

Abstract
Glaucomas are neurodegenerative diseases that cause vision loss, especially in the elderly. The mechanisms initiating glaucoma and driving neuronal vulnerability during normal aging are unknown. Studying glaucoma-prone mice, we show that mitochondrial abnormalities are an early driver of neuronal dysfunction, occurring prior to detectable degeneration. Retinal levels of nicotinamide adenine dinucleotide (NAD+; a key molecule in energy and redox metabolism) decrease with age, and render aging neurons vulnerable to disease-related insults. Oral administration of the NAD+ precursor nicotinamide (vitamin B3), and/or gene therapy (driving expression of Nmnat1, a key NAD+ producing enzyme), was protective both prophylactically and as an intervention. On our highest dose, 93% of eyes did not develop glaucoma. This supports therapeutic use of vitamin B3 in glaucoma and potentially other age-related neurodegenerations.
Glaucoma is a group of complex, multifactorial diseases characterized by the progressive dysfunction and loss of retinal ganglion cells (RGCs), leading to vision loss. Glaucoma is one of the most common neurodegenerative diseases worldwide, affecting over 70 million people (1). High intraocular pressure (IOP) and increasing age are important risk factors for glaucoma (2, 3). However, specific mechanisms rendering retinal ganglion cells more vulnerable to damage with age are unknown. Here, we address how increasing age and high IOP interact to drive neurodegeneration using DBA/2J (D2) mice, a widely used model of chronic, age-related, inherited glaucoma (4).
We utilized RNA-sequencing (RNA-seq) to elucidate age and IOP-dependent molecular changes within RGCs that precede glaucomatous neurodegeneration. We analyzed RGCs of 9 month old (mo) D2 mice (termed early glaucoma - high IOP and molecular changes but lacking neurodegeneration (2)), 4 mo old D2 mice (precedes high IOP) and age, sex and strain matched D2-Gpnmb+ controls (do not develop high IOP or glaucoma (4)) (Fig. 1). RGCs were isolated (Fig. S1) and their RNA sequenced at a depth of 35 million reads per sample. Unsupervised hierarchical clustering (HC) allowed molecular definition of early glaucoma stages among samples that were still morphologically indistinguishable from age-matched D2-Gpnmb+ or young controls. HC identified 4 distinct groups of 9 mo old D2 samples (Groups 1 to 4). Group 1 clustered with all of the control samples and represents D2 RGCs with no detectable glaucoma at a molecular level. Although Groups 2 to 4 were all early stages, increasing group number reflects greater glaucoma progression at a transcriptomic level (Fig. 1A, Fig. S2A and B). As disease progressed, there was an increase in transcript abundance that was most pronounced for mitochondrial reads (Fig. 1B). Emerging evidence suggests that imbalances in the relative proportions of mitochondrial proteins encoded by nuclear and mitochondrial genomes negatively impact mitochondrial function (5). In D2 Groups 2 to 4, differential expression of genes encoding mitochondrial proteins, and significant enrichment of differentially expressed (DE) genes in the mitochondrial dysfunction and oxidative phosphorylation pathways further point to mitochondrial abnormalities (Fig. 1B–G, Fig. S2C–G, Table S1–3). Pathway analysis identified enrichment of eIF2 and mTOR signaling transcripts (Fig. 1C). eIF2 is a key regulator of redox homeostasis and cellular adaptations to stress and was the most enriched pathway in Group 2 (first distinguishable stage from controls). Through mTOR inhibition, it promotes survival in the presence of oxidative stress (6–8) and likely protects from mitochondrial abnormalities in RGCs at this early disease stage. Mitochondrial fission (Fig. 1F) and mitochondrial unfolded protein response (UPRmt) genes were also DE (Fig. 1G) (9–11). Electron microscopy (EM) revealed abnormal mitochondria with reduced cristae volume in the dendrites of D2 RGCs, but not in those of control RGCs (Fig. 1H and I). These mitochondrial EM findings coincide with synapse loss in 9 mo D2 retinas (12), with early decreases in pattern electroretinogram amplitude (PERG) (13), and an increase in retinal cytochrome c levels (Fig. S2D and F). Extending previous studies (14, 15), our data demonstrate that mitochondrial perturbations are among the very first changes occurring within RGCs during glaucoma.
Fig. 1. Mitochondrial dysfunction is associated with progressive RGC damage in glaucoma.
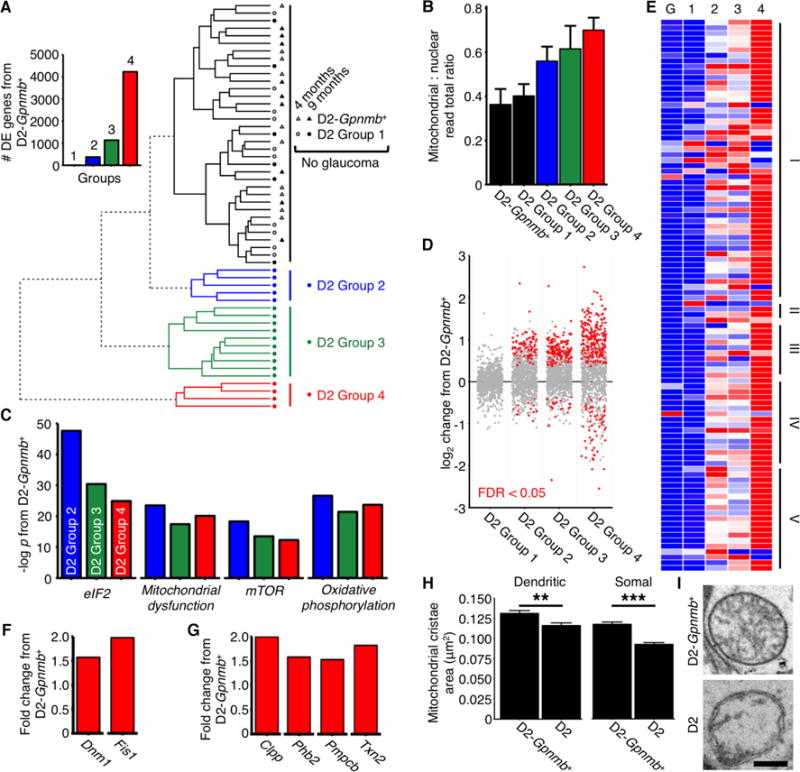
(A) RGC samples were divided into molecularly distinct groups by HC of RNA-seq determined gene expression. Control (D2-Gpnmb+) and young samples were molecularly similar (Spearman’s rho; n = 63 samples). Circles = samples from D2 RGCs, triangles = samples from D2-Gpnmb+ RGCS. Inset: number of DE genes (q < 0.05) between D2-Gpnmb+ and each group. D2 Group 1 and D2-Gpnmb+ represent no glaucoma at a molecular/transcriptomic level. (B) Mitochondrial: nuclear read total ratio increased with increasing HC distance from controls. Colors match key in A. (C) Top significantly enriched pathways between clusters and control based on IPA analysis (there are no differentially expressed pathways in D2 Group 1) (see also Table S3). (D) Transcript expression primarily increased for nuclear encoded mitochondrial proteins with increasing HC distance from controls. Dots represent individual genes, grey = not differentially expressed, red = differentially expressed at q < 0.05. Genes taken from mouse MitoCarta2.0 (28) (E) OXPHOS genes were differentially expressed across all groups. Red = highest expression, blue = lowest expression, I–V = mitochondrial complexes I–V (tabulated in Table S1), G = D2-Gpnmb+, 1–4 = D2 Groups 1–4. (F) RNA-seq identified increased mitochondrial fission gene transcripts early in glaucoma and (G) suggests an early mitochondrial unfolded protein response compared to controls. Data shown is for D2 Group 4. (H and I) Mitochondria of D2 mice have decreased cristae volume (and cristae : total volume ratio, not shown) in RGC somal and dendritic mitochondria (there was no significant difference in total mitochondrial size/volume) (n > 400 mitochondria from 6 retinas/group). Scale bar = 350 nm. All data is at 9 mo unless otherwise stated. ** = P < 0.01, * = P < 0.001 (Student’s t test). See also Table 1, Table 2.
Guided by the above data, we assessed metabolites in retinas with increasing age and disease (D2 and D2-Gpnmb+; 4, 9, and 12 mo). We detected early decreases in metabolites that are central to healthy mitochondrial metabolism and protection from oxidative stress (NAD+ and NADH [total NAD; NAD(t)], GSH and GSSG [total glutathione; glutathione(t)]) (Fig. 2A, Fig. S2H–I). These age-dependent decreases were not a response to IOP-insult(s) as they also occurred in control D2-Gpnmb+ retinas (Fig. S2I). These decreases are expected to sensitize retinal neurons to disease related stresses and mitochondrial dysfunction. Suggesting greater metabolic stress in RGCs than other retinal neurons, HIF-1α (a key metabolic regulator during perturbed redox states (16)) is induced in the ganglion cell layer early in glaucoma (Fig. S3A and B). Our data suggest that RGCs go through a period of mitochondrial stress and metabolite depletion, potentially moving towards fatty acid metabolism (Fig. S4). Fatty acid β-oxidation can increase generation of free radicals/reactive oxygen species (ROS) (17). Both RNA-seq (Fig. S2C) and γ-H2AX immunostaining (Fig. S2J and K) support increased ROS and DNA damage within RGCs early in glaucoma. Providing a link between DNA damage, and increased metabolic stress, PARP activity (NAD consuming) is induced in RGCs with age (Fig. S5A and B).
Fig. 2. Vitamin B3/NAM supplementation protects against glaucoma development in mice.

(A) NAD(t) levels were increased in Vitamin B3/NAM treated D2 retinas as measured by colorimetric assay (n = 22/group). (B and E) NAM intervention protected from optic nerve degeneration as assessed by PPD staining (a sensitive stain for damaged axons). Green = no or early damage (<5% axon loss; NOE), yellow = moderate damage (~30% axon loss; MOD), red = severe (>50% axon loss; SEV) damage. Early start indicates mice that started treatment at 6 mo (prior to IOP elevation in most eyes in our colony, thus prophylactic). Late start indicates mice that started treatment at 9 mo (when the majority of eyes have had continuing IOP elevation, thus interventional). Fisher’s exact test: ** = P < 0.01, *** = P < 0.001. (C and E) NAM protected from RGC soma loss (number of RBPMS+ cells, n = 8/group, the density drop between D2 and D2-Gpnmb+ is due to pressure induced stretching). (D) NAM protected from early loss in PERG amplitude (n > 20/group). (E) NAM protected from RGC soma loss (n = 8/group), retinal NFL and IPL thinning (n = 8/group), optic nerve degeneration (n > 50/group), and loss of anterograde axoplasmic transport (n = 20/group). Corresponding markers and color keys are beneath each column. Scale bars; RBPMS (a specific marker of RGCs; immunofluorescence) = 20 μm, Nissl (a pan-neuronal stain; light microscopy) = 20 μm, PPD (light microscopy) = 20 μm, CT-β = 100 μm (retina; immunofluorescence), 200 μm (LGN, Sup. Col.). ONH = optic nerve head, LGN = lateral geniculate nucleus, Sup. Col. = superior colliculus. White asterisk denotes loss of axonal transport at the site of the ONH. (F) Heatmap of gene expression (all expressed genes) shows that NAM treated RCGs were molecularly similar to controls. (G) Individual gene expression plots show metabolic and DNA damage pathways were returned to normal in NAM treated RGCs. Dots represent individual genes, grey = not differentially expressed, red = differentially expressed at q < 0.05 compared to D2-Gpnmb+ 9 mo control. (B, D, and E) * = P < 0.05, ** = P < 0.01, *** = P < 0.001 (Student’s t test). For boxplots, center hinge represents the mean, and the upper and lower hinges represent the first and third quartiles, whiskers represent 1.5 * interquartile range, values beyond the whiskers are plotted as outliers. See also Fig. S4, Tables 1–3.
Our data support a model where age-dependent declines of NAD+ and glutathione in the retina render RGCs vulnerable to damage from elevated IOP. Thus, increasing NAD levels would be predicted to protect IOP-insulted eyes from glaucomatous changes, by decreasing the probability of metabolic/energetic failure and rendering the RGCs more resilient to IOP-induced stress. Oral supplementation of vitamin B3/nicotinamide (NAM; a precursor of NAD) has been successfully used to correct disturbances in NAD+ metabolism in two mouse models of pre-eclampsia (18). Accordingly, we administered NAM to D2 mice, initially at the same dose (550 mg/kg/d, NAMLo) (Fig. 2). NAM administration in drinking water prevented the decline of NAD levels through to 12 mo (a standard end stage for assessing neurodegeneration in this glaucoma model) (Fig. 2A). Supporting our neuronal vulnerability hypothesis, NAMLo did not alter IOP (Fig. S6), but protected from glaucoma. NAM was protective both prophylactically (starting at 6 mo, prior to IOP elevation in most eyes in our colony) and interventionally (starting at 9 mo, when the majority of eyes have had continuing IOP elevation) (Fig. 2B). NAM significantly reduced the incidence of optic nerve degeneration (Fig. 2B and E), prevented RGC soma loss and retinal nerve fiber layer thinning (Fig 2C and E), and protected visual function as assessed by PERG (13) (Fig. 2D, Fig. S3E). NAM prevented RGC axonal loss, and these axons continued to support anterograde axonal (Fig. 2E). NAM administration was sufficient to inhibit the formation of dysfunctional mitochondria with abnormal cristae (Fig. 2E, Fig. S3G and H) and also limited synapse loss that occurs in this model (12) (Fig. S3C and D). Lipid droplet formation was also prevented in aged D2 retinas (Fig. S3F). NAM also decreased PARP activation, limited levels of DNA damage, and transcriptional induction of HIF-1α (Fig. S3A and B) reflecting less perturbed cellular metabolism. NAM prevented even the earliest molecular signs of glaucoma in most treated eyes as assessed by RNA-seq (Fig. 2F and G, Fig. S7) and prevented the majority of age-related gene expression changes within RGCs (Fig. S8). This highlights the unexpected potency of NAM in decreasing metabolic disruption and prevention of glaucoma.
Attempting to further decrease the probability of glaucoma, we administered a higher dose of NAM (2000mg/kg/d; NAMHi). NAMHi was extremely protective with 93% of treated eyes having no optic nerve damage (Fig. 2B). The degree of protection afforded by administering this single molecule is unprecedented and unanticipated. Although NAMLo demonstrates a clear neuroprotective effect (no effect on IOP), NAMHi lessens the degree of IOP elevation (Fig. S6). This indicates that NAM can protect against age-related pathogenic processes in additional cell types to RGCs (11, 16, 19). Therefore vitamin B3/NAM, a single molecule that protects against both IOP elevation and neural vulnerability, has great potential for glaucoma treatment, however human studies are needed.
NMNAT2 is emerging as an important NAD producing enzyme in axons, protecting from axon degeneration (20). Ongoing stress negatively impacts Nmnat2 expression in RGCs (q < 0.05 in D2 Group 4) (Fig. S7F). This decline of NMNAT2 may induce vulnerability to axon degeneration in glaucoma. NMNAT2 expression is decreased in brains with Alzheimer’s disease and is highly variable in aged postmortem human brains (21). Such variation in expression may contribute to individual differences in vulnerability to various neurodegenerations.
Glaucoma is a complex disease involving multiple insults. Mechanical axon damage and local inflammation are two important contributors in glaucoma (22–24). To more fully assess the general effectiveness of NAM treatment, we tested its efficacy in two models of RGC death that are used to model these glaucomatous insults. We used a tissue culture model of axotomy, and intravitreal injections of soluble murine TNFα which can drive local inflammation as well as mitochondrial dysfunction and is implicated in glaucoma (25). NAM robustly protected cultured retinas from RGC somal degeneration (Fig. S9AB). NAM also protected against a loss of PERG amplitude and cell loss in TNFα injected eyes (Fig. S9C–E). Given these protections against severe acute insults, NAM could have broad implications for treating glaucoma and potentially other age-related neurodegenerative diseases.
Gene therapy is an attractive approach for overcoming compliance issues and improving efficacy. In the eye, gene therapy has proven successful for rare human Mendelian disorders (26). Viral gene therapy allows a large number of cells to be transfected lifelong, delivering a targeted gene product. To date, gene therapy has not been successfully applied to complex human diseases. Given that age is a common risk factor for most glaucomas, protecting from age–related declines in NAD may generally protect many glaucoma patients. Thus, we sought to support NAD+ producing cellular machinery through the overexpression of Nmnat1, a terminal enzyme in NAD+ production, further testing our NAD hypothesis. (Using NAMPT, the rate-limiting enzyme in NAD synthesis, may be complicated both by its cytokine functions and over-production of NMN, which may participate in axon degeneration (27).) D2 eyes were injected once with AAV2.2 containing the Nmnat1 gene and GFP under a CMV promoter expressed as a single transcript (Fig. 3). Nmnat1 expression (as assessed by GFP expression) was robust in RGCs 2 weeks after injection (expressed in >83% of RGCs), and remained robust through to the end stage time point (12 mo) (Fig. S10). Overexpression of Nmnat1 was sufficient to prevent axon and soma loss (Fig. 3A–D), to preserve axoplasmic transport, (Fig. 3B) and to preserve electrical activity in RGCs (PERG) (Fig. 3E). Glaucomatous nerve damage was absent in >70% of treated eyes. Since NMNAT1 catalyzes the terminal step in NAD production, the major protective effects of NAM treatment likely results from driving NAD production in neurons rather than other NAD independent mechanisms (but partial contributions from other mechanism cannot be completely ruled out). We further assessed the effects of combinational therapy of Nmnat1 and NAMLo. This combination afforded significant additional protection over Nmnat1 or NAMLo alone with 84% of eyes having no detectable glaucoma (~4-fold decreased risk of developing glaucoma). Increasing the NAM dose combined with gene therapy may prove even more protective.
Fig. 3. Gene therapy protected eyes from glaucomatous neurodegeneration.
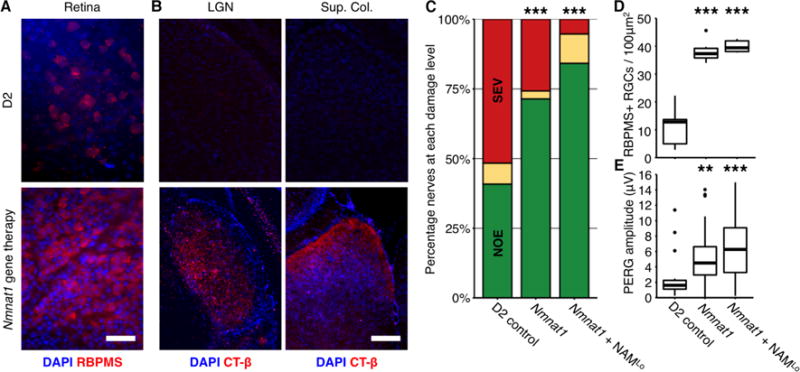
D2 eyes were intravitreally injected at 5.5 mo with AAV2.2 carrying a plasmid to overexpress murine Nmnat1 under a CMV promoter. (A) Nmnat1 overexpression prevented RGC soma loss (red, scale bar = 50 μm) and loss of anterograde axoplasmic transport (n = 10/group) (as demonstrated in Fig. 2.) (B; red, scale bar = 100 μm). Nmnat1 gene therapy also protected D2 eyes with elevated IOP against optic nerve degeneration (n > 40/group; *** = P < 0.001, Fisher’s exact test) (C), soma loss (n = 6/group) (D), and PERG amplitude (n > 20/group) (E). Addition of NAMLo in drinking water afforded additional protection against optic nerve degeneration (Nmnat1 compared to Nmnat1 + NAMLo = P < 0.001, Fisher’s exact test) (C). (D and E) ** = P < 0.01, *** = P < 0.001 (Student’s t test).
In conclusion, we show that dietary supplementation with a single molecule (vitamin B3/nicotinamide), or Nmnat1 gene therapy, significantly reduces vulnerability to glaucoma by supporting mitochondrial health and metabolism. Combined with established medications that lower IOP, NAM treatment (and/or Nmnat1 gene therapy) may be profoundly protective. By providing a new molecular and metabolic link between increased neuronal vulnerability with age and neurodegeneration these findings are of critical importance for glaucoma and possibly other age-related diseases.
Supplementary Material
One Sentence Summary.
Targeting NAD through dietary vitamin B3 or gene therapy prevents IOP-induced metabolic dysfunction and glaucoma.
Acknowledgments
The data reported in this paper are available in the Supplementary Materials. RNA-seq data are available through the Gene Expression Omnibus (accession number GSE90654).
The Authors would like to thank Flow Cytometry, Histology, Gene Expression Services, and Computational Sciences at The Jackson Laboratory, G. Howell and R. Libby for discussion and experiment design, K. Kizhatil for reading of the manuscript, M. de Vries for assistance with diet and organizing, B. Cardozo for colony maintenance and drug changes, and A. Bell for intraocular pressure measurements. The Authors would also like to thank their sources of funding; The Jackson Laboratory Fellowships (PAW, JMH), partial support from EY11721 (SWMJ), the Barbara and Joseph Cohen foundation (SWMJ), and HL49277 (OS). SWMJ is an Investigator of HHMI.
Footnotes
References
- 1.Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howell GR, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. Journal of Clinical Investigation. 2011;121:1429–1444. doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nickells RW, Howell GR, Soto I, John SW. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci. 2012;35:153–179. doi: 10.1146/annurev.neuro.051508.135728. [DOI] [PubMed] [Google Scholar]
- 4.Libby RT, et al. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005;22:637–648. doi: 10.1017/S0952523805225130. [DOI] [PubMed] [Google Scholar]
- 5.Kotiadis VN, Duchen MR, Osellame LD. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim Biophys Acta. 2014;1840:1254–1265. doi: 10.1016/j.bbagen.2013.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gandin V, et al. mTORC1 and CK2 coordinate ternary and eIF4F complex assembly. Nat Commun. 2016;7:11127. doi: 10.1038/ncomms11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 2012;8:e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quirós PM, Mottis A, Auwerx J. Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol. 2016;17:213–226. doi: 10.1038/nrm.2016.23. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mouchiroud L, et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H, et al. NAD⁺ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- 12.Williams PA, et al. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol Neurodegener. 2016;11:26. doi: 10.1186/s13024-016-0091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saleh M, Nagaraju M, Porciatti V. Longitudinal evaluation of retinal ganglion cell function and IOP in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2007;48:4564–4572. doi: 10.1167/iovs.07-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chrysostomou V, Rezania F, Trounce IA, Crowston JG. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol. 2013;13:12–15. doi: 10.1016/j.coph.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Lee S, et al. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp Eye Res. 2011;93:204–212. doi: 10.1016/j.exer.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 16.Gomes AP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schönfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab. 2013;33:1493–1499. doi: 10.1038/jcbfm.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li F, et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc Natl Acad Sci U S A. 2016;47:13450–13455. doi: 10.1073/pnas.1614947113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein LR, Imai S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014;33:1321–1340. doi: 10.1002/embj.201386917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ali YO, et al. NMNAT2:HSP90 Complex Mediates Proteostasis in Proteinopathies. PLoS Biol. 2016;14:e1002472. doi: 10.1371/journal.pbio.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soto I, Howell GR. The complex role of neuroinflammation in glaucoma. Cold Spring Harb Perspect Med. 2014;4 doi: 10.1101/cshperspect.a017269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Križaj D, et al. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res. 2014;39:105–119. doi: 10.3109/02713683.2013.836541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan L, Neufeld AH. Tumor necrosis factor-alpha: a potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia. 2000;32:42–50. [PubMed] [Google Scholar]
- 25.Nakazawa T, et al. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006;26:12633–12641. doi: 10.1523/JNEUROSCI.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bainbridge JW, et al. Long-term effect of gene therapy on Leber's congenital amaurosis. N Engl J Med. 2015;372:1887–1897. doi: 10.1056/NEJMoa1414221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Stefano M, et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015;22:731–742. doi: 10.1038/cdd.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson MG, et al. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nature Genetics. 2002;30:81–85. doi: 10.1038/ng794. [DOI] [PubMed] [Google Scholar]
- 30.Graham LC, et al. Chronic consumption of a western diet induces robust glial activation in aging mice and in a mouse model of Alzheimer's disease. Sci Rep. 2016;6:21568. doi: 10.1038/srep21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27:1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.John SW, et al. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Invest Ophthalmol Vis Sci. 1998;39:951–962. [PubMed] [Google Scholar]
- 36.Chou TH, Bohorquez J, Toft-Nielsen J, Ozdamar O, Porciatti V. Robust mouse pattern electroretinograms derived simultaneously from each eye using a common snout electrode. Invest Ophthalmol Vis Sci. 2014;55:2469–2475. doi: 10.1167/iovs.14-13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savinova OV, et al. Intraocular pressure in genetically distinct mice: an update and strain survey. BMC Genet. 2001;2:12. doi: 10.1186/1471-2156-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith R, John S, Nishina P, Sundberg J. Systematic evaluation of the mouse eye. Anatomy, pathology and biomethods. CRC Press; Boca Raton: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.