentous actin in living cells (see Section 3.1 for more details).
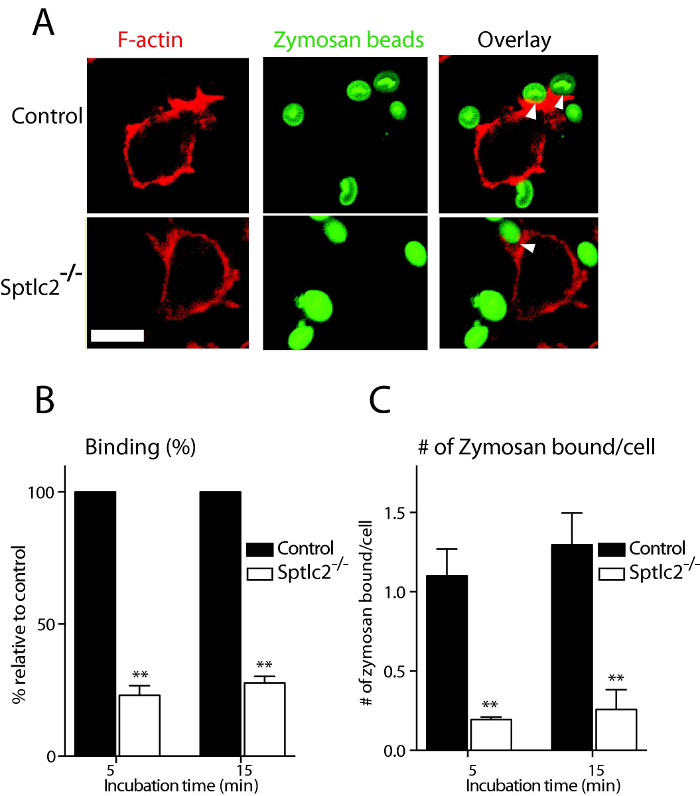 Figure 1: Binding assay of zymosan in DC2.4 cells. (A) Control and Sptlc2-/- DC2.4 cells were incubated with fluorescently conjugated zymosan, and their ability of binding the particulates examined by confocal microscopy. Arrows indicate sites of binding. Scale bar = 100 µm. (B, C) Quantification of the number of bound zymosan particles (B) and the number of zymosan particles bound per cell (C) is shown. Bound particles were quantified and presented as the percentage relative to the control. All graphs display SD of three independent experiments, and at least 200 cells were counted for each experiment. Unpaired t-test was used to analyze the significance of the observed differences. ** p < 0.001. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
Figure 1: Binding assay of zymosan in DC2.4 cells. (A) Control and Sptlc2-/- DC2.4 cells were incubated with fluorescently conjugated zymosan, and their ability of binding the particulates examined by confocal microscopy. Arrows indicate sites of binding. Scale bar = 100 µm. (B, C) Quantification of the number of bound zymosan particles (B) and the number of zymosan particles bound per cell (C) is shown. Bound particles were quantified and presented as the percentage relative to the control. All graphs display SD of three independent experiments, and at least 200 cells were counted for each experiment. Unpaired t-test was used to analyze the significance of the observed differences. ** p < 0.001. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
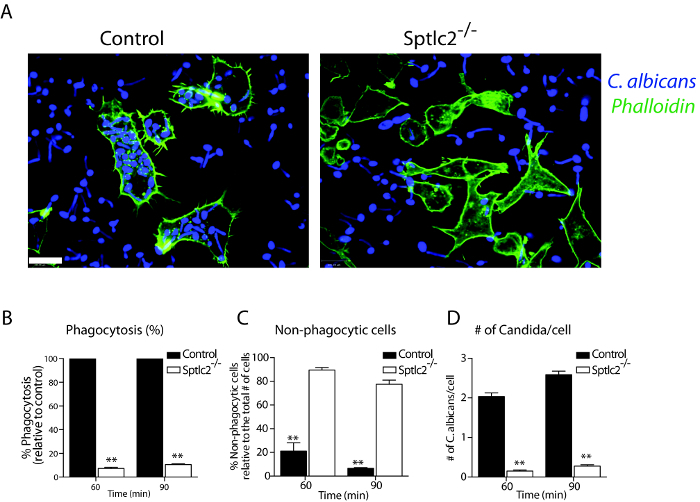 Figure 2: Phagocytosis of C. albicans in DC2.4 cells. (A) Cells were infected with Candida-BFP as described in Step 3.2. At 90 min post infection, cells were fixed and stained with fluorescently conjugated phalloidin and imaged using confocal microscopy. (B-D) Quantification of the number of internalized Candida-BFP, non-phagocytic cells, and the number of Candida-BFP per cell is shown. All graphs display SD of three independent experiments, and at least 200 cells were counted for each experiment. Unpaired t-test was used to analyze the significance of the observed differences. ** p < 0.001. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
Figure 2: Phagocytosis of C. albicans in DC2.4 cells. (A) Cells were infected with Candida-BFP as described in Step 3.2. At 90 min post infection, cells were fixed and stained with fluorescently conjugated phalloidin and imaged using confocal microscopy. (B-D) Quantification of the number of internalized Candida-BFP, non-phagocytic cells, and the number of Candida-BFP per cell is shown. All graphs display SD of three independent experiments, and at least 200 cells were counted for each experiment. Unpaired t-test was used to analyze the significance of the observed differences. ** p < 0.001. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
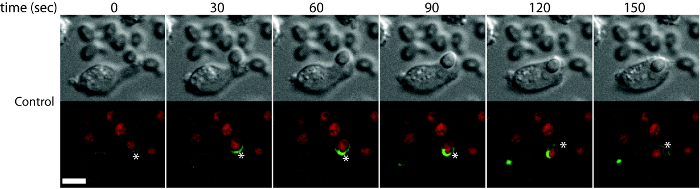 Figure 3: Time-lapse imaging of C. albicans uptake to visualize the early stages of phagocytosis. Wild type DC2.4 cells stably expressing F-actin-mCherry (F-actin) were incubated with Candida-BFP (shown in red) and imaged using confocal microscopy. Images captured at 30 sec intervals are shown. Asterisks show the different stages of actin remodeling during phagocytosis. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
Figure 3: Time-lapse imaging of C. albicans uptake to visualize the early stages of phagocytosis. Wild type DC2.4 cells stably expressing F-actin-mCherry (F-actin) were incubated with Candida-BFP (shown in red) and imaged using confocal microscopy. Images captured at 30 sec intervals are shown. Asterisks show the different stages of actin remodeling during phagocytosis. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
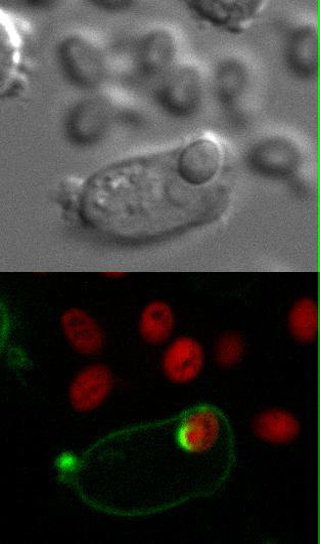 Movie 1: Live imaging. Wild type DC2.4 cells stably expressing F-actin-mCherry (shown in green) were infected with Candida-BFP (shown in red). Live imaging was performed using confocal microscopy as described in Step 3.3. Reprint with permission from Tafesse et al., 2015 8. Please click here to view this video. (Right-click to download.)
Movie 1: Live imaging. Wild type DC2.4 cells stably expressing F-actin-mCherry (shown in green) were infected with Candida-BFP (shown in red). Live imaging was performed using confocal microscopy as described in Step 3.3. Reprint with permission from Tafesse et al., 2015 8. Please click here to view this video. (Right-click to download.)
Discussion
Professional phagocytes, such as macrophages and dendritic cells, engulf and eliminate invading pathogens therefore making phagocytosis an important component of the host defense system. During this process phagocytes undergo extensive membrane reorganization and cytoskeleton rearrangement at their cell surface8,19,20. To better understand this dynamic process, visualization of the different stages of phagocytosis is essential. Here we described a microscope-based method that was used to monitor the various steps of phagocytosis.
The main significance of the live imaging technique is that it provides remarkable capability to monitor the host-pathogen interface at the molecular level. By allowing visualization of the key steps of the microbial infection, live imaging provides critical insight into the fundamental nature of the immune response against pathogens8,10,21,22. In addition to phagocytosis, the techniques described here can be extended to study other types of receptor-mediated endocytosis, including macropinocytosis10. Live imaging is also a valuable approach to study the host-pathogen relationship of intracellular bacteria such as Mycobacterium tuberculosis and Salmonella typhi, as well as parasites including Leishmania and Toxoplasma gondii22,23.
There are several advantages of live cell imaging over the other techniques such as conventional biochemical approaches. Live cell imaging allows us to monitor the spatial and temporal dynamics of cellular processes8. Moreover, live imaging allows us to gain information at a single cell resolution21. Phagocytosis is a dynamic cellular process that involves the clustering of receptors, actin and membrane lipid remodeling in a matter of a few minutes10. Live imaging enables us to capture such information in a time sensitive manner, which is otherwise difficult to accomplish.
The major limitation of this technique is that it requires careful optimization of experimental conditions and microscope setups. There are several key factors crucial for successfully obtaining images. The qualities of the fluorescent protein (or biosensor) utilized to visualize the phagocytes (such as F-actin) as well as the nature of the dye used to label the particulate are critical aspects of imaging. For live imaging, equally important is the equilibration of the environmental chamber of the microscope setup. Depending on the instrument, the equilibration can take from 15 min to several hours. Since CO2 supply is intended to preserve an appropriate pH within the culture media, it is necessary to allow enough runtime prior to setting up the experiment. In cases when the CO2 supply is insufficient and/or uneven, zwitterionic organic chemical buffering agent such as HEPES can be supplemented to the growth media. Long-term exposure of fluorescent particulates and live cells to confocal lasers can lead to phototoxicity and bleaching. Optimizing laser power (the less exposure time the better) and making sure that shutters are closed in between image acquisitions can reduce these problems. Overall, the techniques described here are ideal for studying the dynamic relationship between pathogens and the phagocytes during phagocytosis.
Disclosures
The authors declare no conflicts of interest.
Acknowledgments
We thank Wendy Salmon and Nicki Watson of the Keck facility at the Whitehead Institute of MIT for imaging.
References
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat. Rev. Immunol. 2012;12:492–502. doi: 10.1038/nri3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev. Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Flannagan RS, Harrison RE, Yip CM, Jaqaman K, Grinstein S. Dynamic macrophage "probing" is required for the efficient capture of phagocytic targets. J. Cell Biol. 2010;191:1205–1218. doi: 10.1083/jcb.201007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson JA. Shaping cups into phagosomes and macropinosomes. Nat. Rev. Mol Cell Biol. 2008;9:639–649. doi: 10.1038/nrm2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho RJ, et al. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol. 2000;151:1353–1368. doi: 10.1083/jcb.151.7.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafesse FG, et al. Disruption of Sphingolipid Biosynthesis Blocks Phagocytosis of Candida albicans. PLoS Pathog. 2015;11:e1005188. doi: 10.1371/journal.ppat.1005188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwik J, et al. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. PNAS. 2003;100:13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin R, Grinstein S, Schlam D. Phosphoinositides in phagocytosis and macropinocytosis. BBA. 2015;1851:805–823. doi: 10.1016/j.bbalip.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014;262:193–215. doi: 10.1111/imr.12212. [DOI] [PubMed] [Google Scholar]
- Jutras I, Desjardins M. Phagocytosis: at the crossroads of innate and adaptive immunity. Annu. Rev. Cell. 2005;21:511–527. doi: 10.1146/annurev.cellbio.20.010403.102755. [DOI] [PubMed] [Google Scholar]
- Strijbis K, et al. Bruton's Tyrosine Kinase (BTK) and Vav1 contribute to Dectin1-dependent phagocytosis of Candida albicans in macrophages. PLoS Pathog. 2013;9:e1003446. doi: 10.1371/journal.ppat.1003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. The beta-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe. 2011;10:603–615. doi: 10.1016/j.chom.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban A, et al. Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. PNAS. 2011;108:14270–14275. doi: 10.1073/pnas.1111415108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Meth. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger A, Wittmann T. Fluorescence live cell imaging. Meth. Cell Biol. 2014;123:77–94. doi: 10.1016/B978-0-12-420138-5.00005-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov A, Rieder CL. Imaging the division process in living tissue culture cells. Methods. 2006;38:2–16. doi: 10.1016/j.ymeth.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman SA, et al. Integrins Form an Expanding Diffusional Barrier that Coordinates Phagocytosis. Cell. 2016;164:128–140. doi: 10.1016/j.cell.2015.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaumouille V, et al. Actin cytoskeleton reorganization by Syk regulates Fcgamma receptor responsiveness by increasing its lateral mobility and clustering. Dev. Cell. 2014;29:534–546. doi: 10.1016/j.devcel.2014.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, Gow NA, Erwig LP. Novel insights into host-fungal pathogen interactions derived from live-cell imaging. Sem. Immunopath. 2015;37:131–139. doi: 10.1007/s00281-014-0463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forestier CL. Imaging host-Leishmania interactions: significance in visceral leishmaniasis. Para. Immunol. 2013;35:256–266. doi: 10.1111/pim.12044. [DOI] [PubMed] [Google Scholar]
- John B, Weninger W, Hunter CA. Advances in imaging the innate and adaptive immune response to Toxoplasma gondii. Future Microbiol. 2010;5:1321–1328. doi: 10.2217/fmb.10.97. [DOI] [PMC free article] [PubMed] [Google Scholar]