

Abstract
Background
Fatty-acid-binding proteins (FABPs) are intracellular carriers for endocannabinoids, N-acylethanolamines, and related lipids. Previous work indicates that systemically administered FABP5 inhibitors produce analgesia in models of inflammatory pain. It is currently not known whether FABP inhibitors exert their effects through peripheral or central mechanisms. Here, we examined FABP5 distribution in dorsal root ganglia and spinal cord and examined the analgesic effects of peripherally and centrally administered FABP5 inhibitors.
Results
Immunofluorescence revealed robust expression of FABP5 in lumbar dorsal root ganglia. FABP5 was distributed in peptidergic calcitonin gene-related peptide-expressing dorsal root ganglia and non-peptidergic isolectin B4-expressing dorsal root ganglia. In addition, the majority of dorsal root ganglia expressing FABP5 also expressed transient receptor potential vanilloid 1 (TRPV1) and peripherin, a marker of nociceptive fibers. Intraplantar administration of FABP5 inhibitors reduced thermal and mechanical hyperalgesia in the complete Freund’s adjuvant model of chronic inflammatory pain. In contrast to its robust expression in dorsal root ganglia, FABP5 was sparsely distributed in the lumbar spinal cord and intrathecal administration of FABP inhibitor did not confer analgesic effects. Administration of FABP inhibitor via the intracerebroventricular (i.c.v.) route reduced thermal hyperalgesia. Antagonists of peroxisome proliferator-activated receptor alpha blocked the analgesic effects of peripherally and i.c.v. administered FABP inhibitor while antagonism of cannabinoid receptor 1 blocked the effects of peripheral FABP inhibition and a TRPV1 antagonist blocked the effects of i.c.v. administered inhibitor. Although FABP5 and TRPV1 were co-expressed in the periaqueductal gray region of the brain, which is known to modulate pain, knockdown of FABP5 in the periaqueductal gray using adeno-associated viruses and pharmacological FABP5 inhibition did not produce analgesic effects.
Conclusions
This study demonstrates that FABP5 is highly expressed in nociceptive dorsal root ganglia neurons and FABP inhibitors exert peripheral and supraspinal analgesic effects. This indicates that peripherally restricted FABP inhibitors may serve as a new class of analgesic and anti-inflammatory agents.
Keywords: Fatty-acid-binding protein, endocannabinoid, anandamide, pain
Introduction
Chronic pain affects between 30% and 60% of older adults.1–3 Inflammatory pain is typically treated with non-steroidal anti-inflammatory drugs such as ibuprofen.4 However, prolonged use of non-steroidal anti-inflammatory drugs is associated with significant side-effects including gastrointestinal ulceration and bleeding.4,5 Opioids are another class of highly efficacious analgesics for the treatment of acute pain but lose efficacy with chronic usage.6 Furthermore, opioids are highly addictive and many patients administered chronic opioids develop addiction.7–9 Consequently, there is a pressing need to develop novel efficacious, non-addictive analgesics for the treatment of chronic pain.
Fatty-acid-binding proteins (FABPs) are intracellular lipid chaperones that are expressed in cells of the central and peripheral nervous system.10–12 In addition to fatty acids, FABPs interact with other endogenous lipids including the endocannabinoids, anandamide (AEA) and 2-arachidonoylglycerol (2-AG), and related N-acylethanolamines (NAEs).13–16 FABPs regulate the intracellular delivery of AEA to its catabolic enzyme fatty acid amide hydrolase (FAAH) and FABP inhibition reduces AEA inactivation in vitro and elevates AEA levels in vivo.13–17 Similar to FABP inhibition, FAAH inhibitors elevate tissue AEA levels, which subsequently activates central and peripheral cannabinoid receptor 1 (CB1) to produce analgesia.18–21 However, FAAH inhibition in liver produces hyperglycemia and insulin resistance,22 effects not seen with FABP inhibitors. FABP inhibitors do not produce conditioned place preference in mice,23 suggesting that they are devoid of addictive properties. Consequently, FABP inhibitors may serve as efficacious analgesics and may exhibit a low side-effect profile.
In addition to AEA, FABPs regulate the metabolism of the NAEs palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), which activate peroxisome proliferator-activated receptor alpha (PPARα) centrally and peripherally and produce analgesic and anti-inflammatory effects.24–26 We have previously shown that FABP5 and FABP7 bind to endocannabinoids/NAEs with high affinities and that mice deficient in these proteins exhibit reduced nociception.14,26,27 Furthermore, considering that FABP7 exhibits low expression in adult tissues and the analgesic effects of FABP inhibitors track well with their affinities for FABP515,28 and are lost in FABP5 KO mice (MK, unpublished observations), suggests that FABP5 inhibition likely accounts for the analgesic effects of FABP inhibitors.
We recently reported that the analgesic effects produced by pharmacological or genetic FABP inhibition are mediated by CB1, PPARα, and transient receptor potential vanilloid 1 (TRPV1) receptors.15,26 While CB1 and PPARα receptors produce their analgesic effects centrally and peripherally, TRPV1 activation in primary sensory neurons induces pain while its activation in supraspinal regions leads to analgesia.29–31 The goal of this study was to profile FABP5 expression in anatomical sites that regulate nociception and to determine whether FABP inhibitors exert their analgesic effects through a central and/or peripheral site of action. In particular, we evaluated FABP inhibition using the parent compound SBFI26, which was identified in our laboratories using a dual computational/experimental screening approach and contains a unique truxillic acid scaffold.32 Importantly, truxillic acid-based compounds like SBFI26 are similar to the chemical incarvillateine, a molecule identified as the active component of the traditional Chinese medicine Incarvillea sinensis used to treat pain and inflammation.33
Materials and methods
Chemicals and drugs
PEA, d4-PEA, OEA, d2-OEA, 2-AG, d5-2-AG, and AMG9810 were from Cayman Chemical (Ann Arbor, MI). AEA and d4-AEA were from R&D systems. [14C]AEA (arachidonoyl-[1-14C]ethanolamide) and rimonabant were provided by the Drug Supply Program at the National Institute on Drug Abuse. GW6471 was purchased from Sigma. All other chemicals were obtained from Fisher Scientific.
Synthesis of inhibitors
2,4-Diphenylcyclobutane-1,3-dicarboxylic acid (α-truxillic acid)
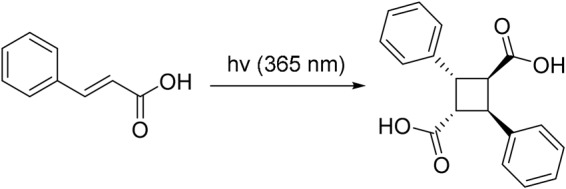
E-Cinnamic acid (2.50 g, mmol) placed in a Pyrex dish and irradiated by a 365-nm mercury lamp (280 mW/cm2) for five days with periodic shaking. The reaction was monitored by 1H NMR. After 87% conversion had been achieved on day 5, the reaction mixture (white solid) was washed with diethyl ether to remove unreacted E-cinnamic acid, and desired α-truxillic acid was obtained as white powder (2.01 g, 80% yield; 92% conversion yield): 1H NMR (300 MHz, acetone-d6) δ 4.00 (dd, J = 10.2, 7.4 Hz, 2H), 4.44 (dd, J = 10.2, 7.4 Hz, 2H), 7.44–7.21 (m, 10H); 13C NMR (75 MHz, acetone-d6) δ 41.6, 46.3, 126.7, 127.7, 128.2, 139.6, 172.3. Data are consistent with the literature values.32
2,4-Diphenylcyclobutane-1,3-dicarboxylic acid mono-1-naphthyl ester (SBFI26)

α-Truxillic acid (297 mg, 1.0 mmol) was suspended in thionyl chloride (3 mL), and one drop of DMF was added to the suspension. The reaction mixture was heated to reflux for 3 h. The excess thionyl chloride and DMF was removed in vacuo to give truxillic acid dichloride, which was used directly in the subsequent reaction. To a solution of truxillic acid dichloride in THF (10 mL) was added dropwise a solution of 1-naphthol (120 mg, 0.84 mmol) in THF (5 mL) and pyridine (0.5 mL), and the reaction mixture was heated to reflux for 3 h. The reaction was quenched by addition of water (2 mL). The resulted solution was diluted with ethyl acetate (15 mL), and the aqueous layer was separated. The organic layer was dried over MgSO4 and concentrated in vacuo. The crude product was purified by flashed column chromatography on silica gel using ethyl acetate/hexanes (10%–25% EtOAc gradient) as eluent to afford SBFI26 as white solid (202 mg, 57% yield): m.p. 192℃–193℃; 1H NMR (300 MHz, acetone-d6) δ 4.15 (dd, J = 10.4, 7.0 Hz, 1H), 4.56 (dd, J = 10.4, 7.0 Hz, 1H), 4.75–4.68 (m, 2H), 6.42 (d, J = 7.5 Hz, 1H), 7.65–7.24 (m, 15H), 7.72 (d, J = 8.2 Hz, 1H); 13C NMR (75 MHz, acetone-d6) δ 41.7, 42.1, 45.4, 46.8, 117.9, 121.4, 125.3, 125.7, 126.3, 126.7, 126.8, 126.9, 127.4, 127.6, 127.8, 128.2, 128.3, 128.5, 128.8, 129.1, 134.5, 139.5, 170.7, 172.1. Data are consistent with the literature values.32
1-Methyl 3 -(1-naphthyl) 2,4-diphenylcyclobutane-1,3-dicarboxylate (SBFI26ME)

To a solution of SBFI26 (60 mg, 0.14 mmol) in acetone (5 mL) was added K2CO3 (39 mg, 0.28 mmol) and Me2SO4 (11 mg, 0.09 mmol), and the reaction mixture was heated to reflux for overnight. The reaction mixture was cooled to room temperature and filtered through Celite. The filtrate was concentrated and the residue was purified by column chromatography on silica gel (EtOAc/hexanes, 0%–10% EtOAc gradient) to afford SBFI26ME as a white solid (62 mg, 99%): m.p. 139℃–140℃; 1H NMR (300 MHz, acetone-d6): δ 3.33 (s, 3H), 4.14 (m, 1H), 4.59 (m, 1H), 4.71 (m, 2H), 6.40 (d, J = 7.5 Hz, 1H), 7.24–7.52 (m, 12H), 7.62–7.74 (m, 3H), 7.87 (d, J = 8.3 Hz, 1H); 13C NMR (100 MHz, acetone-d6): δ 41.7, 41.9, 46.4, 46.7, 50.7, 117.9, 121.4, 125.2, 125.7, 126.8, 127.0, 127.5, 127.6, 128.3, 128.3, 128.8, 134.5, 139.0, 139.3, 146.8, 170.5, 171.7. HRMS (ES) m/z calcd for C29H24NO4 (M + H)+: 437.1747, found 437.1748 (Δ −0.08 ppm).
Animals
Male C57Bl/6 mice (23–35 g, Taconic Farms) were used for all experiments. The animals were housed in groups of three at room temperature and were kept on a 12:12-h light:dark cycle with ad libitum access to food and water. For experiments involving i.c.v. injections or AAV microinjections, mice were singly housed. The experiments were approved by the Stony Brook University Institutional Animal Care and Use Committee (#277150). Euthanasia was commenced with CO2 and subsequent decapitation with the exception of mouse perfusions and endocannabinoid measurements (see below).
Behavioral tests
Mice were habituated to the experimental room for at least two days before baseline measurements and at least 2 h before each experiment. To induce chronic inflammation, mice received unilateral intraplantar injections of 20 µL complete Freund’s adjuvant (CFA) (50% in saline) (#F5881, Sigma). Three to seven days later, thermal and mechanical hyperalgesia was measured, respectively, using the Hargreaves plantar apparatus (Ugo Basile) or graded von Frey filaments as previously reported.15 The experimenter was blinded to the treatment condition of each animal.
Drug administration
For intraplantar drug administration, three to seven days after CFA injections, mice received an injection of 5 µL SBFI26, SBFI26ME, or DMSO using a 30-G needle. Intrathecal injections were performed as described previously with modifications.34 Briefly, mice were anesthetized with 2% isoflurane, the back was shaved, and 2 µL of SBFI26 (50 µg) or DMSO was slowly injected between the L5 and L6 vertebrae using a 10 -µL Hamilton syringe equipped with a 30-G needle. For experiments involving receptor antagonists, the compounds were injected via the intraperitoneal route 30 min before SBFI26 in a volume of 10 µL/g body weight. Rimonabant (3 mg/kg) was dissolved in ethanol:Cremophor-EL:saline (1:1:18), GW6471 (4 mg/kg), and AMG9810 (30 mg/kg) were dissolved in DMSO:Cremophor-EL:saline (1:1:8).
Stereotaxic surgery and inhibitor administration
Adult mice were anesthetized with an i.p. injection of a surgical mixture of ketamine (100 mg/kg)/xylazine (10 mg/kg). The head was shaved and the scalp cleaned with betadine. The animal was subsequently placed in the stereotaxic instrument (BenchMark Digital, Leica Biosystems, Buffalo Grove, IL) and a 1-cm skin incision was made to reveal the skull. After removing the soft tissue from the surface, the skull was cleaned with 3% hydrogen peroxide and was leveled between the bregma and lambda. The placement of the guide cannula was determined in relation to the bregma, a mark was made on the skull surface, and a small hole was drilled through the skull. The stereotaxic coordinates are established from the atlas of Franklin and Paxinos, 3rd edition, 2008. For the i.c.v. injections, the coordinates were AP: −0.34 mm, ML: 1.0 mm, DV: 2.3 mm. For the dorsolateral PAG, the coordinates were AP: −4.6 mm, ML: 0.5 mm, DV: 2.0 mm. For the ventrolateral PAG, the coordinates were AP: −4.6 mm, ML: 0.5 mm, DV: 2.75 mm. For the RVM injections, the coordinates were AP: −5.8 mm, ML: 0.0 mm, DV: 5.5 mm. A low-profile mouse cannula guide (C315GS-4/SPC, Plastics One, Roanoke, VA) with an infusion dummy cannula (C315DCS-4/SPC, Plastics One, Roanoke, VA) was inserted into skull hole and held in place with Loctite gel. The base of the cannula guide was then cemented onto the skull using dental cement (Ortho-Jet Powder, Lang Dental Manufacturing Co. Inc., Wheeling, IL), and the dummy infusion cannula was replaced with a cap. The animal was allowed to recover from the surgery for a week before behavioral testing.
On the day of the drug injection, the mice were sedated with isoflurane, the dummy cap removed and an internal injection cannula (C315IS-4/SPC, Plastics One, Inc., Roanoke, VA) of the proper length for that brain region inserted through the guide cannula. The injection cannula was connected to a 10 -µL syringe (Hamilton Co., Reno, NV) via PE-50 tubing and 0.2 µL of the either the vehicle or the drug solution was injected over a 2-min period using a GenieTouch syringe pump (Lucca Technologies, Harwinton, CT). The injection cannula was allowed to remain for an additional 60 s before withdrawal and the guide cannula cap replaced. Mice were placed back into their home cages to recover before behavioral testing and testing occurred 2 h after drug administration.
AAV-mediated FABP5 knockdown
The AAVs were generated at the Duke University Viral Vector Core. The AAVs (serotype 5) utilized a chicken beta actin promoter, enhanced green fluorescent protein to gauge transduction efficiency, and expressed FABP5 shRNA or scrambled control. The FABP5 shRNA sequence is as follows: TAACCAAAGGAATGATCCT. Mice were anesthetized with 2% isoflurane and 0.2 µL of AAV (titer of 9.3 × 1011/ml) was infused into the dorsolateral PAG through the cannula at a rate of 0.1 µL per minute using a GenieTouch syringe pump (Lucca Technologies). After 21 days, the mice were employed for behavioral experiments or histology.
Enzyme assays
FAAH activity assays were performed as previously described.13,15 Briefly, brain and paw tissues were homogenized in 50 mM Tris-HCl buffer (pH 9) and incubated with 100 µM [14C]AEA at 37℃ to permit ∼10% substrate hydrolysis. The reactions were quenched by the addition of two volumes of 1:1 chloroform:methanol, separated by centrifugation, and the top phase collected and radioactivity quantified using a scintillation counter.
Immunofluorescence
Mice were anesthetized (100 mg/kg ketamine plus 10 mg/kg xylazine, i.p.) and transcardially perfused with saline followed by 4% paraformaldehyde in 0.1 M phosphate buffered saline (PBS), pH 7.4. Brains, spinal cord, and dorsal root ganglia (DRGs) were dissected and immersed in the same fixative overnight at 4℃; this was followed by immersion in 30% sucrose in 0.1 M PB for cryoprotection. The tissues were embedded in a gelatin–albumin mixture (3% gelatin, 30% egg albumin in dH2O) and frozen using a liquid nitrogen-chilled isopentane bath. The blocks were then fixed to a peg and cut using a cryostat at −16℃. The sections of 20 µm in thickness, were thaw-mounted on gelatin/chromium-coated slides, air dried, and stored in a frost-free freezer at −20℃ until further processing. Sections were thawed at room temperature for 20 min, the tissues were fixed onto the slides with 4% PFA for 5 min, rinsed in PBS three times for 10 min, and then incubated with 0.2% Triton X-100 (Sigma-Aldrich Cat# 9002-93-1) in PBS 10 min for permeabilization. A 30-min block was used, consisting of 10% normal donkey serum (NDS, Jackson ImmunoResearch Labs Cat# 017-000-121 RRID:AB_2337258), 2% glycine, and 0.1% Triton X-100 in PBS. Tissues were incubated overnight at 4℃ with primary antibodies diluted in PBS containing 0.3% Triton X-100 and 5% NDS. Primary antisera used were rabbit anti-FABP5 (BioVendor, #RD181060100, RRID:AB_344491) at 1:800 dilution in brain and 1:600 in DRGs, mouse anti-TRPV1 (UC Davis/NIH NeuroMab, #75-254, RRID:AB_11000725) at 1:400 dilution in brain tissue, goat anti-TRPV1 (Neuromics, #GT15129-100, RRID:AB_2209002) at 1:100 dilution in DRGs, mouse anti-peripherin (Millipore, #MAB5380, RRID:AB_2171352) at 1:75 dilution, goat anti-FAAH (LifeSpan Cat# LS-B4126-50 RRID:AB_10720351) at 1:50 in DRGs, and goat anti-CGRP (AbD Serotec Cat# 1720-9007 RRID:AB_2290729) at 1:400 in DRGs. The sections were washed three times for 10 min each in PBS. The slides were incubated for 1 h at room temperature with secondary antibodies diluted in PBS with 0.3% Triton X-100 and 5% NDS. Secondary antibodies used were Alexa Fluor 594 donkey anti-rabbit (Jackson ImmunoResearch Labs, #711-585-152, RRID:AB_2340621) at 1:500 dilution, Cy2-AffiniPure donkey anti-goat IgG (H + L) antibody (Jackson ImmunoResearch Labs, #705-225-147, RRID:AB_2307341) at 1:200 dilution, and Alexa Fluor 488 AffiniPure donkey anti-mouse IgG (H + L) antibody (Jackson ImmunoResearch Labs, #715-545-151, RRID:AB_2341099) at 1:400 dilution. The slides were subsequently washed three times with PBS for 10 min each and immediately mounted with ProLong® Gold antifade mounting medium with DAPI (Life Technologies, #P36931) and stored at 4℃ in the dark. Fluorescent immunoreactivity in cells was observed with a Zeiss Axioplan 2 epifluorescent microscope. Images were obtained using Zeiss AxioCam HRm monochrome digital camera, and AxioVision Rel. 4.6 microscope software. Images were only adjusted for brightness and contrast.
Western blotting
Immunoblots were performed essentially as described.26 The blots were probed with rabbit anti-FABP5 (1:1000, BioVendor #RD181060100, RRID:AB_344491) and mouse anti-beta actin (1:10,000, Abcam #ab6276, RRID:AB_2223210) antibodies followed by HRP-labeled secondary antibodies. The blots were developed using the Clarity Western HRP substrate (Bio-Rad) and scanned using a C-DiGiT scanner (Li-COR). Protein band intensities were quantified and normalized to beta actin intensities.
Lipid quantification
Tissue endocannabinoid levels were quantified as previously described with minor modifications.15 Mice were euthanized by rapid decapitation and brain and paw tissues were flash frozen in liquid nitrogen. The samples were thawed on ice, weighed, spiked with deuterated standards, homogenized, and quantifications performed as described.15 SBFI26 quantification was performed as previously described using an external calibration curve.15 For SBFI26ME quantification, paw tissues were flash frozen in liquid nitrogen and homogenized in 4 ml of 2:1:1 chloroform:methanol:Tris (50 mM, pH 8), the phases separated by centrifugation, and the chloroform phase isolated and dried down under gentle argon stream. The samples were subsequently resuspended in 50% acetonitrile in water and injected into a Thermo TSQ Quantum Access Triple Quadrupole Mass Spectrometer (Thermo-Fisher) at a flow rate of 5 µL/min. Liquid chromatography separation was achieved on a Luna C18 (150 × 2 mm, 5 µm) column. Mobile phase A was composed of 5 mM ammonium acetate while mobile phase B was composed of 100% acetonitrile. A linear gradient was used and started at 50% phase B for 2 min, ramped to 95% phase B in 10 min, and was followed by a 5-min hold at 95% phase B. The system was equilibrated at 50% phase B for 10 min. Quantification was performed in positive ion mode. The following parameters were employed: high voltage was set at 4.5 kV, the sheath pressure was 55 psi, and the capillary was set to 350℃. Multiple reaction monitoring was used with the transition m/z 454 to 131 at 25 eV as the quantitation channel with m/z 454 to 275 at 15 eV serving as the confirmation channel.
Dissociated DRG neurons
Mice were deeply anesthetized with isoflurane and decapitated. The lumbar segments of the spinal cord were removed and placed in a cold Ca2+, Mg2+-free (CMF) Hank’s solution containing (in mM): 137 NaCl, 5.3 KCl, 0.33 Na2HPO4, 0.44 KH2PO4, 5 HEPES, 5.5 glucose, pH = 7.4 with NaOH. The bone surrounding the spinal cord was removed, and DRG (L3, L4, and L5) were exposed and pulled out. After removing the roots, ganglia were chopped in half and incubated for 20 min at 34℃ in Ca2+, Mg2+-free Hank’s solution containing 20 U/ml Papain (Worthington Biochemical, Lakewood, NJ) and 5 mM DL-cysteine. Ganglia were then treated for 20 min at 34℃ with 3 mg/ml collagenase (Type I, Sigma-Aldrich, St. Louis, MO) and 4 mg/ml Dispase II (Boehringer Mannheim, Indianapolis, IN) in Ca2+, Mg2+-free Hank’s solution. Ganglia were then washed with Leibovitz’s L-15 medium (Invitrogen, San Diego, CA) supplemented with 10% fetal calf serum and 5 mM HEPES. Individual cells were dispersed by mechanical trituration using fire-polished Pasteur pipettes with decreasing bore size and plated on glass coverslip treated with 100 µg/ml poly-D-lysine. Cells were incubated in the supplemented L-15 solution at 34℃ (in 5% CO2) and used over the next 4–6 h. Small DRG neurons (diameters < 27 µm) were selected by measuring the diameter from images captured to a computer by a digital camera (ORCA-Flash4.0, Hamamatsu Corporation, Bridgewater, NJ, USA).
Calcium imaging in isolated DRG neurons
Dissociated DRG neurons were loaded with 5 µM fura-2AM and maintained in a modified Tyrode’s solution containing (in mM): 151 NaCl, 2 CaCl2, 1 MgCl2, 2.5 KCl, 10 HEPES, 13 glucose, pH 7.4 with NaOH. DRG neurons loaded with fura-2AM were identified by calcium fluorescence by exciting the fluorophore at 340 nm wavelength illumination (Lambda XL, Sutter Instruments, Novato, CA) for 300 ms. The fluorophore was then excited alternately (300 ms) with 340 and 380 nm wavelength illumination. Images were acquired using the ORCA-Flash4.0 digital camera at a rate of 0.33 Hz. The fluorescence ratio for individual neurons was determined as the intensity of emission during 340 nm excitation (I340) divided by 380 nm emission (I380) and used as an indicator of change in cytoplasmic calcium.35 The I340/I380 ratio was calculated on a pixel-by-pixel basis using the MetaFluor software (Molecular Devices, Sunnyvale, CA, USA). Activation of TRPV1 channels was triggered by 1 µM capsaicin. Capsaicin or the test drug were applied using an array of quartz fiber flow pipes (500 µm internal diameter) positioned about 1 mm away from DRG neurons.
Statistical analysis
Data are presented as means ± SEM. Statistical significance was determined using two-tailed t tests between groups, one-way analysis of variance (ANOVA) followed by Dunnett or Tukey post hoc analysis, or two-way ANOVA followed by Bonferroni post hoc analysis. In all cases, differences of p < 0.05 were considered significant.
Results
Peripheral analgesic effects of FABP inhibitors
Primary sensory neurons innervate the extremities and transmit noxious stimuli to projection neurons in the dorsal horn of the spinal cord.36,37 We examined the expression of FABP5 in DRG of primary sensory neurons. In lumbar DRGs, FABP5 showed robust expression in small and large diameter neurons, consistent with previous results.10 Specifically, FABP5 was expressed in 50.3% of “peptidergic” calcitonin gene-related peptide (CGRP)-positive and in 48.2% of “non-peptidergic” isolectin B4 (IB4)-positive neurons (Figure 1(a); Table 1). CGRP-positive and IB4-positive primary sensory neurons transmit noxious thermal and mechanical stimuli.38–41 In our previous work, we have shown that FABP5 inhibition reduces thermal hyperalgesia.15,26,32 In nociceptors, thermal pain is transduced by the TRPV1 channel.29,30 Consequently, we examined whether FABP5 is co-expressed with TRPV1 in DRG neurons. Indeed, our data demonstrate robust co-expression of FABP5 and TRPV1, with FABP5 expression being present in ∼76% of TRPV1+ DRGs (Figure 1(b); Table 1). Lastly, FABP5 was also expressed in DRGs expressing peripherin, a marker of nociceptors (Figure 1(c)).
Figure 1.

Immunolocalization of FABP5 in lumbar DRGs: (a) sections were probed with FABP5 antibody, CGRP antibody to label peptidergic fibers, IB4 to label non-peptidergic fibers, and DAPI to label nuclei; (b) expression of FABP5 and TRPV1 in lumbar DRGs; (c) immunolocalization of FABP5 and the nociceptor marker peripherin; and (d) lack of FABP5 staining in DRG slices from FABP5 KO mice.
Table 1.
FABP5 distribution in CGRP+, IB4+, and TRPV1+ DRG neurons.
| Marker | % FABP5+/Marker+ |
|---|---|
| CGRP | 50.3 |
| IB4 | 48.2 |
| TRPV1 | 76.2 |
The table indicates the percentage of DRG neurons expressing CGRP, IB4, or TRPV1 that also express FABP5 (n = 682).
The robust expression of FABP5 in sensory neurons suggests that the analgesic effects of FABP inhibitors may be mediated via peripheral FABP inhibition. To examine this directly, we employed the CFA model of chronic inflammatory pain, which results in profound thermal and mechanical hyperalgesia (Figure 2). To inhibit FABP5, we employed the FABP inhibitor SBFI26, which was previously described (Figure (2a)).15,32 Intraplantar injection of SBFI26 dose-dependently reduced mechanical hyperalgesia (Figure 2(b)). Injection of SBFI26 into the contralateral paw did not affect mechanical sensitivity of the CFA-injected ipsilateral paw (Figure 2(c)), arguing against a systemic effect of the compound. In addition to mechanical thresholds, intraplantar administration of SBFI26 likewise reduced thermal hyperalgesia (Figure 2(c)). A time course of SBFI26 analgesic effects indicates that the compound maintains efficacy for 4 h after administration (Figure 2(d) and (e)). We also examined paw tissue levels of SBFI26 after intraplantar injection and observed a rapid decline in SBFI26 1 h after administration and continued reductions thereafter (Figure 2(f)).
Figure 2.

FABP inhibition produces peripherally mediated analgesic effects. (a) Chemical structures of the FABP5 inhibitor SBFI26 and its inactive analog SBFI26ME. (b) Mechanical thresholds in mice before and after CFA in the presence and absence of FABP inhibitors. Intraplantar CFA injection reduces paw withdrawal thresholds (white bars). Intraplantar administration of SBFI26 elevates mechanical thresholds in CFA-injected mice (red bars) (n = 8; ***p < 0.001 versus vehicle controls). Administration of SBFI26 into the contralateral (CL) paw does not alter withdrawal thresholds in the CFA-injected paw. Intraplantar administration of SBFI26ME (gray bar) does not alter withdrawal thresholds. (c) Administration of 50 µg SBFI26 (red bar) reduces thermal hyperalgesia in CFA-injected mice (n = 8; *p < 0.05 versus vehicle controls). (d) Time course of mechanical thresholds in the CFA-injected paw after intraplantar administration of 50 µg SBFI26 (red) or vehicle (black) (n = 8; *p < 0.05 and **p < 0.01 versus vehicle controls). (e) Time course of mechanical thresholds in the contralateral paw after intraplantar administration of 50 µg SBFI26 into the CFA-injected paw (n = 8). (f) Time course of SBFI26 levels in paws after intraplantar administration of 50 µg SBFI26. Inset: Zoomed image depicting plantar SBFI26 levels (n = 3). (g) Levels of SBFI26ME and SBFI26 in paws 2 h after intraplantar administration of 50 µg SBFI26ME (n = 3).
SBFI26ME is a novel congener of SBFI26 wherein the carboxylate is conjugated with a methyl ester (Figure (2a)). After systemic administration, SBFI26ME metabolism converts the compound into SBFI26 (MK, unpublished observations). In contrast, intraplantar injection of SBFI26ME does not lead to appreciable conversion into SBFI26 (Figure 2(g)), suggesting that paw tissue may lack the necessary esterases to hydrolyze the methyl ester moiety. Consistent with its low affinity for FABP5 (Ki > 10 µM), intraplantar administration of SBFI26ME did not affect mechanical thresholds in CFA-treated mice (Figure 2(b)). These data provide strong evidence that inhibition of peripherally expressed FABPs produces analgesic effects.
Central analgesic effects of FABP inhibitors
Sensory neurons that transmit noxious stimuli terminate in lamina I and lamina II of the dorsal horn of the spinal cord.36 Consequently, we examined FABP5 distribution in the lumbar spinal cord and observed uniform distribution of the protein, which was not concentrated in lamina I or lamina II (Figure 3(a)). Additionally, FABP5 was not expressed in the terminals of sensory neurons as indicated by a lack of co-localization with CGRP in the dorsal horn of the spinal cord (Figure 3(b)). Accordingly, intrathecal administration of SBFI26 did not affect thermal or mechanical thresholds in CFA-injected mice (Figure 4(a) and (b)). In addition to the spinal cord, FABP5 is expressed throughout the brain and FABP5 inhibition elevates brain endocannabinoid and NAE levels.15,26 Furthermore, supraspinal sites such as the periaqueductal gray (PAG) influence the descending control of pain42 and activation of CB1 within the PAG produces analgesia.43 Consequently, we examined whether supraspinal FABP inhibition reduces nociception. Intracerebroventricular (i.c.v.) administration of SBFI26 reduced thermal hyperalgesia in CFA-injected mice (Figure 4(c)) but did not affect mechanical thresholds (Figure (4d)).
Figure 3.

Immunolocalization of FABP5 in lumbar spinal cord. (a) Spinal cord sections were probed with FABP5 antibody and DAPI was used to stain nuclei. (b) Dorsal horn of the lumbar spinal cord was probed with FABP5 and CGRP antibodies.
Figure 4.

Supraspinal FABP inhibition reduces thermal hyperalgesia. (a) Thermal withdrawal latencies of mice before and after CFA injection. Administration of 50 µg SBFI26 via the i.t. route does not alter thermal withdrawal latencies (n = 8). (b) Mechanical withdrawal latencies of CFA-injected mice after i.t. administration of 50 µg SBFI26 (n = 8). (c) Thermal withdrawal latencies of CFA-injected mice after administration of 50 µg SBFI26 via the i.c.v. route (n = 8; *p < 0.05 versus vehicle control). (d) Mechanical withdrawal latencies of mice after i.c.v. administration of 50 µg SBFI26 (n = 8).
Anatomically specific engagement of CB1, PPARα, and TRPV1 receptors mediates the analgesic effects of FABP inhibitors
We have previously demonstrated that CB1, PPARα, and TRPV1 receptors are responsible for the analgesic effects of systemically administered FABP inhibitors and in FABP5/7 KO mice.15,26 We next sought to determine the receptor systems that mediate the analgesic effects of FABP inhibitors at each anatomical site. Treatment of mice with the CB1 antagonist rimonabant or the PPARα antagonist GW6471 blocked the analgesic effects of intraplantar SBFI26 (Figure 5(a) and (b)). GW6471 similarly blocked the analgesic effects of i.c.v. administered SBFI26 while rimonabant was without effect (Figure 5(c)). These data indicate that peripheral and central FABP inhibition results in the engagement of distinct receptor systems.
Figure 5.

Effect of CB1, PPARα, and TRPV1 antagonists upon analgesic effects of SBFI26. (a) Intraplantar administration of 50 µg SBFI26 (red bars) reduces thermal hyperalgesia in CFA mice and these effects are reversed by the CB1 inverse agonist rimonabant (3 mg/kg, i.p.) and PPARα antagonist GW6471 (4 mg/kg, i.p.) (n = 9; *p < 0.05 and **p < 0.01). (b) Rimonabant and GW6471 reverse the analgesic effects of 50 µg SBFI26 that was administered via the intraplantar route (n = 9; *p < 0.05). (c) Administration of 50 µg SBFI26 via the i.c.v route reduces thermal hyperalgesia, an effect that is blocked by pretreatment with GW6471 or the TRPV1 antagonist AMG9810 (30 mg/kg, i.p.) (n = 9; *p < 0.05 and **p < 0.01). (d) Injection of AMG9810 (30 mg/kg, i.p.) does not affect thermal hyperalgesia in CFA injected mice.
The inability of rimonabant to block the analgesic effects of i.c.v. administered SBFI26 may have stemmed from the inability of SBFI26 to elevate brain AEA levels. Quantification of brain endocannabinoid/NAE levels revealed that SBFI26 elevated AEA and in addition also raised PEA and OEA levels (Figure 6(a)). We also profiled paw tissue levels of endocannabinoids/NAEs and observed that SBFI26 did not affect PEA, OEA, AEA, or 2-AG levels (Figure 6(b)). This is perhaps not surprising given that previous reports found that inhibition of FAAH, the principal enzyme that hydrolyzes AEA, likewise does not elevate plantar levels of AEA despite producing CB1-mediated analgesic effects.44,45 Importantly, intraplantar and i.c.v. administration of SBFI26 did not inhibit FAAH activity (Figure 6(c) and (d)), confirming that the effects of SBFI26 upon endocannabinoid levels are unlikely to be a result of off-target inhibition of FAAH.
Figure 6.

Endocannabinoid/NAE levels after SBFI26 administration. (a) Levels of PEA, OEA, AEA, and 2-AG in brains 2 h after i.c.v. injection of 50 µg SBFI26 (n = 6; *p < 0.05 versus vehicle controls). (b) Paw levels of PEA, OEA, AEA, and 2-AG after intraplantar administration of 50 µg SBFI26 (n = 6). (c) AEA hydrolysis in brain homogenates of mice receiving i.c.v. injections of 50 µg SBFI26 (n = 6). (d) AEA hydrolysis in paw homogenates of mice receiving 50 µg SBFI26 via the intraplantar route (n = 6).
Previous studies have demonstrated that activation of supraspinal TRPV1 receptors reduces nociception,31,46 and we have recently reported that the analgesic effects observed in FABP5/7 KO mice were mediated by TRPV1.47 AEA is an agonist at TRPV1 and its levels are elevated following i.c.v. administration of SBFI26. Therefore, we examined whether blocking TRPV1 receptors reduces the analgesic effects of i.c.v. SBFI26. Indeed, the TRPV1 antagonist AMG9810 significantly attenuated analgesia produced by i.c.v. SBFI26 (Figure 5(c)). In contrast, AMG9810 did not affect analgesia produced by intraplantar SBFI26 (Figure 5(a)), and it did not significantly alter thermal hyperalgesia when administered alone into CFA-treated animals (Figure 5(d)). To rule out the possibility that SBFI26 directly activates TRPV1, freshly dissociated DRG neurons were incubated with the TRPV1 agonist capsaicin (1 µM) or SBFI26 (10 µM) and TRPV1 activation was examined by calcium imaging. Our results indicate that SBFI26 does not activate TRPV1 (Figure 7). In contrast and as expected, capsaicin robustly activated TRPV1 receptors. Collectively, these data indicate that FABP inhibition potentiates supraspinal TRPV1 activation by elevating the levels of endogenous TRPV1 ligands such as AEA.
Figure 7.

SBFI26 does not activate TRPV1. (a) Left: DRG neurons loaded with Fura 2-AM (arrows) were initially identified by exciting the fluorophore at 340 nm for 300 ms. Right: change in fluorescence in the same DRG neurons upon stimulation with 1 µM capsaicin. (b) Representative fluorimetric traces showing increases in cytoplasmic Ca2+ in the same DRG neuron perfused with 10 µM SBFI26 or 1 µM capsaicin. Data are represented as normalized fluorescence ratios. Arrow indicates the time point at which capsaicin or SBFI26 was applied. (c) Collected results showing increase in cytoplasmic Ca2+ in DRG neurons perfused with 10 µM SBFI26 or 1 µM capsaicin (n = 14; ***p < 0.0001 versus baseline and SBFI26-treated DRGs).
The PAG does not mediate the analgesic effects of supraspinal FABP inhibition
The PAG is an important brain region that regulates the descending control of pain.42 Furthermore, activation of TRPV1 within the dorsolateral and ventrolateral PAG reduces thermal hyperalgesia.31,48–50 Given that the analgesic effects of centrally administered FABP inhibitor were mediated by TRPV1, we examined whether FABP5 and TRPV1 co-localize within the PAG. Indeed, we observed robust expression of FABP5 in the PAG, which co-localized with TRPV1 (Figure 8(a) and (b)).
Figure 8.

Expression of FABP5 and TRPV1 in the PAG. (a) Expression of FABP5 in the PAG. (b) Co-localization of FABP5 and TRPV1 in the PAG.
We subsequently examined whether the PAG mediates the analgesic effects of FABP inhibitors and whether this is mediated via TRPV1 inhibition. To determine whether FABP5 inhibition reduces nociception within the dorsolateral PAG, we knocked down FABP5 expression using adeno-associated viruses (AAVs) bearing shRNA targeting FABP5. Robust transduction within the dorsolateral PAG was observed as indicated by green fluorescent protein (GFP) fluorescence (Figure 9(a) and (b)). AAVs bearing FABP5 shRNAs reduced FABP5 expression in the dorsolateral PAG by ∼55% as indicated by Western blotting (Figure 9(c) and (d)) and elevated AEA levels (Figure 9(e)). Despite the robust FABP5 knockdown and concomitant increase in AEA, thermal hyperalgesia after CFA injection was similar between mice expressing FABP5 shRNA and control mice (Figure 9(f)).
Figure 9.

FABP inhibition in the PAG does not reduce thermal hyperalgesia. (a) Low magnification image of thionine blue stained brain section demonstrating placement of cannula in the dorsolateral PAG. DL: Dorsolateral, L: Lateral, DM: Dorsomedial, Aq: Aqueduct. (b) AAVs bearing FABP5 shRNA or scrambled controls were injected into the dorsolateral PAG and a representative image showing GFP fluorescence is shown. (c) Representative Western blot of FABP5 in the dorsolateral PAG in mice transduced with FABP5 shRNA or scrambled control. Actin serves as a loading control. (d) Quantification of Western blots demonstrated reduced expression of FABP5 in dorsolateral PAG transduced with FABP5 shRNA (n = 3; *p < 0.05). (e) Quantification of PEA, OEA, AEA, and 2-AG levels in dorsolateral PAG transduced with FABP5 shRNA or scrambled control (n = 6; *p < 0.05). (f) Thermal withdrawal latencies at baseline and after CFA injection in mice transduced with FABP5 shRNA or scrambled control (n = 6). (g) Injection of vehicle or 10 µg SBFI26 into the dorsolateral PAG does not alter thermal hyperalgesia in CFA mice (n = 6). (h) Injection of vehicle or 10 µg SBFI26 into the ventrolateral PAG does not affect thermal withdrawal latencies (n = 6).
To eliminate the possibility that the shRNA approach did not produce sufficient knockdown to unmask analgesic effects, we administered 10 µg of SBFI26 into the dorsolateral PAG and examined effects upon thermal hyperalgesia. Similar to the AAV approach, SBFI26 did not affect thermal withdrawal latencies in CFA mice (Figure 9(g)). Lastly, SBFI26 was injected into the ventrolateral PAG and similarly failed to affect thermal hyperalgesia in CFA mice (Figure 9(h)). Collectively, these results indicate that the PAG is unlikely to mediate the analgesic effects resulting from supraspinal FABP inhibition.
In addition to the PAG, recent work indicates that endocannabinoid/NAE modulation of rostral ventromedial medulla (RVM) output can regulate nociception.51 Consequently, we examined whether FABP inhibition within the RVM produces analgesia. SBFI26 (10 µg) or vehicle were injected into the RVM and thermal withdrawal latencies were measured in CFA-treated mice. Similar to the PAG, intra-RVM injection of SBFI26 did not produce antinociceptive effects (Figure 10).
Figure 10.

FABP inhibition in the RVM does not reduce thermal hyperalgesia. (a) Injection site of SBFI26 in the RVM. (b) Thermal withdrawal latencies at baseline and after CFA in mice injected with 10 µg SBFI26 or vehicle (n = 6).
Discussion
Endocannabinoids and NAEs reduce nociception through engagement of central and peripheral CB1 and PPARα receptors25,47 as well as central TRPV1.48 Endocannabinoid inactivation proceeds through cellular uptake followed by intracellular transport by FABPs and subsequent hydrolysis by FAAH.13,52 Consequently, FABP inhibition may serve as a novel strategy for the development of analgesic and anti-inflammatory drugs.15,32 To aid in the design of future FABP inhibitors, an anatomical dissection of site(s) mediating FABP inhibitor-induced analgesia is required and was the goal of the current work.
The robust expression of FABP5 in lumbar DRGs suggests that FABPs may regulate peripheral endocannabinoid metabolism and that the analgesic effects of FABP inhibitors may be peripherally mediated. Indeed, intraplantar administration of the FABP inhibitor SBFI26, but not its inactive analog SBFI26ME, significantly reduced thermal and mechanical hyperalgesia through a mechanism mediated by CB1 and PPARα receptors. This is consistent with our recent work demonstrating that CB1 and PPARα receptors mediate the analgesic effects of systemically administered SBFI26 and in FABP5/7 KO mice.15,26 Similarly, peripheral FAAH inhibition likewise produces CB1 and PPARα-mediated analgesic effects.21 This suggests that FABP5 may regulate the availability of endocannabinoids/NAEs for hydrolysis by FAAH, with FABP inhibition reducing endocannabinoid metabolism. Indeed, we observed robust co-expression of FABP5 and FAAH in lumbar DRGs and FABP inhibition elevated the levels of AEA in DRG neurons (Figure 11).
Figure 11.

FABP5 regulates AEA levels in lumbar DRG. (a) Immunofluorescence demonstrates extensive co-localization between FABP5 and FAAH. (b) Levels of PEA, OEA, AEA, and 2-AG in isolated DRGs incubated for 2 h with vehicle or 20 µM SBFI26. (n = 5; *p < 0.05).
The finding that peripheral FABP inhibition reduces nociception is important from a drug design perspective as it may enable the development of peripherally restricted FABP inhibitors, which may possess improved safety profiles over brain-penetrant compounds. After systemic administration, only a small fraction of SBFI26 permeates into the brain,15 possibly suggesting that its major site of action is in peripheral tissues. Indeed, both systemic and intraplantar SBFI26 administration produces analgesic effects mediated by CB1 and PPARα receptors as is seen with peripherally restricted FAAH inhibitors.21
Our findings uncovered distinct analgesic effects between peripherally and centrally administered FABP inhibitors. FABP5 possesses a diffuse expression pattern in the lumbar spinal cord and its expression was not concentrated in the dorsal horn, the major relay area of the spinal cord that transmits noxious stimuli from primary sensory neurons to higher brain centers.36 Consequently, intrathecal FABP inhibitor administration did not alter nociception. In contrast to the spinal cord, FABP5 is widely expressed in the brain including areas that regulate pain such as the PAG.53 Indeed, administration of SBFI26 via the i.c.v. route reduced thermal hyperalgesia, an effect that was mediated by PPARα and TRPV1, but not CB1 receptors. We also recently reported that FABP5/7 KO mice exhibit analgesic effects mediated by PPARα and TRPV1,26 indicating that the engagement of these receptor systems is not unique to pharmacological FABP inhibition. The inability of centrally administered FABP inhibitors to produce CB1-mediated effects was surprising in light of the elevated AEA levels observed after inhibitor administration (Figure 6). This may suggest that FABPs may not only regulate intracellular endocannabinoid transport and catabolism but may likewise control the delivery of endocannabinoids to CB1 receptors.
We sought to characterize the supraspinal site of action of FABP inhibitors and focused upon the PAG because it is a key brain region that mediates the analgesic effects of exogenous cannabinoids and endocannabinoids.31,43,48 To our surprise, despite elevating AEA levels within the PAG, pharmacological and genetic FABP5 inhibition did not produce analgesic effects. Similar results were observed upon FABP inhibition within the RVM. This suggests that other brain regions may gate analgesia produced by FABP inhibitors such as the prefrontal cortex, wherein endocannabinoids/NAEs can exert analgesic effects.51,54–56
Our results demonstrating peripherally mediated analgesic effects of FABP inhibitors suggest a possible multifactorial involvement of FABP5 in inflammatory pain. In addition to its expression in nociceptors, FABP5 is also widely expressed in cells of the innate immune system.57,58 During inflammation, leukocytes release lipid metabolites such as prostaglandin E2 that sensitize nociceptors and induce pain, while FABP inhibition reduces the inflammatory output of leukocytes.57–59 Therefore, it is likely that in addition to producing acute analgesic effects through modulation of endocannabinoid/NAE signaling in nociceptors, FABP inhibitors dampen the inflammatory output of leukocytes and reduce nociceptor sensitization, which is supported by the anti-inflammatory effects of FABP inhibitors.15,32
Conclusions
FABPs regulate the intracellular transport and subsequently catabolism of endocannabinoids and NAEs. Our findings indicate that FABP inhibitors exert their analgesic effects through peripheral and supraspinal FABP inhibition mediated via activation of overlapping but distinct receptor systems. The peripherally mediated analgesic effects of FABP inhibitors coupled with their previously reported anti-inflammatory effects and low brain penetration15,32 suggests that the design of future peripherally restricted FABP inhibitors should be readily achievable.
Acknowledgments
We would like to thank Robert Rieger at the Stony Brook Proteomics Center for help with mass spectrometry and Yong Lu for help with DRG isolation.
Authors’ contributions
MK conceived and designed the research. XP, KS, MPK, JL, DB, MWE, GC, MJR, MP, and MK conducted the research. XP, KS, MPK, MWE, RCR, HL, DGD, IO, MJR, MP, and MP analyzed data. ST, YHGT, and IO provided compounds used in the manuscript. XP, KS, MPK, MJR, MP, and MK wrote the paper. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Stony Brook University has previously filed a patent application covering SBFI26 and its analogs.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by NIH Grants DA035949 and DA035923.
References
- 1.Patel KV, Guralnik JM, Dansie EJ, et al. Prevalence and impact of pain among older adults in the United States: findings from the 2011 National Health and Aging Trends Study. Pain 2013; 154: 2649–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johannes CB, Le TK, Zhou X, et al. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J Pain 2010; 11: 1230–1239. [DOI] [PubMed] [Google Scholar]
- 3.Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain 2012; 13: 715–724. [DOI] [PubMed] [Google Scholar]
- 4.Ong CK, Lirk P, Tan CH, et al. An evidence-based update on nonsteroidal anti-inflammatory drugs. Clin Med Res 2007; 5: 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ofman JJ, MacLean CH, Straus WL, et al. A metaanalysis of severe upper gastrointestinal complications of nonsteroidal antiinflammatory drugs. J Rheumatol 2002; 29: 804–812. [PubMed] [Google Scholar]
- 6.Fredheim OM, Mahic M, Skurtveit S, et al. Chronic pain and use of opioids: a population-based pharmacoepidemiological study from the Norwegian prescription database and the Nord-Trondelag health study. Pain 2014; 155: 1213–1221. [DOI] [PubMed] [Google Scholar]
- 7.Campbell G, Nielsen S, Larance B, et al. Pharmaceutical opioid use and dependence among people living with chronic pain: associations observed within the pain and opioids in treatment (POINT) cohort. Pain Med 2015; 16: 1745–1758. [DOI] [PubMed] [Google Scholar]
- 8.Vowles KE, McEntee ML, Julnes PS, et al. Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 2015; 156: 569–576. [DOI] [PubMed] [Google Scholar]
- 9.Palmer RE, Carrell DS, Cronkite D, et al. The prevalence of problem opioid use in patients receiving chronic opioid therapy: computer-assisted review of electronic health record clinical notes. Pain 2015; 156: 1208–1214. [DOI] [PubMed] [Google Scholar]
- 10.De Leon M, Welcher AA, Nahin RH, et al. Fatty acid binding protein is induced in neurons of the dorsal root ganglia after peripheral nerve injury. J Neurosci Res 1996; 44: 283–292. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto T, Yamamoto A, Watanabe M, et al. Classification of FABP isoforms and tissues based on quantitative evaluation of transcript levels of these isoforms in various rat tissues. Biotechnol Lett 2009; 31: 1695–1701. [DOI] [PubMed] [Google Scholar]
- 12.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov 2008; 7: 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaczocha M, Glaser ST, Deutsch DG. Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci U S A 2009; 106: 6375–6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaczocha M, Vivieca S, Sun J, et al. Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J Biol Chem 2012; 287: 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaczocha M, Rebecchi MJ, Ralph BP, et al. Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS One 2014; 9: e94200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanson B, Wang T, Sun J, et al. Crystallographic study of FABP5 as an intracellular endocannabinoid transporter. Acta Crystallogr D Biol Crystallogr 2014; 70: 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu S, Levi L, Casadesus G, et al. Fatty acid-binding protein 5 (FABP5) regulates cognitive function both by decreasing anandamide levels and by activating the nuclear receptor peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) in the brain. J Biol Chem 2014; 289: 12748–12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lichtman AH, Shelton CC, Advani T, et al. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain 2004; 109: 319–327. [DOI] [PubMed] [Google Scholar]
- 19.Schlosburg JE, Kinsey SG, Lichtman AH. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J 2009; 11: 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal N, Pacher P, Tegeder I, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci 2007; 10: 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clapper JR, Moreno-Sanz G, Russo R, et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci 2010; 13: 1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godlewski G, Alapafuja SO, Batkai S, et al. Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem Biol 2010; 17: 1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thanos PK, Clavin BH, Hamilton J, et al. Examination of the addictive and behavioral properties of fatty acid-binding protein inhibitor SBFI26. Front Psychiatry 2016; 7: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo Verme J, Fu J, Astarita G, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol 2005; 67: 15–19. [DOI] [PubMed] [Google Scholar]
- 25.D’Agostino G, La Rana G, Russo R, et al. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther 2007; 322: 1137–1143. [DOI] [PubMed] [Google Scholar]
- 26.Kaczocha M, Glaser ST, Maher T, et al. Fatty acid binding protein deletion suppresses inflammatory pain through endocannabinoid/N-acylethanolamine-dependent mechanisms. Mol Pain 2015; 11: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elmes MW, Kaczocha M, Berger WT, et al. Fatty acid-binding proteins (FABPs) are intracellular carriers for delta9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J Biol Chem 2015; 290: 8711–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owada Y, Yoshimoto T, Kondo H. Spatio-temporally differential expression of genes for three members of fatty acid binding proteins in developing and mature rat brains. J Chem Neuroanat 1996; 12: 113–122. [DOI] [PubMed] [Google Scholar]
- 29.Caterina MJ, Leffler A, Malmberg AB, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000; 288: 306–313. [DOI] [PubMed] [Google Scholar]
- 30.Davis JB, Gray J, Gunthorpe MJ, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 2000; 405: 183–187. [DOI] [PubMed] [Google Scholar]
- 31.Starowicz K, Maione S, Cristino L, et al. Tonic endovanilloid facilitation of glutamate release in brainstem descending antinociceptive pathways. J Neurosci 2007; 27: 13739–13749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger WT, Ralph BP, Kaczocha M, et al. Targeting fatty acid binding protein (FABP) anandamide transporters – a novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS One 2012; 7: e50968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura M, Chi YM, Yan WM, et al. Strong antinociceptive effect of incarvillateine, a novel monoterpene alkaloid from Incarvillea sinensis. J Nat Prod 1999; 62: 1293–1294. [DOI] [PubMed] [Google Scholar]
- 34.Njoo C, Heinl C, Kuner R. In vivo SiRNA transfection and gene knockdown in spinal cord via rapid noninvasive lumbar intrathecal injections in mice. J Vis Exp 2014; 22: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paredes RM, Etzler JC, Watts LT, et al. Chemical calcium indicators. Methods 2008; 46: 143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 2010; 11: 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature 2001; 413: 203–210. [DOI] [PubMed] [Google Scholar]
- 38.Hirsch S, Corradini L, Just S, et al. The CGRP receptor antagonist BIBN4096BS peripherally alleviates inflammatory pain in rats. Pain 2013; 154: 700–707. [DOI] [PubMed] [Google Scholar]
- 39.Nieto FR, Clark AK, Grist J, et al. Calcitonin gene-related peptide expressing sensory neurons and spinal microglial reactivity contribute to pain states in collagen-induced arthritis. Arthritis Rheumatol 2015; 67: 1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abrahamsen B, Zhao J, Asante CO, et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 2008; 321: 702–705. [DOI] [PubMed] [Google Scholar]
- 41.McCoy ES, Taylor-Blake B, Street SE, et al. Peptidergic CGRPalpha primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron 2013; 78: 138–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mendell LM. Computational functions of neurons and circuits signaling injury: relationship to pain behavior. Proc Natl Acad Sci U S A 2011; 108: 15596–15601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lichtman AH, Cook SA, Martin BR. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J Pharmacol Exp Ther 1996; 276: 585–593. [PubMed] [Google Scholar]
- 44.Okine BN, Norris LM, Woodhams S, et al. Lack of effect of chronic pre-treatment with the FAAH inhibitor URB597 on inflammatory pain behaviour: evidence for plastic changes in the endocannabinoid system. Br J Pharmacol 2012; 167: 627–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Booker L, Kinsey SG, Abdullah RA, et al. The fatty acid amide hydrolase (FAAH) inhibitor PF-3845 acts in the nervous system to reverse LPS-induced tactile allodynia in mice. Br J Pharmacol 2012; 165: 2485–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mallet C, Barriere DA, Ermund A, et al. TRPV1 in brain is involved in acetaminophen-induced antinociception. PLoS One 2010; 5: e12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piomelli D, Sasso O. Peripheral gating of pain signals by endogenous lipid mediators. Nat Neurosci 2014; 17: 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maione S, Bisogno T, de Novellis V, et al. Elevation of endocannabinoid levels in the ventrolateral periaqueductal grey through inhibition of fatty acid amide hydrolase affects descending nociceptive pathways via both cannabinoid receptor type 1 and transient receptor potential vanilloid type-1 receptors. J Pharmacol Exp Ther 2006; 316: 969–982. [DOI] [PubMed] [Google Scholar]
- 49.Mascarenhas DC, Gomes KS, Nunes-de-Souza RL. Role of TRPV1 channels of the dorsal periaqueductal gray in the modulation of nociception and open elevated plus maze-induced antinociception in mice. Behav Brain Res 2015; 292: 547–554. [DOI] [PubMed] [Google Scholar]
- 50.Palazzo E, de Novellis V, Marabese I, et al. Interaction between vanilloid and glutamate receptors in the central modulation of nociception. Eur J Pharmacol 2002; 439: 69–75. [DOI] [PubMed] [Google Scholar]
- 51.Rea K, Olango WM, Okine BN, et al. Impaired endocannabinoid signalling in the rostral ventromedial medulla underpins genotype-dependent hyper-responsivity to noxious stimuli. Pain 2014; 155: 69–79. [DOI] [PubMed] [Google Scholar]
- 52.Glaser ST, Kaczocha M, Deutsch DG. Anandamide transport: a critical review. Life Sci 2005; 77: 1584–1604. [DOI] [PubMed] [Google Scholar]
- 53.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012; 489: 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okine BN, Rea K, Olango WM, et al. A role for PPARalpha in the medial prefrontal cortex in formalin-evoked nociceptive responding in rats. Br J Pharmacol 2014; 171: 1462–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Novellis V, Mariani L, Palazzo E, et al. Periaqueductal grey CB1 cannabinoid and metabotropic glutamate subtype 5 receptors modulate changes in rostral ventromedial medulla neuronal activities induced by subcutaneous formalin in the rat. Neuroscience 2005; 134: 269–281. [DOI] [PubMed] [Google Scholar]
- 56.de Novellis V, Luongo L, Guida F, et al. Effects of intra-ventrolateral periaqueductal grey palmitoylethanolamide on thermoceptive threshold and rostral ventromedial medulla cell activity. Eur J Pharmacol 2012; 676: 41–50. [DOI] [PubMed] [Google Scholar]
- 57.Reynolds JM, Liu Q, Brittingham KC, et al. Deficiency of fatty acid-binding proteins in mice confers protection from development of experimental autoimmune encephalomyelitis. J Immunol 2007; 179: 313–321. [DOI] [PubMed] [Google Scholar]
- 58.Furuhashi M, Fucho R, Gorgun CZ, et al. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest 2008; 118: 2640–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin CR, Amaya F, Barrett L, et al. Prostaglandin E2 receptor EP4 contributes to inflammatory pain hypersensitivity. J Pharmacol Exp Ther 2006; 319: 1096–1103. [DOI] [PubMed] [Google Scholar]