

Abstract
In the early to mid‐20th century, reductionism as a concept in biology was challenged by key thinkers, including Ludwig von Bertalanffy. He proposed that living organisms were specific examples of complex systems and, as such, they should display characteristics including hierarchical organisation and emergent behaviour. Yet the true study of complete biological systems (for example, metabolism) was not possible until technological advances that occurred 60 years later. Technology now exists that permits the measurement of complete levels of the biological hierarchy, for example the genome and transcriptome. The complexity and scale of these data require computational models for their interpretation. The combination of these – systems thinking, high‐dimensional data and computation – defines systems biology, typically accompanied by some notion of iterative model refinement. Only sequencing‐based technologies, however, offer full coverage. Other ‘omics’ platforms trade coverage for sensitivity, although the densely connected nature of biological networks suggests that full coverage may not be necessary. Systems biology models are often characterised as either ‘bottom‐up’ (mechanistic) or ‘top‐down’ (statistical). This distinction can mislead, as all models rely on data and all are, to some degree, ‘middle‐out’. Systems biology has matured as a discipline, and its methods are commonplace in many laboratories. However, many challenges remain, especially those related to large‐scale data integration.
Keywords: human physiology, metabolism, systems biology
What is systems biology?
Any treatment of systems biology starts with a history of reductionism, the origins of which can be traced to Descartes (1596–1650) (Trewavas, 2006). The essence of the reductionist approach is that one can understand any complex phenomenon by studying its constituent parts and events. Reductionism has been remarkably successful, especially in the physical sciences. In biology, the treatment of living organisms as ‘machines’ – whose behaviour is entirely predictable from an understanding of their components – persisted until the mid‐20th century when it was robustly challenged by thinkers including Williams (1956) and von Bertallanfy, (1968). The latter's seminal book General Systems Theory questioned reductionist thinking and clearly articulated the then novel concept that living organisms were specific examples of complex systems; as such they should share characteristics with other complex systems, such as hierarchical organisation and emergent characteristics (Trewavas, 2006). The book, still in print today, contains diagrams of biochemical pathways and networks that look surprisingly contemporary. If one accepts von Bertalanffy's reasoning, as most now do, then the higher order characteristics and behaviour of organisms cannot necessarily be inferred from a lower order understanding. One must somehow observe, measure and model complete systems in order to capture emergent properties. However, it is worth noting that reductionism and systems thinking are not exclusive or opposed. One needs to understand both the components of a system, and their integrated functioning.
Hence the consideration of living systems as clockwork, uniform, utterly predictable machines, popular at the turn of the 20th century, is no longer tenable. Gene expression is known to be stochastic in nature (Elowitz et al. 2002) and advances in single cell transcriptomics have revealed heterogeneity in gene expression between cells in phenotypically similar populations (Brennecke et al. 2013). Modern biologists think of living organisms as dynamic systems that are chaotic yet organised, and profoundly, astonishingly complex.
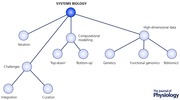
The common approach to the history and origins of systems biology outlined above has led to an unfortunate side effect: the definition of systems biology by exclusion (‘not reductionism’) rather than inclusion (defining what it actually is). Unfortunately this ‘definition by exclusion’ captures all other ‘non‐reductionist’ efforts in biology including (for example) the distinct field of integrative physiology, leading some integrative physiologists to cry foul (Joyner & Saltin, 2008). Yet those who work in systems biology see clear differences with other integrative disciplines, despite their similar philosophy, and it should be possible to provide a more satisfactory definition. A survey of the actual methods of systems biology is a useful point of departure. The core components of the contemporary systems biology workflow are: (1) high‐dimensional data acquisition (genomics, transcriptomics etc.), either experimentally or from the existing literature; (2) sophisticated computational methods for data integration and contextualisation; and (3) the scope for iterative refinement of the computational model via further experiments (termed the ‘model as hypothesis’ (Palsson, 2008)). These components will be discussed individually below. Pragmatic definitions of systems biology (Ideker et al. 2001; Edwards & Thiele, 2013) supplement the non‐reductionist philosophy with the inclusion of the methods used ‘at the coal face’, thus distinguishing systems biology both from integrative physiology as well as (for example) purely method‐driven efforts like high‐content screening. Further, systems biology must now be viewed as a component of a larger life sciences (and medical) ‘informatics ecosystem’ (Fig. 1) that encompasses data science and visualisation, bioinformatics, computational biology, medical/translational informatics, wearable devices (and streaming data) and personalised medicine (to name a few).
Figure 1.
Systems biology – a component of a larger life sciences (and medical) ‘informatics ecosystem’
Metabolic systems biology
Metabolism occupies a special place in systems biology; metabolic networks have been studied and modelled by the community more than any other. There are several possible reasons for this. First, metabolism is the most comprehensively described of any biological network. Since Otto Meyerhoff and others pieced together the first recognized metabolic pathway (glycolysis; Kresge et al. 2005), biochemists have had remarkable success in reconstructing the metabolic networks of cells and tissues. The culmination of these efforts has been the complete reconstruction of the human metabolic network, now in its second revision (Thiele et al. 2013). Second, unlike signalling networks, the substrates and enzymes of metabolism are separated. This means that metabolism can be modelled using analogies with flow. Indeed, it could be argued that the separation of metabolites and enzymes, by allowing straightforward monitoring of metabolite levels as well as the use of tracers (e.g. heavy isotopes), has made the job of elucidating metabolic pathways considerably more straightforward. Signalling networks carry signals (information), encoded in the state of the component protein complexes, which act on each other. This is a more difficult concept to capture (experimentally and mathematically), although fine‐grained modelling of sections of signalling pathways are commonplace, using ordinary differential equations. This separation of enzyme and substrate sets the modelling of signalling networks apart from not only metabolism, but also ion channels (another area where systems biology has had considerable impact). The separation of moieties and enzymes allows modellers to validate their models using physico‐chemical laws such as mass and charge conservation, an option that is otherwise unavailable. Therefore the sheer quantity and completeness of the information regarding metabolic networks is unrivalled elsewhere in biology, while the nature of metabolism is tractable by many mathematical and experimental methods.
Components of the systems biology workflow
High‐dimensional data acquisition
To study an entire system, it is desirable to be able to observe and measure all the components of that system. At present only sequencing‐based technologies offer complete coverage of their targets: DNA or RNA. Hence the genome, transcriptome, miRNome (microRNA‐ome) and methylome can be studied in their entirety (although bias may be present; Risso et al. 2011). This completeness of coverage challenges those who continue to accuse systems biologists of hypothesis‐free ‘fishing’ (Wanjek, 2011): the analogy fails when one catches every fish. However, complete coverage may not even be necessary. Biological networks are densely connected, and few (if any) measures are independent of others. Theoretically therefore, quasi‐complete coverage can be achieved by measuring a carefully chosen subset of ‘sentinel’ molecules. Such an approach is being taken in the L1000 project (The Broad Institute; http://www.lincscloud.org/l1000/): to increase throughput, a subset of 1000 genes have been chosen for measurement, from which 80% of the transcriptome can be reverse‐engineered.
Other ‘omics’ methods show either limited scope or sensitivity (or both). For example, metabolomics using mass spectrometry can be either untargeted or targeted. State‐of‐the‐art targeted metabolomics platforms aim for coverage in the same manner as the L1000 platform (through targeting of a chosen subset of molecules, for example: Metabolon Inc.; http://www.metabolon.com/technology/about‐metabolomics.aspx). Untargeted metabolomics methods are on a continuum: as there is no single optimized method suitable for all classes of molecules, sensitivity increases as scope is reduced. For example, a liquid chromatography‐mass spectrometry (LC‐MS) workflow that has its separation optimized for polar molecules will tend to separate lipids poorly. Further, subsequent identification of molecules is a major bottleneck (Peironcely et al. 2013). Other ‘omics platforms (e.g. proteomics) are subject to similar constraints.
Published (or existing but unpublished) data – the ‘bibliome’ – is a rich source for systems biologists. Whether the data are unstructured (e.g. the academic literature, patents etc.) or structured (i.e. in curated databases), there are many reasons to recommend the re‐use of experimental data, despite the hurdles associated with combining datasets from different sources (see ‘Challenges’ below). Many outstanding systems biology efforts have required no additional experimental data (for example, (Swainston et al. 2016)) and there are now huge repositories of genomic and transcriptomic information, curated databases of pathways, protein–protein interactions, etc; even databases of computational models (Juty et al. 2015).
Computational modelling
At the heart of any systems biology workflow is a computational model. Indeed for some this is the cardinal feature of systems biology (Ideker et al. 2001). It is simply not credible for humans to filter, integrate and gain insight effectively from the vast amounts of information acquired from the sources listed above. There are many mathematical frameworks that are suitable for modelling biological systems and even a cursory treatment is far beyond the scope of this review (although many excellent books exist: Voit, 2012; Ingalls, 2013; Klipp et al. 2016). However, it is important to note that there is no one ‘right way’ to model a biological system. Every approach will yield different views into the underlying biology. Sometimes very simple models can yield surprising insights (Mackey & Glass, 1977).
Systems biology models are often categorized as being either ‘bottom‐up’ or ‘top‐down’. This distinction separates models that have been built to simulate a biological mechanism (bottom‐up), such as models of biochemical pathways, from simple, usually linear, models (plus an error term) that have been fitted to the data (e.g. the general linear model in statistics). Some examples may help to clarify the difference between these concepts. In 1960 Denis Noble used a computational model to predict the interaction between multiple ion channels in the heart and hence the spontaneous emergence of the heartbeat (without resort to a specific ‘heartbeat protein’) (Noble, 1960). This elegant and seminal work was an example of ‘bottom‐up’ modelling, predicting as it did the behaviour of a complex biological system by modelling its mechanism. By contrast, statistical tests of significance fit a model of the distribution of a collection of random events (often a Gaussian or normal distribution) to some data and the practitioner makes inferences based on the likelihood of certain observed events (given a collection of assumptions). No attempt is made in this latter (‘top‐down’) case to extract biological mechanism; the model is of the data, not the biology. Hence perhaps a better distinction would be ‘predictive/mechanistic’ vs. ‘explanatory’.
But as appealing as these categorizations are, they are misleading. All models are based on data and all models are ‘middle‐out’. Even so, predictive/mechanistic models rarely include explicit error terms, but should. Some recognition of this is seen in databases where the level of evidence supporting a biological event is given as a confidence score (e.g. Schellenberger et al. 2010). Further, mixed bottom‐up/top‐down models exist (indeed are common): pharmacokinetic/pharmacodynamics (PK/PD) modellers fit mechanistic models of biology to noisy data by including an error term. Finally, a groundbreaking paper has shown that top‐down modelling of data, at least in principle, can produce a bottom‐up mechanistic model (Schmidt & Lipson, 2009).
Perhaps one of the most often overlooked benefits of building mechanistic/predictive computational or mathematical models is the information gleaned while systematically gathering the data required in a format that can be queried. For example, in the paper describing one of the first two published versions of the human metabolic network reconstruction (the other being the Edinburgh human metabolic network; Ma et al. 2007), the authors were able to produce a heatmap (Fig. 1 in Duarte et al. 2007) illustrating the contrasting levels of human knowledge of each metabolic pathway. The figure identifies important metabolic mechanisms (e.g. the channelling of vitamin C catabolism byproducts into glycolysis) that were poorly understood despite their potential importance. Only by bringing these data together systematically could such an analysis be conducted. Other potential applications include using models to guide which measurements should be made to maximally reduce uncertainty (‘if we only measure one thing, what should it be?’) or systematically inferring ‘missing’ reactions or metabolites (i.e. those that ‘should’ be there but have not yet been discovered; Rolfsson et al. 2011). The first of these hints at a truly integrated systems biology workflow: where the design of experiments is based on a mathematical (or computational) analysis of the biological system under investigation, the data available and the question to be answered. The second provides an excellent example of biological enquiry that would be impossible by other means
Iteration/the ‘model as hypothesis’
Many authors have highlighted the importance of iteration in the systems biology workflow (Kitano, 2002; Zak & Aderem, 2009; Infrastructure for Systems Biology Europe: http://project.isbe.eu/systems‐biology/). This is rarely, if ever, a closed loop and must therefore allow for the incorporation of both external/existing and newly generated information and data. Two prototypical approaches have emerged, characterised by the modelling approaches at their respective cores. In the first, a predictive/mechanistic model of the biology is constructed using already available data from the scientific literature and/or curated databases. This model is then used as a scaffold for new data. The model is refined iteratively and, at least in theory, becomes the centrepiece of all ongoing experimental work.
In the second approach, large scale data acquisition is the first step (for example, transcriptomics). These data are then analysed using relatively simple statistical models before being integrated with existing knowledge using pathway‐mapping tools such as DAVID (database for annotation, visualization and integrated discovery; Huang da et al. 2009) or MetaCore (Thomson Reuters; http://lsresearch.thomsonreuters.com/pages/solutions/1/metacore). Iteration is required to confirm the robustness and generalisability of the statistical model that was fitted to the data. The drawback of this latter approach is clear: due to the relative simplicity of the model used, confirming its robustness yields little significant additional information on the quantitative relationships between species, beyond the magnitude of their change with experimental perturbation. Indeed, this provides a nice example of one of many problems with traditional null hypothesis significance testing: as the precision of the estimates in the statistical model increase (through iteration), rather than challenging the underlying hypothesis (as should be the case), the model merely serves to reinforce it further. Analytical methods have been developed that aim to ‘reverse‐engineer’ the underlying network ((Margolin et al. 2006; Langfelder & Horvath, 2008). A recent DREAM (Dialogue for Reverse Engineering Assessment and Methods) challenge showed that combinations of these methods were especially robust (Marbach et al. 2012).
Challenges
Of the many challenges currently facing systems biologists, perhaps the greatest are in data integration. Although vast amounts of data are readily available, they are of varying quality and much is biased and confounded. Further, data are rarely available on the same subjects at the same time. Generally speaking, data integration takes two forms: horizontal (the collation and integration of a large amount of data of similar type across multiple conditions, species, etc.) and vertical (the integration of data spanning many layers of the biological hierarchy, for a single experimental condition or disease). The first allows the investigator to ask questions of fundamental biological significance (for example, ‘do we see this pattern across multiple species/in many conditions?’). Here, batch effects are especially troublesome. Regarding the second, once again there are bottom‐up and top‐down approaches. Of the bottom‐up approaches, perhaps the best‐known is constraint‐based modelling using a metabolic network reconstruction (Hyduke et al. 2013). Of statistical methods for data integration, similarity network fusion is a promising example (Wang et al. 2014) while network and set‐based methods are useful when batch effects are insurmountable (Ma'ayan et al. 2014). Artificial intelligence (e.g. Deep Learning; LeCun et al. 2015) may provide new and powerful weapons, given sufficient data.
Beyond these tools for integrating the data itself, integrating accompanying metadata is also a significant and critical challenge. Even seemingly simple measurements (for example, total lung volume) can be acquired in a multitude of different ways and called many different things. Although a few datasets can be aligned by hand (with patience) this solution does not scale. New technology that combines machine learning and crowd‐sourcing provides new hope (Held et al. 2015), as do continuing efforts towards consistent ontologies. However, the benefits of integrating multiple datasets are easy to see: as with more traditional meta‐analyses, integrating multiple datasets brings the promise of new insights, increased reliability and better signal‐to‐noise ratio (Ideker et al. 2001).
Closing remarks
The purpose of this primer is to introduce the interested reader to systems biology – its history, definition, practice, opportunities and challenges – with particular emphasis on systems biology's favourite biosystem: metabolism. Systems biology is no longer a young field (perhaps it never was), and much of the early hype (as with so many other innovations) is finally dying away. Meanwhile, its methods have become routine in many laboratories, both in industry and academia. However, although top‐down modelling of high‐dimensional datasets is commonplace, mechanistic models of biology are still regularly met with suspicion, despite their huge potential to codify our knowledge, contextualise disparate data sources and generate new hypotheses for experimental investigation. Systems biologists, and bioinformaticians alike, are regularly encouraged to treat their findings as ‘hypotheses’. Given recent data highlighting poor reproducibility in the scientific literature in general (Begley & Ellis, 2012), perhaps this should be the default setting for us all.
A final note: if one temporarily puts aside the considerations of reductionism and holism and describes systems biology as a cycle of model building, experimental data acquisition, and iterative model refinement, then one is not describing just systems biology, but science in general. It is often useful to remember that all scientists use models (a model being an abstract representation of reality that is always wrong but often useful, to paraphrase George Box), whether they be implicit or explicit. Hypothesis testing is simply a process of establishing experimentally how wrong one's model is.
Additional information
Competing interests
The author is employed by GlaxoSmithKline.
Funding
The author is employed by GlaxoSmithKline. He receives no other funding.
Acknowledgements
Thank you to Adam Taylor and David Hall for their input and advice.
Biography
Lindsay Edwards is Head of Respiratory Data Sciences at GlaxoSmithKline and an Honorary Lecturer in Physiology at King's College London. He came to science relatively late after a successful career as a record‐producer and musician. He completed his doctoral studies at the University of Oxford, where he first became interested in computational modelling of biological systems, and has held faculty posts in Australia and the UK. His interests range from human physiology, through metabolomics, to high‐performance computing and artificial intelligence. He became a Fellow of the Royal Society of Biology in 2015, is married and has a young daughter.

This review was presented at the symposium “New technologies providing insight to human physiological adaptation”, which took place at the meeting of The Biomedical Basis of Elite Performance in Nottingham, UK, 6–8 March 2016.
References
- Begley CG & Ellis LM (2012). Drug development: Raise standards for preclinical cancer research. Nature 483, 531–533. [DOI] [PubMed] [Google Scholar]
- Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC & Heisler MG (2013). Accounting for technical noise in single‐cell RNA‐seq experiments. Nat Meth 10, 1093–1095. [DOI] [PubMed] [Google Scholar]
- Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R & Palsson BØ (2007). Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Nat Acad Sci USA 104, 1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards LM & Thiele I (2013). Applying systems biology methods to the study of human physiology in extreme environments. Extrem Physiol Med 2, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED & Swain PS (2002). Stochastic gene expression in a single cell. Science 297, 1183–1186. [DOI] [PubMed] [Google Scholar]
- Held J, Stonebraker M, Davenport TH, Ilyas I, Brodie ML, Palmer A & Markarian J (2015). Getting Data Right: Tackling The Challenges of Big Data Volume and Variety. O‧ Reilly Media Inc, Sebastopol, CA, USA. [Google Scholar]
- Huang da W, Sherman BT & Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hyduke DR, Lewis NE & Palsson BO (2013). Analysis of omics data with genome‐scale models of metabolism. Mol Biosyst 9, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Dutkowski J & Hood L. Boosting signal‐to‐noise in complex biology: prior knowledge is power. Cell 144, 860–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Galitski T & Hood L (2001). A new approach to decoding life: Systems Biology. Annu Rev Genomics Hum Genet 2, 343–372. [DOI] [PubMed] [Google Scholar]
- Ingalls BP (2013). Mathematical Modeling in Systems Biology: An Introduction. MIT Press, Cambridge, MA, U: SA. [Google Scholar]
- Joyner MJ & Saltin B (2008). Exercise physiology and human performance: systems biology before systems biology! J Physiol 586, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juty N, Ali R, Glont M, Keating S, Rodriguez N, Swat MJ, Wimalaratne SM, Hermjakob H, Le Novere N, Laibe C & Chelliah V (2015). BioModels: content, features, functionality, and use. CPT Pharmacometrics Syst Pharmacol 4, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano H (2002). Computational systems biology. Nature 420, 206–210. [DOI] [PubMed] [Google Scholar]
- Klipp E, Liebermeister W, Wierling C & Kowald A (2016). Systems Biology: A Textbook. Wiley‐Blackwell, New York. [Google Scholar]
- Kresge N, Simoni RD & Hill RL (2005). Otto Fritz Meyerhof and the elucidation of the glycolytic pathway. J Biol Chem 280, e3. [PubMed] [Google Scholar]
- Langfelder P & Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCun Y, Bengio Y & Hinton G (2015). Deep learning. Nature 521, 436–444. [DOI] [PubMed] [Google Scholar]
- Ma H, Sorokin A, Mazein A, Selkov A, Selkov E, Demin O & Goryanin I (2007). The Edinburgh human metabolic network reconstruction and its functional analysis. Mol Syst Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma'ayan A, Rouillard AD, Clark NR, Wang Z, Duan Q & Kou Y (2014). Lean Big Data integration in systems biology and systems pharmacology. Trends Pharmacol Sci 35, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey MC & Glass L (1977). Oscillation and chaos in physiological control systems. Science 197, 287–289. [DOI] [PubMed] [Google Scholar]
- Marbach D, Costello JC, Küffner R, Vega N, Prill RJ, Camacho DM, Allison KR, the DC, Kellis M, Collins JJ & Stolovitzky G (2012). Wisdom of crowds for robust gene network inference. Nat Methods 9, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R & Califano A (2006). ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7 (Suppl. 1), S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D (1960). Cardiac action and pacemaker potentials based on the Hodgkin‐Huxley equations. Nature 188, 495–497. [DOI] [PubMed] [Google Scholar]
- Palsson BO (2008). Systems Biology. Cambridge University Press, Cambridge. [Google Scholar]
- Peironcely JE, Rojas‐Cherto M, Tas A, Vreeken R, Reijmers T, Coulier L & Hankemeier T (2013). Automated pipeline for de novo metabolite identification using mass‐spectrometry‐based metabolomics. Anal Chem 85, 3576–3583. [DOI] [PubMed] [Google Scholar]
- Risso D, Schwartz K, Sherlock G & Dudoit S (2011). GC‐content normalization for RNA‐Seq data. BMC Bioinformatics 12, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfsson O, Palsson BØ & Thiele I (2011). The human metabolic reconstruction Recon 1 directs hypotheses of novel human metabolic functions. BMC Syst Biol 5, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J, Park JO, Conrad TM & Palsson BØ (2010). BiGG: a biochemical genetic and genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M & Lipson H (2009). Distilling free‐form natural laws from experimental data. Science 324, 81–85. [DOI] [PubMed] [Google Scholar]
- Swainston N, Smallbone K, Hefzi H, Dobson PD, Brewer J, Hanscho M, Zielinski DC, Ang KS, Gardiner NJ, Gutierrez JM, Kyriakopoulos S, Lakshmanan M, Li S, Liu JK, Martínez VS, Orellana CA, Quek L‐E, Thomas A, Zanghellini J, Borth N, Lee D‐Y, Nielsen LK, Kell DB, Lewis NE & Mendes P (2016). Recon 2.2: from reconstruction to model of human metabolism. Metabolomics 12, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Swainston N, Fleming RMT, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, Thorleifsson SG, Agren R, Bolling C, Bordel S, Chavali AK, Dobson P, Dunn WB, Endler L, Hala D, Hucka M, Hull D, Jameson D, Jamshidi N, Jonsson JJ, Juty N, Keating S, Nookaew I, Le Novere N, Malys N, Mazein A, Papin JA, Price ND, Selkov Sr E, Sigurdsson MI, Simeonidis E, Sonnenschein N, Smallbone K, Sorokin A, van Beek JHGM, Weichart D, Goryanin I, Nielsen J, Westerhoff HV, Kell DB, Mendes P & Palsson BO (2013). A community‐driven global reconstruction of human metabolism. Nat Biotech 31, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trewavas A (2006). A brief history of systems biology: “Every object that biology studies is a system of systems.” Francois Jacob (1974). Plant Cell 18, 2420–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voit EO (2012). A First Course in Systems Biology. Garland Science, New York. [Google Scholar]
- von Bertallanfy L (1968). General System Theory. Brazillier, New York. [Google Scholar]
- Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M, Haibe‐Kains B & Goldenberg A (2014). Similarity network fusion for aggregating data types on a genomic scale. Nat Meth 11, 333–337. [DOI] [PubMed] [Google Scholar]
- Wanjek C (2011). Systems Biology as defined by NIH. In The NIH Catalyst. https://irp.nih.gov/catalyst/v19i6/systems‐biology‐as‐defined‐by‐nih. [Google Scholar]
- Williams RJ (1956). Biochemical Individuality. The Key for the Genetotrophic Concept. John Wiley & Sons, New York. [Google Scholar]
- Zak DE & Aderem A (2009). Systems biology of innate immunity. Immunol Rev 227, 264–282. [DOI] [PMC free article] [PubMed] [Google Scholar]