

Abstract
Predicting the responsiveness to regular exercise is a topic of great relevance due to its potential role in personalized exercise medicine applications. The present review focuses on cardiorespiratory fitness (commonly measured by maximal oxygen uptake, ), a trait with wide‐ranging impact on health and performance indicators. Gains in demonstrate large inter‐individual variation even in response to standardized exercise training programmes. The estimated heritability of 47% suggests that genomic‐based predictors alone are insufficient to account for the total trainability variance. Candidate gene and genome‐wide linkage studies have not significantly contributed to our understanding of the molecular basis of trainability. A genome‐wide association study suggested that trainability is influenced by multiple genes of small effects, but these findings still await rigorous replication. Valuable evidence, however, has been obtained by combining skeletal muscle transcript abundance profiles with common DNA variants for the prediction of the response to exercise training. Although the physiological determinants of measured at a given time are largely enunciated, what is poorly understood are the details of tissue‐specific molecular mechanisms that limit and related signalling pathways in response to exercise training. Bioinformatics explorations based on thousands of variants have been used to interrogate pathways and systems instead of single variants and genes, and the main findings, along with those from exercise experimental studies, have been summarized here in a working model of trainability.
Abbreviations
- GWAS
genome‐wide association study
- QTL
quantitative trait locus
- SNP
single nucleotide polymorphism
maximal oxygen uptake
Introduction
Since the seminal work of Hill & Lupton (1923), maximal oxygen uptake (), defined as the oxygen uptake attained during maximal exercise intensity, which cannot be increased despite further increase in exercise workload, has been considered a valid indicator of the limits of the cardiorespiratory systems to deliver oxygen to the tissues. The validity of the concept has been confirmed in more recent times by a number of laboratories and most notably by Snell and colleagues (2007). Thus we posit that is a valid indicator of cardiorespiratory fitness with profound health and performance implications.
In this paper, we review the most recently published literature on the genomic and transcriptomic correlates of response levels to endurance exercise training. The overarching question being whether the response of to endurance exercise training can be predicted by common personal characteristics and common physiological traits combined with genomic and transcriptomic signatures. The global theme was reviewed earlier with an emphasis on genetics and genomics (Bouchard et al. 2011a; Bouchard, 2012). The present effort will incorporate other lines of evidence and more recently reported research.
It is commonly recognized that cardiorespiratory fitness along with multiple cardiometabolic risk factors improve as a result of exposure to endurance exercise programmes. However, a series of papers, beginning with two early reports published more than 30 years ago (Bouchard, 1983; Lortie et al. 1984), demonstrated convincingly that there were considerable individual differences among adults of both sexes in the gains in even though all participants were exposed to standardized exercise programmes. In this regard, the findings of many small studies were confirmed, found to be applicable also to other common disease risk factors, and amplified by the larger, multicentre HERITAGE Family Study (Bouchard et al. 1999, 2012; Leon et al. 2000; Wilmore et al. 2001; Katzmarzyk et al. 2003; Boule et al. 2005; Lakka et al. 2005; Blache et al. 2007). Understanding why there is human variability in trainability is of particular importance for personalized exercise medicine applications but also for those who may be motivated by the pursuit of sports performance.
Of particular importance for the present review is the magnitude of the variability in trainability. If we use the gains in in millilitres O2 per minute as reported in the HERITAGE Family Study as a guide, a mean gain of about 400 ml O2 was achieved with a standard deviation (SD) of about 200 ml. Some of the variance in training response can be accounted for by measurement error but also by the day‐to‐day variability in performance. The SD of measured over two or more days provides an approximation of the magnitude of the role of measurement error and daily variability. The HERITAGE programme provided four independent estimates of the SD of repeated measures (2, 3 or 4 tests) and the SDs ranged from 108 to 137 ml O2 (with coefficients of variation (CVs) ranging from 4.1 to 5.0). These SDs for repeated measures accounted for about 15–20% of the group SDs (the latter ranged from 646 to 740 ml O2). If one uses the upper number, the true SD of trainability for the given endurance exercise stimulus of HERITAGE would be reduced to about 160 ml instead of the original 200 ml O2. This new estimate is somewhat higher than two other estimates (123 and 138 ml O2, respectively) obtained by Shephard et al. (2004) on the same dataset but with less input data for the computation of the SD of repeated tests. How can this remaining variability in trainability be accounted for and can the dissection of the unexplained variance illuminate the underlying biology of the responsiveness to exercise programmes?
Trainability of : heritability and correlates
In a series of experiments performed in the 1980s, we showed that the changes in in response to standardized exercise training programmes were highly heterogeneous (Lortie et al. 1984; Prud'homme et al. 1984; Hamel et al. 1986; Simoneau et al. 1986). This was subsequently confirmed and quantified in the large HERITAGE Family Study in which 742 healthy, sedentary adults were deemed ‘completers’ of a standardized, laboratory‐delivered endurance exercise programme lasting 20 weeks (Bouchard et al. 1999). The range of gains extended from no increase to gains of more than 1000 ml O2 (Bouchard et al. 1999; Bouchard & Rankinen, 2001).
As depicted in Fig. 1, baseline , age, sex, ethnicity and initial body weight were all correlated with training response. The association of each of these personal characteristics with Δ was in the range of 2–3%. Since we had the detailed minute by minute heart rates and workloads of each training session for each subject, we explored whether the fluctuations in achieved heart rates and workloads compared to targeted levels for each exercise session were contributing to the variability in training responses. We observed that the fluctuations in workloads accounted for about 6% of the training responses (see Fig. 1). If the percentage variance associated with the technical error and the day‐to‐day variation (a maximum of 20%) is added to the figure, about 40% of the variance in trainability is accounted for by these factors.
Figure 1. A summary of the correlates of the gains in in the HERITAGE Family Study.
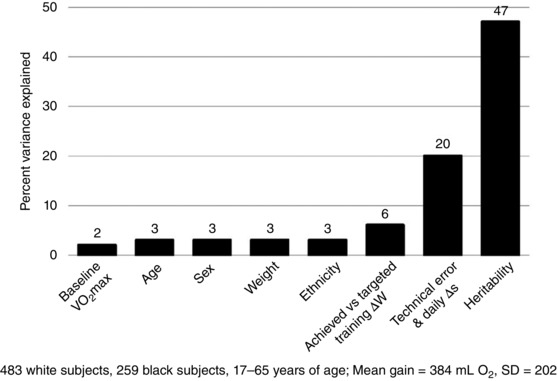
The contributions of various correlates in terms of percentage variance accounted for in response are shown. Details are provided in the text.
Additionally, we know from HERITAGE that the variability in training response aggregates in families. The increase in adjusted for age and sex in 481 individuals from 99 two‐generation families of white subjects showed 2.6 times more variance between families than within families, and a model‐fitting analytical procedure yielded a maximal heritability estimate of 47% (Bouchard et al. 1999). These observations corroborated the results of four experimental exercise studies performed with pairs of identical twins (Prud'homme et al. 1984; Hamel et al. 1986; Simoneau et al. 1986; Bouchard et al. 1994). Intraclass coefficients (computed from the within‐pairs and between‐pairs variances) for the response to training varied from 0.44 to 0.77 across these studies. Using the HERITAGE estimate of trainability (47%), we can see from Fig. 1 that more than 85% of the variance in responsiveness is accounted for by all the factors identified in the figure.
These observations are supported in part by selection experiments performed in rats. In one study, pairs of low and high responders to exercise training were mated and their offspring were later exposed to the same exercise programme. The narrow sense heritability of running performance trainability (as a surrogate for cardiorespiratory fitness) reached 43% (Troxell et al. 2003). More recently, the same group led by Brittton and Koch reported that, after 15 generations of selection (n = 178 rats), low response rats failed to improve in response to 8 weeks of exercise (a decline of 65 m in running distance) while high response rats improved substantially (an increase of 223 m in distance) (Koch et al. 2013). However, the narrow sense heritability for trainability in this model was only 10% (SEM + 0.02), an observation that calls for more studies.
It is clear from the above that less than half of the variance in trainability in humans is potentially explained by genetic differences. Based on the evidence accumulated to date, individual variability in trainability is a normal biological phenomenon, which may in part reflect genetic diversity. The study of individual variability in adaptive potential offers a powerful way to shed light on human physiology (Bennett, 1987) as well as molecular pathways and systems.
Candidate gene studies
A majority of the studies examining genetic predictors of response to endurance exercise training have employed the candidate gene approach. Starting in 2000, a group of researchers began publishing an annual review of the findings for genetic associations with exercise performance and health‐related fitness phenotypes. At the time of its final update in 2006–2007, the human gene map for performance and fitness phenotypes had identified 11 candidate genes (AMPD1, ATP1A2, PPARGC1A, PPARD, HBB, HIF1A, GABPB2, ACE, APOE, CKM and MTND5, a mitochondrial gene) and 12 quantitative trait loci (QTL) associated with the response of cardiorespiratory fitness‐related phenotypes to exercise training, primarily (Bray et al. 2009). However, most of the candidate genes associated with exercise‐response traits were often based on only one study with positive findings, with the variant explaining a small percentage of the total variance in training response phenotypes. The most studied candidate genes in relation to trainability are ACE, APOE and ACTN3, all with discordant or inconclusive results (Bouchard, 2012). Population, sample size and training programme differences are among the factors potentially responsible for the variability of findings in these studies.
Genome‐wide and transcriptome‐wide approaches
Advancements in high‐throughput technologies have allowed for hypothesis‐free, genome‐wide investigations of exercise response phenotypes. As such, several studies employing genome‐wide approaches related to trainability have been published. In the HERITAGE Family Study, genome‐wide linkage scans were initially used to identify gene regions associated with and maximal power output responses to exercise training (Bouchard et al. 2000; Rico‐Sanz et al. 2004). Suggestive evidence of linkages with markers on 1p31, 16q22 and 20q13.1 in black subjects and on 4q27, 7q34 and 13q12 in white subjects were found for training response, while suggestive linkages on 1q22 and 13q11 in black subjects and on 5q23, 1q21, 4p15.1 and 4p13 in white subjects were found for maximal power output training response (Rico‐Sanz et al. 2004).
Genome‐wide approaches also include global skeletal muscle gene expression profiling (i.e. transcriptome‐wide analysis), which can be combined with data on DNA sequence variation. In one report, a combination of genome‐wide muscle gene expression with DNA sequence variation screening was used to identify genes associated with changes in in response to endurance training. RNA expression profiling of pre‐training skeletal muscle samples identified 29 transcripts whose baseline expression levels were associated with training response in 24 subjects (Timmons et al. 2010). The predictive value of the 29 transcripts was confirmed in an independent training sample of 17 subjects. Next, the authors genotyped tagging single nucleotide polymorphisms (SNPs; n = 300) in the 29 genes in the HERITAGE Family Study. A multivariate regression model using the transcriptome‐derived SNPs and a set of SNPs from previous linkage analysis in HERITAGE identified a set of 11 SNPs that explained approximately 23% of the variance in training response. Interestingly, the gene expression levels (i.e. RNA abundance) of the predictor genes was unchanged with exercise training, thereby supporting the concept that the baseline profile of transcript abundance in appropriate tissues in combination with DNA sequence information can potentially serve as predictors of exercise response.
We published the first genome‐wide association study (GWAS) for an exercise‐response trait in 2011, where we examined the association of over 324,000 SNPs with exercise training‐induced changes in in 473 white subjects from the HERITAGE Family Study (Bouchard et al. 2011b). We found that none of the SNPs reached genome‐wide significance, although 39 SNPs were associated with training response at P < 1.5 × 10−4. Regression analysis of these 39 SNPs yielded 21 SNPs explaining 49% of the variance in trainability, a value very similar to the heritability estimate of 47% previously reported in HERITAGE (Bouchard et al. 1999). In the regression model, nine SNPs explained between 2 and 7% of the variance each, while seven SNPs explained between 1 and 2% each (Bouchard et al. 2011b). A predictor score was constructed using these 21 SNPs based on the number of favourable alleles (i.e. high training response alleles) carried across each SNP: low‐response allele homozygote was scored 0, heterozygote scored 1, and homozygote for the high‐response allele was scored 2. These scores were summed across all 21 SNPs resulting in a predictor score that could range from 0 (no favourable alleles) to 42 (two copies of the favourable allele at all 21 SNPs). The observed scores ranged from 7 to 31 and those with the highest scores (19 or more favourable alleles) gained an average of 383 ml O2 min−1 more than those with the lowest scores (9 or less favourable alleles) (Fig. 2).
Figure 2. Adjusted training responses across nine GWAS SNP predictor score categories in HERITAGE white subjects.
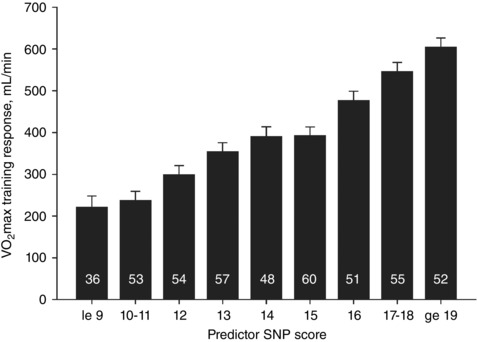
Numbers of subjects within each SNP score category are indicated inside each histogram bar. Mean values adjusted for age, sex and baseline . From Bouchard et al. (2011b). Reproduced with the permission of the American Physiological Society. le, less than or equal to; ge, greater than or equal to.
There was little replication with the top six most strongly associated SNPs in HERITAGE white subjects in the replication cohorts (HERITAGE black subjects, DREW and STRRIDE), with only one SNP showing nominal evidence of replication (Bouchard et al. 2011b). Specifically, rs11715829 (146 kb from ZIC4) was associated with the gains in in STRRIDE subjects (P = 0.02), with the directionality the same as in HERITAGE white subjects (major allele homozygotes gaining 30% less than minor allele carriers). Among the remaining SNPs tested for replication, several positive findings were found with directionality similar to that found in the discovery cohort (i.e. HERITAGE white subjects). In HERITAGE black subjects, the minor alleles of CAMTA1 rs884736 (P = 0.03) and RGS18 rs17581162 (P = 0.03) were associated with lesser gains in compared to major allele homozygotes. In DREW, the minor allele of DAAM1 SNP rs1956197 was associated with greater gains in compared to major allele homozygotes (P = 0.02). Overall, only 5 of the 15 tested SNPs showed partial evidence of replication in one of the replication cohorts, with no consistent findings across studies (Bouchard et al. 2011b). Although HERITAGE black subjects underwent the same exercise programme as HERITAGE white subjects, the population demographics and training programmes of DREW and STRRIDE were quite different and may play a role in the lack of replication. Furthermore, none of the SNPs from the transcriptomic predictor in Timmons et al. (2010) were part of the 21 predictors, although four SNPs from the early panel were associated with changes in HERITAGE white subjects at a P value of 0.008 or better (Bouchard et al. 2011b). This less than satisfactory replication may result from a number of factors, including small sample size, DNA variants or transcript signatures with small effect sizes, heterogeneity of study populations, differences in exercise programmes and timing of measurements, the possibility that the original findings were false positive, and potentially others as well.
Pathways, networks and systems
One of the key characteristics of complex traits is that they are polygenic, i.e. the result of combined contributions from several genes. In contrast to Mendelian traits, it is quite rare for single genes to exert large effects on a complex phenotype. Thus analytical approaches that are capable of interrogating joint contributions have become invaluable for complex trait research. Additionally, with the increasing public availability of large heterogeneous datasets, it has now become possible to conduct bioinformatically driven integrative analysis, incorporating evidence from multiple data types, to construct more powerful hypotheses about gene function. Thus, complementing the genomic and genetic association studies described above, further attempts to elucidate the genetic architecture underlying response to endurance exercise training have involved a combination of bioinformatics and systems biology approaches. These investigations display a balance between the hypotheses‐driven and data‐driven approaches and broadly fall into two categories: (a) identification of biological mechanisms (pathways and networks) that underlie the observed genetic heritability of response, and (b) identification of candidate genes via integrative data analysis.
As described earlier, traditional single marker analysis of the HERITAGE cohort did not yield any SNPs that could be reproducibly associated with changes in exercise‐induced at the genome‐wide significance level (Bouchard et al. 2011b). Consequently, no single genes could be identified that individually explained a significant proportion of the genetic variation in exercise response. Although a 21 SNP‐based genetic risk score could explain 49% of the variance in trainability, it did not adequately inform on the biology driving the response heterogeneity. To address this area, we applied methods drawn from systems biology (Ghosh et al. 2010, 2015; Wang et al. 2010; Zhong et al. 2010; Pemu et al. 2012; Lane et al. 2016) and modelled the response to exercise training as arising from joint contributions of modest effects from several genes. In this scenario, the significance was sought at the ‘gene‐set’ level instead of the ‘gene’ level. These gene‐sets represent collections of functionally or physically interacting genes (e.g. curated pathways such as Kyoto Encyclopedia of Genes and Genomes (KEGG)) (Kanehisa & Goto, 2000), or protein–protein interaction networks (e.g. STRING) (Szklarczyk et al. 2015). Using genetic association data from the HERITAGE GWAS, SNPs were first physically mapped to genes and a gene‐level trait‐association score, based on the association scores of its constituent SNPs, was formulated. These gene scores were used to query KEGG pathways via gene‐set enrichment analysis (GSEA) methods (Nam et al. 2010). This approach allowed for the identification of pathways related to immune function (e.g. type 1 diabetes, graft vs. host disease), cardiomyopathy (dilated and arrhythmogenic right ventricular cardiomyopathy), extracellular matrix regulation (ECM receptor interaction, adherens junction) and metabolism (PPAR signalling, pantothenate and CoA biosynthesis, etc.) as significantly associated to trainability due to the accumulation of moderately associated SNPs in their constituent genes. An alternative approach involving pathway over‐representation analysis (Ingenuity Pathway Analysis) further identified a network of 35 genes, for which 31/35 genes were associated with change in at P < 0.005. These network genes could be further partitioned into pathways related to calcium signalling, nitric oxide signalling and protein kinase A signalling (Ghosh et al. 2013). Together, these findings generated testable hypotheses regarding mechanisms that could influence the exercise‐induced training response through accumulation of constituent gene polymorphisms. Several of these pathways have also been identified in other studies investigating the effects of endurance exercise on skeletal muscle, such as pathways related to calcium signalling, cAMP signalling, AMP kinase activation and PPAR signalling (Hoppeler, 2016). Keeping in mind the increased energetic demands of endurance stress on muscle and other tissues, several of these pathways have been found to further converge on activation of transcriptional programmes directed at energy sensing and mitochondrial biogenesis, such as the transcriptional co‐activator PGC1α (Chan & Arany, 2014).
A second application of how bioinformatics analysis can inform exercise genetics is through the systematic querying and integration of complex heterogeneous databases (e.g. gene expression, phylogeny and sequence conservation, functional annotation, biomedical literature, etc.) for prioritization of candidate genes found to be associated to a phenotype. This is a necessary step because any computational analysis of large‐scale data results in tens or hundreds of candidate hypotheses, which somehow must be narrowed down for further functional testing. Several gene prioritization tools exist (Aerts et al. 2006), some of which are dependent on ‘training’ genes, and others not requiring prior information. In our studies, we used the hypothesis‐free gene prioritization tool CANDID (Hutz et al. 2008) to rank a list of candidate genes found to be moderately associated with changes in in GWAS analysis. A weighted analysis was performed, including text mining, protein domain information, sequence conservation, gene expression, linkage and genetic association results, to score and rank genes mapping to the top 10,000 SNPs (ranked by their association P values to Δ). The largest weight (50%) was assigned to the GWAS association score to ensure that the new results from association analysis would remain the major component of the combined CANDID score for each gene. This approach allowed us to characterize and rank the candidate genes based on their performance across the queried domains; for example, one group of genes (PINX1, CD44, PARK2, RYR2, ADCY5 and SHANK2) displayed overall strong scores across all categories, whereas a second group of genes (KCNQ5, GRIK4, RPTOR, ACVR1C and ACSL1) were strongly associated with some but not all phenotypes. Overall, these integrative analyses allowed us to identify panels of candidate genes using additional context relevant to gene function, instead of depending solely on genetic association scores.
The analyses summarized above, together with other bioinformatics explorations and experimental laboratory‐based studies, have generated useful information on several aspects of the molecular mechanisms that drive adaptations to exercise. The available evidence to date points to pathways related to calcium signalling, energy sensing and partitioning, mitochondrial biogenesis, angiogenesis, immune functions, and regulation of autophagy and apoptosis, among other pathways, as key mechanisms through which the physiological responses of to exercise training are mediated. Additionally, the application of systems biology and bioinformatics approaches have also led to new hypotheses involving novel gene candidates that could further illuminate the mechanisms underlying the response to regular exercise. Newer methodologies, such as those involving next generation sequencing, epigenetic mapping and analysis of non‐coding RNA, are likely to be critical in future research aimed at understanding and predicting individual differences in trainability.***
Trainability of : a working model
It has been recognized for a number of decades that measured during a test leading progressively to exhaustion is determined primarily by the capacity to transport oxygen (stroke volume, haemoglobin concentration and blood volume), the transfer of oxygen to the working muscles (capillary density and microcirculation, membrane permeability, muscle myoglobin content) and oxygen utilization to sustain the generation of ATP for muscle contraction (mitochondrial density and network, carrier, enzyme and protein concentrations). Among this constellation of factors, it is generally accepted that the capacity of the heart and the circulation to transport and deliver oxygen is the most critical determinant of (Blomqvist & Saltin, 1983; Lundby & Robach, 2015; Morales‐Alamo et al. 2015), although other components of the system can be limiting under specific circumstances (Dempsey et al. 2008; Boushel et al. 2011). Consequently, a high trainability of phenotype is unlikely to be achieved if maximal stroke volume and oxygen carrying capacity are not optimally increased by exercise training.
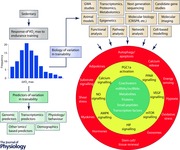
The main reasons for pursuing the investigation of the molecular basis of trainability are to clarify the mechanisms underlying human variation in the ability to respond to regular exercise and to potentially develop biomarkers and predictors of variability in trainability. These two research directions provide the foundation for the model depicted in the Abstract figure.
The model recognizes that if powerful predictors, with appropriate sensitivity and specificity, can be identified, they are likely to include information from genomics and other omics as well as from physiology, behaviour and other personal characteristics. It is, however, uncertain at this stage whether such powerful diagnostics can be derived for trainability of .
The model also defines examples of technologies, experimental models, and research designs and strategies that can be relied on to undertake the dissection of the molecular basis of trainability. Finally, the three adjacent circles of the model depict physiological and metabolic mechanisms, signalling pathways, and subsets of molecules that have been shown to be involved in adaptation to acute or chronic exercise in a variety of research protocols (Hoppeler et al. 2011; Keller et al. 2011; Bouchard et al. 2011a; Egan & Zierath, 2013; several chapters in Bouchard, 2015; Hoppeler, 2016). Although the model does not specify the tissues and organs of interest, the search for molecular transducers driving adaptation to endurance exercise has thus far been highly skeletal muscle centric. It would be important to broaden the search to other tissues and organs, particularly cardiac muscle, blood vessels and endothelium, adipose tissues, liver, pancreas and others as well. Also highlighted in the model are technologies that can be used to probe the extent and nature of individual differences in trainability.
Conclusions
is determined by the capacity to deliver oxygen via the circulation to the working muscle and by the diffusion and utilization of oxygen by these muscles. However, research has shown that there are large individual differences in the gains in even when adults are exposed to a standardized and fully monitored exercise training programme. The estimated heritability of the trainability of reaches 47%. Candidate gene, genome‐wide linkage and GWAS have not elucidated the molecular basis of trainability. On the other hand, interrogating genomic predictors and skeletal muscle transcript abundance profiling has yielded a number of leads on the up‐regulation or down‐regulation of various systems limiting and related signalling pathways particularly in skeletal muscle in response to exercise training. Bioinformatics explorations based on thousands of SNPs have been used to interrogate pathways and systems instead of single variants and genes, and the main findings have been used to develop a working model. Research on the molecular drivers of trainability has the potential to improve our understanding of the molecular transducers of adaptation to regular exercise and could also inform the development of biomarkers and perhaps predictors of the ability to improve cardiorespiratory fitness in response to exercise training.
Additional information
Competing interests
M.A.S. is a consultant for Genetic Direction. The other authors have no conflicts of interest to declare.
Funding
The research reviewed in this paper was partially funded by the US National Institutes of Health (HL‐45670). C.B. is partially funded by the John W. Barton Sr Chair in Genetics and Nutrition.
Biographies
Mark Sarzynski received his PhD from Michigan State University, USA and completed a postdoctoral fellowship at Pennington Biomedical Research Centre, USA. He is now an Assistant Professor at the University of South Carolina, where his research employs an integrated omics approach to examine the effects of exercise on lipids and lipoproteins.

Sujoy Ghosh is an Associate Professor at Duke‐NUS Medical School, Singapore, and Pennington Biomedical Research Centre, USA. He obtained his PhD from the University of Notre Dame, USA, followed by academic and pharmaceutical work experience. His current research applies statistical, bioinformatics and functional approaches to elucidate the genetics of cardiometabolic phenotypes.
Claude Bouchard is the John W. Barton Sr Chair in Genetics and Nutrition at Pennington Biomedical Research Centre. His research focuses on the genetics of obesity and co‐morbidities as well as on the genetics of cardiorespiratory fitness and adaptation to exercise.
This review was presented at the symposium “New technologies providing insight to human physiological adaptation”, which took place at the meeting of The Biomedical Basis of Elite Performance in Nottingham, UK, 6–8 March 2016.
References
- Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, Carmeliet P & Moreau Y (2006). Gene prioritization through genomic data fusion. Nat Biotechnol 24, 537–544. [DOI] [PubMed] [Google Scholar]
- Bennett AF (1987). Interindividual variability: an underutilized resource In New Directions in Ecological Physiology, ed. Feder ME, Bennett A, Burggren W. & Huey R, pp. 147–169. Cambridge University Press, Cambridge, New York. [Google Scholar]
- Blache D, Lussier‐Cacan S, Gagnon J, Leon AS, Rao DC, Skinner JS, Wilmore JH, Rankinen T, Bouchard C & Davignon J (2007). Effect of exercise training on in vitro LDL oxidation and free radical‐induced hemolysis: the HERITAGE Family Study. Antioxid Redox Signal 9, 123–130. [DOI] [PubMed] [Google Scholar]
- Blomqvist CG & Saltin B (1983). Cardiovascular adaptations to physical training. Annu Rev Physiol 45, 169–189. [DOI] [PubMed] [Google Scholar]
- Bouchard C (1983). Human adaptability may have a genetic basis In Health Risk Estimation, Risk Reduction and Health Promotion. Proceedings of the 18th Annual Meeting of the Society of Prospective Medicine, ed. Landry F, pp. 463–476. Canadian Public Health Association, Ottawa. [Google Scholar]
- Bouchard C (2012). Genomic predictors of trainability. Exp Physiol 97, 347–352. [DOI] [PubMed] [Google Scholar]
- Bouchard C (2015). Progress in Molecular Biology and Translational Science, vol. 135 Elsevier/AP, Waltham, MA, USA. [Google Scholar]
- Bouchard C, An P, Rice T, Skinner JS, Wilmore JH, Gagnon J, Perusse L, Leon AS & Rao DC (1999). Familial aggregation of VO2max response to exercise training: results from the HERITAGE Family Study. J Appl Physiol (1985) 87, 1003–1008. [DOI] [PubMed] [Google Scholar]
- Bouchard C, Blair SN, Church TS, Earnest CP, Hagberg JM, Hakkinen K, Jenkins NT, Karavirta L, Kraus WE, Leon AS, Rao DC, Sarzynski MA, Skinner JS, Slentz CA & Rankinen T (2012). Adverse metabolic response to regular exercise: is it a rare or common occurrence? PLoS One 7, e37887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C & Rankinen T (2001). Individual differences in response to regular physical activity. Med Sci Sports Exerc 33 (Suppl.) S452–S443, S446–451 discussion. [DOI] [PubMed] [Google Scholar]
- Bouchard C, Rankinen T, Chagnon YC, Rice T, Perusse L, Gagnon J, Borecki I, An P, Leon AS, Skinner JS, Wilmore JH, Province M & Rao DC (2000). Genomic scan for maximal oxygen uptake and its response to training in the HERITAGE Family Study. J Appl Physiol (1985) 88, 551–559. [DOI] [PubMed] [Google Scholar]
- Bouchard C, Rankinen T & Timmons JA (2011a). Genomics and genetics in the biology of adaptation to exercise. Compr Physiol 1, 1603–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Sarzynski MA, Rice TK, Kraus WE, Church TS, Sung YJ, Rao DC & Rankinen T (2011b). Genomic predictors of the maximal O2 uptake response to standardized exercise training programs. J Appl Physiol (1985) 110, 1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Tremblay A, Després JP, Thériault G, Nadeau A, Lupien PJ, Moorjani S, Prudhomme D & Fournier G (1994). The response to exercise with constant energy intake in identical twins. Obes Res 2, 400–410. [DOI] [PubMed] [Google Scholar]
- Boule NG, Weisnagel SJ, Lakka TA, Tremblay A, Bergman RN, Rankinen T, Leon AS, Skinner JS, Wilmore JH, Rao DC, Bouchard C & Study HF (2005). Effects of exercise training on glucose homeostasis: the HERITAGE Family Study. Diabetes Care 28, 108–114. [DOI] [PubMed] [Google Scholar]
- Boushel R, Gnaiger E, Calbet JA, Gonzalez‐Alonso J, Wright‐Paradis C, Sondergaard H, Ara I, Helge JW & Saltin B (2011). Muscle mitochondrial capacity exceeds maximal oxygen delivery in humans. Mitochondrion 11, 303–307. [DOI] [PubMed] [Google Scholar]
- Bray MS, Hagberg JM, Perusse L, Rankinen T, Roth SM, Wolfarth B & Bouchard C (2009). The human gene map for performance and health‐related fitness phenotypes: the 2006‐2007 update. Med Sci Sports Exerc 41, 35–73. [DOI] [PubMed] [Google Scholar]
- Chan MC & Arany Z (2014). The many roles of PGC‐1α in muscle – recent developments. Metabolism 63, 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey JA, McKenzie DC, Haverkamp HC & Eldridge MW (2008). Update in the understanding of respiratory limitations to exercise performance in fit, active adults. Chest 134, 613–622. [DOI] [PubMed] [Google Scholar]
- Egan B & Zierath JR (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17, 162–184. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Dent R, Harper ME, Gorman SA, Stuart JS & McPherson R (2010). Gene expression profiling in whole blood identifies distinct biological pathways associated with obesity. BMC Med Genomics 3, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Vivar J, Nelson CP, Willenborg C, Segre AV, Makinen VP, Nikpay M, Erdmann J, Blankenberg S, O'Donnell C, Marz W, Laaksonen R, Stewart AF, Epstein SE, Shah SH, Granger CB, Hazen SL, Kathiresan S, Reilly MP, Yang X, Quertermous T, Samani NJ, Schunkert H, Assimes TL & McPherson R (2015). Systems genetics analysis of genome‐wide association study reveals novel associations between key biological processes and coronary artery disease. Arterioscler Thromb Vasc Biol 35, 1712–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Vivar JC, Sarzynski MA, Sung YJ, Timmons JA, Bouchard C & Rankinen T (2013). Integrative pathway analysis of a genome‐wide association study of VO2max response to exercise training. J Appl Physiol (1985) 115, 1343–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel P, Simoneau JA, Lortie G, Boulay MR & Bouchard C (1986). Heredity and muscle adaptation to endurance training. Med Sci Sports Exerc 18, 690–696. [PubMed] [Google Scholar]
- Hill AV & Lupton H (1923). Muscular exercise, lactic acid, and the supply and utilization of oxygen. Q J Med 16, 135–171. [Google Scholar]
- Hoppeler H (2016). Molecular networks in skeletal muscle plasticity. J Exp Biol 219, 205–213. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Baum O, Lurman G & Mueller M (2011). Molecular mechanisms of muscle plasticity with exercise. Compr Physiol 1, 1383–1412. [DOI] [PubMed] [Google Scholar]
- Hutz JE, Kraja AT, McLeod HL & Province MA (2008). CANDID: a flexible method for prioritizing candidate genes for complex human traits. Genet Epidemiol 32, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M & Goto S (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzmarzyk PT, Leon AS, Wilmore JH, Skinner JS, Rao DC, Rankinen T & Bouchard C (2003). Targeting the metabolic syndrome with exercise: evidence from the HERITAGE Family Study. Med Sci Sports Exerc 35, 1703–1709. [DOI] [PubMed] [Google Scholar]
- Keller P, Vollaard NB, Gustafsson T, Gallagher IJ, Sundberg CJ, Rankinen T, Britton SL, Bouchard C, Koch LG & Timmons JA (2011). A transcriptional map of the impact of endurance exercise training on skeletal muscle phenotype. J Appl Physiol (1985) 110, 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch LG, Pollott GE & Britton SL (2013). Selectively bred rat model system for low and high response to exercise training. Physiol Genomics 45, 606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakka TA, Lakka HM, Rankinen T, Leon AS, Rao DC, Skinner JS, Wilmore JH & Bouchard C (2005). Effect of exercise training on plasma levels of C‐reactive protein in healthy adults: the HERITAGE Family Study. Eur Heart J 26, 2018–2025. [DOI] [PubMed] [Google Scholar]
- Lane JM, Vlasac I, Anderson SG, Kyle SD, Dixon WG, Bechtold DA, Gill S, Little MA, Luik A, Loudon A, Emsley R, Scheer FA, Lawlor DA, Redline S, Ray DW, Rutter MK & Saxena R (2016). Genome‐wide association analysis identifies novel loci for chronotype in 100,420 individuals from the UK Biobank. Nat Commun 7, 10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon AS, Rice T, Mandel S, Després JP, Bergeron J, Gagnon J, Rao DC, Skinner JS, Wilmore JH & Bouchard C (2000). Blood lipid response to 20 weeks of supervised exercise in a large biracial population: the HERITAGE Family Study. Metabolism 49, 513–520. [DOI] [PubMed] [Google Scholar]
- Lortie G, Simoneau JA, Hamel P, Boulay MR, Landry F & Bouchard C (1984). Responses of maximal aerobic power and capacity to aerobic training. Int J Sports Med 5, 232–236. [DOI] [PubMed] [Google Scholar]
- Lundby C & Robach P (2015). Performance enhancement: What are the physiological limits? Physiology (Bethesda) 30, 282–292. [DOI] [PubMed] [Google Scholar]
- Morales‐Alamo D, Losa‐Reyna J, Torres‐Peralta R, Martin‐Rincon M, Perez‐Valera M, Curtelin D, Ponce‐Gonzalez JG, Santana A & Calbet JA (2015). What limits performance during whole‐body incremental exercise to exhaustion in humans? J Physiol 593, 4631–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam D, Kim J, Kim SY & Kim S (2010). GSA‐SNP: a general approach for gene set analysis of polymorphisms. Nucleic Acids Res 38, W749–W754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemu PE, Anderson L, Gee BE, Ofili EO & Ghosh S (2012). Early alterations of the immune transcriptome in cultured progenitor cells from obese African‐American women. Obesity (Silver Spring) 20, 1481–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud'homme D, Bouchard C, Leblanc C, Landry F & Fontaine E (1984). Sensitivity of maximal aerobic power to training is genotype‐dependent. Med Sci Sports Exerc 16, 489–493. [DOI] [PubMed] [Google Scholar]
- Rico‐Sanz J, Rankinen T, Rice T, Leon AS, Skinner JS, Wilmore JH, Rao DC & Bouchard C (2004). Quantitative trait loci for maximal exercise capacity phenotypes and their responses to training in the HERITAGE Family Study. Physiol Genomics 16, 256–260. [DOI] [PubMed] [Google Scholar]
- Shephard RJ, Rankinen T & Bouchard C (2004). Test‐retest errors and the apparent heterogeneity of training response. Eur J Appl Physiol 91, 199–203. [DOI] [PubMed] [Google Scholar]
- Simoneau JA, Lortie G, Boulay MR, Marcotte M, Thibault MC & Bouchard C (1986). Inheritance of human skeletal muscle and anaerobic capacity adaptation to high‐intensity intermittent training. Int J Sports Med 7, 167–171. [DOI] [PubMed] [Google Scholar]
- Snell PG, Stray‐Gundersen J, Levine BD, Hawkins MN & Raven PB (2007). Maximal oxygen uptake as a parametric measure of cardiorespiratory capacity. Med Sci Sport Exerc 39, 103–107. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta‐Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ & von Mering C (2015). STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Knudsen S, Rankinen T, Koch LG, Sarzynski M, Jensen T, Keller P, Scheele C, Vollaard NB, Nielsen S, Akerström T, MacDougald OA, Jansson E, Greenhaff PL, Tarnopolsky MA, van Loon LJ, Pedersen BK, Sundberg CJ, Wahlestedt C, Britton SL & Bouchard C (2010). Using molecular classification to predict gains in maximal aerobic capacity following endurance exercise training in humans. J Appl Physiol (1985) 108, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troxell ML, Britton SL & Koch LG (2003). Selected contribution: Variation and heritability for the adaptational response to exercise in genetically heterogeneous rats. J Appl Physiol (1985) 94, 1674–1681. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M & Hakonarson H (2010). Analysing biological pathways in genome‐wide association studies. Nat Rev Genet 11, 843–854. [DOI] [PubMed] [Google Scholar]
- Wilmore JH, Green JS, Stanforth PR, Gagnon J, Rankinen T, Leon AS, Rao DC, Skinner JS & Bouchard C (2001). Relationship of changes in maximal and submaximal aerobic fitness to changes in cardiovascular disease and non‐insulin‐dependent diabetes mellitus risk factors with endurance training: the HERITAGE Family Study. Metabolism 50, 1255–1263. [DOI] [PubMed] [Google Scholar]
- Zhong H, Yang X, Kaplan LM, Molony C & Schadt EE (2010). Integrating pathway analysis and genetics of gene expression for genome‐wide association studies. Am J Hum Genet 86, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]