

Abstract
Background
L-acetylcarnitine, a drug marketed for the treatment of chronic pain, causes analgesia by epigenetically up-regulating type-2 metabotropic glutamate (mGlu2) receptors in the spinal cord. Because the epigenetic mechanisms are typically long-lasting, we hypothesized that analgesia could outlast the duration of L-acetylcarnitine treatment in models of inflammatory and neuropathic pain.
Results
A seven-day treatment with L-acetylcarnitine (100 mg/kg, once a day, i.p.) produced an antiallodynic effect in the complete Freund adjuvant mouse model of chronic inflammatory pain. L-Acetylcarnitine-induced analgesia persisted for at least 14 days after drug withdrawal. In contrast, the analgesic effect of pregabalin, amitryptiline, ceftriaxone, and N-acetylcysteine disappeared seven days after drug withdrawal. L-acetylcarnitine treatment enhanced mGlu2/3 receptor protein levels in the dorsal region of the spinal cord. This effect also persisted for two weeks after drug withdrawal and was associated with increased levels of acetylated histone H3 bound to the Grm2 gene promoter in the dorsal root ganglia. A long-lasting analgesic effect of L-acetylcarnitine was also observed in mice subjected to chronic constriction injury of the sciatic nerve. In these animals, a 14-day treatment with pregabalin, amitryptiline, tramadol, or L-acetylcarnitine produced a significant antiallodynic effect, with pregabalin displaying the greatest efficacy. In mice treated with pregabalin, tramadol or L-acetylcarnitine the analgesic effect was still visible 15 days after the end of drug treatment. However, only in mice treated with L-acetylcarnitine analgesia persisted 37 days after drug withdrawal. This effect was associated with an increase in mGlu2/3 receptor protein levels in the dorsal horns of the spinal cord.
Conclusions
Our findings suggest that L-acetylcarnitine has the unique property to cause a long-lasting analgesic effect that might reduce relapses in patients suffering from chronic pain.
Keywords: Pain, L-acetylcarnitine, metabotropic glutamate receptors, long-lasting analgesia
Introduction
A large body of evidence indicates that L-acetylcarnitine (LAC) is clinically effective in the treatment of neuropathic pain, fibromyalgia, and other types of chronic pain, with an excellent profile of safety and tolerability.1–10 LAC treatment may also cause antidepressant-like effects that may be advantageous in the management of patients suffering from chronic pain.11–14 To our knowledge, LAC is the only drug that causes analgesia via an epigenetic mechanism mediated by acetylation of p65/RelA, a member of the NFκB family of transcription factors. Acetylation of p65/RelA leads to an enhanced expression of type-2 metabotropic glutamate (mGlu2) receptors in the dorsal root ganglia and dorsal horns of the spinal cord with an ensuing reduction of glutamate release from primary afferent sensory fibres.15 In rats subjected to chronic constriction injury (CCI) of the sciatic nerve, repeated administrations of LAC reduced mechanical allodynia after five to seven days, and a single injection of an mGlu2/3 receptor antagonist at the end of LAC treatment was sufficient to abrogate LAC-induced analgesia.16 Acetylation of p65/RelA and induction of mGlu2 receptors by LAC could also be demonstrated in cultured dorsal root ganglia neurons, and the induction of mGlu2 receptors was abrogated by pharmacological inhibition of NFκB.17 Interestingly, drugs that inhibit histone deacetylases (HDACs) mimicked the analgesic action of LAC and were also able to enhance p65/RelA acetylation and mGlu2 receptor expression in the dorsal horns of the spinal cord.18 That epigenetic regulation of mGlu2 expression is critically involved in the control of pain is also demonstrated by the evidence that pharmacological inhibition of histone acetyl transferase (HAT) causes a marked down-regulation of mGlu2 receptors in the spinal cord and a hypoacetylation of histones H3 and H4 in the dorsal root ganglia, thereby preventing the analgesic activity of the mGlu2/3 receptor agonist, LY379268.19 LAC treatment was shown to enhance the amount of acetylated histone H3 acetylation bound to the promoter of the mGlu2 receptor-encoding gene (Grm2 gene) in the prefrontal cortex and hippocampus of spontaneously depressed rats.12 Whether LAC enhances histone acetylation in the spinal cord of mice with chronic pain is unknown.
On the basis of these findings, we predicted that LAC treatment could induce long-lasting changes in the expression of mGlu2 receptors in the spinal cord resulting into a persisting analgesic effect. We now demonstrate that LAC-induced analgesia persists for at least two weeks after drug withdrawal in the complete Freund adjuvant (CFA) mouse model of inflammatory pain and for more than one month in the CCI mouse model of neuropathic pain. In addition, we offer the first evidence that LAC treatment enhances the amount of acetylated histone H3 bound to the Grm2 promoter in the dorsal root ganglia.
Materials and methods
Drugs
LAC was a generous gift from Sigma Tau Laboratories (Pomezia, Italy); N-acetylcysteine (NAC), amitriptyline and ceftriaxone were purchased from Sigma Aldrich (St. Louis, MO); pregabalin was purchased from Tocris Cookson (Avonmouth, Bristol, UK). All these drugs were dissolved into saline. We used a clinical injectable formulation of tramadol HCl (Contramal, 50 mg/ml), which was also dissolved into saline.
Animals
We used adult male C57BL/6 J mice (20–25 g, b.w.) purchased from Charles River (Calco, Italy). All mice were housed five per cage, under a standard 12/12 h light/dark cycle with food and water ad libitum for at least two weeks prior to the experiments. All experiments were carried out according to the European (86/609/EEC) and Italian (D: Lgs. 116/92) guidelines of animal care. The experimental protocol was approved by the Italian Ministry of Health. All efforts were made to minimize animal suffering and the number of animals.
Induction of chronic inflammatory pain and drug treatment design
Chronic inflammatory pain was induced by intraplantar (i.pl.) injections of 20 µl of CFA (F5881 Sigma-Aldrich; 1 mg/ml) in the right hind paw. Control mice received an i.pl. injection of saline. In a first experiment, six groups of six CFA-injected mice were treated intraperitoneally (i.p.) as follows: two groups of mice received a single injection of saline or LAC (100 mg/kg) and pain thresholds were assessed after 1 h; the four remaining groups of mice were treated daily with saline or LAC for either three or seven days, and pain thresholds were assessed 1 h after the last injection. Mice of the first two groups were killed 24 h after a single injection of saline or LAC for the analysis of mGlu2/3 receptor protein levels in the spinal cord. Mice of all the other groups were killed immediately after the assessment of pain thresholds. In a second experiment, four groups of mice were treated as follows: (1) i.pl. injection of saline followed, 1 h later, by i.p. injection of saline (once a day for seven days); (2) i.pl. injection of saline followed by i.p. injection of LAC (100 mg/kg, once a day for seven days); (3) i.pl. injection of CFA followed by i.p. injection of saline (once a day for seven days); and (4) i.pl. injection of CFA followed by i.p. injection of LAC (once a day for seven days). Mechanical pain thresholds were assessed under basal conditions (i.e. before i.pl. injection of CFA or saline) after seven days (i.e. at the end of systemic treatment with LAC or saline), and seven days after drug withdrawal (corresponding to 14 days after i.pl. injection of CFA or saline). Parallel groups of mice injected i.pl. with CFA and treated with saline or LAC for seven days were killed at the end of saline or LAC treatment or seven days later for measurements of mGlu2/3 receptor protein levels in the spinal cord. In a third experiment, two groups of mice were injected i.pl. with CFA and treated for seven days with either saline or LAC. One week after drug withdrawal (i.e. 14 days after CFA injection), mice were killed 1 h after measurements of pain thresholds, and the dorsal root ganglia were used for chromatin immunoprecipitation analysis. In a third experiment, six groups of mice were injected i.pl. with CFA and treated i.p. once a day for seven days with saline, LAC (100 mg/kg), pregabalin (30 mg/kg), amitryptiline (10 mg/kg), ceftriaxone (200 mg/kg), or NAC (100 mg/kg). Pain thresholds were assessed prior to CFA injection (basal), 1 h after the end of drug treatment, and then at seven and 14 days after drug withdrawal (corresponding to 14 and 21 days after CFA injection). One hour after the last injection, three to four mice selected from the groups treated with i.p. with saline, LAC, or pregabalin were killed for measurements of mGlu2/3 receptor protein in the spinal cord.
Induction of neuropathic pain and drug treatment design
Neuropathic pain was induced by CCI of the sciatic nerve using the method described by Bennet and Xie20 modified as follows: mice were anesthetized with isoflurane (5% for induction and 2% for maintenance), the biceps femoris, and gluteus superficialis were separated by blunt dissection, and the left sciatic nerve was exposed for ligation. One ligature was tied loosely around the nerve, until it elicited a brief twitch in the respective hind limb, taking care to preserve epineural circulation. The incision was cleaned and the skin was closed with two to three ligatures of 5-0 dermalon. Mice were then placed on a warmed surface and, following recovery, they were returned to their home cages and checked routinely for 72 h. Five groups of mice (n = 7–8 per group) were treated i.p. daily for 14 days with saline, LAC (100 mg/kg), pregabalin (30 mg/kg), amitryptiline (10 mg/kg), or tramadol (100 mg/kg). Mechanical pain thresholds were measured prior to CCI, and at the end of drug treatment, and then at 7 and 37 days after drug withdrawal. mGlu2/3 Receptor protein levels were measured exclusively in CCI mice treated with saline or LAC.
Measurements of mechanical allodynia
Mechanical allodynia was quantified by measuring the hind paw withdrawal response to von Frey filament stimulation. In brief, mice were placed in a Plexiglas box (20 cm high, 9 cm diameter) with a wire grid bottom through which the von Frey filaments (North Coast Medical, Inc., San Jose, CA, USA; bending force range from 0.008 to 3.5 g) were applied by using a modified version of the up-down paradigm, as reported by Chaplan et al.21 The filaments were applied five times each, in order of increasing forces, and pressed perpendicularly to the plantar surface of the hind paw until they bent. The first filament that evoked at least three responses was assigned as the pain threshold in grams. For the assessment of mechanical sensitivity, mice were placed in individual cages with elevated mesh floor 1 h before all behavioral testing.
Western blot analysis
Tissues (dorsal regions of the lumbar segments of the spinal cord ipsilateral to i.pl. injection of CFA or saline) were homogenized on ice with RIPA buffer containing protease inhibitors cocktail tablet (Santa Cruz Biotechnology, Inc., Temecula, CA, USA). Homogenates were centrifuged at 14,000 rpmin for 10 min and an aliquot was used for protein determinations. Equal amounts of proteins (20 µg) from supernatants were separated by 8% SDS polyacrilamide gel and transferred on Immuno PVDF membranes (Bio-Rad, Milan, Italy) for 7 min using Trans Blot Turbo System (Bio-Rad, Milan, Italy) for the detection of mGlu2/3 receptor. Filters were blocked overnight in blocking buffer (TBS, 0.05% Tween-20 and 5% non-fat milk) at 4℃. Membrane were incubated for 1 h with rabbit polyclonal anti-mGlu2/3 receptors (1:500, Sigma-Aldrich), and secondary peroxidase-coupled anti-rabbit antibodies (1:7000 Calbiochem, Milano, Italy). The blots were re-probed with anti-β-actin monoclonal antibody (1:50,000 Sigma-Aldrich). Immunostaining was revealed by enhanced chemiluminescence luminosity (Amersham Pharmacia Biotech, Arlington Height, IL).
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation assay was performed by standard procedures. Briefly, dorsal root ganglia were cross-linked using 1% formaldehyde at room temperature for 10 min. The cross-linking reaction was stopped by adding glycine to a final concentration of 0.125 M. Tissue was then washed three times with ice-cold PBS supplemented with protease inhibitors (Sigma-Aldrich), homogenized in lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP40, protease inhibitors), and nuclei lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH 8.0, protease inhibitors), and then sonicated on ice using a Covaris S220 ultrasonicator. Following sonication, chromatin fragments of 0.2–0.6 kb in length were obtained. Ten percent of the sonicated lysate was saved and successively used to quantify the total amount of DNA present in different samples before immunoprecipitation (inputs). The chromatin solution was precleared with salmon sperm DNA/protein A-agarose 50% gel slurry for 1 h at 4℃ and immunoprecipitated overnight at 4℃ with antibodies against pan-acetylated-H3 histone (6 µg/µl, Upstate Biotechnology, Billerica, MA, USA) or the respective isotype matched control Ig. After precipitation, the chromatin-antibody complexes were collected using Protein A Sepharose beads and washed. Samples were then eluted with 1% SDS, 100 mM NaHCO3 at room temperature for 15 min, reverse cross-linked with NaCl 100 mM at 65℃ overnight, and treated with proteinase-K. Levels of histone acetylation at the Grm2 gene promoter were expressed as percentage of the input DNA that was immunoprecipitated by the anti-acetyl-histone H3 antibody using the following equation: % (acetDNA-IP/total input) = 2 × [(Ct(10%input) −3.32) −Ct (acetDNA−IP)] ×100%.
Statistical analysis
Statistical analysis was performed by one-way ANOVA or two-way ANOVA for repeated measures followed by Fisher’s LSD or, alternatively, by Student’s t-test, as indicated.
Results
LAC produced long-lasting analgesia and a long-lasting increase in mGlu2/3 receptor protein levels in the CFA model of chronic inflammatory pain
LAC treatment is known to produce analgesia in the CCI model of neuropathic pain after at least five days of daily injections.16 First, we examined whether LAC was also able to produce analgesia in the CFA model with a similar temporal profile. i.pl. CFA injection induced a large reduction in pain thresholds, which persisted for at least two weeks. LAC failed to induce changes in mechanical pain thresholds after a single administration or three days of injections, but produced substantial analgesia after seven days of daily injections (Figure 1(a)). To examine whether the effect of LAC outlasted the end of treatment, we performed a second experiment in which LAC or saline were administered daily for seven days. Pain thresholds were assessed at the end of the treatment and, again, seven days later. LAC treatment produced substantial analgesia in CFA-injected mice at both time points (Figure 1(b)). LAC treatment did not significantly affect mechanical thresholds in control mice that received i.pl. injection of saline instead of CFA (Figure 1(b)). Moving from the evidence that induction of mGlu2 receptors mediates the analgesic action of LAC,16 we measured mGlu2/3 receptor protein levels in the dorsal regions of the lumbar spinal cord ipsilateral to the site of CFA injection. Western blot analysis showed two bands at 100–110 KDa, of which the upper band corresponds to mGlu2/3 receptor monomers, and an additional band at >200 kDa corresponding to the active, dimeric form of mGlu2/3 receptor. LAC had no effect on mGlu2/3 receptor protein levels after a single administration or three days of treatment (Figure 2(a)). In contrast, seven days of treatment with LAC enhanced mGlu2/3 receptor protein levels in the dorsal region of the lumbar spinal cord. This effect persisted after seven days of drug withdrawal (data of the two experiments are similar, and only data of the second experiment are shown in Figure 2(b)).
Figure 1.

Effect of LAC on mechanical pain thresholds in the CFA mouse model of chronic inflammatory pain. (a) Six groups of six CFA-injected mice were treated intraperitoneally (i.p.) as follows: two groups of mice received a single injection of saline or LAC (100 mg/kg) and pain thresholds were assessed after 1 h; the four remaining groups of mice were treated daily with saline or LAC for either three or seven days, and pain thresholds were assessed 1 h after the last injection. Values are means ± S.E.M. of six determinations per group. *p < 0.05 (two-way ANOVA + Fisher’s LSD; F(5,25) = 11.13). Mechanical pain thresholds in mice injected i.pl. with saline or CFA and then treated i.p. with either LAC (100 mg/kg, once a day for seven days) or saline are shown in (b). Thresholds were measured under basal conditions (i.e. prior to i.pl. injections of saline or CFA), at the end of the seven days of treatment with saline or LAC (1 h of withdrawal), and again after seven days of withdrawal. Values are means ± S.E.M. of five to seven determinations. p < 0.01 vs. the corresponding values obtained in mice injected i.pl. and i.p. with saline (*) or vs. the corresponding values obtained in mice injected i.pl. with CFA and i.p. with saline (#) (two-way ANOVA for repeated measures + Fisher’s LSD; treatment x time F(8,24) = 4.975).
CFA: complete Freund adjuvant; LAC: L-acetylcarnitine.
Figure 2.

Repeated injections of LAC induced an increase in mGlu2/3 receptor protein levels that outlasted the end of drug treatment. mGlu2/3 protein levels were measured in the dorsal regions of the lumbar spinal cord ipsilateral to CFA injection. Data in (a) and (b) were obtained by the same groups of mice as in Figure 1(a) and (b), respectively. Mice treated with a single injection of saline or LAC were killed after 24 h. Mice treated with saline or LAC for three or seven days were killed pain assessment 1 h following the last injection. Data at seven days from animals used in Figure 1(a) are not shown. Values are means + S.E.M. of four to five and 5–10 determinations in (a) and (b), respectively. *p < 0.05 vs. the corresponding values of mice treated i.p. with saline (one-way ANOVA + Fisher’s LSD; F(3,23) = 5.884).
Wdt: withdrawal; CFA: complete Freund adjuvant; mGlu2: type-2 metabotropic glutamate.
Increased levels of acetylated histone H3 bound to the Grm2 gene promoter in the dorsal root ganglia one week after LAC treatment in CFA mice
Induction of mGlu2 receptors by LAC in the dorsal root ganglia and spinal cord has been related to an increased acetylation of p65/RelA.17 However, acetylation of a transcription factor may not be sufficient to explain the long-lasting increase in mGlu2/3 receptor protein we have seen in mice treated with LAC. Thus, we examined whether LAC treatment was also able to enhance histone acetylation on the Grm2 gene promoter in the dorsal root ganglia as already observed in other brain regions.12 Two additional groups of mice injected with CFA were treated with saline or LAC for seven days, and killed seven days later. We could confirm in these mice that LAC induced an analgesic effect that outlasted the end of treatment (Figure 3(a)). ChIP analysis showed an increased acetylation of histone H3 bound to the Grm2 promoter in the dorsal root ganglia of LAC-treated mice after seven days of withdrawal, as compared to mice treated with saline (Figure 3(b)).
Figure 3.
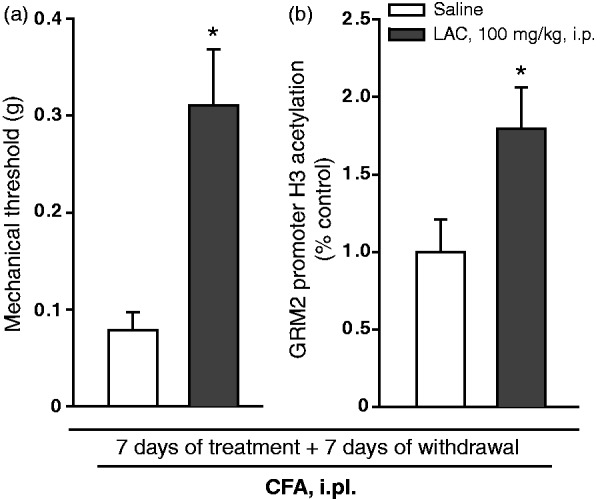
Increased levels of acetylated histone H3 bound to the Grm2 gene promoter in the dorsal root ganglia of mice with inflammatory pain treated with LAC. Mice were injected i.pl. with CFA and then treated with either saline or LAC for seven days. Pain thresholds and levels of acetylated histone H3 bound to the Grm2 gene promoter in the dorsal root ganglia were measured seven days after the end of drug treatment and shown in (a) and (b), respectively. Values are means ± S.E.M. of six to nine determinations for mechanical threshold and five to seven determinations for Grm2 gene promoter. *p < 0.05 (Student’s t-test) vs. the corresponding values obtained in mice treated i.p. with saline (t = −3.384 for mechanical threshold and t = −2.301 for Grm2 gene promoter).
CFA: complete Freund adjuvant.
LAC was the only tested analgesic drug that produced a long-lasting increase in pain thresholds in the CFA model of chronic inflammatory pain
Mice injected i.pl. with CFA were treated for seven days with either saline, LAC (100 mg/kg), pregabalin (30 mg/kg), amitriptyline (10 mg/kg), ceftriaxone (200 mg/kg), or N-acetylcysteine (NAC, 100 mg/kg). All drugs produced similar analgesic effects at the end of treatment, but only mice treated with LAC retained the analgesic at seven or 14 days after drug withdrawal (Figure 4(a)). Selected mice treated with saline, LAC, or pregabalin were also used for measurements of mGlu2/3 receptor protein levels in the dorsal region of the spinal cord after the last determination of pain thresholds. LAC-treated mice showed increased mGlu2/3 receptor protein levels after 14 days of drug withdrawal (Figure 4(b)), whereas a small reduction in mGlu2/3 receptor protein levels was observed in the group of mice treated with pregabalin (Figure 4(c)).
Figure 4.

LAC has the unique property to induce a long-lasting analgesia in the CFA model of chronic inflammatory pain. Mice were injected i.p. with CFA and treated i.p. once a day for seven days with either saline, LAC (100 mg/kg), pregabalin (30 mg/kg), amitryptiline (10 mg/kg), pregabalin (30 mg/kg), ceftriaxone (200 mg/kg) or NAC (100 mg/kg). Pain thresholds measured under basal conditions, at the end of treatments, and then 7 and 14 days after drug withdrawal are shown in (a), where values are means ± S.E.M. of 8 determinations. *p < 0.05 vs. the corresponding values obtained in mice treated i.p. with saline (one-way ANOVA for repeated measures + Fisher’s LSD). F(23,161) = 6.399. mGlu2/3 receptor protein levels in the dorsal region of the ipsilateral lumbar spinal cord of the same groups of mice treated with saline, LAC or pregabalin are shown in (b), where values are means ± S.E.M. of three to four determinations. *p < 0.05 vs. values obtained in mice treated with saline (Student’s t-test; t = 3.199 for pregabalin and t = −24.07 for LAC).
CFA: complete Freund adjuvant; LAC: L-acetylcarnitine; mGlu2: type-2 metabotropic glutamate.
Long-lasting analgesic effect of LAC in the CCI mouse model of neuropathic pain
Mice subjected to CCI of the sciatic nerve were treated daily for 14 days, i.p., with either saline, LAC (100 mg/kg), pregabalin (30 mg/kg), amitriptyline (10 mg/kg), or tramadol (100 mg/kg), and mechanical pain thresholds were measured 1 h after the end of treatments and at 7 and 37 days after drug withdrawal. CCI mice treated with saline showed the expected reduction in mechanical pain threshold as compared to sham-operated mice. At least under our conditions, mechanical allodynia remained stable up to 51 days after CCI in saline-treated mice (Figure 5(a)). At the end of systemic treatments (i.e. 14 days after CCI), a significant increase in pain thresholds was seen in all groups of mice treated with drugs, with pregabalin displaying the greatest analgesic effect (Figure 5(a)). Seven days after the end of treatments, analgesia was still observed in mice treated with pregabalin, tramadol, or LAC, but not in mice treated with amitryptiline (Figure 5(a)). In contrast, only mice treated with LAC still showed a significant increase in pain thresholds 37 days after drug withdrawal (Figure 5(a)). An increase in mGlu2/3 receptor protein levels was observed in the ipsilateral dorsal horns of the spinal cord of the group of mice treated with LAC after 37 days of withdrawal as compared to mice treated with saline (Figure 5(b)).
Figure 5.

Comparative effects of LAC and other analgesic drugs on pain thresholds in the CCI model of neuropathic pain. Mice were subjected to CCI of the sciatic nerve and then treated i.p. once a day for 14 days with either saline, LAC (100 mg/kg), pregabalin (30 mg/kg), tramadol (100 mg/kg), or amitryptiline (10 mg/kg). Pain thresholds were measured under basal conditions, at the end of drug treatments, and then after 7 or 37 days. Data are shown in (a). Basal pain thresholds are not shown and were similar in all experimental groups. Values are means ± S.E.M. of seven to eight determinations. *p < 0.05 (one-way ANOVA for repeated measures + Fisher’s LSD; F(14,89) = 8.984). mGlu2/3 protein levels in the ipsilateral dorsal region of the lumbar spinal cord of mice treated with saline or LAC at the end of experiment (i.e. after 37 days of withdrawal) are shown in (b). Values are means ± S.E.M. of seven determinations. Two samples from sham-operated rats (S.O., 51 days after surgery) are also shown in one of the blots. *p < 0.05 (Student’s t-test; t = −3.89) if compared to CCI mice treated with saline.
CCI: chronic constriction injury; LAC: L-acetylcarnitine; mGlu2: type-2 metabotropic glutamate.
Discussion
A number of recent reviews highlight the role of epigenetic mechanisms in the pathophysiology and treatment of chronic pain.22–26 Changes in histone acetylation have been consistently found in the pain neuraxis of mice developing chronic inflammatory or neuropathic pain. CFA-induced inflammatory pain is associated with an increased expression of class-II HDACs in the dorsal horns of the spinal cord.27 Spinal nerve ligation causes a reduced acetylation of histone H3 bound to the promoter of the gene encoding for the GABA-synthesizing enzyme, glutamate decarboxylase-65, in the nucleus raphe magnus, resulting into a disinhibition of serotonergic neurons projecting to the spinal cord.28 In models of neuropathic pain, changes in acetylation mechanisms affect the expression of genes encoding for proteins that critically regulate pain thresholds, such as µ opiate receptors, Kv4.3 potassium channels, Nav1.8 voltage-sensitive sodium channels, and brain-derived neurotrophic factor.28–31 Drugs that enhance acetylation of histones or transcription factors, such as HDAC inhibitors, consistently cause analgesia in models of inflammatory and neuropathic pain, although histone acetyl-transferase inhibitors may also relieve pain.24 The acetylating agent, LAC, causes analgesia by enhancing the expression of mGlu2 receptors in the dorsal root ganglia and dorsal horns of the spinal cord.15–17 At least a component of the analgesic action of LAC is mediated by acetylation of NFκB p65/RelA, which enhances its transcriptional activity at the Grm2 gene promoter.17 This mechanism is shared by at least two HDAC inhibitors, which were shown to cause analgesia in the second phase of the formalin test by up-regulating mGlu2 receptors.18
LAC is a safe drug that is currently marketed for the treatment of chronic pain (see Introduction section and references section therein). However, LAC is not considered as a first-line drug in the treatment of inflammatory, mixed, or neuropathic pain32 in spite of its widespread use in the medical practice. Our data demonstrate that LAC has the unique property to produce an analgesic effect that outlasts by several days or weeks the end of treatment in models of chronic inflammatory and neuropathic pain. This strengthens the value of LAC as an analgesic drug and supports the hypothesis that epigenetic mechanisms are valuable candidate drug targets in the treatment of chronic pain. As active comparators, we used drugs that are known to cause analgesia by targeting mechanisms that regulate pain threshold in the spinal cord and other region of the pain neuraxis. Pregabalin binds to the α2δ subunit of voltage-sensitive Ca2+ channels, thereby restraining neurotransmitter release at the synapses between primary afferent fibres and spinal cord neurons.33 Pregabalin also prevents the increased transport of the α2δ subunit from dorsal root ganglia to the spinal cord associated with neuropathic pain,34 thus weakening the molecular machinery that allows Ca2+ entry in the presynaptic terminals of primary sensory fibres. Although pregabalin is a first-line drug in the treatment of neuropathic pain,32 it also shows efficacy in the CFA model of chronic inflammatory pain.35 Amiptryptiline is a tricyclic antidepressant that is also used as a first-line drug in the treatment of neuropathic pain32 and acts primarily by inhibiting noradrenaline and serotonin uptake from presynaptic terminals. However, amitryptiline exerts pleiotropic effects that widen its action to different types of pain, such as inhibition of TrpV1 channels36 and induction of endogenous opioid release.37 Tramadol is a µ opioid receptor agonist that can also inhibit serotonin and noradrenaline uptake and is widely used in the treatment of different types of pain including neuropathic pain.32 Ceftriaxone is a β-lactam antibiotic that induces the expression of the glial glutamate transporter, GLT-1, thereby causing analgesia in experimental animal models and humans.38–40 NAC is an activator of the cystine-glutamate antiporter that causes analgesia by enhancing the endogenous activation of mGlu2 receptors.41,42 We used all these drugs at a single dose, selected in conformity with previous reports.16,35,40–44 In the CFA model of chronic inflammatory pain, all drugs were equally effective in increasing mechanical pain thresholds, but only in mice treated with LAC the analgesic effect was still observed after one or two weeks of drug withdrawal. In the CCI model, pregabalin and amitryptiline displayed the greatest efficacy in causing analgesia, as expected by their first-line indication in the treatment of neuropathic pain.32 Interestingly, the analgesic effect of pregabalin and tramadol persisted after one week of drug withdrawal, whereas the effect of amitryptiline was not durable. Pregabalin-induced analgesia outlasted the end of treatment in mice with neuropathic pain but not in mice with chronic inflammatory pain. In mice with neuropathic pain, pregabalin may cause a prolonged analgesia by preventing the increased trafficking of the α2δ subunit from the dorsal root ganglia to the spinal cord.34 LAC was less efficacious than pregabalin in enhancing mechanical pain thresholds in CCI mice, but its effect persisted after five weeks of drug withdrawal, when the effect of all other drugs was no longer visible.
The long-lasting effects of LAC in the CFA and CCI models of chronic pain suggest that the drug causes persistent changes in the molecular machinery that controls pain threshold in the CNS. LAC is known to induce the expression of mGlu2 receptors by enhancing the acetylation of the transcription factor, p65/RelA.15–17 This mechanism has been clearly demonstrated in in vitro and in vivo models,11,12,17 but it is insufficient to explain the induction of mGlu2 receptors we have seen in the spinal cord of CFA and CCI mice after weeks of drug withdrawal. At least in CFA mice, we have demonstrated that levels of acetylated histone H3 bound to Grm2 promoter were elevated in LAC-treated animals after two weeks of withdrawal. This suggests that LAC-induced acetylation is not restricted to transcription factors, and that durable changes in chromatin structure caused by histone acetylation may underlie an analgesic effect that outlasts by several days or weeks the end of LAC treatment. It is likely that LAC enhanced histone acetylation acting as an acetyl donor at the Grm2 promoter. However, we cannot exclude that LAC acts indirectly to enhance H3 histone acetylation at the Grm2 promoter. For example, LAC might acetylate a transcription factor that represses HDAC expression, thereby enhancing histone acetylation.
An increased histone acetylation by LAC has already been found in the hippocampus and prefrontal cortex of spontaneously depressed Flinders Sensitive Line rats, where LAC was shown to induce a long-lasting antidepressant-like effect.12 It is therefore possible that acetylation of both p65/Rel and histones contributes to Grm2 induction in response to LAC, and that histone acetylation in particular accounts for the long-lasting increase in mGlu2 receptor expression and analgesia in models of chronic pain. A direct correlation between histone acetylation and mGlu2/3 receptor expression in the spinal cord has been demonstrated in a model of stress-induced irritable bowel disorder, in which HDAC inhibition attenuates visceral hypersensitivity by enhancing the levels of acetylated histone H3 bound to Grm2 and Grm3 promoters.45
mGlu2 and mGu3 receptors are expressed in all stations of the pain neuraxis, and their activation consistently produces analgesia in a variety of pain models.46–56 At least in the formalin model of inflammatory pain, mGlu2 rather than mGlu3 receptors appear to regulate pain threshold and mediate the analgesic activity of mGlu2/3 receptor agonists.57 Thus, our findings support the hypothesis15 that targeting the epigenetic mechanisms that regulate mGlu2 receptor expression may be a valuable strategy for an efficient and durable treatment of chronic pain.
Conclusions
We have shown that LAC, a drug marketed for the treatment of chronic pain, produces a long-lasting analgesia in mouse models of inflammatory and neuropathic pain. Analgesia outlasted by several days or weeks the end of treatment. This property was unique to LAC in mice with chronic inflammatory pain, and was also shared by pregabalin and tramadol in mice with neuropathic pain. However, at a late time point after drug withdrawal, only mice with neuropathic pain treated with LAC retained the analgesic effect. An increase in histone acetylation at Grm2 gene promoter may contribute to the long-lasting analgesic effect of LAC. These findings suggest that LAC may relieve pain for a long time causing a persistent up-regulation of mGlu2 receptors, which are key players in the regulation of pain threshold. These findings strengthen the value of mGlu receptors as drug targets for the treatment of pain.58–62 The peculiar mechanism of action of LAC is promising from a clinical standpoint considering that the benefit achieved with current analgesic drugs in patients suffering of neuropathic pain and other types of chronic pain is suboptimal and a large percentage of patients is resistant to medication.63 Our data encourage clinical studies with LAC in patients suffering from chronic inflammatory or neuropathic pain in which the relapse rate at different time intervals after drug withdrawal is included as a primary endpoint.
Author Contributions
SN and GM contributed equally to this article. SN, GM, MB, CZ, PS, MC, TI: performed experiments and analyzed data; RG, GB, VB, FN: designed experiments, supervised research and wrote the manuscript. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Italian Ministry of Health (project code: RF-2011-02352582).
References
- 1.Onofrj M, Fulgente T, Melchionda D, et al. L-acetylcarnitine as a new therapeutic approach for peripheral neuropathies with pain. Int J Clin Pharmacol Res 1995; 15: 9–15. [PubMed] [Google Scholar]
- 2.Sima AA, Calvani M, Mehra M, et al. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care 2005; 28: 89–94. [DOI] [PubMed] [Google Scholar]
- 3.Flatters SJ, Xiao WH, Bennett GJ. Acetyl-L-carnitine prevents and reduces paclitaxel-induced painful peripheral neuropathy. Neurosci Lett 2006; 397: 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evans JD, Jacobs TF, Evans EW. Role of acetyl-L-carnitine in the treatment of diabetic peripheral neuropathy. Ann Pharmacother 2008; 42: 1686–1691. [DOI] [PubMed] [Google Scholar]
- 5.Memeo A, Loiero M. Thioctic acid and acetyl-L-carnitine in the treatment of sciatic pain caused by a herniated disc: a randomized, double-blind, comparative study. Clin Drug Investig 2008; 28: 495–500. [DOI] [PubMed] [Google Scholar]
- 6.Xiao WH, Bennett GJ. Chemotherapy-evoked neuropathic pain: abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitine. Pain 2008; 135: 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janiri L, Martinotti G, Tonioni F, et al. Acetyl-L-carnitine in the management of pain during methadone withdrawal syndrome. Clin Neuropharmacol 2009; 32: 35–40. [DOI] [PubMed] [Google Scholar]
- 8.Valcour V, Yeh TM, Bartt R, Clifford D, et al. Acetyl-l-carnitine and nucleoside reverse transcriptase inhibitor-associated neuropathy in HIV infection. HIV Med 2009; 10: 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leombruni P, Miniotti M, Colonna F, et al. A randomised controlled trial comparing duloxetine and acetyl L-carnitine in fibromyalgic patients: preliminary data. Clin Exp Rheumatol 2015; 33: S82–S85. [PubMed] [Google Scholar]
- 10.Li S, Li Q, Li Y, et al. Acetyl-L-carnitine in the treatment of peripheral neuropathic pain: a systematic review and meta-analysis of randomized controlled trials. PLoS One 2015; 10: e0119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cuccurazzu B, Bortolotto V, Valente MM, et al. Upregulation of mGlu2 receptors via NF-κB p65 acetylation is involved in the Proneurogenic and antidepressant effects of acetyl-L-carnitine. Neuropsychopharmacology 2013; 38: 2220–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nasca C, Xenos D, Barone Y, et al. L-acetylcarnitine causes rapid antidepressant effects through the epigenetic induction of mGlu2 receptors. Proc Natl Acad Sci USA 2013; 110: 4804–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanardi R, Smeraldi E. A double-blind, randomised, controlled clinical trial of acetyl-L-carnitine vs. amisulpride in the treatment of dysthymia. Eur Neuropsychopharmacol 2006; 16: 281–287. [DOI] [PubMed] [Google Scholar]
- 14.Bersani G, Meco G, Denaro A, et al. L-Acetylcarnitine in dysthymic disorder in elderly patients: a double-blind, multicenter, controlled randomized study vs. fluoxetine. Eur Neuropsychopharmacol 2013; 23: 1219–1225. [DOI] [PubMed] [Google Scholar]
- 15.Chiechio S, Copani A, Zammataro M, et al. Transcriptional regulation of type-2 metabotropic glutamate receptors: an epigenetic path to novel treatments for chronic pain. Trends Pharmacol Sci 2010; 31: 153–160. [DOI] [PubMed] [Google Scholar]
- 16.Chiechio S, Caricasole A, Barletta E, et al. L-Acetylcarnitine induces analgesia by selectively up-regulating mGlu2 metabotropic glutamate receptors. Mol Pharmacol 2002; 61: 989–996. [DOI] [PubMed] [Google Scholar]
- 17.Chiechio S, Copani A, De Petris L, et al. Transcriptional regulation of metabotropic glutamate receptor 2/3 expression by the NF-kappaB pathway in primary dorsal root ganglia neurons: a possible mechanism for the analgesic effect of L-acetylcarnitine. Mol Pain 2006; 2: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiechio S, Zammataro M, Morales ME, et al. Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol Pharmacol 2009; 75: 1014–1020. [DOI] [PubMed] [Google Scholar]
- 19.Zammataro M, Sortino MA, Parenti C, et al. HDAC and HAT inhibitors differently affect analgesia mediated by group II metabotropic glutamate receptors. Mol Pain 2014; 10: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988; 33: 87–107. [DOI] [PubMed] [Google Scholar]
- 21.Chaplan SR, Bach FW, Pogrel JW, et al. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994; 53: 55–63. [DOI] [PubMed] [Google Scholar]
- 22.Kurita M, Moreno JL, Holloway T, et al. Repressive epigenetic changes at the mGlu2 promoter in frontal cortex of 5-HT2A knockout mice. Mol Pharmacol 2013; 83: 1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Descalzi G, Ikegami D, Ushijima T, et al. Epigenetic mechanisms of chronic pain. Trends Neurosci 2015; 38: 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Géranton SM, Tochiki KK. Could targeting epigenetic processes relieve chronic pain states? Curr Opin Support Palliat Care 2015; 9: 138–146. [DOI] [PubMed] [Google Scholar]
- 25.Géranton SM, Tochiki KK. Regulation of gene expression and pain states by epigenetic mechanisms. Prog Mol Biol Transl Sci 2015; 131: 147–183. [DOI] [PubMed] [Google Scholar]
- 26.Liang L, Lutz BM, Bekker A, et al. Epigenetic regulation of chronic pain. Epigenomics 2015; 7: 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai G, Wei D, Zou S, et al. Inhibition of class II histone deacetylases in the spinal cord attenuates inflammatory hyperalgesia. Mol Pain 2010; 6: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, Cai YQ, Zou F, et al. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med 2011; 17: 1448–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uchida H, Ma L, Ueda H. Epigenetic gene silencing underlies C-fiber dysfunctions in neuropathic pain. J Neurosci 2010; 30: 4806–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida H, Matsushita Y, Ueda H. Epigenetic regulation of BDNF expression in the primary sensory neurons after peripheral nerve injury: implications in the development of neuropathic pain. Neuroscience 2013; 240: 147–154. [DOI] [PubMed] [Google Scholar]
- 31.Uchida H, Sasaki K, Ma L, et al. Neuron-restrictive silencer factor causes epigenetic silencing of Kv4.3 gene after peripheral nerve injury. Neuroscience 2010; 166: 1–4. [DOI] [PubMed] [Google Scholar]
- 32.Attal N, Cruccu G, Baron R, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. European Federation of Neurological Societies. Eur J Neurol 2010; 17: 1113–e88. [DOI] [PubMed] [Google Scholar]
- 33.Dooley DJ, Taylor CP, Donevan S, et al. Ca2+ channel alpha2delta ligands: novel modulators of neurotransmission. Trends Pharmacol Sci 2007; 28: 75–82. [DOI] [PubMed] [Google Scholar]
- 34.Bauer CS, Nieto-Rostro M, Rahman W, et al. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2 delta ligand pregabalin. J Neurosci 2009; 29: 4076–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maciel IS, Silva RB, Morrone FB, et al. Synergistic effects of celecoxib and bupropion in a model of chronic inflammation-related depression in mice. PLoS One 2013; 8: e77227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oláh Z, Jósvay K, Pecze L, et al. Anti-calmodulins and tricyclic adjuvants in pain therapy block the TRPV1 channel. PLoS One 2007; 2: e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gray AM, Spencer PS, Sewell RD. The involvement of the opioidergic system in the antinociceptive mechanism of action of antidepressant compounds. Br J Pharmacol 1998; 124: 669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin Y, Tian G, Roman K, et al. Increased glial glutamate transporter EAAT2 expression reduces visceral nociceptive response in mice. Am J Physiol Gastrointest Liver Physiol 2009; 296: G129–G134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, He Y, Wang ZJ. The beta-lactam antibiotic, ceftriaxone, inhibits the development of opioid-induced hyperalgesia in mice. Neurosci Lett 2012; 509: 69–71. [DOI] [PubMed] [Google Scholar]
- 40.Macaluso A, Bernabucci M, Trabucco A, et al. Analgesic effect of a single preoperative dose of the antibiotic ceftriaxone in humans. J Pain 2013; 14: 604–612. [DOI] [PubMed] [Google Scholar]
- 41.Bernabucci M1, Notartomaso S, Zappulla C, et al. N-Acetyl-cysteine causes analgesia by reinforcing the endogenous activation of type-2 metabotropic glutamate receptors. Mol Pain 2012; 8: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Truini A, Piroso S, Pasquale E, et al. N-acetyl-cysteine, a drug that enhances the endogenous activation of group-II metabotropic glutamate receptors, inhibits nociceptive transmission in humans. Mol Pain 2015; 11: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jesse CR, Wilhelm EA, Nogueira CW. Depression-like behavior and mechanical allodynia are reduced by bis selenide treatment in mice with chronic constriction injury: a comparison with fluoxetine, amitriptyline, and bupropion. Psychopharmacology 2010; 212: 513–522. [DOI] [PubMed] [Google Scholar]
- 44.Mika J, Jurga AM, Starnowska J, et al. Effects of chronic doxepin and amitriptyline administration in naïve mice and in neuropathic pain mice model. Neuroscience 2015; 294: 38–50. [DOI] [PubMed] [Google Scholar]
- 45.Cao DY, Bai G, Ji Y, et al. Histone hyperacetylation modulates spinal type II metabotropic glutamate receptor alleviating stress-induced visceral hypersensitivity in female rats. Mol Pain 2016; 12: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neugebauer V, Chen PS, Willis WD. Groups II and III metabotropic glutamate receptors differentially modulate brief and prolonged nociception in primate STT cells. J Neurophysiol 2000; 84: 2998–3009. [DOI] [PubMed] [Google Scholar]
- 47.Popik P, Kozela E, Pilc A. Selective agonist of group II glutamate metabotropic receptors, LY354740, inhibits tolerance to analgesic effects of morphine in mice. Br J Pharmacol 2000; 130: 1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simmons RM, Webster AA, Kalra AB, et al. Group II mGluR receptor agonists are effective in persistent and neuropathic pain models in rats. Pharmacol Biochem Behav 2002; 73: 419–427. [DOI] [PubMed] [Google Scholar]
- 49.Yang D, Gereau RW., 4th Peripheral group II metabotropic glutamate receptors mediate endogenous anti-allodynia in inflammation. Pain 2003; 106: 411–417. [DOI] [PubMed] [Google Scholar]
- 50.Yang D, Gereau RW., 4th Peripheral group II metabotropic glutamate receptors (mGluR2/3) regulate prostaglandin E2-mediated sensitization of capsaicin responses and thermal nociception. J Neurosci 2002; 22: 6388–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jones CK, Eberle EL, Peters SC, et al. Analgesic effects of the selective group II (mGlu2/3) metabotropic glutamate receptor agonists LY379268 and LY389795 in persistent and inflammatory pain models after acute and repeated dosing. Neuropharmacology 2005; 49: 206–218. [DOI] [PubMed] [Google Scholar]
- 52.Du J, Zhou S, Carlton SM. Group II metabotropic glutamate receptor activation attenuates peripheral sensitization in inflammatory states. Neuroscience 2008; 154: 754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carlton SM, Du J, Zhou S. Group II metabotropic glutamate receptor activation on peripheral nociceptors modulates TRPV1 function. Brain Res 2009; 1248: 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou HY, Chen SR, Chen H, et al. Functional plasticity of group II metabotropic glutamate receptors in regulating spinal excitatory and inhibitory synaptic input in neuropathic pain. J Pharmacol Exp Ther 2011; 336: 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asseri KA, Puil E, Schwarz SK, et al. Group II metabotropic glutamate receptor antagonism prevents the antiallodynic effects of R-isovaline. Neuroscience 2015; 293: 15–156. [DOI] [PubMed] [Google Scholar]
- 56.Kiritoshi T, Neugebauer V. Group II mGluRs modulate baseline and arthritis pain-related synaptic transmission in the rat medial prefrontal cortex. Neuropharmacology 2015; 95: 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zammataro M, Chiechio S, Montana MC, et al. mGlu2 metabotropic glutamate receptors restrain inflammatory pain and mediate the analgesic activity of dual mGlu2/mGlu3 receptor agonists. Mol Pain 2011; 7: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neugebauer V. Peripheral metabotropic glutamate receptors: fight the pain where it hurts. Trends Neurosci 2001; 24: 550–552. [DOI] [PubMed] [Google Scholar]
- 59.Neugebauer V. Metabotropic glutamate receptors – important modulators of nociception and pain behavior. Pain 2002; 98: 1–8. [DOI] [PubMed] [Google Scholar]
- 60.Goudet C, Magnaghi V, Landry M, et al. Metabotropic receptors for glutamate and GABA in pain. Brain Res Rev 2009; 60: 43–56. [DOI] [PubMed] [Google Scholar]
- 61.Montana MC, Gereau RW. Metabotropic glutamate receptors as targets for analgesia: antagonism, activation, and allosteric modulation. Curr Pharm Biotechnol 2011; 12: 1681–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brumfield S, Korakas P, Silverman LS, et al. Synthesis and SAR development of novel mGluR1 antagonists for the treatment of chronic pain. Bioorg Med Chem Lett 2012; 22: 7223–7226. [DOI] [PubMed] [Google Scholar]
- 63.Eisenstein M. Neuropathy: a name for their pain. Nature 2016; 535: S10–S11. [DOI] [PubMed] [Google Scholar]