

Summary
De novo copy number variants (dnCNVs) arising at multiple loci in a personal genome have usually been considered to reflect cancer somatic genomic instabilities. We describe a multiple dnCNV (MdnCNV) phenomenon in which individuals with genomic disorders carry five to ten constitutional dnCNVs. These CNVs originate from independent formation incidences, are predominantly tandem duplications or complex gains, exhibit breakpoint junction features reminiscent of replicative repair, and show increased de novo point mutations flanking the rearrangement junctions. The active CNV mutation shower appears to be restricted to a transient peri-zygotic period. We propose that a defect in the CNV formation process is responsible for the ‘CNV-mutator state’, and that this state is dampened after early embryogenesis. The constitutional MdnCNV phenomenon resembles chromosomal instability in various cancers. Investigations of this phenomenon may provide unique access to understanding genomic disorders, structural variant mutagenesis, human evolution and cancer biology.
Introduction
Copy number variants (CNV) contribute to polymorphic variations, pathological conditions and evolution (Lupski, 2015). It is estimated that for a given locus, spontaneous de novo CNVs (dnCNVs) can occur at a rate that is hundreds or even thousands of fold higher than that of de novo single nucleotide variants (dnSNV) (Lupski, 2007; Turner et al., 2008). Depending on the different size thresholds being used, studies have estimated that large (> 100 Kb) human dnCNVs arise at a rate ranging from 0.003 to 0.03 per meiosis (Conrad et al., 2010; McRae et al., 2015).
Most of the de novo structural variants (dnSVs) observed as constitutional events in a human subject, i.e. occurring within all cells of the organism, can be traced to a singular mutational event. This single event can affect the copy number of one specific genomic region, such as a simple deletion as a result of a nonallelic homologous recombination; a single event can also interweave multiple genomic loci by a catastrophic mutational assault, such as the recently described “one-off” mutational phenomenon termed chromothripsis or chromoanasynthesis (Kloosterman et al., 2012; Liu et al., 2011; Stephens et al., 2011; Zhang et al., 2013). In a germ line setting, no matter what degree of genomic instability may be sculpted by a dnSV, the mutational process is believed to be a single event corresponding to a constitutional, non-evolving, and spontaneous occurrence.
When multiple independent dnCNVs coincide, spontaneity is no longer the culprit. In fact, such a phenomenon is well known in cancers. Multiple changes of chromosome structures and numbers, or chromosomal instability, contribute to a majority of the genomic instability observed in cancers (Negrini et al., 2010). It is only in recent years that the origins, patterns, and mechanisms of chromosome instability are beginning to be understood (Hanahan and Weinberg, 2011; Macheret and Halazonetis, 2015; Negrini et al., 2010). A similar phenomenon has been mimicked using in vitro cell line experiments, where replication stress triggered by an environmental stimulus has been shown to be responsible for the emergence of dnCNVs (Arlt et al., 2009; Arlt et al., 2012). Chromosomal instability in cancer patients is thus a process that involves multiple complex structural changes, somatic mosaicism, evolution and selection over time, and presumably a driver mutation that is responsible for the accumulation of the observed rearrangements.
Here, we report a novel type of constitutional chromosomal instability that differs from somatic cell mutagenesis exemplified by cancer associated genomic instability. This novel phenomenon is found in pediatric patients being evaluated for various developmental disorders by genomic studies. It is characterized by multiple dnCNVs (MdnCNV) arising at multiple genomic loci through independent molecular events. The excessive number of dnCNVs in each patient cannot be explained as simply the extreme tail of the same spontaneous mutation process observed in the general population. These CNVs present uniform and unique properties in that they are predominantly copy number gains spanning hundreds to thousands of kilobases and that the breakpoint junction features are suggestive of replicative mechanism for formation. dnSNVs are observed near the breakpoints, even though the burst of dnCNVs does not seem to escalate the emergence of dnSNVs observed genome wide. This MdnCNV phenomenon likely occurs in a transient period before or after fertilization, perhaps prior to the synthesis or elevated concentration of genome stability factors, and does not seem to persist over time. Together, these findings suggest that the MdnCNV phenomenon is a genome-wide rearrangement pattern that is distinct from the other spontaneous dnCNVs, and may represent a CNV mutator phenotype (Liu et al., 2012). Investigation into this phenomenon is a rare opportunity to understand fundamental processes driving dnSVs in genomic disorders and evolution and to help understand the natural history of genomic instability in cancers.
Results
Definition and identification of individuals with MdnCNV
In order to define the threshold for the number of dnCNVs that qualifies in the excessive range, we analyzed the genome-wide CNV data of the personal genomes from a large cohort of clinical samples submitted for chromosomal microarray analysis (CMA). This cohort consists of patients with a mixture of various clinically relevant neurodevelopmental issues that is not selected for any specific phenotype. This cohort is expected to have a higher dnCNV rate compared to the general population given selection for a clinical phenotype prompting personal genome analyses. The number of CNVs may be underestimated because the clinical arrays tend to focus interrogating oligonucleotides on disease associating regions with relatively lower coverage in non-disease associated regions of the human genome. Results from 1479 samples were randomly selected to evaluate the distribution of the number of dnCNVs per individual. When manually counting dnCNVs in each subject, we considered mutational events rather than affected CNV regions. For example, a complex rearrangement may result in two duplications (DUP) separated by a copy number neutral interval (NML). This complex genomic rearrangement (CGR), revealed by an array CGH observed duplication-normal-duplication pattern, designated DUP-NML-DUP (Carvalho and Lupski, 2016; Carvalho et al., 2013; Gu et al., 2015), is considered as one mutational event rather than two events. As expected, there are very few individuals with a large number of dnCNVs. There are 1437 individuals with 1 dnCNV, 39 with 2, and 1 with 3. The number of dnCNVs observed in each patient does not appear to correlate with parental ages (Figure S1). Interestingly, 2 of the 1479 individuals appear to be outliers. They are found to have four and seven dnCNVs. Based on the above observations, we empirically defined individuals with four or more independent events of greater than 100 kb as presenting with the MdnCNV phenomenon (Poisson probability of ~3x10−8 based on a background dnCNV rate of 0.03).
Among 60,000 individuals with genomic DNA samples submitted to clinical CMA testing, we identified five subjects fulfilling the MdnCNV criteria. These individuals do not share any common genetic background or phenotypic similarities. Their only apparent common feature is the MdnCNV molecular profile. Four of the five subjects have complete parental studies confirming the de novo occurrence of the CNVs reported; one subject (mCNV3) lacks parental studies, but had eight large CNVs that are not found in public databases. It is unlikely that all the eight CNVs are inherited. In addition, the genomic rearrangements observed in this subject exhibited similar rearrangement features as the other four samples, which will be discussed below. We therefore classified this subject as one of the “MdnCNV individuals”.
Mutational signatures of the MdnCNV phenomenon
To capture the full spectrum of dnCNVs, the five subjects with dnCNVs initially ascertained by clinical chromosomal microarray studies, and parental samples when available, were analyzed on a series of high-resolution genome-wide microarrays including the Agilent SurePrint G3 Unrestricted CGH 1x1M array, the Roche NimbleGen Human CGH 4.2M Whole Genome Tiling Array, and SNP genotyping arrays including the Illumina HumanOmni2.5–8 Array, the Illumina HumanOmni1-Quad Array, the Affymetrix Genome-Wide Human SNP Array 6.0, and the Affymetrix CytoScan HD Array (Table 1). All the candidate regions with dnCNV were validated by (1) a customized region specific Agilent array CGH with ~ 150 bp average probe spacing and (2) breakpoint PCR analysis, if feasible (Table S1). In addition, to uncover CNVs beyond the detection resolution of array CGH and to aid breakpoint mapping, whole genome sequencing was performed for two trios (BAB3097 and BAB3596) with an average depth-of-coverage of 30.3 to 29.1 in the probands. These integrated genome analyses approaches revealed a suite of dnCNVs that exhibit unique patterns that are shared amongst the five individuals (Figure 1 and Tables 1 and S1). The mutational signatures identified include: i) involvement of multiple chromosomes, ii) predominance of copy number gains, iii) most copy number gains arranged as tandem duplications, iv) sequence microhomeology at breakpoint junctions, v) involvement of relatively long genomic segments (e.g. >100 kb) for individual dnCNVs, and vi) dnSNVs at breakpoints, surrounded by apparently iterative short template switches.
Table 1. Detection of dnCNVs from BAB3097 and BAB3596 by various array platforms and WGS; related to Data S1, S2 and Tables S1.
CNVs above the double line are from BAB3097; CNVs below the double line are from BAB3596. Dup, duplication; Trp, triplication; IDD, insertional double duplication; BD, BreakDancer.
| Locus | Size | Type | Array detection | WGS detection | Parent of Origin | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 180k | 1M | 1M (cell*) | 4.2M | SNP 1M/2.6M | BD | Pindel | novo Break | ||||
| 1p36 | 6.4 Mb | Dup | Y | Y | Y | Y | Y | Y | N | Y | Pat |
| 3q13q21 | 956 kb | Dup | Y | Y | Y | Y | Y | Y | N | Y | Pat |
| 5p12 | 440 kb | Dup | N | Y | Y | Y | Y | Y | N | Y | Mat |
| 5q33q34 | 5.8 Mb | IDD | Y | Y | Y | Y | Y | Y | Y | Y | Pat |
| 9p13 | 1.2 Mb | Trp | Y | Y | Y | Y | Y | Y | N | Y | Mat |
| 17p11p12 | 6.0 Mb | Dup | Y | Y | Y | Y | Y | Y | N | Y | Mat |
| 22q13 | 309 kb | IDD | N | Y | Y | Y | Y | Y | Y | Y | Mat |
| 1p34p35 | 1.7 Mb | Dup | Y | Y | Y | Y | Y | Y | N | Y | Mat |
| 3p14p21 | 4.2 Mb | Dup | Y | Y | Y | Y | Y | Y | N | Y | Mat |
| 8q24 | 4.5 Mb | Dup | Y | Y | Y | Y | Y | Y | Y | Y | Mat |
| 10q24q25 | 4.7 Mb | Dup | Y | Y | Y | Y | Y | Y | N | Y | Mat |
| 16p11 | 322 kb | IDD | Y | Y | Y | Y | Y | Y | N | Y | Pat |
| 16q23 | 4.2 Mb | IDD | Y | Y | Y | Y | Y | Y | N | Y | Mat |
| 16q24 | 312 kb | Dup | Y | Y | Y | Y | Y | Y | N | Y | Mat |
| 19q13 | 4.3 Mb | Dup | Y | Y | Y | Y | Y | Y | N | Y | Mat |
| Xp11 | 214 kb | Dup | N | Y | Y | Y | Y | Y | Y | Y | Mat |
DNA was extracted from cultured lymphoblastoid cell lines of the individuals and analyzed on the microarray; for the other columns under “Array detection”, DNA was extracted from blood directly.
Figure 1. The landscape of dnCNVs in the five subjects; related to Data S1, S2 and Tables S1, S2.

A. The subject ID is shown at the top of each panel. The X-axis indicates the genome view for chromosomes 1–22, X, and Y. The Y-axis indicates the proband versus control log2 ratio of the array CGH result, with duplications at 0.58, triplications at 1, and heterozygous deletions at −1. Each dot in the graph represents a dnCNV. The color of the dot indicates the parental origin of the dnCNV, with orange, blue, and black designating maternal, paternal, and unknown origin. The number above the dot denotes the CNV length in megabase. Numbers highlighted in red indicate that the corresponding CNV involves a complex rearrangement. B. A representative array CGH raw data showing the de novo duplication on chromosome 17 in subject BAB3097.
The majority of CNVs identified are copy number gains
The 34 dnCNV identified in the 5 personal genomes ranged in size from 6.5 kb to 11 Mb (Figure 1, Tables 1 and S1). There is a strikingly skewed distribution of copy number gains versus losses. Four of the five subjects carry exclusively copy number gains (BAB3097, N=6; BAB3596, N=8; mCNV3, N=8; mCNV4, N=7); a heterozygous loss of 6.5 kb was identified in only one subject (mCNV7), despite four other copy number gains in the same subject. As a comparison, subjects ascertained by the same clinical arrays carrying only one dnCNV displayed a relative higher proportion of losses (losses:gains = 741:253 or 1:0.34), which significantly deviates from the distribution in the five subjects with excessive number of dnCNVs (losses:gains = 1:33, two-tailed Fisher’s exact test, p= 8.1x10−17). Among the copy number gains in the five subjects, an apparent triplication is observed in BAB3097 (Data S1D). The preponderance of copy number gains and finding of triplication suggest that the underlying DNA repair mechanism is synthetic in nature, for example, the replication based mechanisms such as fork stalling and template switching (FoSTeS) and microhomology-mediated break-induced replication (MMBIR) (Hastings et al., 2009; Lee et al., 2007).
Most of the dnCNV demonstrate a tandem duplication configuration
Breakpoint sequencing and array CGH results demonstrate that the majority of dnCNVs are tandem duplications. In this analysis, a dnCNV duplicated with a gross head-to-tail configuration, with or without short breakpoint complexities (<200 bp), is considered as a tandem duplication. Among all the dnCNVs, at least 26 out of 34 (76%) are tandem duplications (Figure 2A); if we exclude the 2 dnCNVs in which a definite rearrangement structure could not be determined, the proportion of tandem duplication becomes 26/32 or 81%. The high proportion of tandem duplication configurations in MdnCNV patients, which is similar to the observation made in patients with singular de novo duplications (Newman et al., 2015; Weckselblatt and Rudd, 2015), favors the hypothesis that the individual CNV events arose independently from each other in an MdnCNV setting. In contrast, gains in chromothripsis are almost always insertional duplications, part of an inversion, or in other forms of complex rearrangements, but rarely present as tandem duplications (Liu et al., 2011; Stephens et al., 2011).
Figure 2. Unique features are shared by dnCNVs identified from the five subjects; related to Data S1 and Table S5.

(A) Proportions of tandem duplication (TD), insertional double duplication (IDD), complex triplication (CT), deletion (DEL), and unresolved rearrangement structure (N/A) among MdnCNV events. (B) A representative microhomeology from breakpoint BAB3097-chr5-1 (Data S1C and the first row in Table 2). The junction sequence is aligned with two reference sequences, with the red and blue colors indicating their origins from the colored rectangles above them. Microhomeology is highlighted in yellow. The junction sequence shares perfect identity to the blue reference, but not to the red reference, suggesting that MMBIR initiates from the blue end and invades into the red end. (C) Density plots illustrating that dnCNVs identified in this study tend to be large in size.
Even though CGRs consistent with multiple iterative template switches only contribute to a small proportion of all the dnCNVs (5/34), they are still observed in 3 out of the 5 subjects, providing an important clue to the mechanism of MdnCNV. Two specific patterns of CGR observed are insertional double duplications and triplications. Insertional double duplications are observed in BAB3097, BAB3596 and mCNV4, where two seemingly independent duplications are detected by array CGH but subsequent breakpoint analysis reveals that the extra copy of one of the duplications is inserted in between the two copies of the other duplication (Figure 2A and Data S1F, K, X, and Z). The gained genomic material in the two duplications can map to two different chromosomes (chr5 and chr22 in BAB3097, Data S1F), on short and long arms of one chromosome (chr16p and chr16q in BAB3596, Data S1K), or within one chromosomal location separated by various distances [or DUP-NML-DUP (Carvalho and Lupski, 2016; Carvalho et al., 2013; Gu et al., 2015), 2.2 kb away on chr1 and 112 kb away on chr3, both in mCNV4, Data S1X and Z, respectively].
A triplication is observed in BAB3097. Breakpoint sequencing revealed that one copy of the segment is inserted in an inverted orientation in between the other two copies (Figure 2A and Data S1D); a DUP–TRP/INV-DUP pattern (Carvalho et al., 2011). Based on a replicative mechanism hypothesis, the gross structure of this triplication can be formed by two template switches to the opposite DNA strand during DNA replication. Additional complexities are observed in proximity to one of these two template switches (Data S1D).
Sequence microhomeologies are identified at the breakpoint junction
Previous studies that systematically interrogated DNA sequence characteristics at rearrangement breakpoint junctions in various organisms frequently revealed homologous or microhomologous sequences acting to facilitate the rearrangement process (Carvalho and Lupski, 2016). It was also noted that highly identical but still divergent sequences, or homeologous sequences, can be observed at breakpoint junctions (Anand et al., 2014; Mezard et al., 1992). Human genome Alu repetitive sequences, even when from divergent families, mediate Alu-Alu disease associated rearrangements (Boone et al., 2014), including CGR (Boone et al., 2011; Gu et al., 2015), and they provide excellent substrates in a yeast template switch replicative repair assay (Mayle et al., 2015). The sequence characteristics of recombinant product, chimeric Alu repeats, found at Alu-Alu breakpoint junctions are often consistent with potential homeology facilitating template switching.
In the MdnCNV individuals, comparison of reference sequence pairs that correspond to each breakpoint frequently revealed short intervals of 2 to 24 base pairs with highly identical but yet sometimes imperfect sequence matches, a breakpoint junction sequence hallmark which we term “microhomeology” (Figure 2B and Data S1). Furthermore, for the base pairs that differ between the two reference sequences of a breakpoint, the actual breakpoint junction always shows exclusive identity to one of the two reference sequences. This observation is consistent with an MMBIR model underlying these rearrangements, wherein the end of the breakpoint with perfect sequence match to the junction acts as the donor site of template switching, and the end with imperfect matches serves as the target site of template switch invasion (Figure 2B). Among the 45 breakpoint junctions with sequence information, 7 had perfect identity between donor and recipient sequences, and 26 had 50–99% sequence identity. Seventeen of 26 breakpoint microhomeologies were 6–24 bases in length with 71–92% sequence identity (Table 2 and Data S1), a percentage of homeology similar to that shared amongst Alu repetitive sequences that are members of different Alu families. One breakpoint directly involved an AluS-AluY mediated rearrangement (Data S1HH).
Table 2. Microhomeologous sequences at the breakpoint junctions; related to Data S1 and Table S5.
Breakpoint junctions with perfect matching microhomology or with a matching identity below 70% are not listed in this table but can be found in Data S1. The sequence of the actual breakpoint junction is the same as the donor reference sequence except when in rare situations de novo mutations occurred and altered the sequence from the reference donor; when de novo mutations are present at the breakpoint junction, it is shown as lower case letters and included in the donor sequence.
| Sample | Total # of nucleotides | # of matching nucleotides | Identity matching | Putative donor sequence (top) and putative target sequence (bottom) | Parent of origin of donor/target sequence |
|---|---|---|---|---|---|
| BAB3097 | 12 | 10 | 83% |
|
mat/mat |
| BAB3097 | 24 | 18 | 75% |
|
mat/mat |
| BAB3097 | 8 | 6 | 75% |

|
mat/mat |
| BAB3097 | 7 | 5 | 71% |

|
pat/pat |
| BAB3097 | 9 | 7 | 78% |

|
mat/pat |
| BAB3596 | 11 | 8 | 73% |
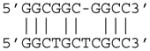
|
pat/mat |
| BAB3596 | 10 | 8 | 80% |

|
mat/pat |
| mCNV3 | 12 | 11 | 92% |
|
NA |
| mCNV3 | 7 | 6 | 86% |

|
NA |
| mCNV3 | 9 | 7 | 78% |

|
NA |
| mCNV3 | 7 | 6 | 86% |

|
NA |
| mCNV3 | 13 | 10 | 77% |
|
NA |
| mCNV3 | 20 | 15 | 75% |
|
NA |
| mCNV4 | 16 | 12 | 75% |
|
mat/mat |
| mCNV4 | 8 | 7 | 88% |

|
mat/mat |
| mCNV7 | 16 | 14 | 88% |
|
pat/pat |
| mCNV7 | 6 | 5 | 83% |
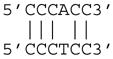
|
pat/pat |
| mCNV7 | 300 | 249 | 80% | AluS-AluY | NA |
To analyze the significance of microhomeology observed in our breakpoints, we adopted a gapped alignment analysis approach and simulations to determine a reference distribution of alignment scores. To create a comparison, we used the data from our prior paper (Campbell et al., 2014), in which patients ascertained by the same clinical arrays but carried only one dnCNV were selected for analysis; we also made a comparison to a group of germline chromothripsis-like events with massive balanced rearrangements (Chiang et al., 2012). Of the 45 sequences in the MdnCNV group, 30/45=66.7% were observed to have significant microhomology or microhomeology by the 0.05 p-value criteria (Table S5). When only microhomeologies are considered (i.e. perfect matching microhomologies are excluded as they may also be products of other rearrangement mechanisms such as nonhomologous end joining), 25/45=55.6% breakpoints remain significant. For the single dnCNV group and the chromothripsis-like group, 11/67=16.4% and 26/141=18.4% of sequences had significant microhomeology. The rate of microhomeology in our study was significantly higher, with an odds ratio of 6.36 and 5.53 and p-values of 1.8X10−5 and 3.3X10−6. The enrichment of breakpoint microhomeology in the MdnCNV group suggests that the specific rearrangement mechanism used in the MdnCNV phenomenon frequently involves imperfect template priming.
Both microhomology and microhomeology are capable of priming strand invasion of a single ended single strand overhang into an ectopic site, resulting in a genomic rearrangement (Sakofsky et al., 2015). If template switching occurs more than once in a single MMBIR mutational event, it should be expected that the iterative template switches may occur over a uniform direction and that the imperfect alignments within each microhomeology should always be found at the target side of each breakpoint. As hypothesized, for each of the six complex rearrangements for which microhomeology could be identified at the breakpoint, analyses of all junction versus reference sequences suggest a uniform replication direction within each complex event (Data S1).
Additional breakpoint sequence features that potentially support the MMBIR model include no involvement of long stretches of homologous sequences at the breakpoints, an architectural feature observed with NAHR-mediated homology driven events, and sequence insertions potentially copied from either close by (<20 bp, Data S1D, Q, S, and U) or distant from (50Mb, Data S1D) the breakpoint.
We also examined the breakpoints’ locations in association with replication timing (Figure S2), replication rate, segmental duplications, and aphidicolin-inducible common fragile sites. No clustering was evident, though it should be noted that these analysis may be limited by the sample size.
Individual dnCNVs involve significantly more genomic material than inherited CNVs
Previous studies have shown that patients with single dnCNVs carry a higher burden of larger events than inherited CNVs (Girirajan et al., 2011; Itsara et al., 2010; Sanders et al., 2015). We sought to explore the size distribution of events within MdnCNV individuals, as usually only one or two dnCNVs in one MdnCNV profile are under the pressure of selection (reason that prompted the clinical CMA analysis), leaving the status of the remaining dnCNVs unknown. In MdnCNV samples, the lengths of most of the genomic regions involved tend to be large, ranging from 6.5 kb to 11 Mb, with 97% (33/34) of the events measuring between 100 kb and 10 Mb. These data suggest long distance template switching. High-density array CGH and whole genome sequencing can readily detect CNVs as small as 1–10 kb. This can be illustrated by the fact that most of the inherited CNVs are within the range of 1–10 kb (Figure 2C). From the array CGH and WGS results, we attempted to validate all the candidate dnCNVs smaller than 10 kb by custom array CGH and breakpoint PCR. However, validation results suggest that all of the smaller dnCNVs candidates are false calls, with the vast majority consisting of false positive calls in the child, indicating that the dnCNVs in the five subjects are restricted to larger lengths. This is in sharp contrast with the previous report regarding the length distribution of deletions in an individual genome (most of which are presumably inherited rather than de novo), where the frequency of events escalates rapidly as the length decreases from 1 kb to 2 bp (Wheeler et al., 2008). Thus, the MdnCNV phenomenon appears to preferentially favor the formation of large (>100kb) CNV. At the same time, we do not rule out the possibility that dnCNVs below the array detection resolution (~2.8 kb for the Nimblegen 4.2M array and ~0.6 for the Agilent targeted array) may evade the analysis.
The timing of MdnCNV mutations in the context of human embryological development
Constitutional new mutations were thought to arise from germline mutational events; however, recent data suggest postzygotic new mutations contribute to disease (Acuna-Hidalgo et al., 2015; Campbell et al., 2014; Rahbari et al., 2016; Robberecht et al., 2012). Specific data have shown active CNV mutations in human cleavage-stage embryos (Vanneste et al., 2009). To infer the timing within the organismal life cycle of the de novo events, we attempted to track the parent of origin for the DNA segments resultant from the dnCNV. The dnCNVs were phased onto the paternal or the maternal chromosomes using informative SNPs from WGS and SNP genotyping arrays (Figure 1, Table S1, and Data S2). For subjects BAB3097, BAB3596 and mCNV4, there is a mix of paternal and maternal de novo events. In particular, for the complex insertional double duplications from BAB3097 and BAB3596, both cases have the donor and the recipient duplications phased onto opposing parental chromosomes. These data strongly indicate the presence of post-zygotic mutational events in the three subjects. However, we cannot exclude the possibility that the process of CNV mutations was initiated in germ cells and persisted until the post-zygotic period. Of note, the CNV mutations in the three subjects mentioned above demonstrated excessive maternal contributions (maternal:paternal = 4:3, 8:1, 6:2, respectively), which hints at commencement of CNV mutations potentially during oogenesis.
A different pattern of parent of origin for dnCNVs is observed in mCNV7. The four duplications originated from the paternal chromosomes, whereas the deletion does not contain enough informative SNPs for phasing. These data favor that the CNV mutations in mCNV7 occurred during spermatogenesis, but do not exclude the possibility that a subset or all of the CNVs in this subject arose after fertilization. For subject mCNV3, parental DNAs were not available for phasing genotypes and therefore origins of dnCNVs could not be inferred.
If a copy number gain was produced pre-zygotically, there is a possibility that the gained material could be acquired from the non-transmitted chromosome - either the rearrangement process itself directly involved the non-transmitted chromosome, or the rearrangement was restricted within the transmitted chromosome, but crossing-over during meiosis introduced segments from the non-transmitted chromosome. To explore these possibilities, we selected all the SNP variants within each CNV that are informative for distinguishing whether the gained material is from the transmitted or the non-transmitted chromosome. This analysis suggests that none of the CNVs involve the non-transmitted chromosome (Data S2).
For the MdnCNVs that exhibit a postzygotic component, the formation of dnCNVs seems to potentially persist into cleavage stage embryo, and stop thereafter. This is supported by the observation that no obvious mosaicism is evident for any of the dnCNVs in this study as evidenced by the comparative signal intensities obtained from array CGH results, read depth coverage obtained from the whole genome sequencing data, and high resolution SNP microarray analysis (Table S2). The apparent homogenous constitutional genome profile of MdnCNV can be concluded by a mutational event in a blastomere from the early cleavage stage embryo, which drove the other lineages of blastomere(s) to extinction by negative selective processes (Vanneste et al., 2009).
Taken together, the aggregate data support the idea that the MdnCNV phenomenon occurs during a peri-zygotic timing of the human life cycle, i.e. between the period of gametogenesis to post fertilization early cleavage stage embryo.
The driver for the transient CNV mutator phenotype is lost or silenced
The accumulated features from our observational studies that are presented by the MdnCNV phenomenon in multiple subjects are reminiscent of a “mutator phenotype”. The mutator phenotype was initially described in drosophila and bacteria (Miyake, 1960; Plough, 1941; Treffers et al., 1954). It has recently been widely appreciated in cancers (Loeb, 2001). Mutations in a “mutator” are usually driven by a force that is intrinsic to the subject itself; for example, a defective mismatch repair gene (Kinzler and Vogelstein, 1996; Nicolaides et al., 1998; Palles et al., 2013). As a result, the mutations are expected to evolve over time. This will result in new mutations accumulating in a somatic mosaic state. However, as discussed in the previous section, no evidence for mosaicism was found for any of the dnCNVs in any MdnCNV subject (Table S2).
We collected a second DNA sample from blood of subjects BAB3097 and BAB3596 half a year after the first DNA collection, and assessed their new CNV profile using the same array CGH platforms. The new profiles appear identical to the initial observations – the existing CNVs remained stable and no new CNVs arose. Additionally, we generated lymphoblastoid cell lines for these two subjects and repeated array CGH using cultured cells. Again, the same CNV profile was obtained. Note that the two approaches above will not detect new CNVs that arose from single cells but failed to expand clonally. The collective evidence suggests that the “mutator” phenotype is no longer operating after early post-zygotic embryo divisions.
The CNV mutator phenotype seems to be restricted within a short time interval in the organismal life cycle, which likely terminated after the division of the zygote. Therefore, we hypothesize that the driving force for these mutations is silenced or lost after that time period. To uncover such a potential genetic driver, trio whole exome sequencing was performed on two subjects, BAB3097 and BAB3596, and their parents. We specifically focused on both the proband and the maternal specific variants (because of the high maternal contribution of dnCNVs). However, no specific gene variants, either SNV alleles or intragenic CNV, were found that could potentially drive the MdnCNV phenotype. The absence of a candidate mutation from these two trios suggests either that the causal mutation resides in an unknown disease gene, noncoding/regulatory region of a critical gene, or that the driver mutation is lost or suppressed after triggering a “transient” CNV mutator phenotype.
dnCNV are accompanied by dnSNV formation in proximity to breakpoint junctions
Chromosomal instabilities and point mutations are two prominent imprints in progressing cancer genomes; these two hallmarks are sometimes found concomitantly in certain cancers (Nik-Zainal et al., 2012). To explore whether the generation of dnSNVs is influenced by MdnCNV, we investigated the number, distribution, and context of dnSNVs in the subjects with MdnCNVs.
We surveyed the entire genomes of two trios (BAB3097 and BAB3596) with whole genome sequencing analysis. These numbers of dnSNV (62 from BAB3097 and 82 from BAB3596 including INDELs) do not appear to be elevated compared with the general population when matched for the father’s age at conception (Crow, 2000; Goldmann et al., 2016; Iossifov et al., 2014; Kong et al., 2012; Rahbari et al., 2016). As is theoretically expected (Crow, 2000) as well as previous empirical studies have shown (Michaelson et al., 2012; Rahbari et al., 2016), a majority of dnSNVs were phased onto the paternal chromosomes (15/20 for BAB3097 and 22/28 for BAB3596). In addition to the number of mutations, the distribution of mutation types in the two trios also appears to be similar to the control population (Rahbari et al., 2016) (data not shown).
The spatial distribution of dnSNVs on a genome-wide scale does not show obvious clustering of mutations or loci with evidence for mutational showers (data not shown). However, when searching for dnSNVs in close proximity to the breakpoints among all five MdnCNV individuals, 5 dnSNVs are found at 4 out of 45 breakpoints, which are all within a 10-bp distance to the junctions (Table 3 and S3). The dnSNVs are frequently found in a context of homonucleotide runs (Table 3). Two of these four breakpoints involve complex rearrangements, including a complex triplication (underlined nucleotides in Data S1D) and a complex double duplication (Data S1F), whereas the other two breakpoints involve complex replication slippages as part of an apparently simple tandem duplication (Data S1Q and S1U). An additional striking similarity shared by the five dnSNVs is that they all precede or reside in short reiterative template switches. The dnSNV in the first breakpoint precedes three short segment insertions at the breakpoint, each involving 13 bp, 24 bp, and 7146 bp sequences, respectively (Data S1D). The dnSNV of the second breakpoint is situated within a short sequence insert of 23 bp, which is followed by another sequence insert of 20 bp (Data S1F). The remaining dnSNVs are located within two breakpoints involving replication slippages of 14 bp and 18 bp (Data S1Q and S1U). These observations corroborate the recent finding of elevated point mutation rates at CNV breakpoint junctions, especially in the sequence context of homonucleotide runs, suggesting a reduced fidelity polymerase utilized for replicative repair, at the site of rearrangements associated with MMBIR (Carvalho et al., 2013; Carvalho et al., 2015; Deem et al., 2011). The close association with reiterative short template switches suggests that the specific polymerase used also is of low processivity.
Table 3.
| Mutation type | Reference | Mutation (lower case) | Rearrangement context |
|---|---|---|---|
| Di-nucleotide substitution | 5′-TTTTGGGGGATTG-3′ | 5′-TTTTGGGGccTTG-3′ | Complex triplication |
| SNV | 5′-ATTATTATTTATAATAATATAT-3′ | 5′-ATTTATTATTcATAATAATATAT-3′ | Complex double duplication |
| SNV | 5′-GTGGTTGC-3′ | 5′-GTGGTTtC -3′ | Replication slippage |
| Single nucleotide insertion | 5′-GTGCAAT-3′ | 5′-GTGCaAAT-3′ | Replication slippage |
| Single nucleotide deletion | 5′-AAAAACTGTG-3′ | 5′-AAAA-CTGTG-3′ | Replication slippage |
Taken together, these data suggest that in the MdnCNV subjects, the CNV mutations does not appear to affect genome-wide point mutations, but can show evidence for reduced replication fidelity coupled with low processivity in close proximity to the CNV breakpoint junctions.
Discussion
Several features of the MdnCNV phenomenon suggest that these mutations arose due to a faulty replicative repair process. The presence of CGRs, microhomeology, templated insertion at the breakpoints, and dnSNVs in proximity to breakpoint junctions are all characteristics of the FoSTeS/MMBIR mechanism (Carvalho and Lupski, 2016; Hastings et al., 2009; Lee et al., 2007). It is interesting to note that almost all the dnCNVs observed in this study are copy number gains. FoSTeS/MMBIR favors the formation of copy number gains over losses, especially when complex rearrangements are involved (Liu et al., 2012). This is because replicative mechanisms introduce structural errors by synthesizing DNA segments; the more template switches there may be, the more likely that the genomic DNA will be over-synthesized. Despite these considerations, it is still striking to observe such a skewed distribution of gains versus losses (33:1). Part of this could be a result of selection and clonal expansion following the burst of CNV mutations, which is common in human cleavage stage embryos (Vanneste et al., 2009). Gains are in general less detrimental than losses and sometimes can confer an advantage for cell growth. Multiple changes involving more than one loss may be incompatible with cellular life and conducive to apoptotic death and selected against both on a single cell level and on an individual organismal level. If this occurs during early development while cells are still pluripotent, it may have little effect on development. Alternatively, the primary CNV mutation mechanism in these subjects may be one that we do not fully understand; perhaps favoring template switching to a position behind the advancing fork. This mechanism is of a replicative nature, favors the production of tandem duplications, can be frequently mediated by microhomeologies, and allows emergence of complex rearrangements.
What is the origin of the MdnCNVs? This remains without a definitive answer at this point. Our results suggest that the “CNV mutator” phenotype is apparently “transient”, which makes environmental stress a plausible candidate driving force as it only strikes the genome once. However, the dnCNV breakpoints do not cluster around common fragile sites. The predominance of tandem duplications would also require the environmental stimulus to be one specific kind that creates such mutational signatures.
Can a single factor be responsible for the observed CNV mutator phenotype? The transient nature of the phenomenon implies that the putative primary defect responsible for the CNV mutator phenotype is only limited to a short time interval in early embryonic development, serving as a genomic stability factor for this specific timing – perhaps maternally contributed in the oocyte. One possibility is that the defect originates as a form of epigenetic silencing, and it is subsequently reverted after the early embryonic timing. The timing of the parental specific epigenetic reorganization events in the zygote, such as the replacement of protamines in the condensed spermatic genome and the asynchronous DNA replication in two pronuclei, is at least partly in line with the occurrence of MdnCNV events. These differences of epigenetic biology between paternal and maternal chromosomes may play a role in explaining the maternal:paternal ratio of dnCNV events in a ‘CNV mutator zygote’.
We postulate that an alternative genetic factor theory arguing for a novel inheritance mode can provide an equally plausible explanation, in which a mutation arising in the germ cells facilitated the emergence of dnCNVs but the mutated genetic material is subsequently segregated into the non-transmitted gamete after meiosis. Candidate proteins include those genes pivotal to the MMBIR process (e.g. translesion polymerases) or the BIR process. This model predicts that the causative mutation occurs before the end of meiosis II. If the mutation occurred in the father, the dnCNVs are expected to be exclusively of paternal origin, which fits the observations for mCNV7. If the mutation occurred in the mother, the phenotypic outcome becomes controlled by the maternal cytoplasmic effect in the zygote (Figure 3). After the defective genetic material is lost in meiosis, the defective mRNA remains in the cytoplasm and exerts its abnormal functions until it is cleared at the time of zygote genome activation in 4- or 8- cell stage. Thus, the cumulative dnCNVs may be a mixture of paternal and maternal events; the maternal contribution is expected to be higher if the causative mutation occurred earlier during oogenesis. The model does not exclude the possibility that the causative mutation occurs after fertilization or that the mutation can be transmitted to the zygote. However, both of these possibilities will result in the “mutator” not being “turned off” and the consequent genomic changes too devastating to be compatible with a live birth. The timeframe of the MdnCNV phenomenon predicts that the dnCNVs occurring before the embryo 2-cell stage are constitutional by default; dnCNVs that occur in the one or two divisions that follow will also likely to be constitutional after the process of selection on the level of blastomeres (Vanneste et al., 2009). It is readily apparent that additional MdnCNV samples be studied to refine our understanding of the biological timing of the mutational events, to provide more statistical power to estimate the extent of maternal effect in different individuals, and to test this proposed model. If evidence accumulates to support this model, perhaps the most significant implication is not which gene can serve as the driver, but rather the idea that hyper- CNV mutation can be switched on and off by the elegant balance of replicative based rearrangement formation mechanism and selection during gametogenisis/embryogenesis – a potential rapid way for a multicellular organism with a long life cycle (e.g. humans with a 25 year generation) to respond to dramatic environmental or genetic changes occurring over a short time interval.
Figure 3. Model for the CNV mutator phenotype restricted to early development.

(A) A genetically normal oogonium synthesizing wild type mRNA (black curved lines). (B) A mutation (blue cross) on a gene that is critical for CNV formation occurs in the process of a primary oocyte development. Mutant mRNAs (red curved lines) are consequently synthesized, activating the CNV mutator phenotype, as is shown by the accumulation of dnCNVs (green bars) on chromosomes. (C) After meiosis I, the chromosome harboring the mutation is lost to the first polar body, but the mutant mRNA is still present in the plasma of the oocyte at a high concentration. Meanwhile, a subset of the newly formed dnCNVs may also be segregated into the first polar body. (D) As the sperm enters the oocyte, meiosis II is initiated. The second polar body is extruded. (E) Maternal and paternal DNAs replicate separately, which is accompanied by generation of additional dnCNVs. (F) A zygote is formed after the fusion of the two pronuclei. (G) In the early cleavage stages, the cellular concentration of the mutant mRNA decreases as the cell divides. Meanwhile, dnCNVs keep accumulate. The inter-chromosomal dnCNVs involving both parental chromosomes observed in this study can be formed during this time interval. (H) As the mutant mRNA is removed by maternal clearance (usually in 4- or 8- cell stage in humans) and the zygote transcription is activated, the number of dnCNV becomes stable. (I) Schematic diagram showing the fluctuation of number of mutant DNA, mRNA and dnCNV in the MdnCNV phenomenon. The X-axis is in proportion to the timeline shown above in (A) to (H).
Genomic instability has been proposed as a hallmark of cancer (Hanahan and Weinberg, 2011; Negrini et al., 2010). Recent evidence suggests that oncogene-induced replication stress propels genomic instability (Macheret and Halazonetis, 2015). This model is particularly attractive in explaining certain sporadic cancers. We propose that the transient MdnCNV phenomenon shares intrinsically the same molecular mechanism with certain sporadic cancers. Interestingly, in an effort to understand the somatic rearrangement landscape in various breast cancer genomes, a subgroup of subjects were found to have the majority of the somatic rearrangements as tandem duplications (Stephens et al., 2009). Recently, a novel phenomenon termed Tandem Duplicator Phenotype (TDP) was reported in triple negative breast, ovarian, endometrial, and liver cancers (Menghi et al., 2016; Ng et al., 2012). This TDP phenomenon is characterized by numerous tandem duplications and breakpoint features suggestive of replicative mechanisms. These findings suggest a common mechanism and origin for some cancer CNVs as the germ line MdnCNV phenomenon reported here. Rearrangement mechanisms are usually difficult to dissect in cancer genomes, where somatic heterogeneity obscures rearrangement mechanisms; however, the MdnCNV phenotype provides us with a clean homogeneous snapshot of a representation of a cancer genome. Investigations into the MdnCNV phenomenon, as well as other extremely rare constitutionally mutated genomic profiles, such as chromoanasnythesis (Liu et al., 2011) and CGR coupled with regions of absence of heterozygosity (Carvalho et al., 2015), lend themselves a unique access to understanding the mutational nature of cancer genomes.
STAR Methods
KEY RESOURCES TABLE (provided as a separate file)
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for raw data, analysis details, and DNA samples may be directed to, and will be fulfilled by the lead author Pengfei Liu (pengfei.liu@bcm.edu). Whole genome and exome sequencing data are not deposited in a repository because the Institutional Review Board protocol used in this study restricted the deposition of such data from minors into public or controlled-access databases.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The study has been approved by the Institutional Review Board of Baylor College of Medicine. The subjects discussed in this study are treated as anonymized individuals. Subjects BAB3097 and mCNV7 are female. Subjects BAB3596, mCNV3, and mCNV4 are male. The authentication of the lymphoblastoid cell lines used in this study was conducted by performing the Agilent SurePrint G3 Unrestricted CGH 1x1M microarrays on DNA samples extracted from the cell lines and the blood sample of the same individual, and matching the private variants between them.
METHOD DETAILS
Estimating the normal distribution of the number of dnCNVs per individual
We utilized data from a cohort of patients who have gone through clinical CMA analysis to understand the normal distribution of number of dnCNVs per individual. We selected individuals with complete parental studies who were found to carry at least one dnCNV. As described in the main text, we consolidated different CNV regions that likely originate from a common event in an individual. The interpretation of attributing multiple CNV regions in one sample to a single origin versus independent origins is sometimes facilitated by clinically available karyotyping and FISH analyses results, which is frequently part of the clinical work up of a patient with suspected genomic disorders. When such information is not available, multiple CNV regions mapping to the same chromosome band are empirically considered as a single event.
Microarray analysis
DNAs were isolated from blood samples or cultured Epstein-Barr virus transformed lymphoblastoid cell lines. DNAs from each subject and their parents were initially analyzed on both the Agilent SurePrint G3 Unrestricted CGH 1x1M (G4824A) array and the Roche NimbleGen Human CGH 4.2M Whole Genome Tiling Array following the manufacturer’s protocols. The same DNA samples were subsequently analyzed on SNP genotyping arrays including the Illumina HumanOmni2.5–8 Array, the Illumina HumanOmni1-Quad Array, the Affymetrix Genome-Wide Human SNP Array 6.0, and the Affymetrix CytoScan HD Array. The constellation of array results enabled dnCNV and SNP haplotyping analyses.
Candidate regions of CNV from the subjects were compiled from the above-mentioned array results. These candidate regions were manually examined in the parental arrays to search for potential dnCNVs. Candidate de novo regions were compiled and interrogated on a custom designed Agilent targeted high-density array. The average probe spacing of these regions in the custom array is ~150 bp, with higher probe coverage in intervals surrounding the suspected breakpoint region. The trios were studied using these targeted arrays to validate dnCNVs. Manual review of these array results refined the list of candidate dnCNVs.
We relied primarily on the two high-density genome wide arrays, Agilent 1M and Nimblegen 4.2M, and the Agilent region specific targeted array to construct our core dnCNV list. The average probe spacing for these three arrays are ~3 kb, ~0.7 kb, and ~0.15 kb. Assuming that the smallest CNV call requires five probes, these three arrays have a minimum CNV detection size threshold of ~12 kb, ~2.8 kb, and ~0.6 kb.
Breakpoint junction amplification and sequencing analysis
To amplify the breakpoint junctions, we designed outward-facing primers for presumed tandem duplications and used inward-facing primers for deletions. Different orientations of primers were tested for complex rearrangements. Long-range PCR was conducted using the TaKaRa LA Taq polymerase. PCR products that potentially contained breakpoint junctions were subject to Sanger dideoxy sequencing. DNA sequences were analyzed by comparing to reference sequences using the UCSC Genome Browser. For junctions that are refractory to the long-range PCR method, an alternative method of capture and breakpoint approaching sequencing was used (Xiao et al., 2014). All primers used are listed in Data S1. After the multi-step microarray analyses, candidate dnCNVs can still be shown to be false positive by break point PCR analysis. Examples include situations where a small triplication is identified in the subject and the same region is apparently copy number neutral in both parents. However, PCR demonstrated that the “triplication” is actually a homozygous duplication; both parents are heterozygous for the exact same duplication and they are both heterozygous for the reciprocal deletion. Therefore, such an event is not a dnCNV. Events proven to be false positives by PCR are excluded from the dnCNV list. CNVs with high quality microarray results supporting its de novo nature but no successful breakpoint junction amplification were still considered de novo events.
For the majority of the CNV junctions (45/50), breakpoint PCR successfully amplified the unique breakpoint junction from the subject, with subsequent Sanger sequencing confirmation across the junction. In such situations, the same PCR assay is repeated in the parental DNAs. This is to rule out the possibility that the parents are of low-level mosaicism for the breakpoint junction in the blood (Campbell et al., 2014), which makes the CNV event not truly a “de novo” mutation in the subject.
CNV size analysis
The size of a dnCNV is calculated by the difference between the start and end coordinates of the CNV interval based on the reference genome, using either the mapped precise breakpoint coordinates or the estimates from array CGH if the breakpoints are not mapped to the nucleotide resolution. The length of a CGR involving CNVs on different chromosome bands or different chromosomes is calculated by the sum of the length of each simplex CNV. For inherited CNVs, calls from the NimbleGen array data are collected to examine for the presence in the subjects and parents. A call is only included in this analysis if there is over 80% reciprocal overlap for the CNV observed in the subject and one parent. Variants seen in both parents are excluded because they likely reflect systematic errors or CNV from the comparative control individual used in the CGH experiment. Data from the trios of BAB3097 and BAB3096, for which WGS data are available, were used for the inherited CNV analysis.
Characterization of microhomeology and directionality of MMBIR events
The breakpoint junction sequence is aligned with the two reference sequences from the proximal and distal end of the breakpoint. At the transition point when the junction sequence is matched to one of reference sequence versus the other, short homologies may be identified that are similar between all three aligned sequences. The following empirical criteria are employed to identify microhomology/microhomeology in this study. For homologies with 1–4 nucleotides, a 100% matching is required. For homologies with 5 or more nucleotides, two matching identity thresholds are used: a minimum of 50% identity is used as a lower threshold, and a 70% cutoff is used as a more stringent threshold. A maximum of 2-nucleotide gap is allowed within each imperfect breakpoint sequence alignment.
To analyze the significance of imperfect alignments observed in our breakpoints, we adopted a gapped alignment analysis approach and simulations to determine a reference distribution of alignment scores. See the “QUANTIFICATION AND STATISTICAL ANALYSIS” section below for details of the statistical analysis of this approach. When a microhomeology with at least one mismatch is identified at the breakpoint, directionality of the MMBIR event can be inferred. It is proposed that replication stalls at the end of the breakpoint with perfect matching. Then, the broken end of the replication disassociates with the original strand and invades into a new locus. The invasion is primed by a possibly imperfect match between the 3′ overhang of the broken replication end and the target site. Therefore, the end of the breakpoint with a mismatch in the microhomeology is inferred as the target site. The concept of microhomeology and MMBIR directionality is illustrated in Figure 2B. The actual conclusions for each breakpoint are summarized in Data S1.
Characterization of replication timing and rate for dnCNV breakpoints
Genome-wide replication timing data are obtained from Koren et al (Koren et al., 2012) and replication rate data are obtained from Chen et al (Chen et al., 2015). For all the dnCNVs that have their breakpoint boundary defined at the nucleotide level in this study, their genomic coordinates are queried against the above-mentioned reference databases to obtain the replication timing and rate that corresponds to each breakpoint locus. As a comparison, 100 CNVs are randomly simulated throughout the human genome, with each of them spanning 1 Mb genomic interval, and the replication timing and rate for these random CNVs are obtained likewise (Figure S2).
Whole genome sequencing and discovery for dnCNV
DNAs from blood samples of two subjects (BAB3097 and BAB3596) and their parents were prepared and sequenced on the Illumina HiSeq platform following standard protocols. The paired-end sequencing reads were aligned to the GRCh37 reference genome using standard procedures. The total mean autosomal sequencing read depth-of-coverage ranged from 24.8 to 38.3 per sample.
Structural variants were called with BreakDancer (Chen et al., 2009), Pindel (Ye et al., 2009) and novoBreak (Chong et al., 2016) with default parameters. novoBreak is a tool used for SV detection in cancer genomes, especially somatic ones. It employed a k-mer targeted local assembly algorithm to detect structural variation breakpoints at single base pair resolution. Based on experiments using both synthetic and real patients’ data, novoBreak outperformed the state-of-the-art methods to detect chromosomal rearrangements and won the ICGC-TCGA Somatic Mutation Calling DREAM Challenge SV sub-challenge. The detection of dnCNVs in the trio data is similar to detection of somatic SVs in tumor-normal paired cancer samples. We treated the proband as the “tumor” sample and we merged the parental bam files as “normal”. The novoBreak pipeline was directly applied to the proband and merged parents WGS data. Finally, we manually checked and confirmed the dnCNVs reported by novoBreak by inspecting the bam alignments.
When comparing CNVs between subjects and their parents to judge for their de novo or inherited nature, two CNVs are considered to be identical if they have reciprocal overlap of over 50% intervals. High quality calls are filtered from raw software output. The empirical filtering criteria are as follows. For de novo discovery in BreakDancer: supporting reads >4, score >80; for inherited discovery in BreakDancer: supporting reads >2, score >90. For de novo discovery in Pindel: deletion, length >100 bp; short insertion, length > 50 bp; inversion, score >100; tandem duplication, length > 50bp and score > 200; long insertion, no filtering; unknown type, supporting reads > 5. For inherited discovery in Pindel, only filter for deletions and tandem duplications with supporting reads > 2. For dnCNV discovery, the variant has to pass the above-mentioned criteria and there has to be no supporting reads from both parents. The candidate variants are visualized in IGV to manually rule out obvious false positives. The remaining candidates are subjected to breakpoint PCR amplification. All the variants not verified by PCR are excluded. novoBreak and BreakDancer detected all the dnCNVs (7/7 in BAB3097 and 9/9 in BAB3596) identified from arrays, showing a sensitivity of 100% for dnCNV discovery, whereas Pindel showed a sensitivity of 25% (2/7 in BAB3097 and 2/9 in BAB3596) (Table 1). These sensitivity numbers are calculated using the dnCNV set obtained from the different array platforms used, which is only applicable to the CNVs above the array minimum detection size limit (~2.8 kb for the Nimblegen 4.2M array and ~0.6 kb for the Agilent targeted array). For dnCNVs below this size threshold, the sensitivities of the WGS CNV analysis algorithms are unknown. The total number of dnCNVs predicted by the callers are: BreakDancer BAB3097=380, BAB3596=77, novoBreak BAB3097=80, BAB3596=57, Pindel BAB3097=107, BAB3596=37.
For inherited CNVs, the variants fulfilling the above-mentioned criteria with over 50% subject-parent reciprocal overlap are included in the analysis from Figure 2C without further PCR confirmation. Those variants that are seen in both parents are excluded to rule out the possibility of false positive. Among the 166 and 135 inherited CNVs detected by the Nimblegen microarray from BAB3097 and BAB3596, concordant calls from BreakDancer were 34 and 31, respectively. Since the paired-end (BreakDancer) and split reads (Pindel) methods are not sensitive for copy number gains, we used a read depth analysis of the WGS data as a supplementary method to validate a wide range of different sizes of inherited CNVs that were discovered using Nimblegen microarrays. For each event, due to the potentially inaccurate junctions, we extended the event to more than 5kb of both left and right coordinates. Then we plotted the read depth of the region using an R script. An obvious read depth fall indicates a copy number loss, while a read depth rise indicates a copy number gain. By combining BreakDancer, Pindel and read depth analysis, we confirmed all the Nimblegen microarray inherited results.
Discovery for dnSNV from whole genome sequencing
We called SNV likelihoods from the sequencing data using Bcftools and dnSNVs using De Novo Gear (DNG) version 0.5 (Ramu et al., 2013) using default parameters. DNG output consisted of around 12,700 candidate sites in both individuals. We filtered this output based on our previous experience with dnSNV calling and arrived at 228 and 246 sites respectively that were of sufficiently high quality to validate. The two distinct and complementary validation methods we used were capillary sequencing and Illumina MiSeq sequencing.
DNG identified 12,602 potential dnSNVs in one patient and 12,732 in the other. From this set, we removed candidate sites using a series of filters. Sites only passed if the putative dnSNV had been identified in the child, less than 10% of sequencing reads in either parent support the dnSNV, if the minimum sequencing coverage in any trio member at the candidate site is not below 10x and the maximum sequencing coverage in any trio member is not above 2 times the average coverage. After manual inspection of candidate de novo sites in IGV, we made the decision to exclude a number of SNV candidates that were likely to be false positives or inherited and to include a number of indel candidates that had not passed automated filtering. We designed primers 150–215 bp upstream and downstream of each candidate site that had passed filtering using Primer 3 so that the mutation lay in the centre of a 300–430 bp PCR product. We split the resulting amplification products into two halves. One half was sequenced on the Illumina MiSeq platform (250 bp paired end reads) and the other half by capillary sequencing. Thus, the sequencing data from the two platforms was complementary. Whilst there was generally good overlap between the capillary and MiSeq validation data, in a few cases capillary sequencing revealed clear evidence of the variant being present in one or both of the parents, changing its classification from de novo to inherited. In other cases, capillary sequencing failed and we had to rely on MiSeq data alone for validation. We identified a total of 56 de novo point mutations and 6 de novo INDELS for BAB3097, and 80 de novo point mutations and 2 de novo INDELs for BAB3596. The validated high-quality dnSNVs are listed in Tables S4A and S4B. The father’s age of conception for BAB3097 and BAB3596 are 29 and 41 years old, respectively.
dnSNVs that occur in close proximity to breakpoint junctions are also examined. The mapability of these dnSNVs may be impaired by the sequence arrangement at the breakpoint. To circumvent this potential issue, we utilized sequences from two sources to ensure that we capture the events in the allele that maps to the junction. The first source derives from Sanger sequence traces obtained from the same amplicon as the breakpoint junction. The second source derives from manually assembled sequences from WGS that contain the allele from the breakpoint junction. The segments from these two sources are manually examined for dnSNVs, and the numbers of dnSNVs as well as the length of the merged segment examined are listed in Table S3.
Calculation of mosaic ratios for the dnCNVs
For each dnCNV, the copy number is calculated by two ways. One way is through the average log2 ratio from array CGH comparative signal intensity. The second way is through comparative estimation from the average read depth within the dnCNV versus the average read depth of the flanking 1 Mb region of neutral copy number. After the two copy number estimates are obtained, the mosaic ratio is calculated based on the hypothesis that the entire population of the cell is only composed of two sub-populations, one with the dnCNV and the other with neutral copy number in the same region. As an additional approach, Affymetrix CytoScan HD arrays were performed for all MdnCNV samples. CNV mosaicism analysis was performed using the manufacture’s software. CNVs that are known to be inherited from a parent, and thus should have an expected mosaic ratio of 100%, are also included in this analysis to demonstrate the range of experimental variation.
Phasing the dnSNVs onto parental chromosomes and distribution of dnSNVs
We determined the parent of origin of validated point mutations by using the phasing functionality of the DNG software (Ramu et al., 2013). DNG identifies inherited SNV in the proximity of the dnSNV and determines whether they are observed on the same paired-end read as the mutation. We were able to determine the parent of origin for 11 mutations in one trio and for 27 mutations in the other trio.
The expected distance to the next dnSNV was modelled by randomly rearranging the position of dnSNVs whilst keeping the number of dnSNVs per chromosome constant. The expected distance of each dnSNV to the closest other dnSNV was computed as the mean of 1,000 rearrangements.
The expected distance to the next breakpoint site was modelled by randomly rearranging the position of dnSNVs, again keeping the position of CNV breakpoints and the number of dnSNVs per chromosome constant. The average dnSNV-breakpoint distance of each dnSNV was computed as the mean of 1,000 rearrangements.
Phasing dnCNVs onto parental chromosomes
SNP genotype information was obtained using a series of arrays including the Illumina Infinium HD assay platform with HumanOmni1-Quad BeadChip (Illumina Inc.), Affymetrix CytoScan™ HD Array, and Affymetrix Genome-Wide Human SNP Array 6.0. The experiments were performed according to the manufacturer’s instructions. Additional SNP information was obtained for the two trios analyzed by whole genome sequencing (BAB3097 trio and BAB3596 trio). The genotypes for the SNPs within the dnCNVs were obtained for the subjects and their parents. The genotypes for the subjects had to be recalculated based on a tri-allelic or tetra-allelic model depending on the actual copy number status of the region involved, which can be obtained from the array CGH analysis. Homozygous variants that are different between the parents are selected, for example, AA in the mother and BB in the father. The genotypes of all the variants within the same dnCNV in the child should uniformly indicate the parent of origin, for example, AAB illustrating the maternal origin for the duplication. After determining the parent of origin of all the dnCNVs in a subject, the overall mutation timing (pre- or post- zygotic) is inferred. If dnCNVs from an individual are determined to be partly paternal and the other part maternal, it is indicated that at least a number of the dnCNVs from this individual arose postzygotically.
Whole exome sequencing
We applied trio WES on the families of BAB3097 and BAB3596 at Baylor College of Medicine Human Genome Sequencing Center through the Baylor-Hopkins Center for Mendelian Genomics research initiative. Samples underwent targeted whole-exome capture using the BCM HGSC Core design followed by Illumina HiSeq massively parallel sequencing. Raw sequence data were mapped and aligned to the reference human genome sequence GRCh37/Hg19 using the BWA algorithm. The coverage of the six samples ranges from 101X to 140X, and the percent of regions with over 20X coverage ranges from 90.89 to 93.28. Variants were called and annotated using an in-house-developed bioinformatics pipeline.
QUANTIFICATION AND STATISTICAL ANALYSIS
To analyze the significance of imperfect alignments observed in our breakpoints, we adopted a gapped alignment analysis approach and simulations to determine a reference distribution of alignment scores. For each observed sequence from the MdnCNV breakpoints, we performed a gapped alignment using the R implementation of pairwise sequence alignment from the Biostrings package. We used the global alignment approach (Needleman-Wunch algorithm). We set the match score to 1, the mismatch penalty to 0, the gap opening penalty to 0.5 and the gap extension penalty to 1. This choice of parameterization was observed to recapitulate the alignments we manually generated on the sequences. For each breakpoint sequence, we then randomly sampled 5000 sequences from hg19 genome build, making sequence draws of the same length as the microhomeology domain and occurring within 100,000 bases of each observed breakpoint. We performed pairwise alignment on these randomly drawn human genome sequences using the same method. We recorded the alignment score of the true sequences and each pair of random sequences. For each breakpoint we determined a p-value for the observed microhomeology as the number of random alignments where the random sequence alignment scores equal or exceeded that of the observed breakpoint microhomeology. We also applied the same method described above to two alternate sets of breakpoints as described in the main text and we again computed p-values for each breakpoint in this set by counting the number of times randomly drawn genomic sequences had gapped alignment similarity scores meeting or exceeding the microhomeology of the breakpoint.
To compare the rate of significant microhomeology between two sets of break points, we counted the number of breakpoints in each study that had significant microhomeology with imperfect match (identity < 100%) according to the simulation based gapped alignment procedure (i.e. simulation based p-value<0.05). We used Fisher’s exact test to compare the rates of significant microhomeology.
To test whether the number of dnCNVs is related to parental age, we plotted distribution of parental ages for patients with one, two, or multiple dnCNVs from our cohort, and analyzed whether there is a biased distribution using the Wilcoxon signed-rank test (Figure S1).
Distribution of replication timing and rate between the MdnCNV group and the random control CNV group are compared using the Wilcoxon signed-rank test.
DATA AND SOFTWARE AVAILABILITY
The accession number for the microarray data reported in this paper is NCBI GEO: GSE87915.
Supplementary Material
Additional Data S1. Individual dnCNV events from the MdnCNV phenomenon; related to Figures 1–2, Tables 1–3.
By comparing the B-allele frequency data of the patient and the parents, origins of the dnCNVs can be inferred. For each dnCNV, two graphs are plotted. The left side graph depicts all the informative SNPs that reveal the parent of origin of the dnCNV, with red SNPs indicating maternal origin and blue SNPs indicating paternal origin. The right side graph depicts all the informative SNPs that reveal chromosome of origin of the dnCNV, with the orange SNP indicating intrachromosomal origin and green SNPs indicating interchromosomal origin (i.e. rearrangement material derived from the non-transmitted parental chromosome). The SNP B-allele frequency data for BAB3097 and BAB3596 are obtained from whole genome sequencing, and those for mCNV4 and mCNV7 are obtained from Illumina genotyping array. All dnCNVs are shown except for one CNV in mCNV7, which is too small to contain enough informative SNPs.
“QUANTIFICATION AND STATISTICAL ANALYSIS”. The distribution of age of conception of fathers and mothers, when available, are plotted and compared with each other. The number of data points are as follows, father of patients with one dnCNV (N=1368), two dnCNV (N=38), MdnCNV (N=4), mother of patients with one dnCNV (N=1388), two dnCNV (N=38), MdnCNV (N=4). The distribution of parental ages is not significantly different between patients with two or more dnCNVs compared to those with one (Wilcoxon rank sum test, p>0.05).
The X-axis values indicate the standard deviation of replication timing deviating from the genome-wide mean (X=0), with positive values indicating early and negative values late replication.
Acknowledgments
We thank all the family members and collaborators who participated in this study. This work was supported in part by grants from the US National Institute of Neurological Disorders and Stroke (http://www.ninds.nih.gov/, R01NS058529), the National Human Genome Research Institute (NHGRI https://www.genome.gov/, U54HG003273), a joint NHGRI (https://www.genome.gov/) / National Heart Blood and Lung Institute (http://www.nhlbi.nih.gov/) grant (U54HG006542) to the Baylor Hopkins Center for Mendelian Genomics, and the BCM Intellectual and Developmental Disabilities Research Center, IDDRC Grant Number 5P30HD024064-23, from the Eunice Kennedy Shriver National Institute of Child Health & Human Development. The work was also partially supported by the WellcomeTrust (WT098051). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development, the Wellcome Trust or the National Institutes of Health.
Footnotes
Author Contributions
PL and JRL conceived the study and wrote the manuscript. PL, BY, CMBC, AW, KWa, LZ, VG, SL, and MW performed the experiments. PL, BY, CMBC, AW, KWa, LZ, TG, ZCh, IMC, ZCo, CGJ, FZ, KC, MEH, and JRL analyzed the data. KWr, CAB, BR, SWC, JS, AB, CAS, AP and JRL contributed to anonymized bulk patient molecular data. PL, JW, JS, PS, BR, SWC, JS, AB, CAS, AP and JRL contributed to microarray data and analysis. SNJ, DMM, RAG, MEH, and JRL supervised exome and genome sequencing analyses. All contributing co-authors have read, edited, and agreed to the contents of the manuscript.
Disclosures
Baylor College of Medicine (BCM) and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of the Baylor Genetics (BG), which performs clinical microarray analysis and clinical exome sequencing. PL, CAB, JSc, PS, SWC, JS, AB, CS, AP and JRL are employees of BCM and derive support through a professional services agreement with the BG. JRL serves on the Scientific Advisory Board of the BG. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, has stock options in Lasergen, Inc. and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. RAG reports consulting fees from GE-Clarient. AW is an employee of Genentech Inc.
References
- Acuna-Hidalgo R, Bo T, Kwint MP, van de Vorst M, Pinelli M, Veltman JA, Hoischen A, Vissers LE, Gilissen C. Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. Am J Hum Genet. 2015;97:67–74. doi: 10.1016/j.ajhg.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand RP, Tsaponina O, Greenwell PW, Lee CS, Du W, Petes TD, Haber JE. Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev. 2014;28:2394–2406. doi: 10.1101/gad.250258.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Mulle JG, Schaibley VM, Ragland RL, Durkin SG, Warren ST, Glover TW. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet. 2009;84:339–350. doi: 10.1016/j.ajhg.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlt MF, Rajendran S, Birkeland SR, Wilson TE, Glover TW. De novo CNV formation in mouse embryonic stem cells occurs in the absence of Xrcc4-dependent nonhomologous end joining. PLoS Genet. 2012;8:e1002981. doi: 10.1371/journal.pgen.1002981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Liu P, Zhang F, Carvalho CM, Towne CF, Batish SD, Lupski JR. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet Med. 2011;13:582–592. doi: 10.1097/GIM.0b013e3182106775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Yuan B, Campbell IM, Scull JC, Withers MA, Baggett BC, Beck CR, Shaw CJ, Stankiewicz P, Moretti P, et al. The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. Am J Hum Genet. 2014;95:143–161. doi: 10.1016/j.ajhg.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, Nagamani SC, Erez A, Bartnik M, Wisniowiecka-Kowalnik B, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. 2014;95:173–182. doi: 10.1016/j.ajhg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Pehlivan D, Ramocki MB, Fang P, Alleva B, Franco LM, Belmont JW, Hastings PJ, Lupski JR. Replicative mechanisms for CNV formation are error prone. Nat Genet. 2013;45:1319–1326. doi: 10.1038/ng.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Pfundt R, King DA, Lindsay SJ, Zuccherato LW, Macville MV, Liu P, Johnson D, Stankiewicz P, Brown CW, et al. Absence of heterozygosity due to template switching during replicative rearrangements. Am J Hum Genet. 2015;96:555–564. doi: 10.1016/j.ajhg.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Ramocki MB, Pehlivan D, Franco LM, Gonzaga-Jauregui C, Fang P, McCall A, Pivnick EK, Hines-Dowell S, Seaver LH, et al. Inverted genomic segments and complex triplication rearrangements are mediated by inverted repeats in the human genome. Nat Genet. 2011;43:1074–1081. doi: 10.1038/ng.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhou W, Zhang C, Lupski JR, Jin L, Zhang F. CNV instability associated with DNA replication dynamics: evidence for replicative mechanisms in CNV mutagenesis. Hum Mol Genet. 2015;24:1574–1583. doi: 10.1093/hmg/ddu572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. 2012;44:390–397. S391. doi: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Z, Ruan J, Gao M, Zhou W, Chen T, Fan X, Ding L, Lee AY, Boutros P, Chen J, et al. novoBreak: local assembly for breakpoint detection in cancer genomes. Nat Methods. 2016 doi: 10.1038/nmeth.4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A. Break-induced replication is highly inaccurate. PLoS Biol. 2011;9:e1000594. doi: 10.1371/journal.pbio.1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Brkanac Z, Coe BP, Baker C, Vives L, Vu TH, Shafer N, Bernier R, Ferrero GB, Silengo M, et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 2011;7:e1002334. doi: 10.1371/journal.pgen.1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann JM, Wong WS, Pinelli M, Farrah T, Bodian D, Stittrich AB, Glusman G, Vissers LE, Hoischen A, Roach JC, et al. Parent-of-origin-specific signatures of de novo mutations. Nat Genet. 2016;48:935–939. doi: 10.1038/ng.3597. [DOI] [PubMed] [Google Scholar]
- Gu S, Yuan B, Campbell IM, Beck CR, Carvalho CM, Nagamani SC, Erez A, Patel A, Bacino CA, Shaw CA, et al. Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Hum Mol Genet. 2015;24:4061–4077. doi: 10.1093/hmg/ddv146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, London SJ, Eichler EE. De novo rates and selection of large copy number variation. Genome Res. 2010;20:1469–1481. doi: 10.1101/gr.107680.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Tavakoli-Yaraki M, van Roosmalen MJ, van Binsbergen E, Renkens I, Duran K, Ballarati L, Vergult S, Giardino D, Hansson K, et al. Constitutional chromothripsis rearrangements involve clustered double-stranded DNA breaks and nonhomologous repair mechanisms. Cell reports. 2012;1:648–655. doi: 10.1016/j.celrep.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren A, Polak P, Nemesh J, Michaelson JJ, Sebat J, Sunyaev SR, McCarroll SA. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am J Hum Genet. 2012;91:1033–1040. doi: 10.1016/j.ajhg.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- Liu P, Carvalho CM, Hastings PJ, Lupski JR. Mechanisms for recurrent and complex human genomic rearrangements. Current opinion in genetics & development. 2012;22:211–220. doi: 10.1016/j.gde.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA. A mutator phenotype in cancer. Cancer research. 2001;61:3230–3239. [PubMed] [Google Scholar]
- Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Structural variation mutagenesis of the human genome: Impact on disease and evolution. Environmental and molecular mutagenesis. 2015;56:419–436. doi: 10.1002/em.21943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annual review of pathology. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, Ira G. DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science. 2015;349:742–747. doi: 10.1126/science.aaa8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae AF, Visscher PM, Montgomery GW, Martin NG. Large Autosomal Copy-Number Differences within Unselected Monozygotic Twin Pairs are Rare. Twin research and human genetics : the official journal of the International Society for Twin Studies. 2015:1–6. doi: 10.1017/thg.2014.85. [DOI] [PubMed] [Google Scholar]
- Menghi F, Inaki K, Woo X, Kumar PA, Grzeda KR, Malhotra A, Yadav V, Kim H, Marquez EJ, Ucar D, et al. The tandem duplicator phenotype as a distinct genomic configuration in cancer. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1520010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezard C, Pompon D, Nicolas A. Recombination between similar but not identical DNA sequences during yeast transformation occurs within short stretches of identity. Cell. 1992;70:659–670. doi: 10.1016/0092-8674(92)90434-e. [DOI] [PubMed] [Google Scholar]
- Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake T. Mutator Factor in Salmonella Typhimurium. Genetics. 1960;45:11–14. doi: 10.1093/genetics/45.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nature reviews Molecular cell biology. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Newman S, Hermetz KE, Weckselblatt B, Rudd MK. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet. 2015;96:208–220. doi: 10.1016/j.ajhg.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CK, Cooke SL, Howe K, Newman S, Xian J, Temple J, Batty EM, Pole JC, Langdon SP, Edwards PA, et al. The role of tandem duplicator phenotype in tumour evolution in high-grade serous ovarian cancer. The Journal of pathology. 2012;226:703–712. doi: 10.1002/path.3980. [DOI] [PubMed] [Google Scholar]
- Nicolaides NC, Littman SJ, Modrich P, Kinzler KW, Vogelstein B. A naturally occurring hPMS2 mutation can confer a dominant negative mutator phenotype. Mol Cell Biol. 1998;18:1635–1641. doi: 10.1128/mcb.18.3.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plough HH. Spontaneous mutability in Drosophila. Cold Spring Harb Symp Quant Biol. 1941;9:127–137. [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–133. doi: 10.1038/ng.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramu A, Noordam MJ, Schwartz RS, Wuster A, Hurles ME, Cartwright RA, Conrad DF. DeNovoGear: de novo indel and point mutation discovery and phasing. Nat Methods. 2013;10:985–987. doi: 10.1038/nmeth.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht C, Voet T, Utine GE, Schinzel A, de Leeuw N, Fryns JP, Vermeesch J. Meiotic errors followed by two parallel postzygotic trisomy rescue events are a frequent cause of constitutional segmental mosaicism. Molecular cytogenetics. 2012;5:19. doi: 10.1186/1755-8166-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Ayyar S, Deem AK, Chung WH, Ira G, Malkova A. Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol Cell. 2015;60:860–872. doi: 10.1016/j.molcel.2015.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treffers HP, Spinelli V, Belser NO. A Factor (or Mutator Gene) Influencing Mutation Rates in Escherichia Coli. Proc Natl Acad Sci U S A. 1954;40:1064–1071. doi: 10.1073/pnas.40.11.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DJ, Miretti M, Rajan D, Fiegler H, Carter NP, Blayney ML, Beck S, Hurles ME. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneste E, Voet T, Le Caignec C, Ampe M, Konings P, Melotte C, Debrock S, Amyere M, Vikkula M, Schuit F, et al. Chromosome instability is common in human cleavage-stage embryos. Nature medicine. 2009;15:577–583. doi: 10.1038/nm.1924. [DOI] [PubMed] [Google Scholar]
- Weckselblatt B, Rudd MK. Human Structural Variation: Mechanisms of Chromosome Rearrangements. Trends Genet. 2015;31:587–599. doi: 10.1016/j.tig.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Xiao J, Zhang L, Wang J, Jiang Y, Jin L, Lu J, Jin L, Zhong C, Xu X, Zhang F. Rearrangement structure-independent strategy of CNV breakpoint analysis. Molecular genetics and genomics : MGG. 2014;289:755–763. doi: 10.1007/s00438-014-0850-4. [DOI] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Leibowitz ML, Pellman D. Chromothripsis and beyond: rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013;27:2513–2530. doi: 10.1101/gad.229559.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Data S1. Individual dnCNV events from the MdnCNV phenomenon; related to Figures 1–2, Tables 1–3.
By comparing the B-allele frequency data of the patient and the parents, origins of the dnCNVs can be inferred. For each dnCNV, two graphs are plotted. The left side graph depicts all the informative SNPs that reveal the parent of origin of the dnCNV, with red SNPs indicating maternal origin and blue SNPs indicating paternal origin. The right side graph depicts all the informative SNPs that reveal chromosome of origin of the dnCNV, with the orange SNP indicating intrachromosomal origin and green SNPs indicating interchromosomal origin (i.e. rearrangement material derived from the non-transmitted parental chromosome). The SNP B-allele frequency data for BAB3097 and BAB3596 are obtained from whole genome sequencing, and those for mCNV4 and mCNV7 are obtained from Illumina genotyping array. All dnCNVs are shown except for one CNV in mCNV7, which is too small to contain enough informative SNPs.
“QUANTIFICATION AND STATISTICAL ANALYSIS”. The distribution of age of conception of fathers and mothers, when available, are plotted and compared with each other. The number of data points are as follows, father of patients with one dnCNV (N=1368), two dnCNV (N=38), MdnCNV (N=4), mother of patients with one dnCNV (N=1388), two dnCNV (N=38), MdnCNV (N=4). The distribution of parental ages is not significantly different between patients with two or more dnCNVs compared to those with one (Wilcoxon rank sum test, p>0.05).
The X-axis values indicate the standard deviation of replication timing deviating from the genome-wide mean (X=0), with positive values indicating early and negative values late replication.