

Abstract
Key points
A classic unresolved issue in human integrative physiology involves the role of exercise intensity, duration and volume in regulating skeletal muscle adaptations to training.
We employed counterweighted single‐leg cycling as a unique within‐subject model to investigate the role of exercise intensity in promoting training‐induced increases in skeletal muscle mitochondrial content.
Six sessions of high‐intensity interval training performed over 2 weeks elicited greater increases in citrate synthase maximal activity and mitochondrial respiration compared to moderate‐intensity continuous training matched for total work and session duration.
These data suggest that exercise intensity, and/or the pattern of contraction, is an important determinant of exercise‐induced skeletal muscle remodelling in humans.
Abstract
We employed counterweighted single‐leg cycling as a unique model to investigate the role of exercise intensity in human skeletal muscle remodelling. Ten young active men performed unilateral graded‐exercise tests to measure single‐leg and peak power (W peak). Each leg was randomly assigned to complete six sessions of high‐intensity interval training (HIIT) [4 × (5 min at 65% W peak and 2.5 min at 20% W peak)] or moderate‐intensity continuous training (MICT) (30 min at 50% W peak), which were performed 10 min apart on each day, in an alternating order. The work performed per session was matched for MICT (143 ± 8.4 kJ) and HIIT (144 ± 8.5 kJ, P > 0.05). Post‐training, citrate synthase (CS) maximal activity (10.2 ± 0.8 vs. 8.4 ± 0.9 mmol kg protein−1 min−1) and mass‐specific [pmol O2•(s•mg wet weight)−1] oxidative phosphorylation capacities (complex I: 23.4 ± 3.2 vs. 17.1 ± 2.8; complexes I and II: 58.2 ± 7.5 vs. 42.2 ± 5.3) were greater in HIIT relative to MICT (interaction effects, P < 0.05); however, mitochondrial function [i.e. pmol O2•(s•CS maximal activity)−1] measured under various conditions was unaffected by training (P > 0.05). In whole muscle, the protein content of COXIV (24%), NDUFA9 (11%) and mitofusin 2 (MFN2) (16%) increased similarly across groups (training effects, P < 0.05). Cytochrome c oxidase subunit IV (COXIV) and NADH:ubiquinone oxidoreductase subunit A9 (NDUFA9) were more abundant in type I than type II fibres (P < 0.05) but training did not increase the content of COXIV, NDUFA9 or MFN2 in either fibre type (P > 0.05). Single‐leg was also unaffected by training (P > 0.05). In summary, single‐leg cycling performed in an interval compared to a continuous manner elicited superior mitochondrial adaptations in human skeletal muscle despite equal total work.
Keywords: exercise intensity, high‐intensity interval training, muscle fibre
Key points
A classic unresolved issue in human integrative physiology involves the role of exercise intensity, duration and volume in regulating skeletal muscle adaptations to training.
We employed counterweighted single‐leg cycling as a unique within‐subject model to investigate the role of exercise intensity in promoting training‐induced increases in skeletal muscle mitochondrial content.
Six sessions of high‐intensity interval training performed over 2 weeks elicited greater increases in citrate synthase maximal activity and mitochondrial respiration compared to moderate‐intensity continuous training matched for total work and session duration.
These data suggest that exercise intensity, and/or the pattern of contraction, is an important determinant of exercise‐induced skeletal muscle remodelling in humans.
Abbreviations
- BIOPS
biopsy preservation solution
- CS
citrate synthase
- CI
complex I
- CII
complex II
- COXIV
cytochrome c oxidase subunit IV
- ETS capacity (or E)
electron transfer system capacity
- HIIT
high‐intensity interval training
- JO2
oxygen flux
- MiR05
mitochondrial respiration medium
- MFN2
mitofusin 2
- MICT
moderate‐intensity continuous training
- MHCI
myosin heavy chain I
- MHCIIa
myosin heavy chain IIa
- NDUFA9
NADH:ubiquinone oxidoreductase subunit A9
- PBST
PBS with 0.025% Tween
- SIT
sprint interval training
- TBST
Tris‐buffered saline with Tween
peak oxygen uptake
- Wpeak
peak power output
Introduction
Skeletal muscle mitochondrial content impacts fuel use and endurance capacity (Holloszy and Coyle, 1984), as well as aspects of metabolic health and ageing (Joseph et al. 2012; Goodpaster, 2013). Mitochondrial content increases after aerobic‐based exercise training (Holloszy, 1967; Morgan et al. 1971) and is markedly greater in the skeletal muscle of endurance‐trained compared to untrained individuals (Hoppeler et al. 1973; Jacobs and Lundby, 2013). Short‐term training programmes, involving moderate‐ to vigorous‐intensity exercise and performed in a continuous or intermittent manner, rapidly increase mitochondrial content in untrained humans (Saltin et al. 1976; MacDougall et al. 1998; Talanian et al. 2006; Burgomaster et al. 2008). Relatively few studies have compared work‐matched programmes that differ in exercise intensity, and the precise role of exercise intensity in mediating mitochondrial adaptations to training in humans is equivocal (Bishop et al. 2014).
Exercise intensity mediates many acute responses to aerobic exercise, with higher intensities typically inducing greater neuromuscular fatigue (Theurel and Lepers, 2008), greater type II muscle fibre recruitment (Kristensen et al. 2015) and augmenting the activation of molecular pathways linked to mitochondrial biogenesis (Egan et al. 2010; Di Donato et al. 2014; Kristensen et al. 2015). Skeletal muscle adaptations to exercise training may therefore be linked to relative work intensity, although surprisingly few data are available to directly address this hypothesis. High‐intensity interval training (HIIT) and sprint interval training (SIT) elicit adaptations similar to moderate‐intensity continuous training (MICT) despite lower total work (Gibala et al. 2006; Burgomaster et al. 2008); however, the few studies that have compared MICT, HIIT and/or SIT protocols matched for total work have yielded inconsistent findings, with higher exercise intensities eliciting greater adaptations in some comparisons (Daussin et al. 2008a; Granata et al. 2016) but not others (Henriksson and Reitman, 1976; Saltin et al. 1976; Granata et al. 2016). Most training studies have exclusively examined adaptations in whole muscle; however, training‐induced mitochondrial adaptations might occur in a fibre type‐dependent manner (Henriksson and Reitman, 1976). For example, relative to continuous exercise, the greater AMP‐kinase activity induced by a single bout of interval exercise was also fibre type‐dependent (Kristensen et al. 2015) and, in response to 6 weeks of MICT, type II fibres demonstrated greater increases in mitochondrial volume than type I fibres (Howald et al. 1985). Furthermore, most of the previous studies have progressively increased the duration and/or intensity of the exercise training, complicating any interpretation of the results. Accordingly, an examination of the short‐term effect of aerobic exercise intensity on human skeletal muscle mitochondrial content is warranted.
Single‐leg cycling permits the comparison of training adaptations to two different exercise training protocols within the same subject, controlling for individual variability in training responsiveness and increasing statistical power. The addition of a counterweight to the contralateral pedal assists with the upstroke phase of the revolution, making the perceptual ‘feel’ of counterweighted single‐leg cycling similar to double‐legged cycling (Burns et al. 2014; Bini et al. 2015). There is no evidence to suggest that mitochondrial adaptations to single‐leg cycling transfer to the non‐exercising leg, meaning that the contralateral limb can be trained in a different manner or serve as a non‐exercise control (Saltin et al. 1976; Henriksson, 1977). Furthermore, the adaptations induced by single‐leg cycling are potentially greater than those induced by double‐legged cycling, as a result of increased relative workloads (Abbiss et al. 2011).
The present study aimed to compare mitochondrial adaptations in human skeletal muscle following six sessions of HIIT or MICT, matched for total work and session duration. The legs of subjects were randomly assigned to one of the two training interventions and exercised separately but consecutively on six training sessions over a 2 week period. Resting skeletal muscle needle biopsies were collected from each leg before and after training to measure mitochondrial content. We hypothesized that HIIT would elicit greater mitochondrial adaptations than MICT as a result of the cumulative effect of greater metabolic stress induced over the course of the training sessions.
Methods
Participants and ethical approval
Ten healthy young men were recruited for the study (age 23 ± 1 years; body mass index 25 ± 1 kg m–2). All subjects were habitually active but not specifically training for any sport. The subjects completed a Physical Activity Readiness Questionnaire and provided their written informed consent prior to their participation. The Hamilton Integrated Research Ethics Board approved the protocol, and the study conformed with the Declaration of Helsinki (2013).
Pre‐training procedures
Subjects initially performed a standard (double‐legged) ramp test to exhaustion on an electronically‐braked cycle ergometer (Excalibur Sport, version 2.0; Lode, Groningen, The Netherlands) to determine whole‐body peak oxygen uptake () and peak power output (W peak). Following a 1 min warm‐up at 50 W, workload was increased 1 W every 2 s until the subject reached volitional exhaustion or cadence decreased below 60 rpm. Expired gases were analysed using an online gas collection system (Moxus modular oxygen uptake system; AEI Technologies, Pittsburgh, PA, USA) and the was determined from the greatest 30 s average of . The and Wpeak of subjects were 46 ± 2 mL kg−1 min−1 and 302 ± 14 W, respectively.
At least 48 h following the double‐legged ramp test, subjects were familiarized with a single‐leg cycling technique modelled after previous work (Abbiss et al. 2011; Burns et al. 2014). Briefly, one crank on an electronically‐braked cycle ergometer (Velotron; RacerMate, Seattle, WA, USA) was fitted with a custom‐machined pedal that held an 11.4 kg counterweight. Subjects pedalled using one leg, with the non‐exercising leg resting on a stationary platform. The counterweight assisted with the upstroke phase of the revolution, eliminating the need to pull up on the pedal. Using this set‐up, subjects performed an incremental exercise test to volitional exhaustion with each leg. The single‐leg tests were similar to the double‐legged tests, except the rate at which the workload increased was reduced by half (i.e. 1 W every 4 s). The left leg was tested 10 min after the right leg, given previous data showing that fatigue does not transfer to the non‐exercising leg (Elmer et al. 2012). The (2.5 ± 0.2 vs. 2.6 ± 0.2 l min−1, P = 0.26) and power output (151 ± 9 W vs. 155 ± 9 W, P = 0.28) were similar for the left and right legs. Pre‐training, single‐leg (average of both legs) was 73 ± 1.0 % of the double‐legged value. On a separate day, each leg completed a brief familiarization session that involved half of the total work to be performed during the main training sessions. The goal of familiarization was to determine whether the prescribed workload was tolerable (see Training Intervention).
At least 3 days after the final baseline testing session, resting muscle needle biopsies were collected from the vastus lateralis of the left and right legs of each subject. Subjects refrained from exercise, alcohol and food for a minimum of 48, 24 and 10 h, respectively, and subjects recorded a 24 h diet log before the first biopsy to repeat this diet for the post‐training biopsy. Samples were collected under local anaesthesia (1% xylocaine) using a Bergström needle modified for suction, as described previously (Tarnopolsky et al. 2011). Muscle biopsies were immediately sectioned, with one piece placed in ice‐cold buffer for the measurement of mitochondrial respiration and the remaining pieces frozen in liquid nitrogen and stored at –80°C until analysis.
Training intervention
All training was performed on the same cycle ergometer adapted for single‐leg cycling as that used for baseline testing. Using an allocation concealment procedure, each leg was randomly assigned to complete six sessions of work‐ and duration‐matched HIIT or MICT over 2 weeks (essentially every second day). Training began 3–4 days after the muscle biopsy procedures.
Exercise prescriptions were based on the average W peak obtained during the two single‐leg tests. Legs in the HIIT group performed four 5 min bouts of cycling at 65% average W peak, each followed by a 2.5 min recovery period at 20% of average W peak. Legs in the MICT group performed 30 min of cycling at 50% W peak to match the total work [total work (kJ) = average power (W) × time (s)/1000] of the HIIT group. For two subjects who could not complete the prescribed HIIT protocol, the loads were reduced to 60% and 15% of average W peak for HIIT and 45% of average W peak for MICT. All training sessions were preceded by a 5 min warm‐up at 25 W. Subjects were instructed to cycle at the same cadence (∼80 rpm) throughout each session. The legs of each subject were trained consecutively on the same day, following a 10 min rest period, with the order alternating each day. Training intensities were held constant for the entire study.
To determine acute responses to each protocol, heart rate was measured continuously (Polar Electro, Kempele, Finland) and ratings of perceived exertion and dyspnoea were measured periodically (Borg Category‐Ratio Scale, 0–10). Heart rate data were averaged for the entire session and for each of the 5 min intervals (or the corresponding period of the MICT protocol), whereas subjective ratings of exertion and dyspnoea were recorded following each interval (or the corresponding period of the MICT protocol). Blood lactate was measured via finger prick during the final 2 min of the first session of each training programme only (Lactate Plus; Nova Biomedical, Mississauga, ON, Canada).
Post‐training procedures
A resting muscle needle biopsy was obtained from each leg 72 h after the final training session to avoid potential acute effects of exercise (Leek et al. 2001). The single‐leg incremental tests to exhaustion were repeated 72 h later, as described above.
Muscle analysis
Citrate synthase (CS) maximal activity
One piece of muscle was homogenized for the determination of CS maximal activity. Briefly, ∼25 mg of muscle was homogenized in 20 volumes of buffer (70 mm sucrose, 220 mm mannitol, 10 mm Hepes and 1 mm EGTA, supplemented with protease inhibitors; Complete Mini®; Roche Applied Science, Laval, PQ, Canada) using Lysing Matrix D tubes (MP Biomedicals, Solon, OH, USA) and the FastPrep‐24 Tissue and Cell Homogenizer (MP Biomedicals). Enzyme activity was determined by measuring the formation of the thionitrobenzoate anion at a wavelength of 412 nm using a spectrophotometer (Cary Bio‐300; Varion, Inc., Palo Alto, CA, USA), as previously described (Carter et al. 2001). CS maximal activity was expressed relative to total protein measured with a BCA Assay Kit (Pierce, Rockford, IL, USA).
Mitochondrial respiration in permeabilized muscle fibres
Mitochondrial respiration was simultaneously measured in permeabilized muscle from the left and right legs of subjects using the two chambers of an Oxygraph‐2K respirometer (Oroboros, Innsbruck, Austria). The protocol is based on the general protocol described by Pesta and Gnaiger (2011). After being sectioned from the biopsy sample, ∼10 mg of muscle tissue was immediately placed in ice‐cold biopsy preservation solution (BIOPS) (10 mm Ca‐EGTA buffer, 0.1 μm free calcium, 20 mm imidazole, 20 mm taurine, 50 mm K‐MES, 0.5 mm DTT, 6.56 mm MgCl2, 5.77 mm ATP and 15 mm phosphocreatine, pH 7.1). Within 1 h of the biopsy procedure, bundles of muscle fibres were suspended in an aliquot of BIOPS and mechanically separated under a microscope using jeweler's forceps. Samples were then transferred to BIOPS containing saponin (50 μg ml−1) and incubated on a rocker at 4°C for 30 min to permeabilize muscle fibres. Samples were then transferred to ice‐cold mitochondrial respiration medium (MiR05; 0.5 mm EGTA, 3 mm MgCl2•6H20, 60 mm K‐lactobionate, 20 mm taurine, 10 mm KH2PO4, 20 mm Hepes, 110 mm sucrose and 1 g l−1 bovine serum albumin, pH 7.1) and washed on a rocker at 4°C for 15 min. Finally, samples were blotted dry with filter paper, and a piece of muscle, with a wet weight of between 1.5 and 2 mg (1.8 ± 0.2 mg), was added to the chamber of the respirometer, which contained 2 ml of MiR05 with blebbistatin to inhibit contractions. All measurements were performed at 37°C and at an oxygen concentration between 100 and 200 nmol ml−1.
A substrate uncoupler inhibitor titration protocol was used to examine mitochondrial respiration in permeabilized fibres. First, malate (2 mm) and glutamate (10 mm) were added consecutively in the absence of ADP to measure leak respiration. ADP (5 mm) was added to measure oxidative phosphorylation (oxidative phosphorylation capacity; P) through complex I (PCI) followed by succinate (10 mm) to measure oxidative phosphorylation through complexes I and II (PCI&CII). The electron transfer system capacity (E) was assessed through the serial addition of 1 μl aliquots (in steps of 0.5 μm) of the protonophore, carbonyl cyanide p‐(trifluoromethoxy) phenyl‐hydrazone. The addition of rotenone (0.5 μm), an inhibitor of complex I, allowed for the measurement of ECII. Cytochrome c (10 μm) was then added to ensure the functional integrity of the outer mitochondrial membrane and, finally, anti‐mycin A (2.5 μm), which is an inhibitor of complex III, was added to measure the residual oxygen consumption that remains when electron flow through complexes I, II, and III is inhibited.
For analysis of mitochondrial respiration, oxygen flux (J O2) was calculated from the derivative of the oxygen concentration of the chamber, using DatLab 4.1.08 (Oroboros) and expressed as mass‐specific J O2 (pmol O2•[s•mg wet weight]−1). All values were corrected by subtracting the corresponding residual oxygen consumption value for the sample. To address the possibility that exercise training can increase J O2 independent of mitochondrial content, J O2 was normalized to CS activity (pmol O2•[s•CS]−1) to yield mitochondria‐specific J O2 (Jacobs and Lundby, 2013; Jacobs et al. 2013). Data are reported for PCI, PCI&CII and E.
Whole muscle western blotting
Frozen pieces of whole muscle (12 ± 5.0 mg) were cut into 10 μm sections at –20°C, placed in loading buffer, vortexed, incubated at room temperature for 1 h and frozen at –80°C until analysis. All samples were run on 4–15% Criterion TGX Stain‐Free protein gels (BioRad, Hercules, CA, USA) at 200 V for 45 min. A protein ladder (Fermentas PageRuler Prestained Ladder, ThermoFisher Scientific, Waltham, MA, USA) and a calibration curve (e.g. 2, 4, 8, 16 μl) (Fig. 1) of pooled whole muscle homogenates were run on every gel. The total protein loaded was visualized using ultraviolet (UV) activation of the gel and analysed with Image Lab 5.2.1 (Bio‐Rad, Hercules, CA, USA). Proteins were then wet‐transferred to nitrocellulose at 100 V for 30 min in circulating 4°C transfer buffer (25 mm Tris, 192 mm glycine, 0.1% SDS and 20% methanol, pH 8.3). Proper transfer was visualized with UV activation of the gel and membrane post‐transfer (StainFree Imager; Bio‐Rad). Membranes were treated with Miser solution (Pierce) and placed in blocking buffer (5% skim milk in Tris‐buffered saline‐Tween; TBST) for 2 h at room temperature. After rinsing in TBST, sections of membranes were placed in solutions of primary antibodies (see below) and then incubated for 2 h at room temperature and overnight at 4°C on rockers. After washing in blocking buffer, membranes were incubated in goat anti‐mouse IgG HRP secondary antibody (PIE31430; dilution 1:20 000 in blocking buffer; ThermoFisher Scientific) or goat anti‐rabbit IgG HRP (PIE31460; dilution 1:60 000 in blocking buffer; ThermoFisher Scientific) for 1 h at room temperature. Following a final wash in TBST, membranes were exposed to Supersignal West Femto (Pierce), imaged (ChemiDoc MP; Bio‐Rad) and analysed (ImageLab, version 5.2.1; Bio‐Rad). When required, membranes were washed in TBST for 1 h (but were not stripped) before being re‐probed with another primary antibody. The abundance of proteins of interest were normalized to total protein for analysis, using the calibration curve from each gel (Mollica et al. 2009).
Figure 1. Representative western blot images for whole muscle and pooled type I and type II muscle fibres.
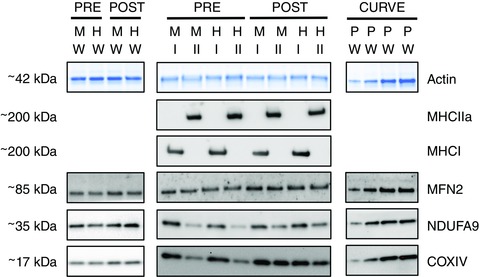
Samples from pre‐ and post‐training muscle biopsies collected from the MICT and HIIT groups at rest were run on the same gels. Whole muscle (W) samples were run on separate gels from type I and type II muscle fibres (I and II). Protein content was normalized to total protein loaded in each lane, although only a representative image of the actin band is displayed. A calibration curve (e.g. 1, 2, 4, 8 μl) comprised of pooled (P) whole muscle samples was run on each gel. The presence and absence of MHCI and MHCIIa were used to confirm the purity of pooled muscle fibre types. [Colour figure can be viewed at wileyonlinelibrary.com]
All primary antibodies were diluted in 1% BSA in PBS with 0.025% Tween (PBST). The proteins of interest were cytochrome c oxidase subunit IV (COXIV) (rabbit polyclonal IgG; dilution 1:1000; 4844, lot no. 0003; Cell Signalling Technology, Danvers, MA, USA), NADH:ubiquinone oxidoreductase subunit A9 (NDUFA9; rabbit polyclonal IgG; dilution 1:1000; as described previously; Stroud et al. 2013) and mitofusin 2 (MFN2; rabbit polyclonal IgG; dilution 1:500; verified with MFN2‐knockout tissue in our laboratory). Professor Michael Ryan (Monash University, Australia) kindly provided the latter two antibodies. Representative blots are shown in Fig. 1.
Single‐fibre collection and western blotting
Muscle samples (21 ± 6.5 mg) were freeze‐dried for 48 h to permit the collection of segments of individual muscle fibres, as described by Murphy (2011). Briefly, freeze‐dried pieces of muscle (4.2 ± 1.5 mg) were brought to room temperature and segments of individual fibres were removed using jeweller's forceps under a microscope. Each segment was added to a tube containing 10 μl of loading buffer, vortexed, incubated at room temperature for 1 h, and frozen at –80°C until analysis. Between 24 and 78 (31 ± 12) fibres were collected from each sample.
Dot blotting was performed to determine the fibre type of each muscle fibre segment. Briefly, polyvinylidene fluoride membrane was activated with 95% ethanol and equilibrated in transfer buffer, and then 1 μl of each sample was applied to the wet membrane and allowed to dry. The dry membrane was then reactivated with 95% ethanol, equilibrated in transfer buffer, washed in TBST for 5 min, and then placed in blocking buffer for 5 min. The presence of myosin heavy chain type II a (MHCIIa; mouse monoclonal IgG, clone A4.74, Developmental Studies Hybridoma Bank [DSHB]; dilution 1:200 in 1% BSA/PBST) protein was determined using a western blotting procedure similar to that described above (beginning after the Miser solution step). After analysis, MHCIIa antibodies were removed from the membrane with stripping buffer (Pierce) prior to incubation in antibodies for myosin heavy chain type I (MHCI; mouse monoclonal IgM, clone A4.840, DSHBl dilution 1:200 in 1%BSA/PBST). For each sample, unambiguous type I (7.0 ± 2.9) and type II (9.0 ± 5.2) muscle fibre segments were pooled prior to electrophoresis, which followed the same protocol as that used for whole muscle samples. The presence/absence of MHCI and MHCIIa was used to confirm the fibres were pooled correctly (Fig. 1). The protein contents of COXIV, MFN2 and NDUFA9 were determined for each pooled sample following the protocol already described for whole muscle samples. Representative blots are shown in Fig. 1.
Statistical analysis
Data are presented as the mean ± SE unless otherwise stated. CS maximal activity, J O2, whole muscle protein content, pooled type I and type II muscle fibre content, and Wpeak were measured using a two‐way, within‐subject ANOVA, with group (HIIT and MICT) and time (pre‐ and post‐training) as the two factors. To examine the possibility of fibre‐type differences in protein content (Albers et al. 2015; Christensen et al. 2015), pre‐training pooled type I and type II muscle fibres, averaged across HIIT and MICT, were compared with paired t tests. Variables measured during each training session (heart rate and ratings of perceived exertion and dyspnoea) were averaged across the six sessions and analysed using a two‐way, within‐subject ANOVA, with group and bout (1, 2, 3, 4) as the within‐subject factors. Other data (blood lactate concentrations, cadence, total work) were compared across groups with paired Student's t tests. Post hoc testing (Holm–Sidak test) was performed only when statistically significant interactions were detected with ANOVA. P ≤ 0.05 was considered statistically significant. All data analyses were performed with Prism, version 6.0 (GraphPad Software, La Jolla, CA, USA).
Results
Exercise data and cardiorespiratory response to training
Mean power was 98 ± 6 and 31 ± 2 W, respectively, during the work and recovery periods in HIIT, and 75 ± 5 W during MICT. The total work performed each session, including warm up, was similar in HIIT and MICT (143 ± 8.4 vs. 144 ± 8.5 kJ; P = 0.44), although pedal cadence was slightly higher in MICT compared to HIIT (79 ± 1 vs. 77 ± 1 rpm; P = 0.02). Heart rate and ratings of perceived exertion and dyspnoea increased during the sessions and were greater during HIIT compared to MICT (Table 1). Blood lactate was also higher for HIIT compared to MICT (8.0 ± 0.8 vs. 5.5 ± 0.7 mm; P < 0.001).
Table 1.
The heart rate and ratings of perceived exertion and dyspnoea for each bout of the single‐leg MICT and HIIT protocols, averaged across the six sessions
| Timea | ||||||
|---|---|---|---|---|---|---|
| Variable | Group | Bout 1 | Bout 2 | Bout 3 | Bout 4 | P valuesb |
| Heart rate (bpm) | MICT | 114 ± 2.1 | 122 ± 2.7 | 125 ± 2.7 | 125 ± 2.8 | <0.001, 0.001, 0.814 |
| HIIT | 122 ± 2.6* | 131 ± 2.9* | 133 ± 3.0* | 134 ± 2.9* | ||
| Exertion (0–10) | MICT | 2.8 ± 0.4 | 3.5 ± 0.4 | 3.8 ± 0.4 | 3.9 ± 0.4 | <0.001, <0.001, 0.001 |
| HIIT | 3.8 ± 0.5* | 4.7 ± 0.5* | 5.3 ± 0.6* | 5.6 ± 0.6* | ||
| Dyspnoea (0–10) | MICT | 2.0 ± 0.3 | 2.4 ± 0.4 | 2.8 ± 0.4 | 2.8 ± 0.4 | <0.001, 0.001, <0.001 |
| HIIT | 2.4 ± 0.4* | 3.2 ± 0.5* | 3.7 ± 0.5* | 3.9 ± 0.6* | ||
*Significant difference between groups at this time point (P < 0.05).
For MICT, ‘bouts’ refer to the time periods corresponding to the interval periods of the HIIT protocol.
P values from the two‐way ANOVA are listed in the order: main effect of bout, main effect of group, and the interaction between bout and group.
Values are the mean ± SEM (n = 10).
With training, single‐leg W peak increased from 150 ± 9.3 to 163 ± 10.0 W in the HIIT group and from 155 ± 8.4 to 163 ± 8.3 W in the MICT group (P = 0.003, main effect of training), although the change was similar between groups (P = 0.08, interaction effect). Training did not increase single‐leg in the HIIT (2.5 ± 0.2 to 2.5 ± 0.2 l min−1) or MICT groups (2.6 ± 0.1 to 2.5 ± 0.2 l min−1; P = 0.81).
CS maximal activity and mitochondrial respiration
There was a significant interaction between group and training for both CS maximal activity (HIIT: +39%, MICT: +11%; P = 0.02) and mass‐specific J O2 for PCI (HIIT: +22% MICT: –7%; P = 0.03) and PCI&CII (HIIT: +22% MICT: –9%; P = 0.05) such that the training‐induced increases were greater in HIIT compared to MICT (Figs 2 and 3). Training did not increase E (P = 0.31), although E was 17% greater in HIIT than MICT when averaged across time points (P = 0.03) (Fig. 3). Mitochondrial‐specific JO2 was unaffected by training or group (Fig. 3).
Figure 2. Maximal CS activity in resting skeletal muscle biopsies in response to six sessions of MICT (white bars) and HIIT (black bars).
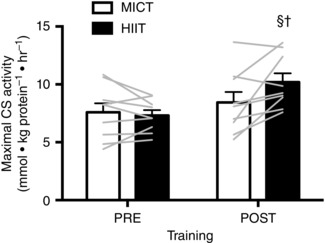
The bars represent the mean data pre‐ and post‐training, whereas individual data are displayed as grey lines. Symbols indicate a significant difference from the within‐group, pre‐training mean (§) and a significant difference between groups at the post‐training mean (†). Error bars represent the SEM (n = 9 subjects).
Figure 3. Mass‐specific and mitochondria‐specific J O2 in resting permeabilized muscle fibres in response to six sessions of MICT (white bars) and HIIT (black bars).
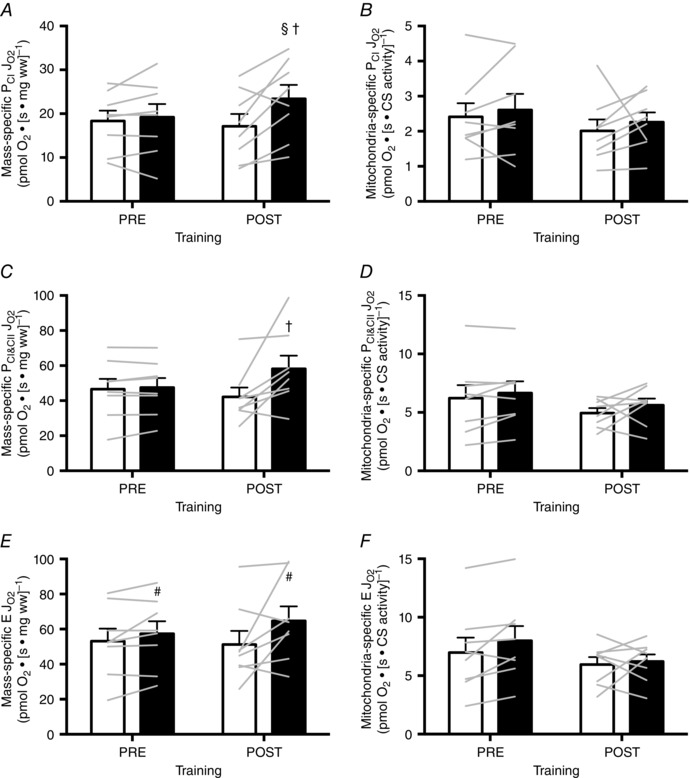
Bars in (A), (C) and (E) show the mean values for mass‐specific PCI, PCI&CII and E respectively, whereas the bars in (B), (D) and (F) show the mean values for mitochondria‐specific PCI, PCI&CII and E, respectively. Individual subjects are shown as grey lines. Symbols indicate a significant effect of group (#), a significant difference from the within‐group, pre‐training mean (§) and a significant difference between groups at the post‐training mean (†). Error bars represent the SEM (n = 8 subjects).
Whole muscle protein content
The protein content of COXIV (+24%, P < 0.01), NDUFA9 (+11%, P = 0.05) and MFN2 (+16%; P = 0.02) increased after training, although there were no differences between groups (P > 0.05) (Fig. 4).
Figure 4. Protein content in resting whole muscle samples, pooled type I muscle fibres and pooled type II muscle fibres in response to six sessions of MICT (white bars) and HIIT (black bars).
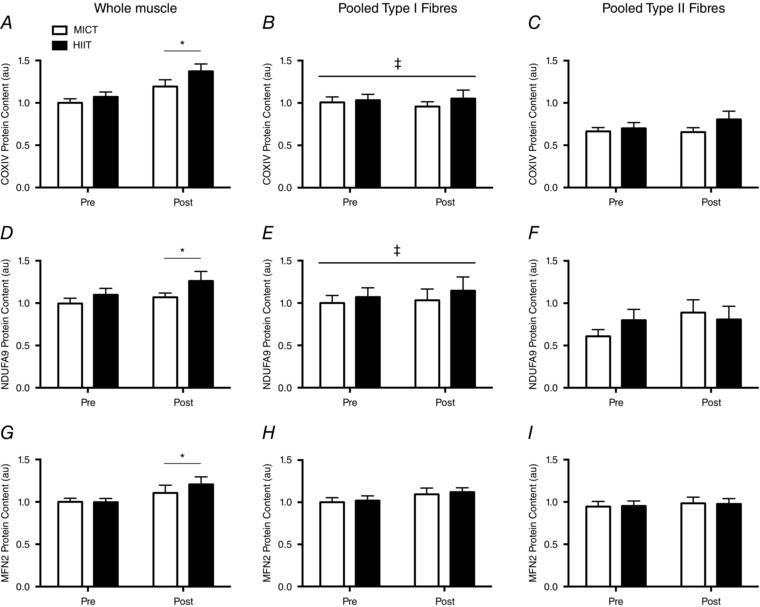
COXIV (A–C), NDUFA9 (D–F) and MFN2 (G–I) are displayed separately. Whole muscle samples were normalized to the pre‐training MICT samples, whereas pooled muscle fibres were normalised to the pre‐training, type I fibre, MICT samples. Symbols indicate a significant main effect of training (*); and a significant fibre‐type difference (‡). Representative blots are displayed in Fig. 1. Error bars represent the SEM (n = 10 subjects).
Single‐fibre protein content
Pre‐training, type I muscle fibres had greater abundances of COXIV (49%; P < 0.001) and NDUFA9 (47%; P = 0.006) relative to type II muscle fibres, although the two fibre types had similar abundances of MFN2 (P = 0.23) (Fig. 4). Training did not affect COXIV (P = 0.80; P = 0.24), NDUFA9 (P = 0.67; P = 0.08) or MFN2 (P = 0.21; P = 0.51) protein content in type I or type II muscle fibres, and there were no significant training by group interactions (P > 0.05) (Fig. 4).
Discussion
The major novel finding of the present study was that interval compared to continuous single‐leg cycling, matched for total work and session duration, elicited greater increases in mitochondrial content in human skeletal muscle. These data suggest an important role for exercise intensity, and/or the pattern of contraction, in determining skeletal muscle adaptations to aerobic exercise training. Building on recent work (Abbiss et al. 2011; Vincent et al. 2015) and analogous to single‐limb training studies that have examined skeletal muscle responses to resistance training (Chesley et al. 1992; Burd et al. 2012; Mitchell et al. 2012), the present study demonstrates the utility of single‐leg cycling as a model to investigate adaptations to different exercise training protocols within the same subject.
We measured a series of mitochondrial content biomarkers to comprehensively assess aerobic exercise training adaptations. Whole muscle COXIV protein content, CS maximal activity and mass‐specific J O2 in permeabilized muscle fibres are associated with mitochondrial volume density (Larsen et al. 2012) and represent a hierarchical assessment of mitochondrial content (i.e. protein content, enzyme activity, organelle function, respectively). In the present study, 2 weeks of HIIT induced greater increases in CS maximal activity and mass‐specific J O2 (PCI and PCI&CII) relative to MICT, whereas the training‐induced increase in whole muscle COXIV protein content was similar between groups. Of note, CS maximal activity and mass‐specific J O2 are more strongly related to mitochondrial volume density than is ETS complex 4 protein content (Larsen et al. 2012). These results agreed with our hypothesis, which was based on higher exercise intensities augmenting the acute activation of processes related to mitochondrial biogenesis relative to lower intensities, when work is matched (Egan et al. 2010; Di Donato et al. 2014; Kristensen et al. 2015). The divergent adaptations elicited by HIIT and MICT in the present study probably reflect acute differences in the activation of pathways that regulate mitochondrial content that compound with training (Coffey and Hawley, 2007; Perry et al. 2010). Blood lactate concentration, heart rate, and ratings of perceived exertion and dyspnoea were also greater with HIIT relative to MICT, suggesting that HIIT provided a greater metabolic stress despite the equivalent total work performed.
The present study is the first to use a single‐leg cycling model to compare HIIT and MICT. Our within‐subject, parallel‐group design provided adequate statistical power to demonstrate that HIIT induced significantly greater increases in multiple markers of mitochondrial content relative to MICT. To our knowledge, three other studies have compared mitochondrial adaptations to work‐matched HIIT and MICT in humans. Using a cross‐over design, Daussin et al. (2008a) demonstrated that an 8 week HIIT intervention, involving four to seven 1 min intervals (90% W peak) per session, elicited a greater increase in mass‐specific JO2 (PCI) relative to a MICT protocol that involved cycling at ∼60% W peak. By contrast, Granata et al. (2016) recently reported that mitochondrial content (CS maximal activity, mass‐specific J O2 and COXII protein content) was unchanged after 4 weeks of MICT or HIIT. The observation that their HIIT protocol, which involved four to seven 4 min intervals (∼80–90% W peak) per session, did not elicit a training effect is contrary to the present data, as well as previous studies of short‐term HIIT (Talanian et al. 2006; Little et al. 2010; Jacobs et al. 2013; Cochran et al. 2015a). Finally, Henriksson and Reitman (1976) reported similar increases in succinate dehydrogenase activity following 8 weeks of HIIT (+27%) and MICT (+22%), although the sample size was very small (n = 4 and n = 5, respectively) and comparisons were made between subjects. The present findings and the results reported by Daussin et al. (2008a), which each used a within‐subject design, suggest that intensity, and/or the pattern of contraction (see further discussion below), are important determinants of mitochondrial adaptations to short‐term exercise training.
The ETS proteins, COXIV and NDUFA9, were chosen to assess mitochondrial content (Larsen et al. 2012), whereas MFN2 was chosen to assess mitochondrial fusion (Yan et al. 2012). Although exercise is known to increase mitochondrial content, less is known about its effects on mitochondrial dynamics (Perry et al. 2010; Yan et al. 2012). The content of these proteins increased in response to training at the whole muscle level, suggesting increased mitochondrial content and potentially greater rates of mitochondrial fusion. Because we only measured MFN2, more data are needed to understand the effects of exercise and exercise intensity on aspects of mitochondrial dynamics.
We hypothesized that exercise‐induced adaptations in type II muscle fibres would be greater in response to HIIT relative to MICT as a result of greater skeletal muscle activation/fatigue (Theurel and Lepers, 2008; Kristensen et al. 2015); however, the effect of training appeared to be depressed in pooled fibres: the response of COXIV protein content in type I and type II muscle fibres followed the same pattern as the whole muscle data, except with smaller (non‐significant) mean differences between groups. We have shown that pooling as few as four muscle fibres is sufficient to obtain a representative sample of the muscle (Christiansen D, MacInnis MJ, Zacharewicz E, Frankish BP & Murphy RM; unpublished data), which suggests that inter‐fibre variation in protein content was probably not responsible for the absence of a training effect. Both COXIV and NDUFA9 were more abundant in type I compared to type II muscle fibres, reflecting the well known fibre‐type difference in mitochondrial content of the human vastus lateralis muscle fibres (Howald et al. 1985) and agreeing with previous studies of mitochondrial protein content measured in pooled individual fibres (Albers et al. 2015; Christensen et al. 2015). MFN2 was expressed similarly in type I and type II fibres. Although we were able to identify fibre type‐specific differences in protein content, it is possible that more muscle fibres would need to be pooled to detect the effect of training, which was smaller than the observed fibre type differences for COXIV and NDUFA9.
The present study assessed mitochondrial adaptations to a relatively short training period. Mitochondrial content is known to increase rapidly and plateau with aerobic‐based exercise training. For example, increases in mitochondrial adaptations to exercise stabilized after three to seven sessions of HIIT (Perry et al. 2010) or MICT (Egan et al. 2013). The six sessions of exercise used in the present study (at least with respect to HIIT) were sufficient to elicit relatively large increases in CS maximal activity (39%), various states of mass‐specific J O2 (12–22%) and COXIV protein content (24%) that were comparable to previous 2 week studies of HIIT (Hood et al. 2011; Jacobs et al. 2013; Cochran et al. 2015a).
An important attribute of the present study was that training intensity and duration were not altered during the experiment. Green et al. (2012) reported similar metabolic adaptations to short‐term training for 30 or 60 min day−1 when exercising at 60% ; however, Green et al. (2013) also demonstrated augmented metabolic adaptations in response to longer training sessions when the exercise intensity was higher (i.e. 70% and 86% of ). This pair of studies suggests that exercise duration does affect skeletal muscle remodelling, although this effect is intensity‐dependent. Such findings are in agreement with an early study in rats (Fitts et al. 1975); however, there is also evidence that increasing the exercise duration does not affect mitochondrial content in humans (Rosenkilde et al. 2015) or rats (Dudley et al. 1982). Ultimately, given that the effect of exercise duration is potentially intensity‐dependent, altering the duration of training sessions during an experiment might hinder the comparisons of different exercise intensities.
In support of the role of exercise intensity in mediating human skeletal muscle adaptations to exercise (Gibala et al. 2012), SIT typically elicits similar (Saltin et al. 1976; Gibala et al. 2006; Burgomaster et al. 2008) or greater (Granata et al. 2016) skeletal muscle adaptations relative to MICT, even when less work is performed (Gibala et al. 2006; Burgomaster et al. 2008; Granata et al. 2016). In one of the few work‐matched comparison of SIT and MICT (Saltin et al. 1976), the progression in the training stimuli could explain the discordant results: the intensity of MICT, which was relatively high to begin with (i.e. 75% of ), was increased as necessary to maintain target heart rate, whereas it appears that the duration of the SIT protocol was increased to match total work because the intensity began at 150% of , and it is difficult to increase the intensity of ‘all‐out’ exercise (Cochran et al. 2015b).
The available data are unclear regarding the potential for exercise training to increase mitochondrial function (i.e. mitochondria‐specific J O2). Mitochondrial content (Hoppeler et al. 1973; Jacobs and Lundby, 2013) and mitochondrial‐specific J O2 (Daussin et al. 2008b; Jacobs and Lundby, 2013) are greater in the muscle of aerobically trained individuals relative to less trained individuals, suggesting that training might increase mitochondrial content and function (Bishop et al. 2014). Although short‐term aerobic exercise training has been shown to increase mitochondrial function (Starritt et al. 1999), most studies do not support this result (Wibom et al. 1992; Tonkonogi et al. 2000; Jacobs et al. 2013). In the present study, mitochondrial‐specific J O2 was unaffected by training, which is in agreement with these latter studies. A recent comparison of HIIT and MICT also demonstrated no effect of training on mitochondrial function (Granata et al. 2016). In that same study, mitochondrial‐specific J O2 increased in the SIT group; however, the non‐significant increase in CS maximal activity (to which mass‐specific J O2 was normalized) was lower than the changes reported in previous SIT studies (Burgomaster et al. 2006; Little et al. 2010; Cochran et al. 2015b). Mass‐specific J O2 values (PCI, PCI&CII and E) normalized to COXIV or NDUFA9 protein content (i.e. alternative measures of mitochondrial function) were also unaffected by training (data not reported).
The aim of the present study was to compare work‐ and duration‐matched aerobic exercise training programmes differing in intensity, necessitating an interval format for the high‐intensity exercise protocol. Thus, we cannot exclude the possibility that the interval style of exercise per se is partly responsible for the divergent mitochondrial adaptations observed between HIIT and MICT. An acute session of moderate‐intensity exercise performed in a series of 30 intervals of 1 min in duration was demonstrated to elicit greater acute molecular responses in skeletal muscle compared to 30 min of continuous exercise performed at the same intensity (Combes et al. 2015); however, it is unclear whether these acute differences would be apparent with fewer intervals, as in the present study, or translate to divergent chronic adaptations. We hypothesize that the augmented response observed following HIIT compared to MICT is a result of greater cellular stress incurred by exercising at a higher intensity. In general, higher intensities of exercise use more glycogen, produce more lactate and augment the Cr:PCr and AMP:ATP ratios compared to lower intensities of exercise (Gollnick et al. 1974; Vøllestad et al. 1985; Howlett et al. 1998; Egan et al. 2010; Kristensen et al. 2015). Downstream of these metabolic differences, the activity of AMPK (Wojtaszewski et al. 2000; Kristensen et al. 2015), an energy‐sensing molecule that regulates mitochondrial biogenesis, and other signalling pathways related to mitochondrial content, including the expression of peroxisome proliferator‐activated receptor gamma coactivator 1α (Egan et al. 2010), is also greater at higher intensities of exercise. In the present study, blood lactate was greater following HIIT compared to MICT; however, we did not assess any acute skeletal muscle responses, preventing conclusions on the metabolic basis of the observed differences in response to 2 weeks of HIIT and MICT.
Short‐term HIIT generally increases (Talanian et al. 2006; Perry et al. 2008; Jacobs et al. 2013); however, the six sessions of single‐leg cycling HIIT in the present study were insufficient to increase single‐leg . This finding is potentially a result of the relatively lower cardiorespiratory stimulus associated with single‐leg cycling; however, has been increased with longer periods of single‐leg cycling training (Saltin et al. 1976; Rud et al. 2012). The reduced cardiorespiratory demand allows single‐leg cycling to be used as a training stimulus for patients with chronic obstructive pulmonary disease (Dolmage and Goldstein, 2006; Dolmage, 2008), as well as trained cyclists (Abbiss et al. 2011). For the latter group, 3 weeks of single‐leg cycling HIIT augmented mitochondrial adaptations relative to double‐legged cycling HIIT, probably as a result of the greater relative intensity (Abbiss et al. 2011). That the oxidative capacity of skeletal muscle increased markedly in the present study (at least with HIIT), whereas the single‐leg was unchanged, supports the concept of skeletal muscle mitochondrial capacity exceeding, and thus not restricting, aerobic capacity (Boushel et al. 2011).
Unilateral exercise training is frequently used to investigate skeletal muscle remodelling in response to resistance exercise (Chesley et al. 1992; Burd et al. 2012; Mitchell et al. 2012), although comparisons of aerobic exercise training protocols typically use between‐subject designs (Gibala et al. 2006; Burgomaster et al. 2008; Granata et al. 2016). Unlike cross‐over designs (Daussin et al. 2008a), single‐leg cycling permits within‐subject designs that have a parallel‐group design, minimizing the influence of individual differences and confounding factors at the same time as also eliminating the need for washout periods. If a between‐subject design had been used in the present study, a much larger number of subjects would have been needed to detect statistically differences in markers of mitochondrial content (i.e. ∼70 subjects for CS maximal activity).
In conclusion, this experiment compared two short‐term, exercise‐training protocols that were matched for work and session duration. The main finding was that interval compared to continuous single‐leg cycling elicited superior mitochondrial adaptations in human skeletal muscle, as indicated by higher CS maximal activity and mass‐specific J O2 (PCI and PCI&CII) after training. By contrast, mitochondrial function was unaffected by either training protocol, suggesting that the observed adaptations reflect an increase in mitochondrial content with short‐term training. The present data suggest that exercise intensity, and/or the pattern of contraction, is an important determinant of exercise‐induced skeletal muscle remodelling in humans.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
MM, MG and RM conceived the study. MM, EZ, MH, BM, LS, MT, RM and MG collected samples, performed analyses and/or interpreted data. MM and MG wrote the first draft of the manuscript. All authors critically revised the manuscript for intellectual content. All authors approved of the final manuscript and agreed to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This project was supported by an operating grant from the Natural Sciences and Engineering Research Council (NSERC; grant number RGPIN‐2015‐04632) to MG. MM was supported by an NSERC Postdoctoral Fellowship, and LS was supported by an NSERC Vanier Canada Graduate Scholarship.
Acknowledgements
The authors wish to thank Dr Jenna Gillen, Dr Lauren MacNeil and Ms. Heidy Latchman for assistance with laboratory procedures, as well as Dr Jim Martin for providing the customized pedals that allowed the cycle ergometer to be adapted for single‐leg cycling.
References
- Abbiss CR, Karagounis LG, Laursen PB, Peiffer JJ, Martin DT, Hawley JA, Fatehee NN & Martin JC (2011). Single‐leg cycle training is superior to double‐leg cycling in improving the oxidative potential and metabolic profile of trained skeletal muscle. J Appl Physiol 110, 1248–1255. [DOI] [PubMed] [Google Scholar]
- Albers PH, Pedersen AJT, Birk JB, Kristensen DE, Vind BF, Baba O, Nøhr J, Højlund K & Wojtaszewski JFP (2015). Human muscle fiber type‐specific insulin signaling: impact of obesity and type 2 diabetes. Diabetes 64, 485–497. [DOI] [PubMed] [Google Scholar]
- Bini RR, Jacques TC, Lanferdini FJ & Vaz MA (2015). Comparison of kinetics, kinematics, and electromyography during single‐leg assisted and unassisted cycling. J Strength Cond Res 29, 1534–1541. [DOI] [PubMed] [Google Scholar]
- Bishop DJ, Granata C & Eynon N (2014). Can we optimise the exercise training prescription to maximise improvements in mitochondria function and content? Biochim Biophys Acta 1840, 1266–1275. [DOI] [PubMed] [Google Scholar]
- Boushel R, Gnaiger E, Calbet JAL, Gonzalez‐Alonso J, Wright‐Paradis C, Sondergaard H, Ara I, Helge JW & Saltin B (2011). Muscle mitochondrial capacity exceeds maximal oxygen delivery in humans. MITOCH 11, 303–307. [DOI] [PubMed] [Google Scholar]
- Burd NA, Andrews RJ, West DWD, Little JP, Cochran AJR, Hector AJ, Cashaback JGA, Gibala MJ, Potvin JR, Baker SK & Phillips SM (2012). Muscle time under tension during resistance exercise stimulates differential muscle protein sub‐fractional synthetic responses in men. J Physiol 590, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgomaster KA, Heigenhauser GJF & Gibala MJ (2006). Effect of short‐term sprint interval training on human skeletal muscle carbohydrate metabolism during exercise and time‐trial performance. J Appl Physiol 100, 2041–2047. [DOI] [PubMed] [Google Scholar]
- Burgomaster KA, Howarth KR, Phillips SM, Rakobowchuk M, Macdonald MJ, McGee SL & Gibala MJ (2008). Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J Physiol 586, 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KJ, Pollock BS, LaScola P & McDaniel J (2014). Cardiovascular responses to counterweighted single‐leg cycling: implications for rehabilitation. Eur J Appl Physiol 114, 961–968. [DOI] [PubMed] [Google Scholar]
- Carter SL, Rennie C & Tarnopolsky MA (2001). Substrate utilization during endurance exercise in men and women after endurance training. Am J Physiol Endocrinol Metab 280, E898–E907. [DOI] [PubMed] [Google Scholar]
- Chesley A, MacDougall JD, Tarnopolsky MA, Atkinson SA & Smith K (1992). Changes in human muscle protein synthesis after resistance exercise. J Appl Physiol 73, 1383–1388. [DOI] [PubMed] [Google Scholar]
- Christensen PM, Gunnarsson TP, Thomassen M, Wilkerson DP, Nielsen JJ & Bangsbo J (2015). Unchanged content of oxidative enzymes in fast‐twitch muscle fibers and kinetics after intensified training in trained cyclists. Physiol Rep 3, e12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran AJ, Myslik F, MacInnis MJ, Percival ME, Bishop D, Tarnopolsky MA & Gibala MJ (2015a). Manipulating carbohydrate availability between twice‐daily sessions of high‐intensity interval training over two weeks improves time‐trial performance. Int J Sport Nutr Exerc Metab 25, 463–470. [DOI] [PubMed] [Google Scholar]
- Cochran AJ, Percival ME, Thompson S, Gillen JB, MacInnis MJ, Potter MA, Tarnopolsky MA & Gibala MJ (2015b). β‐alanine supplementation does not augment the skeletal muscle adaptive response to 6 weeks of sprint interval training. Int J Sport Nutr Exerc Metab 25, 541–549. [DOI] [PubMed] [Google Scholar]
- Coffey VG & Hawley JA (2007). The molecular bases of training adaptation. Sports Med 37, 737–763. [DOI] [PubMed] [Google Scholar]
- Combes A, Dekerle J, Webborn N, Watt P, Bougault V & Daussin FN (2015). Exercise‐induced metabolic fluctuations influence AMPK, p38‐MAPK and CaMKII phosphorylation in human skeletal muscle. Physiol Rep 3, e12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daussin FN, Zoll J, Dufour SP, Ponsot E, Lonsdorfer‐Wolf E, Doutreleau S, Mettauer B, Piquard F, Geny B & Richard R (2008a). Effect of interval versus continuous training on cardiorespiratory and mitochondrial functions: relationship to aerobic performance improvements in sedentary subjects. Am J Physiol Regul Integr Comp Physiol 295, R264–R272. [DOI] [PubMed] [Google Scholar]
- Daussin FN, Zoll J, Ponsot E, Dufour SP, Doutreleau S, Lonsdorfer E, Ventura‐Clapier R, Mettauer B, Piquard F, Geny B & Richard R (2008b). Training at high exercise intensity promotes qualitative adaptations of mitochondrial function in human skeletal muscle. J Appl Physiol 104, 1436–1441. [DOI] [PubMed] [Google Scholar]
- Di Donato DM, West DWD, Churchward‐Venne TA, Breen L, Baker SK & Phillips SM (2014). Influence of aerobic exercise intensity on myofibrillar and mitochondrial protein synthesis in young men during early and late postexercise recovery. Am J Physiol Endocrinol and Metab 306, E1025–E1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmage TE (2008). Effects of one‐legged exercise training of patients with COPD. Chest 133, 370. [DOI] [PubMed] [Google Scholar]
- Dolmage TE & Goldstein RS (2006). Response to one‐legged cycling in patients with COPD. Chest 129, 325–332. doi: 10.1378/chest.129.2.325 [DOI] [PubMed] [Google Scholar]
- Dudley GA, Abraham WM & Terjung RL (1982). Influence of exercise intensity and duration on biochemical adaptations in skeletal muscle. J Appl Physiol Respir Environ Exerc Physiol 53, 844–850. [DOI] [PubMed] [Google Scholar]
- Egan B, Carson BP, Garcia‐Roves PM, Chibalin AV, Sarsfield FM, Barron N, McCaffrey N, Moyna NM, Zierath JR & O'Gorman DJ (2010). Exercise intensity‐dependent regulation of peroxisome proliferator‐activated receptor γ coactivator‐1α mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol 588, 1779–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, O'Connor PL, Zierath JR & O'Gorman DJ (2013). Time course analysis reveals gene‐specific transcript and protein kinetics of adaptation to short‐term aerobic exercise training in human skeletal muscle. PLoS ONE 8, e74098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmer SJ, Amann M, McDaniel J, Martin DT & Martin JC (2012). Fatigue is specific to working muscles: no cross‐over with single‐leg cycling in trained cyclists. Eur J Appl Physiol 113, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitts RH, Booth FW, Winder WW & Holloszy JO (1975). Skeletal muscle respiratory capacity, endurance, and glycogen utilization. Am J Physiol 228, 1029–1033. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, Little JP, MacDonald MJ & Hawley JA (2012). Physiological adaptations to low‐volume, high‐intensity interval training in health and disease. J Physiol 590, 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibala MJ, Little JP, van Essen M, Wilkin GP, Burgomaster KA, Safdar A, Raha S & Tarnopolsky MA (2006). Short‐term sprint interval versus traditional endurance training: similar initial adaptations in human skeletal muscle and exercise performance. J Physiol 575, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollnick PD, Piehl K & Saltin B (1974). Selective glycogen depletion pattern in human muscle fibres after exercise of varying intensity and at varying pedalling rates. J Physiol, 241, 45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodpaster BH (2013). Mitochondrial deficiency is associated with insulin resistance. Diabetes 62, 1032–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata C, Oliveira RSF, Little JP, Renner K & Bishop DJ (2016). Training intensity modulates changes in PGC‐1α and p53 protein content and mitochondrial respiration, but not markers of mitochondrial content in human skeletal muscle. FASEB J 30, 959–970. [DOI] [PubMed] [Google Scholar]
- Green HJ, Burnett M, Carter S, Jacobs I, Ranney D, Smith I & Tupling S (2013). Role of exercise duration on metabolic adaptations in working muscle to short‐term moderate‐to‐heavy aerobic‐based cycle training. Eur J Appl Physiol 113, 1965–1978. [DOI] [PubMed] [Google Scholar]
- Green HJ, Burnett M, Jacobs I, Ranney D, Smith I & Tupling S (2012). Adaptations in muscle metabolic regulation require only a small dose of aerobic‐based exercise. Eur J Appl Physiol 113, 313–324. [DOI] [PubMed] [Google Scholar]
- Henriksson J (1977). Training induced adaptation of skeletal muscle and metabolism during submaximal exercise. J Physiol 270, 661–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson J & Reitman JS (1976). Quantitative measures of enzyme activities in type I and type II muscle fibres of man after training. Acta Physiol Scand 97, 392–397. [DOI] [PubMed] [Google Scholar]
- Holloszy JO (1967). Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242, 2278–2282. [PubMed] [Google Scholar]
- Holloszy JO & Coyle EF (1984). Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol Respir Environ Exerc Physiol 56, 831–838. [DOI] [PubMed] [Google Scholar]
- Hood MS, Little JP, Tarnopolsky MA, Myslik F & Gibala MJ (2011). Low‐volume interval training improves muscle oxidative capacity in sedentary adults. Med Sci Sports Exerc 43, 1849–1856. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Lüthi P, Claassen H, Weibel ER & Howald H (1973). The ultrastructure of the normal human skeletal muscle. A morphometric analysis on untrained men, women and well‐trained orienteers. Pflügers Arch – Eur J Physiol 344, 217–232. [DOI] [PubMed] [Google Scholar]
- Howald H, Hoppeler H, Claassen H, Mathieu O & Straub R (1985). Influences of endurance training on the ultrastructural composition of the different muscle fiber types in humans. Pflügers Arch – Eur J Physiol 403, 369–376. [DOI] [PubMed] [Google Scholar]
- Howlett RA, Parolin ML, Dyck DJ, Hultman E, Jones NL, Heigenhauser GJ & Spriet LL (1998). Regulation of skeletal muscle glycogen phosphorylase and PDH at varying exercise power outputs. Am J Physiol Regul Interg Comp Physiol 275, R418–R25. [DOI] [PubMed] [Google Scholar]
- Jacobs RA, Flück D, Bonne TC, Bürgi S, Christensen PM, Toigo M & Lundby C (2013). Improvements in exercise performance with high‐intensity interval training coincide with an increase in skeletal muscle mitochondrial content and function. J Appl Physiol 115, 785–793. [DOI] [PubMed] [Google Scholar]
- Jacobs RA & Lundby C (2013). Mitochondria express enhanced quality as well as quantity in association with aerobic fitness across recreationally active individuals up to elite athletes. J Appl Physiol 114, 344–350. [DOI] [PubMed] [Google Scholar]
- Joseph A‐M, Adhihetty PJ, Buford TW, Wohlgemuth SE, Lees HA, Nguyen LMD, Aranda JM, Sandesara BD, Pahor M, Manini TM, Marzetti E & Leeuwenburgh C (2012). The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high‐ and low‐functioning elderly individuals. Aging Cell 11, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen DE, Albers PH, Prats C, Baba O, Birk JB & Wojtaszewski JFP (2015). Human muscle fibre type‐specific regulation of AMPK and downstream targets by exercise. J Physiol 593, 2053–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F & Hey‐Mogensen M (2012). Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 590, 3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek BT, Mudaliar SR, Henry R, Mathieu‐Costello O & Richardson RS (2001). Effect of acute exercise on citrate synthase activity in untrained and trained human skeletal muscle. Am J Physiol Reg Integr Comp Physiol 280, R441–R447. [DOI] [PubMed] [Google Scholar]
- Little JP, Safdar A, Wilkin GP, Tarnopolsky MA & Gibala MJ (2010). A practical model of low‐volume high‐intensity interval training induces mitochondrial biogenesis in human skeletal muscle: potential mechanisms. J Physiol 588, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDougall JD, Hicks AL, MacDonald JR, McKelvie RS, Green HJ & Smith KM (1998). Muscle performance and enzymatic adaptations to sprint interval training. J Appl Physiol 84, 2138–2142. [DOI] [PubMed] [Google Scholar]
- Mitchell CJ, Churchward‐Venne TA, West DWD, Burd NA, Breen L, Baker SK & Phillips SM (2012). Resistance exercise load does not determine training‐mediated hypertrophic gains in young men. J Appl Physiol 113, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica JP, Oakhill JS, Lamb GD & Murphy RM (2009). Analytical Biochemistry. Anal. Biochem. 386, 270–275. [DOI] [PubMed] [Google Scholar]
- Morgan TE, Cobb LA, Short FA, Ross R & Gunn DR (1971). Effects of Long‐Term Exercise on Human Muscle Mitochondria, in: Advances in Experimental Medicine and Biology, pp. 87–95. Springer, Boston, MA. [Google Scholar]
- Murphy RM (2011). Enhanced technique to measure proteins in single segments of human skeletal muscle fibers: fiber‐type dependence of AMPK‐alpha1 and ‐beta1. J Appl Physiol 110, 820–825. [DOI] [PubMed] [Google Scholar]
- Perry CGR, Heigenhauser GJF, Bonen A & Spriet LL (2008). High‐intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl Physiol Nutr Metab 33, 1112–1123. [DOI] [PubMed] [Google Scholar]
- Perry CGR, Lally J, Holloway GP, Heigenhauser GJF, Bonen A & Spriet LL (2010). Repeated transient mRNA bursts precede increases in transcriptional and mitochondrial proteins during training in human skeletal muscle. J Physiol 588, 4795–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesta D & Gnaiger E (2011). High‐resolution respirometry: oxphos protocols for human cells and permeabilized fibers from small biopsies of human muscle In Methods in Molecular Biology, Methods in Molecular Biology, ed. Carlos M Palmeira & Antonio J Moreno, pp. 25–58. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Rosenkilde M, Reichkendler MH, Auerbach P, Bonne TC, Sjödin A, Ploug T & Stallknecht BM (2015). Changes in peak fat oxidation in response to different doses of endurance training. Scand J Med Sci Sports 25, 41–52. [DOI] [PubMed] [Google Scholar]
- Rud B, Foss O, Krustrup P, Secher NH & Hallén J (2012). One‐legged endurance training: leg blood flow and oxygen extraction during cycling exercise. Acta Physiol (Oxf) 205, 177–185. [DOI] [PubMed] [Google Scholar]
- Saltin B, Nazar K, Costill DL, Stein E, Jansson E, Essén B & Gollnick D (1976). The nature of the training response; peripheral and central adaptations of one‐legged exercise. Acta Physiol Scand 96, 289–305. [DOI] [PubMed] [Google Scholar]
- Starritt EC, Angus D & Hargreaves M (1999). Effect of short‐term training on mitochondrial ATP production rate in human skeletal muscle. J Appl Physiol 86, 450–454. [DOI] [PubMed] [Google Scholar]
- Stroud DA, Formosa LE, Wijeyeratne XW, Nguyen TN & Ryan MT (2013). Gene knockout using transcription activator‐like effector nucleases (TALENs) reveals that human NDUFA9 protein is essential for stabilizing the junction between membrane and matrix arms of complex I. J Biol Chem 288, 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talanian JL, Galloway SDR, Heigenhauser GJF, Bonen A & Spriet LL (2006). Two weeks of high‐intensity aerobic interval training increases the capacity for fat oxidation during exercise in women. J Appl Physiol 102, 1439–1447. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Pearce E, Smith K & Lach B (2011). Suction‐modified Bergström muscle biopsy technique: experience with 13,500 procedures. Muscle Nerve 43, 717–725. [DOI] [PubMed] [Google Scholar]
- Theurel J & Lepers R (2008). Neuromuscular fatigue is greater following highly variable versus constant intensity endurance cycling. Eur J Appl Physiol 103, 461–468. [DOI] [PubMed] [Google Scholar]
- Tonkonogi M, Walsh B, Svensson M & Sahlin K (2000). Mitochondrial function and antioxidative defence in human muscle: effects of endurance training and oxidative stress. J Physiol 528, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent G, Lamon SV, Gant N, Vincent PJ, MacDonald JR, Markworth JF, Edge JA & Hickey AJR (2015). Changes in mitochondrial function and mitochondria associated protein expression in response to 2‐weeks of high intensity interval training. Front Physiol 6, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vøllestad NK & Blom PC (1985). Effect of varying exercise intensity on glycogen depletion in human muscle fibres. Acta Physiol Scand 125, 395–405. [DOI] [PubMed] [Google Scholar]
- Wibom R, Hultman E, Johansson M, Matherei K, Constantin‐Teodosiu D & Schantz PG (1992). Adaptation of mitochondrial ATP production in human skeletal muscle to endurance training and detraining. J Appl Physiol 73, 2004–2010. [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA & Kiens B (2000). Isoform‐specific and exercise intensity‐dependent activation of 5′‐AMP‐activated protein kinase in human skeletal muscle. J Physiol 528, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Lira VA & Greene NP (2012). Exercise training‐induced regulation of mitochondrial quality. Exerc Sport Sci Rev 40, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]