

Abstract
Optimising training and performance through nutrition strategies is central to supporting elite sportspeople, much of which has focused on manipulating the relative intake of carbohydrate and fat and their contributions as fuels for energy provision. The ketone bodies, namely acetoacetate, acetone and β‐hydroxybutyrate (βHB), are produced in the liver during conditions of reduced carbohydrate availability and serve as an alternative fuel source for peripheral tissues including brain, heart and skeletal muscle. Ketone bodies are oxidised as a fuel source during exercise, are markedly elevated during the post‐exercise recovery period, and the ability to utilise ketone bodies is higher in exercise‐trained skeletal muscle. The metabolic actions of ketone bodies can alter fuel selection through attenuating glucose utilisation in peripheral tissues, anti‐lipolytic effects on adipose tissue, and attenuation of proteolysis in skeletal muscle. Moreover, ketone bodies can act as signalling metabolites, with βHB acting as an inhibitor of histone deacetylases, an important regulator of the adaptive response to exercise in skeletal muscle. Recent development of ketone esters facilitates acute ingestion of βHB that results in nutritional ketosis without necessitating restrictive dietary practices. Initial reports suggest this strategy alters the metabolic response to exercise and improves exercise performance, while other lines of evidence suggest roles in recovery from exercise. The present review focuses on the physiology of ketone bodies during and after exercise and in response to training, with specific interest in exploring the physiological basis for exogenous ketone supplementation and potential benefits for performance and recovery in athletes.
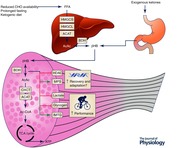
Keywords: acetoacetate, β‐hydroxybutyrate, ketosis, performance, substrate
Abbreviations
- AcAc
acetoacetate
- AcAc‐CoA
acetoacetyl CoA
- ACAT
acetyl‐CoA acetyltransferase
- βHB
β‐hydroxybutyrate
- BDH
3‐hydroxybutyrate dehydrogenase
- CHO
carbohydrate
- CPT1
carnitine palmitoyltransferase
- FFA
free‐fatty acid
- HDAC
histone deacetylase
- HMG‐CoA
hydroxymethylglutaryl‐CoA
- HMGCL
HMG‐CoA lyase
- HMGCS
HMG CoA synthase
- KB
ketone body
- KE
(R)‐3‐hydroxybutyl (R)‐3‐hydroxybutyrate ketone monoester
- MCT
monocarboxylate transporter
- OXCT
succinyl‐CoA:3‐oxoacid CoA transferase
- PDH
pyruvate dehydrogenase
- PEK
post‐exercise ketosis
- PFK
phosphofructokinase
- PGC‐1
peroxisome proliferator‐activated receptor gamma coactivator 1
- SLC
solute ligand carrier
- TCA
tricarboxylic acid
Introduction
Over the past century, exercise physiologists have appreciated the role of carbohydrate (CHO) and fat in energy provision to exercising skeletal muscle. Much of the work examining the metabolic response to exercise and the impact of exercise on metabolic regulation and adaptive responses to training has focused on the relative contribution of these fuels (Egan & Zierath, 2013). Optimising training and nutrition strategies by manipulating the relative intakes of these macronutrients is central to supporting elite sports performance (Cermak & van Loon, 2013; Bartlett et al. 2015; Burke, 2015). An alternative fuel source to CHO and fat are ketone bodies (KBs), namely acetoacetate (AcAc), acetone, and β‐hydroxybutyrate (βHB), which are produced in the liver during physiological states and nutritional manipulations that result in reduced CHO availability, most commonly during prolonged fasting, starvation, and ketogenic (very low CHO (∼5%), low protein (∼15%), high fat (∼80%)) diets (Robinson & Williamson, 1980; Laffel, 1999). This relative glucose deprivation and concomitant elevation in circulating free‐fatty acids (FFAs) results in the production of KBs to replace glucose as the primary fuel for peripheral tissues such as the brain, heart and skeletal muscle in these states.
Aside from a role as an alternative fuel source, KBs exert a range of metabolic effects including attenuating glucose utilisation in peripheral tissues, anti‐lipolytic effects on adipose tissue, and potential attenuation of proteolysis in skeletal muscle (Robinson & Williamson, 1980). KBs are utilised by working muscle during exercise (Fery & Balasse, 1986, 1988), and the capacity to take up and oxidise KBs during exercise is higher in exercise‐trained skeletal muscle (Winder et al. 1975). Despite these observations, in addition to a glucose sparing action (Maizels et al. 1977) and potential to lower the exercise‐induced rise in plasma [lactate] (Fery & Balasse, 1988), the potential performance benefits of KBs when provided as an exogenous fuel source has received little attention, but has been postulated (Cox & Clarke, 2014; Pinckaers et al. 2017). Apart from a role as an alternative fuel source, KBs may act as signalling molecules to regulate gene expression and adaptive responses (Shimazu et al. 2013; Zou et al. 2016). Moreover, therapeutic roles for KBs have long been proposed in a variety of disease states including aberrant glucose metabolism, genetic myopathies, hypoxic states and neurodegenerative pathologies (Veech, 2004). For therapeutic effects, exogenous ketones are ingested in the form of βHB salts or ketone esters to produce acute (∼0.5 to 6 h) nutritional ketosis (Clarke et al. 2012; Kesl et al. 2016), but a surge in interest in KBs as a performance aid for athletes arose when ketone ester supplementation was confirmed in professional cycling (Abraham, 2015; Pinckaers et al. 2017). Moreover, a recent report provides the first evidence for acute nutritional ketosis achieved by ketone ester ingestion to alter the metabolic response to exercise and enhance exercise performance (Cox et al. 2016). Aspects of ketogenic diets, ketogenesis and ketone body metabolism have been reviewed elsewhere (Robinson & Williamson, 1980; Laffel, 1999; Paoli et al. 2013), so the present review will focus on the physiology of ketone bodies during and after exercise and in response to training, with specific interest in exploring the physiological basis for exogenous supplementation and potential benefits for performance and recovery in athletes.
Overview of ketone body metabolism
Ketone bodies in circulation
Plasma [KB] reflects the balance between hepatic production (‘ketogenesis’) and peripheral breakdown and utilisation (‘ketolysis’) in extra‐hepatic tissues, both of which are under various levels of control as detailed in previous reviews (Robinson & Williamson, 1980; Laffel, 1999). Ketogenesis is an evolutionarily conserved adaptive response playing a critical role in survival during an energy crisis by providing a substrate for brain, which cannot utilise FFAs as a fuel source. AcAc, acetone, and βHB comprise the KBs, although βHB is not technically a ketone because the ketone moiety has been reduced to a hydroxyl group. AcAc and βHB are short‐chain, four carbon organic acids that act as FFA‐derived circulating substrates to provide energy to extra‐hepatic tissues, whereas the contribution of acetone, readily generated by the spontaneous decarboxylation of AcAc, to energy provision is negligible. Plasma [KB] is <0.1 mm in the postprandial state, whereas hyperketonaemia is accepted as [KB] exceeding 0.2 mm (Robinson & Williamson, 1980). Various states of CHO restriction, depletion and dysregulation produce hyperketonaemia to different degrees (Fig. 1).
Figure 1. Changes in [βHB] under various physiological states.
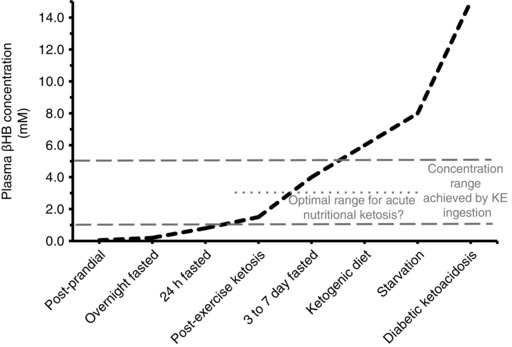
Plasma [KB] is <0.1 mm in the postprandial state when consuming high CHO or high protein meals, and rises upward after an overnight fast and with ketogenic dieting, prolonged fasting, starvation, and pathological states of ketoacidosis. After prolonged aerobic exercise, post‐exercise ketosis (0.3 to 2.0 mm) may ensue depending on intensity and duration of exercise, aerobic fitness and nutrition status. The circulating KB ratio of βHB:AcAc is generally ∼1:1 to 3:1, but during the aforementioned nutritional states can rise six‐ to tenfold, such that [KB] primarily reflects changes in [βHB]. An optimal concentration range for βHB to improve performance after exogenous ketone ingestion is proposed as ∼1 to 3 mm, with concentrations ranging from ∼1 to 5 mm reported after ketone ester (KE) ingestion. See text for further details.
Ketogenesis
The primary substrate for ketogenesis is FFAs liberated from adipose tissue. Ketogenic amino acids, namely leucine, lysine, phenylalanine, isoleucine, tryptophan, and tyrosine also serve ketogenesis, but are likely contribute to less than 5% of circulating KBs (Thomas et al. 1982). The rise in FFAs is consequent to the stimulation of lipolysis as a result of declines in plasma glucose and insulin that are characteristic of reduced CHO availability. Factors stimulating ketogenesis include an elevated glucagon‐to‐insulin ratio and decline in hepatic glycogen concentration, while reduced blood flow to the liver or elevations in [KBs] suppress ketogenesis (Robinson & Williamson, 1980; Laffel, 1999). Ketogenesis involves a series of sequential reactions beginning with acetyl CoA (Ac‐CoA) and acetoacetyl CoA (AcAc‐CoA), and ending with the liberation of AcAc (Fig. 2). Some AcAc is exported, but the majority is reduced to βHB in an NAD+–NADH‐coupled near equilibrium reaction catalysed by 3‐hydroxybutyrate dehydrogenase (BDH), in which the equilibrium constant favours βHB formation. These KBs are transported into the circulation via the solute ligand carrier (SLC) protein 16A (SLC16A) family of monocarboxylate transporters (MCTs) in mitochondrial and sarcolemmal membranes.
Figure 2. Metabolic pathways of ketone body metabolism in liver and skeletal muscle.
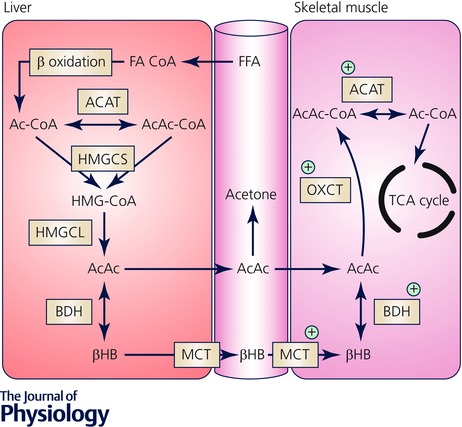
Ketogenesis: FFAs are converted to fatty acyl CoA (FA‐CoA), enter hepatic mitochondria via CPT1‐mediated transport and undergo β‐oxidation to acetyl CoA. Sequential reactions of condensation of Ac‐CoA molecules to acetoacetyl CoA (AcAc‐CoA) by mitochondrial thiolase activity of Ac‐CoA acetyltransferase (ACAT), generation of hydroxymethylglutaryl‐CoA (HMG‐CoA) by hydroxymethylglutaryl CoA synthase (HMGCS), and decomposition of HMG‐CoA, liberating AcAc and Ac‐CoA, in a reaction catalysed by HMG‐CoA lyase (HMGCL). AcAc is the central KB, and some will be exported to the circulation but the majority is reduced to βHB in an NAD+–NADH‐coupled near equilibrium reaction catalysed by BDH, in which the equilibrium constant favours βHB formation. Ketolysis: The only metabolic fate of βHB is inter‐conversion with AcAc, and upon entry into peripheral tissues it is re‐oxidised to AcAc. Covalent activation of AcAc by CoA is catalysed by succinyl‐CoA:3‐oxoacid CoA transferase (OXCT) resulting in generation of AcAc‐CoA. This near equilibrium reaction exchanges CoA between succinate and AcAc, with succinyl‐CoA acting as a CoA donor. Because the free energy released by hydrolysis of AcAc‐CoA is greater than that of succinyl‐CoA, the equilibrium of this reaction thermodynamically favours the formation of AcAc. Two molecules of Ac‐CoA are liberated by thiolytic cleavage of AcAc‐CoA by ACAT, after which Ac‐CoA is incorporated into the TCA cycle. Protein content and enzyme activity that are higher in exercise‐trained skeletal muscle are indicated by the green cross (+).
Ketolysis in extra‐hepatic tissues
In peripheral tissues, KBs, primarily in the form of βHB, enter the mitochondrial matrix again via MCT1‐mediated transport. βHB is re‐oxidised to AcAc via BDH after which sequential reactions result in the generation of two molecules of Ac‐CoA (Fig. 2). These are incorporated into the TCA cycle via citrate synthase for terminal oxidation and production of ATP, which in skeletal muscle contributes to fuelling muscular work (Fery & Balasse, 1986, 1988). Succinyl‐CoA:3‐oxoacid CoA transferase (OXCT) is essential for ketolysis in extra‐hepatic tissues, with very low abundance in hepatocytes explaining the lack of ketolytic activity in these cells (Robinson & Williamson, 1980).
Activity of OXCT is highest in heart and kidney, followed by skeletal muscle and the brain (Robinson & Williamson, 1980), but because skeletal muscle accounts for ∼40% of body mass in adult humans, this organ accounts for the highest fraction of total KB metabolism at rest (Balasse & Fery, 1989; Laffel, 1999). Beginning almost 50 years ago, models using various durations of fasting, and combined with primed constant infusion of radiolabelled either AcAc or βHB tracers and arteriovenous difference measures to quantify KB turnover, established that skeletal muscle is a major site of ketolysis at rest (Hagenfeldt & Wahren, 1968; Owen & Reichard, 1971; Wahren et al. 1984; Elia et al. 1990; Mikkelsen et al. 2015). Skeletal muscle has a high affinity to KBs, but because of low circulating concentrations under normal conditions, the contribution to energy provision in muscle is less than 5%, and FFAs are the main source of energy provision in the post‐absorptive state. The relationship between ketone oxidation and [KB] is curvilinear such that contribution to energy provision in skeletal muscle rises to ∼10% after an overnight fast (Hagenfeldt & Wahren, 1968; Owen & Reichard, 1971), 20% to 50% after 72 h of fasting (Owen & Reichard, 1971; Elia et al. 1990), but declines to ∼15% after 24 days of starvation (Owen & Reichard, 1971). Thus, skeletal muscle demonstrates saturation kinetics for the KB concentration–oxidation relationship, with saturation likely between 1 and 2 mm as demonstrated by fasting of various durations (compiled in Balasse & Fery, 1989) or step‐wise βHB infusion (Mikkelsen et al. 2015).
Effect of aerobic exercise training on enzymes of ketogenesis and ketolysis
Adaptations to exercise training reduce perturbations to homeostasis during subsequent bouts of exercise, and thereby enhance resistance to fatigue. Central to these effects are enhanced respiratory capacity and contractile parameters, and importantly adaptations that contribute towards maximising delivery and utilisation of circulating substrates (reviewed in Egan & Zierath, 2013). Therefore, if KBs make a meaningful contribution to energy provision during exercise, it is pertinent to explore analogous regulation in skeletal muscle. Training‐induced changes in expression and activities of enzymes of ketolysis in skeletal muscle have not been described in humans, but differences in KB metabolism during and after exercise between trained and untrained individuals have been reported (Johnson et al. 1969; Johnson & Walton, 1972; Rennie et al. 1974; Rennie & Johnson, 1974a). The general pattern is for attenuation in trained individuals of the post‐exercise rise in [KB], but this is influenced by nutritional manipulation and relative exercise intensity, the latter of which has often been poorly controlled (see later sections).
Nevertheless, circulating concentrations reflect the balance between ketogenesis and ketolysis, these differences may be explained by the factors influencing one or both. For ketogenesis, data are limited but suggest that in exercise‐trained rodents enzymatic activity of BDH or ACAT (Winder et al. 1974), or HMGCS (Askew et al. 1975) is unaltered in liver, and, in fact, the overall activity of the ketogenic pathway may be lower (El Midaoui et al. 2006) compared to untrained rodents. In these rodent models of intense aerobic exercise training, the activities of the ketolytic enzymes BDH, OXCT and ACAT are higher in trained skeletal muscle (Winder et al. 1974, 1975; Askew et al. 1975; Beattie & Winder, 1984). This coincides with two‐ to threefold higher ex vivo rates of βHB and AcAc oxidation in gastrocnemius muscle homogenates presented with concentrations of both βHB and AcAc at 0.1 and 0.5 mm (Winder et al. 1973, 1975).
In terms of muscle fibre type, enzymatic activities of BDH, OXCT and ACAT are all highest in type I fibres, intermediate in type IIA fibres, and lowest in type IIB fibres of rats (Winder et al. 1974). BDH is essentially undetectable in type IIB muscle fibres, and across the fibre types BDH activity is much lower than activities of OXCT and ACAT (Winder et al. 1974). Although OXCT is essential for ketolysis, BDH activity is, therefore, potentially rate limiting in skeletal muscle. When rats performed 12 weeks of treadmill running, compared to sedentary rats BDH activity was almost threefold higher in type I fibres, but sixfold higher in type IIA fibres of trained skeletal muscle, resulting in levels comparable to the type I fibres (Winder et al. 1974). OXCT activity was 26% higher in type I, and approximately twofold higher in type IIA and IIB fibres, whereas ACAT activity was 40% to 45% higher in all three fibre types in trained skeletal muscle (Winder et al. 1974). Similarly, in skeletal muscle from mice with 8 weeks of access to running wheels, the difference compared to sedentary mice was greater for BDH mRNA expression (∼twofold higher than sedentary) compared to differences in OXCT and ACAT mRNA expression (∼30% to 50% higher) (Svensson et al. 2016). These changes in ketolytic enzymes are localised to the working muscle given the absence of change after training in the heart (Askew et al. 1975), kidney and brain (Winder et al. 1974).
In terms of KB transport into skeletal muscle, similarly to the ketolytic enzymes, MCT1 protein expression is highest in type I fibres, poorly expressed in type II fibres, and correlates well with muscle oxidative capacity (Bonen, 2001). Elevated MCT1 protein expression after exercise training is well‐established for human skeletal muscle, and increases occur in an intensity‐dependent manner (Thomas et al. 2012). Using a rodent perfused hindlimb model, the capacity for uptake of KBs in skeletal muscle at 1 mm each of βHB and AcAc was higher in an aerobically trained group of rats, with uptake of total KB, AcAc and βHB 33%, 27% and 53% higher, respectively, compared to untrained rats (Ohmori et al. 1990). Similarly, βHB clearance during a βHB tolerance test is higher in mice given 8 weeks of running wheel access, or with enhanced oxidative capacity consequent to skeletal muscle overexpression of PGC‐1α, a transcriptional co‐activator and master regulator of mitochondrial biogenesis in adaptive responses such as exercise training (Svensson et al. 2016). In both conditions, this coincides with elevated expression of MCT1 and the ketolytic enzymes in skeletal muscle. Therefore, the uptake and utilisation of KBs in skeletal muscle is likely to be greatest in those individuals that are highly trained with a high proportion of type I muscle fibres and a high oxidative capacity in skeletal muscle.
Ketone body metabolism during exercise
The existing literature on fuel selection during exercise has focused almost exclusively on utilisation of CHO and fat, but skeletal muscle has the ability to resynthesize ATP from other substrates including protein, lactate and KBs (Fery & Balasse, 1986, 1988; Mazzeo et al. 1986; Wagenmakers et al. 1991). With increasing exercise intensity, the contribution of substrates to energy provisions shifts from blood‐borne FFAs and glucose to increased reliance on intramuscular fuel stores, namely intramuscular triglyceride (IMTG) and muscle glycogen, such that at moderate to high intensities (>75% ) of exercise, muscle glycogen is the main source of energy provision (van Loon et al. 2001). This pattern is readily altered by nutritional manipulation such as CHO loading and acute CHO ingestion resulting in increased CHO utilisation (Bosch et al. 1996), glycogen depletion resulting in increased contribution of protein to energy provision (Wagenmakers et al. 1991), and habitual high fat consumption resulting in increased contribution of fat to energy provision (Volek et al. 2016). Clearly, skeletal muscle is a major site of ketolysis under fasting conditions, but central to the rationale for exogenous ketone supplementation must be the observations that ketolysis increases during exercise, makes a meaningful contribution to energy provision, and can alter patterns of substrate utilisation.
The pioneering work of Hagenfeldt, Wahren and colleagues (Hagenfeldt & Wahren, 1968, 1971; Wahren et al. 1984) and Fery, Balasse and colleagues (Balasse et al. 1978; Fery & Balasse, 1983, 1986, 1988) established that KB disposal into human skeletal muscle is elevated as much as fivefold during exercise. This is generally reflected by a drop in [KB] soon after the onset of exercise, primarily βHB, concomitant with increases in KB oxidation in skeletal muscle and elevated metabolic clearance rate (MCR). MCR is a measure of the ability of tissues to remove ketones from the blood, analogous to arteriovenous difference per unit time, but when measured during exercise is taken to represent an index of the ability of exercise to stimulate the capacity of working muscles to extract and utilise ketones (Fery & Balasse, 1983; Balasse & Fery, 1989). Because the stoichiometry of KB oxidation yields respiratory quotients of 1.00 and 0.89 for AcAc and βHB, respectively (Frayn, 1983), calculation of oxidation rates for KBs from whole‐body gas exchange data has not been routinely performed using methods that determine the relative contribution of CHO and fat oxidation. However, a recent attempt has been made (Cox et al. 2016) based on methods and assumptions described for KB utilisation during ketogenesis (Frayn, 1983). Previous to this, oxidation rates for KBs have historically been derived from arteriovenous differences of radiolabelled KBs across working muscles with rates calculated as a fraction of O2 consumption or CO2 production (Hagenfeldt & Wahren, 1968; Balasse et al. 1978).
Like CHO and fat utilisation, KB metabolism during exercise is influenced by a variety of factors including metabolic status (Wahren et al. 1984; Fery & Balasse, 1986), training status (Johnson & Walton, 1972; Rennie et al. 1974; Beattie & Winder, 1985), and the intensity of exercise (Cox et al. 2016). Given the aforementioned fibre type‐specific differences for activities of ketolytic enzymes, the muscle fibre type profile of the working muscle is also likely to be an important determinant of ketolysis during exercise. However, the most important determinant of KB metabolism during exercise is the degree of ketonaemia, and the method by which this is achieved, i.e. of endogenous or exogenous origin.
Ketone body metabolism during exercise under conditions of endogenous ketosis
Like KB metabolism in resting skeletal muscle, the relationship between concentration and oxidation or MCR is curvilinear (reviewed in Balasse & Fery, 1989). At low ketonaemia (<1.0 mm) such as that produced by an overnight fast, resting MCR is as much as fourfold greater than during prolonged fasting (Fery & Balasse, 1983). During prolonged exercise of low‐to‐moderate intensity after an overnight fast, MCR increases by 50% to 75% (Fery & Balasse, 1983, 1986), which indicates that working muscle has an increased capacity to extract ketones from blood compared to rest. However, when ketonaemia exceeds 2.5 mm such as that achieved by greater than 72 h of fasting, the exercise‐induced rise in MCR is abolished (Fery & Balasse, 1986). Therefore, when ketosis is achieved by prolonged (>72 h) fasting there is a negligible contribution of KB oxidation to energy provision (Hagenfeldt & Wahren, 1971; Fery & Balasse, 1986), but after an overnight fast, the contribution ranges from 2 to 10% (Balasse et al. 1978; Fery & Balasse, 1983; Wahren et al. 1984). Under these conditions, the majority of energy provision in working muscle is from CHO and fat as classically described (van Loon et al. 2001). Moreover, unlike CHO and fat, there is progressive attenuation of the oxidation of KBs with rising ketonaemia, and thus the mobilisation of KBs is not the factor limiting oxidation in skeletal muscle. This attenuation of exercise‐stimulated MCR suggests either that above a threshold concentration the capacity for skeletal muscle to oxidise KBs becomes saturated, and/or that hyperketonaemia itself is a self‐inhibitory factor (Balasse & Fery, 1989). Mechanistically, this is likely to be mediated either through the inhibition of OXCT by elevated AcAc, and/or via FFA‐mediated inhibition of ketolysis (Robinson & Williamson, 1980). This regulation is critical in the starvation response because the capacity of the liver to produce KBs closely matches the requirements of the brain to utilise KBs as an energy source (Robinson & Williamson, 1980). Therefore, excessive oxidation by working muscle would threaten survival, whereas its inhibition spares circulating substrate for the brain (Hagenfeldt & Wahren, 1971; Fery & Balasse, 1983).
Methods of exogenous ketone supplementation producing acute nutritional ketosis
Investigating effects of ketosis on skeletal muscle metabolism has been typically achieved by endogenous ketosis using fasting of various durations (Balasse & Fery, 1989), or by exogenous ketosis produced by either ketone salt ingestion (Johnson & Walton, 1972), or infusion of AcAc or βHB (Fery & Balasse, 1988; Mikkelsen et al. 2015). Endogenous ketosis may also be achieved by CHO restriction, particularly by ketogenic diets (Paoli et al. 2013). The practical relevance for athletes seeking performance gains of metabolic responses generated from prolonged fasting is negligible, whereas benefits of ketogenic dieting for performance with a high intensity component are equivocal (Burke, 2015). This has led to the exploration of exogenous ketone ingestion as a means to achieve acute nutritional ketosis. Importantly, because endogenous ketosis results in concomitant elevations in FFAs and alterations in glucose, insulin and counter‐regulatory hormones, isolating the metabolic effects specific to KBs has proved challenging. Therefore, exogenous ketone supplementation is a means to address these questions and explore potential for performance and therapeutic benefits.
Oral administration of KBs in their free acid form is expensive and ineffective at producing ketosis, so buffering the free acid form with sodium/potassium/calcium salts has been explored and these compounds are commercially available. These too are relatively ineffective at increasing [βHB], but may be improved by co‐ingestion with medium chain triglycerides (C:8, C:10), at least in rats (Kesl et al. 2016). However, ingestion of large quantities of KB salts is impractical due to resulting gastrointestinal distress, and potentially undesirable consequences of cation overload or acidosis (Veech, 2004).
The development of ketone esters provides an alternative method to increase [βHB], which is well‐tolerated in rodents and humans (Clarke et al. 2012; Cox et al. 2016; Kesl et al. 2016). Two prominent ketone esters in the published literature are the R,S‐1,3‐butanediol acetoacetate diester (Kesl et al. 2016) and the (R)‐3‐hydroxybutyl (R)‐3‐hydroxybutyrate ketone monoester (Clarke et al. 2012; Cox et al. 2016). Acute ingestion of either ester can result in short‐term (∼0.5 to 6 h) nutritional ketosis indicated by [βHB] >1 mm (Clarke et al. 2012; Kesl et al. 2016). For the ketone monoester, ingestion at a dose of 573 mg (kg body mass (BM))−1 resulted in [βHB] of ∼3 mm after 10 min and rising to ∼6 mm 30 min after ingestion (Cox et al. 2016). Nutritional ketosis is therefore achieved without the impracticality of prolonged fasting or ketogenic dieting.
Ketone body metabolism during exercise under conditions of exogenous ketosis
The aforementioned self‐inhibitory effect of rising ketonaemia underscores a key methodological issue when considering KB metabolism in skeletal muscle, namely the method of achieving ketosis. While fasting of various durations is a widely used model of ketosis, acute nutritional ketosis relevant to sports performance would be achieved with replete glycogen stores, and in the absence of prolonged elevations in FFAs and [KB] that would be likely to impair KB oxidation rates through these mechanisms. To our knowledge, only two studies have addressed this convincingly by examining effects of exercise on KB metabolism without interference from the various hormonal and metabolic perturbations associated with prolonged fasting or diabetes (Fery & Balasse, 1988; Cox et al. 2016).
In the former study (Fery & Balasse, 1988), infusion of sodium AcAc after an overnight fast achieved [KB] of ∼6 mm (βHB ∼3.5 mm, AcAc ∼2.5 mm) at the onset of 2 h of exercise at ∼52% . Notably, AcAc did not change during exercise whereas βHB declined throughout exercise, to be reduced by ∼2 mm at the end of exercise. This coincided with a progressive rise in MCR throughout exercise, peaking at ∼75% higher than rest at the end of exercise. In contrast, this effect was abolished with similar ketonaemia in 3–5 day fasted participants. Importantly, although the inhibition of KB oxidation by hyperketonaemia is present during exogenous ketosis, an ‘auto‐amplification’ was noted that is not present in fasting ketosis, i.e. the initial rise in MCR induced by exercise causes a reduction in concentration which, in turn, provokes a further rise in MCR and so on. Additionally, the threshold concentration at which hyperketonaemia inhibits MCR was higher in exogenous ketosis than in fasting ketosis. However, in terms of contribution to energy provision, this ultimately only resulted in a 2% contribution over the 2 h exercise bout. Nevertheless, plasma [lactate] did not rise during exercise after AcAc infusion compared to a ∼1 mm rise in the fasted participants, which suggests that despite a modest contribution to energy provision, exogenous ketosis can impact on metabolic processes during exercise.
Despite this promise, these data remained largely isolated for almost 30 years with the exception of a couple of obscure reports that admittedly did recapitulate the effects of βHB to alter the metabolic response to very intense exercise in rats (Kamysheva & Ostrovskaia, 1980), and ischaemic exercise in humans (Lestan et al. 1994). The latter report, in fact, supported the ability of a modest elevation in βHB (∼0.5 mm) via infusion of sodium βHB to reduce the plasma lactate response to exercise in an ischaemic forearm model. However, with the development of the (R)‐3‐hydroxybutyl (R)‐3‐hydroxybutyrate ketone monoester (KE), a comprehensive investigation of substrate metabolism in highly trained athletes in the presence of acute nutritional ketosis has recently been published (Cox et al. 2016).
In one of a series of experiments, ingestion of KE resulted in acute nutritional ketosis indicated by [βHB] of ∼3 mm after 10 min and rising to ∼6 mm 30 min after ingestion. During exercise lasting 45 min at either 40% or 75%Wmax, [βHB] was ∼2 and 3 mm, respectively, lower than ketosis produced after ingestion at rest. This provided the first evidence of intensity‐dependent disposal of βHB during exercise. Moreover, based on expired air analysis adjusted for oxidation of KBs, βHB oxidation contributed 18% and 16% of oxygen consumption to energy provision at the respective intensities. The larger than previously reported contribution of βHB oxidation probably reflects the fact that the participants in these experiments were highly trained cyclists, therefore with a greater capacity of skeletal muscle to uptake and oxidise KBs. Moreover, this model is markedly different to the fasting‐induced ketonaemia and the associated self‐inhibitory regulation so making direct comparisons are difficult. In a separate exercise bout lasting 60 min at 75% and with similar [βHB] after KE ingestion, the rise in plasma [lactate] was blunted by ∼2 to 3 mm (∼50% reduction) compared to ingestion of an isocaloric CHO drink. Subsequent experiments with ingestion of the KE demonstrated inhibition of glycolytic metabolism, sparing of muscle glycogen, reduced deamination of branched‐chain amino acids, and increased reliance on IMTG during exercise (Cox et al. 2016). Lastly, after a 60 min pre‐load at 75% , cycling performance in a 30 min time‐trial was improved by 2% (411±162 m; mean ± SEM, n = 8) with KE + CHO compared to isocaloric CHO ingestion. The KE + CHO fuelling strategy combined KE (40%; 573 mg (kg BM)−1) with CHO (60%) and elevated [βHB] to between ∼1.5 and 3 mm throughout. Importantly, the KE + CHO condition provided CHO at a minimum rate of 1.2 g min−1, consistent with an optimal CHO‐based fuelling strategy (Burke, 2015). Taken together, these data suggest that acute nutritional ketosis by consumption of exogenous ketones has dramatic effects on skeletal muscle metabolism during exercise, and can confer a performance benefit to elite athletes (Fig. 3). The positive findings notwithstanding, potential adverse effects should be considered for any performance aid prior to adoption. Side‐effects of KE ingestion have been reported in humans (Clarke et al. 2012). Specifically, in a repeated dose design over 5 days, adverse effects such as flatulence, nausea, diarrhoea and dizziness were reported in five out of twenty‐four participants at doses ranging from 420 to 1071 mg (kg BM)−1. Such issues were prevalent in almost all participants when the dose was increased to 2142 mg (kg BM)−1 per day, indicating a possible upper limit of tolerability in adults (Clarke et al. 2012). Therefore, these data combined with the dosing strategy associated with exercise performance benefits should be used to guide future investigations on ergogenic potential.
Figure 3. βHB as a metabolic regulator and signalling metabolite.
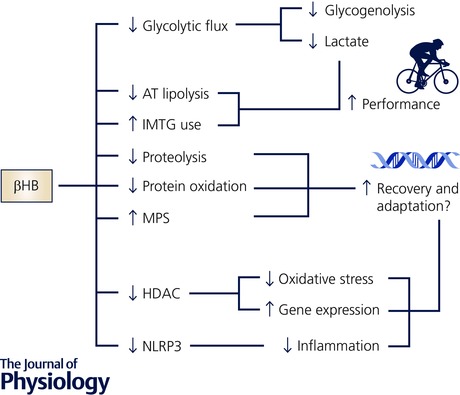
Effects of elevating βHB through acute nutritional ketosis may be mediated by acute regulation of substrate utilisation that may enhance performance, and/or possibly through regulation of recovery and adaptive processes related to inflammation, oxidative stress and changes in gene expression. See text for further discussion. AT, adipose tissue; HDAC, histone deacetylase; IMTG, intramuscular triglyceride; MPS, muscle protein synthesis.
Ketone body metabolism after exercise: post‐exercise ketosis
Despite the aforementioned decline in [KB] at the onset of exercise, this pertains to situations where exercise has begun during hyperketonaemia (Balasse et al. 1978; Fery & Balasse, 1983, 1988; Cox et al. 2016). In the post‐absorptive state, the pattern generally observed is for [KB] to rise gradually during prolonged exercise up to 0.2 to 0.4 mm, after which time post‐exercise ketosis (PEK) of 0.3 to 2.0 mm is observed for several hours into recovery (Koeslag, 1982). Explained in terms of plasma kinetics, at cessation of exercise, the rate of appearance of KBs increases coincident with a decrease in MCR relative to rates present during exercise. MCR remains above resting values for several hours after exercise, but ketogenesis exceeds ketolysis during this period.
On a mechanistic level, regulation probably resides at several sites including malonyl CoA‐mediated regulation of fat transport into hepatocytes via CPT‐1 in addition to availability of Ac‐CoA for ketogenesis, and oxaloacetate for the TCA cycle as classically described for ketogenic regulation. Because oxaloacetate is a product of pyruvate formed during glycolysis, reductions in glycolytic flux with low glycogen content after intense exercise result in oxaloacetate moving to cytoplasm for preferential use in gluconeogenesis, which allows diversion of Ac‐CoA towards ketogenesis during the post‐exercise period rather than to citrate synthesis for the TCA cycle. Additionally, the actions of insulin and glucagon exert a strong influence through activation and inhibition, respectively, of Ac‐CoA carboxylase (ACC), which catalyses the synthesis of malonyl CoA from Ac‐CoA. When liver glycogen becomes depleted and glucagon:insulin ratio is elevated, the synthesis of malonyl CoA is reduced, thereby relieving the inhibition of fat transport into hepatocytes, and resulting in elevated levels of Ac‐CoA. These regulatory mechanisms are acutely sensitive to nutrient manipulations before and after exercise and to aerobic exercise training, given their respective influences on substrate availability and utilisation during exercise.
Modulation of post‐exercise ketosis by aerobic exercise training and nutrition intervention
An attenuation of, or abolished, post‐exercise ketosis has been consistently observed in rodents and humans in response to aerobic exercise in trained versus untrained individuals (Johnson et al. 1969; Johnson & Walton, 1972; Rennie et al. 1974), or after a period of exercise training (Rennie & Johnson, 1974a; Beattie & Winder, 1984, 1985; Adams & Koeslag, 1988, 1989; Ohmori et al. 1990). The aforementioned enhanced ketolytic capacity and downregulation of ketogenic capacity by training may play a role in these observations, but the majority of this work has been performed in comparisons, with the absolute exercise intensity and duration being the same for comparisons (reviewed in Koeslag, 1982). This is problematic because the relative exercise intensity is the key determinant of the metabolic and hormonal response to acute exercise, e.g. catecholamine responses, FFA mobilisation and glycogen utilisation among others. When trained and untrained participants have performed exercise at a similar relative intensity, PEK is blunted but not abolished in trained individuals (Rennie et al. 1974). Moreover, in rodents when exercise is completed to exhaustion, i.e. the trained rats exercise for longer than untrained, [βHB] is ∼twofold higher at the exercise cessation in the trained group (Askew et al. 1975). These divergent findings are likely to be due to the degree of liver glycogen depletion that occurs (Adams & Koeslag, 1988), inasmuch as higher levels of resting liver glycogen and attenuated rates of depletion are a consequence of training (Baldwin et al. 1975).
Therefore, PEK is strongly influenced by nutrition manipulation. High CHO feeding prior to exercise attenuates PEK regardless of training status (Rennie & Johnson, 1974b; Askew et al. 1975; Koeslag et al. 1980), and CHO restriction increases PEK (Impey et al. 2016). Glucose ingestion at 2 h into recovery (Koeslag et al. 1982; Carlin et al. 1987) and alanine during recovery (Koeslag et al. 1980, 1985; Carlin et al. 1987) attenuate PEK, but the glucose effect is not seen when glucose is ingested immediately after exercise. Alanine ingestion increases mitochondrial [oxaloacetate] in liver, thereby allowing condensation with Ac‐CoA and diversion away from ketogenesis. This suggests that the early PEK response is determined by the extent of liver glycogen depletion and reduced glycolytic flux, whereas several hours into recovery it is under regulation by insulin and [FFA] related to nutrition intake.
Metabolic consequences of post‐exercise ketosis during recovery: a role for exogenous ketones as a recovery aid?
The physiological role for PEK is likely to favour the replenishment of muscle glycogen, consistent with classically described metabolic actions of ketosis in the sparing of protein and CHO stores during times of low CHO availability. During the post‐exercise recovery period, in contrast to the reliance on CHO metabolism during exercise, muscle glycogen resynthesis has a high metabolic priority and is facilitated by an increase in fat oxidation and sparing of CHO sources for energy provision (Kiens & Richter, 1998). A priority for muscle glycogen resynthesis over liver glycogen resynthesis is suggested to occur because in ancestral terms, a depleted liver is less of a hindrance to intense exertion than depleted muscle (Adams & Koeslag, 1988). To this end, the priority for muscle glycogen resynthesis is observed even during CHO restriction (Adams & Koeslag, 1989), and is achieved through non‐CHO sources such as lactate and alanine being used for hepatic gluconeogenesis and redistribution to skeletal muscle (Fournier et al. 2002). The contribution of PEK may be via the ability of KBs to inhibit glycolysis and increase the conversion of glucose to glycogen as demonstrated in rat skeletal muscle in vitro (Maizels et al. 1977), and a perfused heart model in dogs (Laughlin et al. 1994). This effect is likely to be mediated by inhibition of PDH and phosphofructokinase (PFK) by elevations in Ac‐CoA and citrate formation, respectively, as a consequence of metabolism of AcAc in mitochondria (Randle et al. 1964; Maizels et al. 1977; Laughlin et al. 1994; Kashiwaya et al. 1997).
This raises the possibility that an optimal post‐exercise recovery milieu exists that includes both CHO and ketones to enhance recovery of muscle glycogen. This is not possible by conventional nutrition strategies because elevations in glucose, lactate and alanine ultimately limit ketogenesis and PEK. The suggestion is that the co‐ingestion of exogenous ketones and CHO in a recovery protocol can confer a metabolic advantage. This hypothesis remains to be tested rigorously, but a preliminary report describes a 33% increase in glucose disposal and 50% increase in muscle glycogen content after 2 h of recovery when nutritional ketosis (∼5 mm βHB) is superimposed on a hyperglycaemic (10 mm glucose) clamp in well‐trained military servicemen (Holdsworth et al. 2016).
Repletion of muscle glycogen is only one component of post‐exercise recovery, and nutrition strategies for recovery include protein ingestion, with the aim to limit muscle protein breakdown and enhance muscle protein synthesis (MPS). KBs have protein sparing effects in skeletal muscle as indicated by reduced alanine release during starvation (Sherwin et al. 1975), and reduced leucine oxidation (Nair et al. 1988). In the latter study, this coincided with a 10% increase in MPS measured by fractional synthesis rate and occurred with [βHB] of ∼2 mm achieved via sodium βHB infusion. This raises the possibility that acute nutritional ketosis can complement current strategies for optimising MPS in the post‐exercise period. Additionally, because low CHO stores during exercise lead to elevated rates of protein oxidation (Wagenmakers et al. 1991), exogenous ketone supplementation may provide both a fuel source and contribute to protein sparing and recovery during training in CHO‐restricted states commonly practiced by athletes (reviewed in Bartlett et al. 2015). Together with the preliminary data for muscle glycogen resynthesis, this suggests that post‐exercise recovery is another application where elite athletes may benefit from exogenous ketone supplementation, and where future research is warranted.
Effects beyond fuelling: βHB as a HDAC inhibitor
As investigative techniques in molecular biology evolve, so too does our appreciation of how complex integrative signalling networks regulate skeletal muscle adaptation in response to stimuli such as nutrient manipulation and exercise training (Egan & Zierath, 2013). Previously considered relatively inert outside their primary metabolic function, numerous substrates and metabolites are emerging as important regulators of intracellular signalling and tissue adaptation (Hashimoto et al. 2007; Gao et al. 2009; Morton et al. 2009; Roberts et al. 2014). Noteworthy for the present review is the recent identification of AcAc as a regulator of skeletal muscle satellite cell proliferation and muscle regeneration (Zou et al. 2016), and βHB as an inhibitor of HDACs (Shimazu et al. 2013) and the NLRP3 inflammasome (Youm et al. 2015). The latter observations are a consequence of βHB, in essence, acting as a signalling metabolite to regulate gene expression and metabolic processes (Fig. 3).
Histone acetyltransferases (HATs) and HDACs are enzymes that facilitate the addition or removal, respectively, of acetyl moieties from specific lysine residues on histones and target proteins (McKinsey et al. 2001). In general, hyperacetylation of histone tails induces transcriptional activation while hypoacetylation is associated with transcriptional repression. Class IIa HDACs (HDAC4, ‐5, ‐7 and ‐9) are highly expressed in skeletal muscle (McKinsey et al. 2001) and their function is responsive to both aerobic endurance exercise in humans (McGee et al. 2009; Egan et al. 2010) and nutritional intervention in rodents (Gao et al. 2009; Shimazu et al. 2013). An acute bout of aerobic exercise increases class IIa HDAC phosphorylation and subsequent nuclear exclusion, thus inhibiting HDAC‐mediated repression of specific exercise‐responsive genes such as GLUT4 and PGC‐1α (McGee & Hargreaves, 2004; McGee et al. 2009; Egan et al. 2010). This suggests that compounds that inhibit or disrupt HDAC inhibition could be used to mimic or enhance adaptations to exercise.
Regulation of HDAC activity by nutrients including butyrate and βHB has also been established (Gao et al. 2009; Shimazu et al. 2013). Butyrate, a short chain fatty acid formed via the fermentation of indigestible dietary fibres by microbial species in the gut, is a potent inhibitor of HDAC activity (Gao et al. 2009). Mice supplemented with sodium butyrate are resistant to diet‐induced obesity, and have elevations in markers of skeletal muscle mitochondrial biogenesis analogous to exercise effects (Gao et al. 2009). βHB is structurally similar to butyrate, and although not as potent as butyrate, also inhibits HDAC class I and II activity in a dose‐dependent manner and supressed oxidative stress responses (Shimazu et al. 2013). Importantly, HDAC inhibition by βHB both in vitro and in vivo is evident at physiologically relevant concentrations of βHB, i.e. 1 to 4 mm, which is similar to those attained during fasting, PEK and exogenous ketone ingestion (Fig. 1; Clarke et al. 2012; Kesl et al. 2016). However, although the inhibitory effects were observed in multiple tissues, they remain to be confirmed in skeletal muscle. If confirmed, it will be intriguing to explore whether, apart from the aforementioned ergogenic effects, exogenous ketone supplementation complements exercise‐mediated adaptive changes associated with modulating HDAC function (Fig. 3).
Exogenous ketone supplementation for athletes: cautionary notes and future directions
Despite a strong physiological basis for a variety of benefits for performance and recovery, the relatively recent availability of exogenous ketones and thus far only one peer‐reviewed paper examining exercise metabolism, performance and nutritional ketosis, means that much more research remains to be performed (Pinckaers et al. 2017). The central tenet is that the combination of fuel sparing and improved energetic efficiency during acute nutritional ketosis confers performance benefits (Fig. 3). Alterations in fuel selection during steady‐state exercise have been demonstrated, which indicate reduced glycolytic flux, sparing of CHO and increased contribution of IMTG and βHB to energy provision (Cox et al. 2016). Whether this sparing of CHO, in fact, manifests as impaired CHO utilisation remains to be determined. The mechanistic basis for CHO sparing by exogenous ketones is presently proposed as inhibition of glycolytic flux via inhibition of PDH and PFK by increases in NADH:NAD+, acetyl‐CoA:CoA ratio or citrate. In theory, this could be problematic for sports that rely heavily on contributions from glycolytic pathways, or a range of sports that are intermittent and/or require periods of high intensity ‘bursts’ on a moderate intensity background. This is analogous to the lack of performance benefits for most athletes undertaking low CHO, high fat diets (Burke, 2015). In fact, impaired performance during high intensity efforts has been observed under such conditions (Havemann et al. 2006), and may be explained by sustained attenuation of PDH activity (Stellingwerff et al. 2006). Whether the same effects are observed with acute nutritional ketosis given that this is a very different metabolic milieu, especially in the context of exercise, remains to be explored.
The metabolic consequences of inhibition of adipose tissue lipolysis by KBs also warrants further exploration, given that this process is an important contributor to circulating FFAs, and therefore to the contribution of fat oxidation to energy provision during long duration, submaximal exercise. Nutritional ketosis achieved by either AcAc infusion (Fery & Balasse, 1988) or KE ingestion (Cox et al. 2016) inhibits the lipolytic effect of exercise, i.e. the amount of lipid‐derived substrates available for working muscle is reduced. In the latter study, this did not manifest as increased glycogenolysis and/or glucose utilization, despite these usually being accelerated by the inhibition of FFA availability (van Loon et al. 2005). In fact, glycogenolysis was attenuated and IMTG utilisation was increased in the KE experiments (Cox et al. 2016), suggesting differential regulation to that achieved by nicotinic acid administration (van Loon et al. 2005). However, in each of the experimental conditions with KE, the duration of exercise was between 45 and 120 min at moderate intensity (Cox et al. 2016). Recently, the inhibition of lipolysis via nicotinic acid impaired cycling time‐trial performance in long (120 min), but not shorter (60 and 90 min) duration efforts (Torrens et al. 2016). Thus, even in events with high CHO dependence (∼80 to 95% of energy provision), inhibition of lipolysis may impair endurance performance, particularly in long duration activities analogous to professional cycling or triathlon. Clearly, the many nodes of metabolic regulation influencing skeletal muscle fuel selection that are altered by nutritional ketosis need to be fully elucidated before sports‐specific ergogenic strategies can be advised.
Improved energetic efficiency is an often‐cited potential benefit of acute nutritional ketosis (Veech, 2004; Cox & Clarke, 2014). In this model, exogenous ketones may provide thermodynamic advantages over CHO and fat, because the available free energy to perform work, the free energy of ATP hydrolysis (ΔG′ATP), is greater with KBs, and require less oxygen per mole of carbon to oxidise. Support for this hypothesis comes from a perfused working rat heart model where adding KBs to the perfusate supressed glycolytic flux, and increased hydraulic efficiency (expressed as work in J (mol O2 consumed)−1) by 28% (Sato et al. 1995; Kashiwaya et al. 1997). In practical terms, if the same effect occurs in skeletal muscle, this would translate as a higher power output for the same oxygen consumption (i.e. improved muscular efficiency) during exercise with nutritional ketosis, but this remains unexplored at present.
Because KBs serve as a substrate for the brain, and therapeutic uses for KBs have been proposed for cognitive enhancement and neurodegenerative pathologies (Veech, 2004), the central nervous system (CNS) may be another target for performance‐enhancing effects of nutritional ketosis. Although speculative at present, effects related to motor recruitment, perceived exertion, pacing strategies, skill execution, reaction time, and decision‐making will be interesting for future research, in addition to the proposed role for the CNS in regulating performance beyond effects related to skeletal muscle metabolism (Noakes, 2011).
As with any ergogenic aid or nutrition strategy, optimising dosing strategies including quantity and timing will be important. Given the saturation kinetics of KB oxidation by skeletal muscle and curvilinear relationship between oxidation and plasma concentrations, it is likely that there is an optimal range for performance benefits. At present, we speculate that this exists between 1 and 3 mm βHB. As with many ergogenic acids, more is unlikely to be better and may even be deleterious given the potential for acidosis at higher [KB], and aforementioned gastrointestinal distress and other side‐effects sometimes observed with KE, so careful consideration should be given to these issues.
In conclusion, although data are preliminary, acute nutritional ketosis achieved by exogenous ketone supplementation has the potential to alter fuel selection during exercise and confer performance benefits. This is most likely to be the case in trained individuals who have a greater capacity to take up and oxidise KBs during exercise as a result of training. Additionally, a strong physiological basis exists that suggests potential benefits for supporting training and recovery. While much work remains to be performed, particularly in relation to sport‐specific strategies, this promises to be an exciting topic for scientists, practitioners and athletes alike for the coming years.
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
B.E. conceived the review and drafted the outline. M.E., K.E.C. and B.E. drafted the initial manuscript, revised and finalised the content. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
M.E. is supported by funding through a UCD Institute for Sport and Health Student Bursary. K.E.C. is supported by funding from Food for Health Ireland (FHI).
Biographies
Mark Evans and Karl Cogan are graduate students at the Institute for Sport and Health, University College Dublin, Ireland. Mark received his MSc in Sport Nutrition from Liverpool John Moores University in 2015. Karl received his MSc in Biotechnology from University College Dublin in 2013. Their research explores optimising nutrition strategies for performance and recovery in athletes with specific interest in ketone bodies and protein hydrolysates, respectively.

Brendan Egan PhD is Senior Lecturer in Sport and Exercise Physiology at Dublin City University's School of Health and Human Performance, and Visiting Associate Professor at University College Dublin. His research group investigates the molecular regulation of skeletal muscle function, adaptation and performance across the life course with special interest in the synergy between nutrition and exercise interventions ranging from athletes to older adults. All three authors are accomplished sportsmen in their own right, and currently involved in the provision of sports science support to team sport athletes.
References
- Abraham R (2015). Ketones: Controversial new energy drink could be next big thing in cycling. Cycling Weekly. [Google Scholar]
- Adams JH & Koeslag JH (1988). Carbohydrate homeostasis and post‐exercise ketosis in trained and untrained rats. J Physiol 407, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JH & Koeslag JH (1989). Glycogen metabolism and post‐exercise ketosis in carbohydrate‐restricted trained and untrained rats. Q J Exp Physiol 74, 27–34. [DOI] [PubMed] [Google Scholar]
- Askew EW, Dohm GL & Huston RL (1975). Fatty acid and ketone body metabolism in the rat: response to diet and exercise. J Nutr 105, 1422–1432. [DOI] [PubMed] [Google Scholar]
- Balasse EO & Fery F (1989). Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev 5, 247–270. [DOI] [PubMed] [Google Scholar]
- Balasse EO, Fery F & Neef MA (1978). Changes induced by exercise in rates of turnover and oxidation of ketone bodies in fasting man. J Appl Physiol Respir Environ Exerc Physiol 44, 5–11. [DOI] [PubMed] [Google Scholar]
- Baldwin KM, Fitts RH, Booth FW, Winder WW & Holloszy JO (1975). Depletion of muscle and liver glycogen during exercise. Protective effect of training. Pflugers Arch 354, 203–212. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Hawley JA & Morton JP (2015). Carbohydrate availability and exercise training adaptation: too much of a good thing? Eur J Sport Sci 15, 3–12. [DOI] [PubMed] [Google Scholar]
- Beattie MA & Winder WW (1984). Mechanism of training‐induced attenuation of postexercise ketosis. Am J Physiol Regul Integr Comp Physiol 247, R780–R785. [DOI] [PubMed] [Google Scholar]
- Beattie MA & Winder WW (1985). Attenuation of postexercise ketosis in fasted endurance‐trained rats. Am J Physiol Regul Integr Comp Physiol 248, R63–R67. [DOI] [PubMed] [Google Scholar]
- Bonen A (2001). The expression of lactate transporters (MCT1 and MCT4) in heart and muscle. Eur J Appl Physiol 86, 6–11. [DOI] [PubMed] [Google Scholar]
- Bosch AN, Weltan SM, Dennis SC & Noakes TD (1996). Fuel substrate kinetics of carbohydrate loading differs from that of carbohydrate ingestion during prolonged exercise. Metabolism 45, 415–423. [DOI] [PubMed] [Google Scholar]
- Burke LM (2015). Re‐examining high‐fat diets for sports performance: did we call the ‘nail in the coffin’ too soon? Sports Med 45 (Suppl. 1), S33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin JI, Olson EB Jr, Peters HA & Reddan WG (1987). The effects of post‐exercise glucose and alanine ingestion on plasma carnitine and ketosis in humans. J Physiol 390, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak NM & van Loon LJ (2013). The use of carbohydrates during exercise as an ergogenic aid. Sports Med 43, 1139–1155. [DOI] [PubMed] [Google Scholar]
- Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa‐Veloso K, Ho M, Roberts A, Robertson J, Vanitallie TB & Veech RL (2012). Kinetics, safety and tolerability of (R)‐3‐hydroxybutyl (R)‐3‐hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol 63, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox PJ & Clarke K (2014). Acute nutritional ketosis: implications for exercise performance and metabolism. Extrem Physiol Med 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B, West J, McLure SW, King MT, Dodd MS, Holloway C, Neubauer S, Drawer S, Veech RL, Griffin JL & Clarke K (2016). Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab 24, 256–268. [DOI] [PubMed] [Google Scholar]
- Egan B, Carson BP, Garcia‐Roves PM, Chibalin AV, Sarsfield FM, Barron N, McCaffrey N, Moyna NM, Zierath JR & O'Gorman DJ (2010). Exercise intensity‐dependent regulation of peroxisome proliferator‐activated receptor γ coactivator‐1 α mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol 588, 1779–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B & Zierath JR (2013). Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17, 162–184. [DOI] [PubMed] [Google Scholar]
- El Midaoui A, Chiasson JL, Tancrede G & Nadeau A (2006). Physical training reverses the increased activity of the hepatic ketone body synthesis pathway in chronically diabetic rats. Am J Physiol Endocrinol Metab 290, E207–E212. [DOI] [PubMed] [Google Scholar]
- Elia M, Wood S, Khan K & Pullicino E (1990). Ketone body metabolism in lean male adults during short‐term starvation, with particular reference to forearm muscle metabolism. Clin Sci (Lond) 78, 579–584. [DOI] [PubMed] [Google Scholar]
- Fery F & Balasse EO (1983). Ketone body turnover during and after exercise in overnight‐fasted and starved humans. Am J Physiol Endocrinol Metab 245, E318–E325. [DOI] [PubMed] [Google Scholar]
- Fery F & Balasse EO (1986). Response of ketone body metabolism to exercise during transition from postabsorptive to fasted state. Am J Physiol Endocrinol Metab 250, E495–E501. [DOI] [PubMed] [Google Scholar]
- Fery F & Balasse EO (1988). Effect of exercise on the disposal of infused ketone bodies in humans. J Clin Endocrinol Metab 67, 245–250. [DOI] [PubMed] [Google Scholar]
- Fournier PA, Brau L, Ferreira LD, Fairchild T, Raja G, James A & Palmer TN (2002). Glycogen resynthesis in the absence of food ingestion during recovery from moderate or high intensity physical activity: novel insights from rat and human studies. Comp Biochem Physiol A Mol Integr Physiol 133, 755–763. [DOI] [PubMed] [Google Scholar]
- Frayn KN (1983). Calculation of substrate oxidation rates in vivo from gaseous exchange. J Appl Physiol 55, 628–634. [DOI] [PubMed] [Google Scholar]
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT & Ye J (2009). Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenfeldt L & Wahren J (1968). Human forearm muscle metabolism during exercise. 3. Uptake, release and oxidation of beta‐hydroxybutyrate and observations on the beta‐hydroxybutyrate/acetoacetate ratio. Scand J Clin Lab Invest 21, 314–320. [DOI] [PubMed] [Google Scholar]
- Hagenfeldt L & Wahren J (1971). Human forearm muscle metabolism during exercise. VI. Substrate utilization in prolonged fasting. Scand J Clin Lab Invest 27, 299–306. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Hussien R, Oommen S, Gohil K & Brooks GA (2007). Lactate sensitive transcription factor network in L6 cells: activation of MCT1 and mitochondrial biogenesis. FASEB J 21, 2602–2612. [DOI] [PubMed] [Google Scholar]
- Havemann L, West SJ, Goedecke JH, Macdonald IA, St Clair Gibson A, Noakes TD & Lambert EV (2006). Fat adaptation followed by carbohydrate loading compromises high‐intensity sprint performance. J Appl Physiol (1985) 100, 194–202. [DOI] [PubMed] [Google Scholar]
- Holdsworth D, Cox PJ & Clarke K (2016). Oral ketone body supplementation accelerates and enhances glycogen synthesis in human skeletal muscle following exhaustive exercise [Abstract] In Proceedings of the Physiological Society. London, UK. [Google Scholar]
- Impey SG, Hammond KM, Shepherd SO, Sharples AP, Stewart C, Limb M, Smith K, Philp A, Jeromson S, Hamilton DL, Close GL & Morton JP (2016). Fuel for the work required: a practical approach to amalgamating train‐low paradigms for endurance athletes. Physiol Rep 4, e12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RH & Walton JL (1972). The effect of exercise upon acetoacetate metabolism in athletes and non‐athletes. Q J Exp Physiol Cogn Med Sci 57, 73–79. [DOI] [PubMed] [Google Scholar]
- Johnson RH, Walton JL, Krebs HA & Williamson DH (1969). Metabolic fuels during and after severe exercise in athletes and non‐athletes. Lancet 2, 452–455. [DOI] [PubMed] [Google Scholar]
- Kamysheva VA & Ostrovskaia RU (1980). [Effect of sodium hydroxybutyrate on the ammonia level in the rat muscles under physical exercise]. Biull Eksp Biol Med 89, 25–27. [PubMed] [Google Scholar]
- Kashiwaya Y, King MT & Veech RL (1997). Substrate signaling by insulin: a ketone bodies ratio mimics insulin action in heart. Am J Cardiol 80, 50a–64a. [DOI] [PubMed] [Google Scholar]
- Kesl SL, Poff AM, Ward NP, Fiorelli TN, Ari C, Van Putten AJ, Sherwood JW, Arnold P & D'Agostino DP (2016). Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague‐Dawley rats. Nutr Metab (Lond) 13, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiens B & Richter EA (1998). Utilization of skeletal muscle triacylglycerol during postexercise recovery in humans. Am J Physiol Endocrinol Metab 275, E332–E337. [DOI] [PubMed] [Google Scholar]
- Koeslag JH (1982). Post‐exercise ketosis and the hormone response to exercise: a review. Med Sci Sports Exerc 14, 327–334. [PubMed] [Google Scholar]
- Koeslag JH, Levinrad LI, Lochner JD & Sive AA (1985). Post‐exercise ketosis in post‐prandial exercise: effect of glucose and alanine ingestion in humans. J Physiol 358, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeslag JH, Noakes TD & Sloan AW (1980). Post‐exercise ketosis. J Physiol 301, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeslag JH, Noakes TD & Sloan AW (1982). The effects of alanine, glucose and starch ingestion on the ketosis produced by exercise and by starvation. J Physiol 325, 363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffel L (1999). Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev 15, 412–426. [DOI] [PubMed] [Google Scholar]
- Laughlin MR, Taylor J, Chesnick AS & Balaban RS (1994). Nonglucose substrates increase glycogen synthesis in vivo in dog heart. Am J Physiol Heart Circ Physiol 267, H219–H223. [DOI] [PubMed] [Google Scholar]
- Lestan B, Walden K, Schmaltz S, Spychala J & Fox IH (1994). β‐Hydroxybutyrate decreases adenosine triphosphate degradation products in human subjects. J Lab Clin Med 124, 199–209. [PubMed] [Google Scholar]
- McGee SL, Fairlie E, Garnham AP & Hargreaves M (2009). Exercise‐induced histone modifications in human skeletal muscle. J Physiol 587, 5951–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee SL & Hargreaves M (2004). Exercise and myocyte enhancer factor 2 regulation in human skeletal muscle. Diabetes 53, 1208–1214. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL & Olson EN (2001). Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev 11, 497–504. [DOI] [PubMed] [Google Scholar]
- Maizels EZ, Ruderman NB, Goodman MN & Lau D (1977). Effect of acetoacetate on glucose metabolism in the soleus and extensor digitorum longus muscles of the rat. Biochem J 162, 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzeo RS, Brooks GA, Schoeller DA & Budinger TF (1986). Disposal of blood [1‐13C]lactate in humans during rest and exercise. J Appl Physiol (1985) 60, 232–241. [DOI] [PubMed] [Google Scholar]
- Mikkelsen KH, Seifert T, Secher NH, Grondal T & van Hall G (2015). Systemic, cerebral and skeletal muscle ketone body and energy metabolism during acute hyper‐D‐beta‐hydroxybutyratemia in post‐absorptive healthy males. J Clin Endocrinol Metab 100, 636–643. [DOI] [PubMed] [Google Scholar]
- Morton JP, Croft L, Bartlett JD, Maclaren DP, Reilly T, Evans L, McArdle A & Drust B (2009). Reduced carbohydrate availability does not modulate training‐induced heat shock protein adaptations but does upregulate oxidative enzyme activity in human skeletal muscle. J Appl Physiol 106, 1513–1521. [DOI] [PubMed] [Google Scholar]
- Nair KS, Welle SL, Halliday D & Campbell RG (1988). Effect of beta‐hydroxybutyrate on whole‐body leucine kinetics and fractional mixed skeletal muscle protein synthesis in humans. J Clin Invest 82, 198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noakes TD (2011). Time to move beyond a brainless exercise physiology: the evidence for complex regulation of human exercise performance. Appl Physiol Nutr Metab 36, 23–35. [DOI] [PubMed] [Google Scholar]
- Ohmori H, Kawai K & Yamashita K (1990). Enhanced ketone body uptake by perfused skeletal muscle in trained rats. Endocrinol Jpn 37, 421–429. [DOI] [PubMed] [Google Scholar]
- Owen OE & Reichard GA Jr (1971). Human forearm metabolism during progressive starvation. J Clin Invest 50, 1536–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoli A, Rubini A, Volek JS & Grimaldi KA (2013). Beyond weight loss: a review of the therapeutic uses of very‐low‐carbohydrate (ketogenic) diets. Eur J Clin Nutr 67, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinckaers PJ, Churchward‐Venne TA, Bailey D & van Loon LJ (2017). Ketone bodies and exercise performance: the next magic bullet or merely hype? Sports Med 47, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle PJ, Newsholme EA & Garland PB (1964). Regulation of glucose uptake by muscle. 8. Effects of fatty acids, ketone bodies and pyruvate, and of alloxan‐diabetes and starvation, on the uptake and metabolic fate of glucose in rat heart and diaphragm muscles. Biochem J 93, 652–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennie MJ, Jennett S & Johnson RH (1974). The metabolic effects of strenuous exercise: a comparison between untrained subjects and racing cyclists. Q J Exp Physiol Cogn Med Sci 59, 201–212. [DOI] [PubMed] [Google Scholar]
- Rennie MJ & Johnson RH (1974a). Alteration of metabolic and hormonal responses to exercise by physical training. Eur J Appl Physiol Occup Physiol 33, 215–226. [DOI] [PubMed] [Google Scholar]
- Rennie MJ & Johnson RH (1974b). Effects of an exercise‐diet program on metabolic changes with exercise in runners. J Appl Physiol 37, 821–825. [DOI] [PubMed] [Google Scholar]
- Roberts LD, Bostrom P, O'Sullivan JF, Schinzel RT, Lewis GD, Dejam A, Lee YK, Palma MJ, Calhoun S, Georgiadi A, Chen MH, Ramachandran VS, Larson MG, Bouchard C, Rankinen T, Souza AL, Clish CB, Wang TJ, Estall JL, Soukas AA, Cowan CA, Spiegelman BM & Gerszten RE (2014). β‐Aminoisobutyric acid induces browning of white fat and hepatic β‐oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab 19, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AM & Williamson DH (1980). Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev 60, 143–187. [DOI] [PubMed] [Google Scholar]
- Sato K, Kashiwaya Y, Keon CA, Tsuchiya N, King MT, Radda GK, Chance B, Clarke K & Veech RL (1995). Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J 9, 651–658. [DOI] [PubMed] [Google Scholar]
- Sherwin RS, Hendler RG & Felig P (1975). Effect of ketone infusions on amino acid and nitrogen metabolism in man. J Clin Invest 55, 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr, de Cabo R, Ulrich S, Akassoglou K & Verdin E (2013). Suppression of oxidative stress by β‐hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellingwerff T, Spriet LL, Watt MJ, Kimber NE, Hargreaves M, Hawley JA & Burke LM (2006). Decreased PDH activation and glycogenolysis during exercise following fat adaptation with carbohydrate restoration. Am J Physiol Endocrinol Metab 290, E380–E388. [DOI] [PubMed] [Google Scholar]
- Svensson K, Albert V, Cardel B, Salatino S & Handschin C (2016). Skeletal muscle PGC‐1α modulates systemic ketone body homeostasis and ameliorates diabetic hyperketonemia in mice. FASEB J 30, 1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Bishop DJ, Lambert K, Mercier J & Brooks GA (2012). Effects of acute and chronic exercise on sarcolemmal MCT1 and MCT4 contents in human skeletal muscles: current status. Am J Physiol Regul Integr Comp Physiol 302, R1–R14. [DOI] [PubMed] [Google Scholar]
- Thomas LK, Ittmann M & Cooper C (1982). The role of leucine in ketogenesis in starved rats. Biochem J 204, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrens SL, Areta JL, Parr EB & Hawley JA (2016). Carbohydrate dependence during prolonged simulated cycling time trials. Eur J Appl Physiol 116, 781–790. [DOI] [PubMed] [Google Scholar]
- van Loon LJ, Greenhaff PL, Constantin‐Teodosiu D, Saris WH & Wagenmakers AJ (2001). The effects of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol 536, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loon LJ, Thomason‐Hughes M, Constantin‐Teodosiu D, Koopman R, Greenhaff PL, Hardie DG, Keizer HA, Saris WH & Wagenmakers AJ (2005). Inhibition of adipose tissue lipolysis increases intramuscular lipid and glycogen use in vivo in humans. Am J Physiol Endocrinol Metab 289, E482–E493. [DOI] [PubMed] [Google Scholar]
- Veech RL (2004). The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids 70, 309–319. [DOI] [PubMed] [Google Scholar]
- Volek JS, Freidenreich DJ, Saenz C, Kunces LJ, Creighton BC, Bartley JM, Davitt PM, Munoz CX, Anderson JM, Maresh CM, Lee EC, Schuenke MD, Aerni G, Kraemer WJ & Phinney SD (2016). Metabolic characteristics of keto‐adapted ultra‐endurance runners. Metabolism 65, 100–110. [DOI] [PubMed] [Google Scholar]
- Wagenmakers AJ, Beckers EJ, Brouns F, Kuipers H, Soeters PB, van der Vusse GJ & Saris WH (1991). Carbohydrate supplementation, glycogen depletion, and amino acid metabolism during exercise. Am J Physiol Endocrinol Metab 260, E883–E890. [DOI] [PubMed] [Google Scholar]
- Wahren J, Sato Y, Ostman J, Hagenfeldt L & Felig P (1984). Turnover and splanchnic metabolism of free fatty acids and ketones in insulin‐dependent diabetics at rest and in response to exercise. J Clin Invest 73, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW, Baldwin KM & Holloszy JO (1973). Exercise‐induced adaptive increase in rate of oxidation of β‐hydroxybutyrate by skeletal muscle. Proc Soc Exp Biol Med 143, 753–755. [DOI] [PubMed] [Google Scholar]
- Winder WW, Baldwin KM & Holloszy JO (1974). Enzymes involved in ketone utilization in different types of muscle: adaptation to exercise. Eur J Biochem 47, 461–467. [DOI] [PubMed] [Google Scholar]
- Winder WW, Baldwin KM & Holloszy JO (1975). Exercise‐induced increase in the capacity of rat skeletal muscle to oxidize ketones. Can J Physiol Pharmacol 53, 86–91. [DOI] [PubMed] [Google Scholar]
- Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D'Agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S, Horvath TL, Fahmy TM, Crawford PA, Biragyn A, Alnemri E & Dixit VD (2015). The ketone metabolite β‐hydroxybutyrate blocks NLRP3 inflammasome‐mediated inflammatory disease. Nat Med 21, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Meng J, Li L, Han W, Li C, Zhong R, Miao X, Cai J, Zhang Y & Zhu D (2016). Acetoacetate accelerates muscle regeneration and ameliorates muscular dystrophy in mice. J Biol Chem 291, 2181–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]