

Summary
Response to immune checkpoint blockade in mesenchymal tumors is poorly characterized, but immunogenomic dissection of these cancers may inform immunotherapy mediators. We identified a treatment-naïve patient with metastatic uterine leiomyosarcoma who experienced complete tumor remission for >2 years on anti-PD-1 (pembrolizumab) monotherapy. We analyzed primary tumor, the sole treatment-resistant metastasis, and germline tissue to explore mechanisms of immunotherapy sensitivity and resistance. Both tumors stained diffusely for PD-L2, with sparse PD-L1 staining. PD-1+ cell infiltration significantly decreased in the resistant tumor (p=0.02). Genomically, the treatment-resistant tumor uniquely harbored biallelic PTEN loss and had reduced expression of two neoantigens that demonstrated strong immunoreactivity with patient T cells in vitro, suggesting long-lasting immunological memory. In this near-complete response to PD-1 blockade in a mesenchymal tumor, we identified PTEN mutation and reduced expression of genes encoding neoantigens as potential mediators of resistance to immune checkpoint therapy.
Keywords: immune checkpoint, sarcoma, neoantigen, whole exome sequencing, whole transcriptome sequencing, exceptional response, immunotherapy
eTOC Blurb
George et al. report an exceptional responder to anti-PD-1 monotherapy in uterine leiomyosarcoma and propose mediators of treatment sensitivity and resistance. Neoantigen-directed immunoreactivity was associated with sensitivity to anti-PD-1 monotherapy, whereas resistance was associated with reduction in neoantigen expression consistent with immune evasion, and biallelic PTEN loss associated with induction of an immunosuppressive microenvironment.

Introduction
Immune checkpoint blockade therapies, including monoclonal antibodies targeting programmed death 1 (PD-1) and cytotoxic T lymphocyte associated antigen-4 (CTLA-4), yield durable clinical benefit in multiple tumor types (Sharma and Allison, 2015), but response in some cancers, including mesenchymal tumors (Maki et al., 2013), is rare. Developing predictive biomarkers of clinical benefit from immune checkpoint therapy is an ongoing challenge, and could help identify patients with immune checkpoint-responsive disease in largely immunotherapy-resistant tumor types.
Immunohistochemical staining for PD-1 ligands (PD-L1 and PD-L2) has positive prognostic value for response, but patients with PD-L1-negative tumors can still respond (Philips and Atkins, 2015). In genomic analyses of pre-treatment tumors from immune-checkpoint-treated patients in non-small cell lung cancer (NSCLC) and melanoma, total tumor burden of nonsynonymous mutations, as well as burden of tumor-specific immunogenic peptides generated from these mutations (neoantigens), are associated with patient benefit (McGranahan et al., 2016; Rizvi et al., 2015; Snyder et al., 2014; Van Allen et al., 2015), though identification of the specific neoantigens mediating a strong anti-tumor response occurs in only a subset of cases (Rizvi et al., 2015; Snyder et al., 2014; van Rooij et al., 2013). Certain tumor-immune microenvironment gene expression signatures developed from whole transcriptome sequencing (RNA-seq) on pre-treatment patient tumors also correlate with response (Hugo et al., 2016; Van Allen et al., 2015). However, all of these factors imperfectly stratify responders and nonresponders prospectively, and additional studies in independent patient cohorts are needed.
Furthermore, acquired tumor resistance mechanisms important in determining overall patient benefit have yet to be deeply explored. In case reports and case series, genetic alterations that delete genes encoding tumor-specific neoantigens, disrupt neoantigen presentation on major histocompatibility (MHC) class I alleles, or interfere with downstream interferon-γ signaling have been associated with acquired resistance to cancer immunotherapies (Anagnostou et al., 2017; Tran et al., 2016; Verdegaal et al., 2016; Zaretsky et al., 2016). In the preclinical space, loss of the tumor suppressor PTEN in melanoma models leads to immunoresistance via induction of VEGF and other immunosuppressive cytokines (Peng et al., 2016), and Pten-null prostate tumors similarly suppress anti-tumor immune activity by activating the Jak2/Stat3 pathway (Toso et al., 2014). Thus, preclinical and clinical studies suggest tumor immune evasion via alteration of antigen presentation machinery, loss of neoantigens, and disruption of immune-related signaling are all potential mechanisms of acquired resistance to cancer immunotherapies.
Analysis of immune checkpoint blockade response and resistance mediators has predominantly focused on tumor types with established clinical benefit to these agents, such as melanoma and NSCLC. In this study, we report a case of a treatment-naïve patient with uterine leiomyosarcoma (ULMS) who received anti-PD-1 monotherapy (pembrolizumab) for metastatic disease and experienced complete disease remission following resection of one treatment-resistant tumor. Sarcoma generally harbors few targetable genetic alterations (Barretina et al., 2010), and while the first reported results on anti-CTLA-4 therapy in synovial sarcoma have been disappointing (Maki et al., 2013), multiple clinical trials are ongoing to explore whether anti-PD-1 or anti-PD-L1 agents may be more promising (e.g. NCT02609984). We hypothesized that a combination of tumor-intrinsic genomic alterations and changes in neoantigen-immune microenvironment interactions mediated response and selective resistance in this case. Thus, we analyzed immunohistochemistry, RNA-seq, and whole exome sequencing (WES) to examine this patient’s changing tumor-immune dynamics over time.
Results
Clinical course and immunohistochemistry
A treatment-naïve woman with uterine leiomyosarcoma was enrolled in a multi-disease phase II study of pembrolizumab monotherapy (10 mg/kg every 2 weeks) (KEYNOTE-028) (Figure 1A, Supplemental Information). After four doses of pembrolizumab, marked tumor regression was observed at numerous locations, with continued, albeit slowed, growth of a single tumor (Figure 1B), which was removed after 9 months of treatment. More than 2 years after resection of the sole treatment-resistant metastasis, the patient remains disease-free on anti-PD-1 monotherapy.
Figure 1. Histologic and radiographic findings in a treatment-naïve patient with metastatic uterine leiomyosarcoma receiving anti-PD-1 monotherapy.
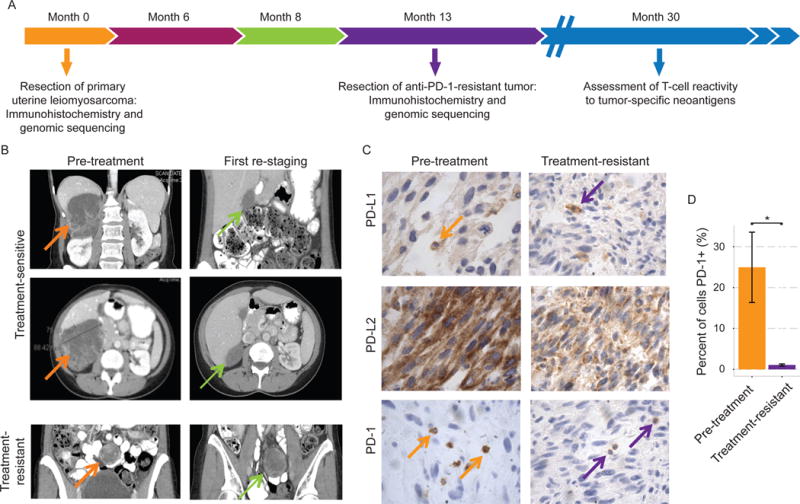
(A) Clinical course and tissue collection for immunohistochemical assessment and whole exome and whole transcriptome sequencing. (B) Computerized tomography (CT) imaging of treatment-responsive tumors and the sole treatment-resistant lesion. (C) Immunohistochemical staining of the primary and treatment-resistant tumors for PD-1, PD-L1, and PD-L2. (D) Quantification from representative tumor sections showing decrease in PD-1+ cell infiltration in the treatment-resistant lesion (p=0.039; Student’s t test). *p < 0.05.
The pre-treatment tumor stained weakly for PD-L1 in infiltrating mast cells (<5% of overall cellularity) and demonstrated diffuse cytoplasmic staining for PD-L2, with about 25% infiltration of PD-1+ cells (Figure 1C). Patterns of PD-L1 and PD-L2 staining persisted in the treatment-resistant tumor, but PD-1 positivity diminished significantly to <1% of overall cellularity (p=0.02; Student’s t test) (Figure 1C,D). Thus, WES and RNA-seq were pursued on pre-treatment and treatment-resistant tumors, along with WES on whole blood for germline comparison, to assess genomic explanations for the diminished PD-1+ cell infiltration and associated treatment-resistance, as well as to explore genomic features associated with otherwise exceptional sensitivity to anti-PD-1 therapy in this patient (Figure 2A).
Figure 2. Pre-treatment and post-treatment exomic features in uterine leiomyosarcoma in comparison to TCGA.
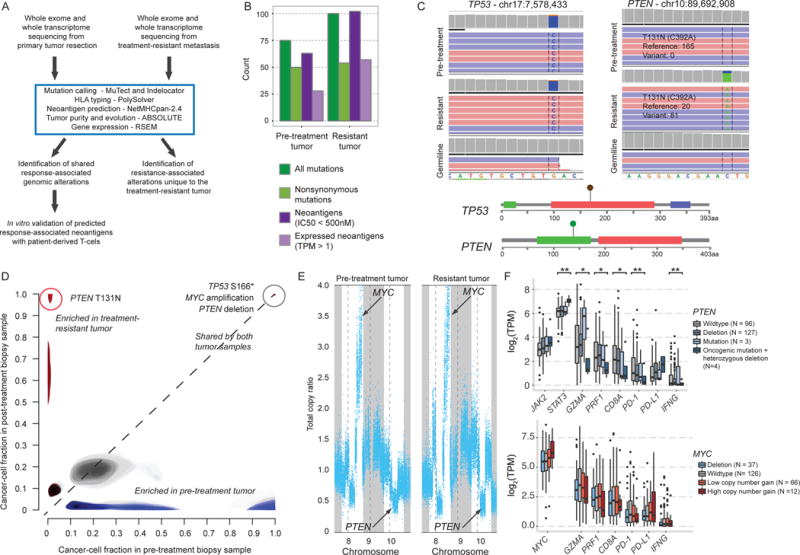
(A) Computational workflow for whole exome and transcriptome analysis and neoantigen prediction. (B) Pre-treatment and resistant tumors had similar neoantigen and mutational loads. See also Table S1. (C) Integrated Genomics Viewer (IGV_2.3.57) (Thorvaldsdottir et al., 2013) and lollipop plots showing the TP53 S166* and PTEN T131N mutations in pre-treatment and treatment-resistant tumors. (D) Comparison of the estimated proportion of cancer cells harboring specific mutations in the pre-treatment (x-axis) versus resistant (y-axis) biopsy samples, with shared clonal mutations in the upper right (grey), mutations exclusive to the pre-treatment tumor in the lower right (blue), and mutations exclusive to the treatment-resistant tumor in the upper left (red). Lighter shading indicates mutations with cancer cell fraction distributions with high uncertainty. (E) Copy number plots show amplification of chromosome 8 and deletion of chromosome 10 affecting MYC and PTEN, respectively, in both tumors. See also Figure S1. (F) Expression of genes related to JAK/STAT and immune inhibitory signaling in untreated sarcoma tumors from the TCGA by PTEN (top) and MYC (bottom) mutational and copy number status. *p<0.05; **p<0.005
Genomic features of the pre-treatment and treatment-resistant tumors
Pre-treatment and treatment-resistant tumors had modest overall mutational loads (50 and 54 nonsynonymous mutations per exome, respectively) (Figure 2B, Table S1A–C). No mutations conferring microsatellite instability or DNA repair defects were detected, and no events were observed in JAK1, JAK2, JAK3, or B2M (encoding β2-microglobulin), despite adequate sequencing coverage to detect these events (Table S1D). Phylogenetic analysis of both tumors for events in genes implicated in oncogenesis (Brastianos et al., 2015; Futreal et al., 2004) revealed a shared clonal nonsense mutation in TP53 (S166*; Figure 2C–D), a focal amplification in chromosome 8q containing the oncogene MYC, and a heterozygous large deletion in chromosome 10q containing the tumor suppressor PTEN (Figure 2E). Additional somatic alterations and copy number events (Table S1B, Figure S1) were of uncertain clinical significance.
Most mutations (n = 38) were shared between the pre-treatment and treatment-resistant tumors, though 12 nonsynonymous mutations were lost from the primary tumor (not seen in the treatment-resistant tumor) and 16 (all missense) were exclusive to the resistant tumor (Figure 2D, Table S1B–C). Of these 16, the only nonsynonymous alteration in a gene previously associated with oncogenic signaling (Brastianos et al., 2015; Futreal et al., 2004) was a clonal missense mutation in PTEN, with evidence of heterozygous loss of the wild-type copy (81 variant reads out of 101 total reads) (Figure 2C). This alteration occurred within a 3D hotspot (Gao et al., 2017) in the tyrosine phosphatase domain of PTEN (Figure 2C) at the same locus as mutations observed in glioblastoma (Brennan et al., 2013) and colorectal cancer (Giannakis et al., 2016), suggesting a functional role. It was not detected in the pre-treatment tumor despite 165 reads at this locus (Figure 2C). Though differences in degree of stromal admixture from nearby normal tissue between the two tumor samples precluded drawing strong conclusions about tumor-specific gene expression alterations, we observed an increase in expression of VEGFA in the treatment-resistant tumor (353 to 1103 transcripts per million (TPM), Table S1E), consistent with a previously described role of PTEN loss in immune resistance in a murine model (Peng et al., 2016) (Figure S2). Transcripts encoding all three HLA alleles and β2-microglobulin were expressed at high levels in both tumor samples (Figure S2).
Immune gene expression by MYC and PTEN copy number status in mesenchymal tumors
To further investigate possible phenotypic consequences of MYC amplification and PTEN loss in this patient, we drew on matched mutation, copy number, and RNA-seq data from 241 untreated primary sarcomas available through The Cancer Genome Atlas (TCGA). Compared to tumors that were wildtype and copy number neutral at the PTEN locus on chromosome 10q23.1, sarcomas that harbored both heterozygous deletion and oncogenic mutation of PTEN – similar to the genotype observed in this patient – had significantly higher levels of STAT3 expression, consistent with activation of the Jak2/Stat3 pathway in the Pten-null state (Toso et al., 2014) (p < 0.005; Student’s t test), while tumors with either mutation of PTEN or copy-number loss over chromosome 10q23.1 but not both did not differ significantly from wildtype (Figure 2F). In addition, tumors with biallelic PTEN loss had significantly lower levels of mRNA expression of PDCD1, CD8A, IFNG, PRF1, and GZMA compared to PTEN-wildtype tumors (p < 0.05 for all) (Figure 2F). Tumors harboring high MYC amplifications trended towards increased mRNA expression of MYC (p=0.06), but did not differ in expression of any of the previously mentioned immune genes (Figure 2F).
Predicted neoantigen presentation in pre-treatment and treatment-resistant tumors
To identify tumor-specific peptides that mediate immune recognition of malignant cells in the context of immune checkpoint therapy, we predicted neoantigens from WES with in silico techniques (Hoof et al., 2009; Shukla et al., 2015; Van Allen et al., 2015). As observed with mutational loads, neoantigen loads were moderate in both pre-treatment and treatment-resistant tumors (82 and 107, respectively) (Figure 2B, 3A; Table S1F).
Figure 3. Predicted and expressed neoantigens in pre-treatment and treatment-resistant tumors.

(A) Workflow of neoantigen analysis, with Venn diagram showing prioritization of putatively response-associated neoantigens. (B) Line plot showing expression of genes containing predicted neoantigens over time. Each line represents a unique neoantigen, with those synthesized for in vitro testing in green, neoantigens in MB21D2 and QKI in dark green, and all other neoantigens in gray. (C) IGV showing mRNA transcripts of mutations generating neoantigens in QKI and MB21D2 in the pre-treatment and treatment-resistant tumors. See also Figure S1, Figure S2, and Table S2.
None of the predicted neoantigens was previously described as clinically T cell-reactive (Fritsch et al., 2014; Robbins et al., 2013; Snyder et al., 2014; van Rooij et al., 2013; Verdegaal et al., 2016; Walter et al., 2012), nor were they seen in HLA-matched responders to immune checkpoint therapy (Hugo et al., 2016; Rizvi et al., 2015; Snyder et al., 2014; Van Allen et al., 2015) (Table S2). Thus, predicted neoantigens were prioritized using matched gene expression (RNA-seq) data (Figure 3A, Table S1F). Four putative neoantigens were predicted to be unique to the pre-treatment sample, but none of these occurred in expressed genes. Two predicted neoantigens derived from clonal somatic mutations in MB21D2 and QKI were expressed at low levels in the resistant compared to pre-treatment sample (Figure 3A–C).
In vitro evaluation of neoantigen recognition by patient-derived T cells
To evaluate the immunogenicity of these neoantigens, peripheral blood mononuclear cells (PBMCs) obtained from the patient following resection of the treatment-resistant tumor were incubated in vitro with neoantigens of interest from MB21D2 and QKI. Five other neoantigens that demonstrated more constant expression throughout the clinical course or were present only in the treatment-resistant setting, and thus not expected to be involved in mediating treatment response, were also evaluated (Figure 3B, Figure S3, Table S1G). In these experiments, patient CD8+ T cells were analyzed for interferon-γ release following exposure to tumor-specific peptides versus wildtype peptide analogs (Figure S4, Figure 4A–B). Secretion of interferon-γ was substantially higher following incubation with tumor-specific compared to wildtype peptides, with significant differences noted for neoantigens in QKI and MB21D2 (p=0.004 and p=0.05, respectively; Student’s t test) (Figure 4C). In addition, patient CD8+ T cells exhibited a stronger interferon-γ response to mutant peptides than CD8+ T cells from an unrelated healthy donor, again with the most drastic differences in QKI and MB21D2 (Figure 4D). Thus, circulating T cells were able to recognize tumor-specific antigens months after surgical removal of metastatic disease, suggesting involvement of immunological memory in durable responses to cancer immunotherapy, while acquired resistance to cancer immunotherapies may occur via decreased expression of genes encoding immunogenic clonal tumor-specific mutations.
Figure 4. In vitro validation of patient T cell reactivity to predicted immunogenic tumor-specific peptides.
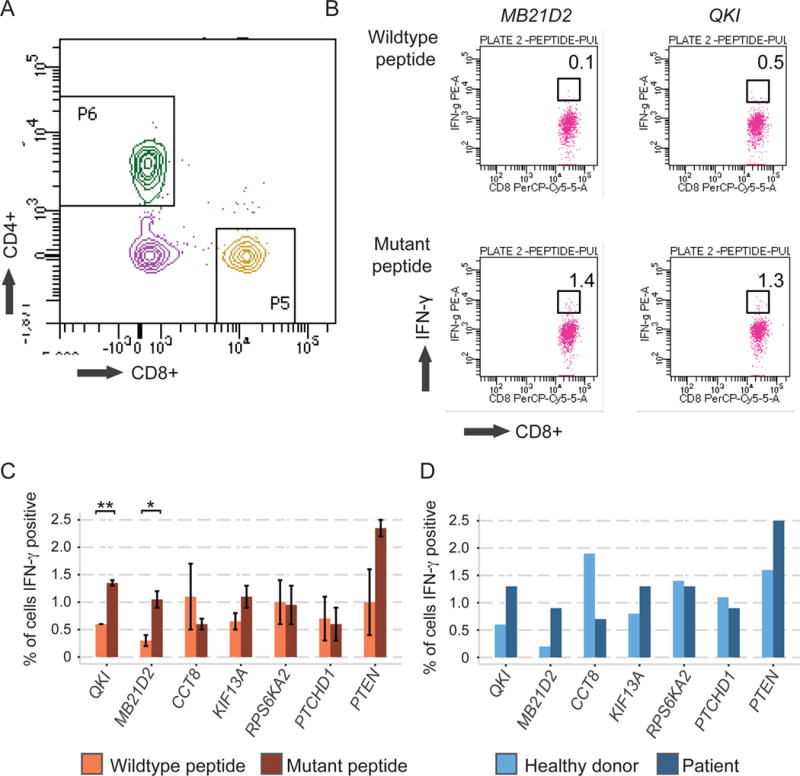
(A) Representative CD4+ versus CD8+ dot plot from viable CD3+ T cells after cell culture. (B) Representative dot plots showing proportions of interferon γ-producing CD8+ T cells following peptide incubation. (C) Bar chart showing proportions of interferon-γ-secreting CD8+ T cells following incubation with tumor-specific mutant peptides (dark orange) or corresponding wildtype control peptides (light orange). Error bars show standard error (SE) above and below the mean. (D) Comparison of reactivity of healthy donor PBMCs (light blue) to patient PBMCs (dark blue) to tumor-specific mutant peptides. See also Experimental Procedures, Supplemental Information, and Figure S4. *p<0.05; **p<0.005
Discussion
This report of a chemotherapy-naïve patient with rapidly-progressive metastatic uterine leiomyosarcoma who had an exceptional clinical response to pembrolizumab supports the need for biologically comprehensive investigation of immune checkpoint inhibitors in sarcoma. Oncogenic alterations in this patient’s cancer included MYC amplification and TP53 loss-of-function mutation in both tumors; meanwhile, biallelic PTEN loss and changes in neoantigen presentation exclusive to treatment-resistant tumor may have contributed to acquired immune resistance.
In melanoma cell lines, PTEN loss increases production of immunosuppressive cytokines, especially VEGF, which decrease cytolytic activity and T cell infiltration in the tumor microenvironment in a PD-L1-independent manner (Peng et al., 2016). In melanoma clinical samples, negative PTEN immunohistochemical staining is associated with reduced efficacy of adoptive cell transfer and anti-PD-1 therapies (Peng et al., 2016). Pten-deficient prostate tumors similarly induce an immunosuppressive tumor microenvironment by upregulating PTPN11 and inducing activity of the Jak2/Stat3 pathway (Toso et al., 2014), but genetic alterations in PTEN have not yet been linked to intrinsic or acquired resistance to immune checkpoint therapies in melanoma or NSCLC (Rizvi and Chan, 2016).
One major role of JAK/STAT signaling is controlling the interferon-γ response, which is commonly disrupted in melanoma tumors intrinsically resistant to immune checkpoint therapy targeting CTLA-4 (Gao et al., 2016). Our observations of increased VEGFA expression accompanied by reduced PD-1+ T cell infiltration in a treatment-resistant mesenchymal tumor with biallelic PTEN loss, as well as increased STAT3 expression in untreated sarcomas with similar PTEN genotypes, tie together these preclinical and clinical findings in support of a PTEN-mediated mechanism of immune resistance to anti-PD-1 therapy in this patient. These findings suggest that addition of JAK/STAT or VEGF inhibitors in clinical cases of PTEN-mediated acquired resistance to immune checkpoint monotherapy could help further control tumor growth (Peng et al., 2016; Toso et al., 2014).
We additionally identified two neoantigens with reduced expression in the treatment-resistant compared to pre-treatment tumor and high patient T cell reactivity in vitro, indicating that a subset of neoantigens encoded by tumor-specific mutations were capable of stimulating a T cell-mediated anti-tumor response, and their reduced expression in the treatment-resistant tumor may have contributed to immune escape. Other patient-specific neoantigens also stimulated T cell effector cytokine release, though not significantly more than non-mutated normal peptides, and whether an effective cytotoxic T cell response, presumably induced by PD-1 blockade, relies on the presence of a small number of highly immunogenic tumor antigens or a large array of moderately reactive peptides deserves further study.
Prior studies in immune checkpoint therapy-treated patients show disruptions in genes involved in neoantigen presentation are key in tumor immune evasion. For example, loss of expression of MHC class I molecules necessary for neoantigen presentation via frameshift deletion of β2-microglobulin (Zaretsky et al., 2016) or deletion of HLA alleles (Tran et al., 2016) is associated with acquired resistance to cancer immuunotherapies, as is loss of neoantigens themselves via DNA-level loss-of-heterozygosity events that eliminate neoantigen-producing mutations (Anagnostou et al., 2017; Verdegaal et al., 2016). By combining whole exome-based mutation calls with RNA-seq, this study adds loss of neoantigens via reduced of expression of mutated genes as an additional mechanism contributing to acquired resistance to immune checkpoint therapy.
In summary, immunohistochemical, whole exome, and whole transcriptome analyses of pre-treatment and treatment-resistant tumors from one patient identified PTEN loss and changes in neoantigen expression as potential clinical mechanisms of acquired resistance to immune checkpoint therapy, though further experimental work in mesenchymal tumors is needed. Prospective integrated assessment of large immunotherapy-treated cohorts in tumor types not typically considered responsive to immune checkpoint blockade may further inform recurrent features associated with response and resistance.
Experimental Procedures
Patient history and oversight
A previously healthy 48-year-old woman developed metastatic disease three months after primary tumor resection of uterine leiomyosarcoma and was enrolled in a multi-disease phase II study of pembrolizumab monotherapy (10 mg/kg every 2 weeks) (KEYNOTE-028). She received no adjuvant systemic or radiation therapies prior to pembrolizumab. After four doses of pembrolizumab over two months, marked tumor regression was observed at a large right retroperitoneal subhepatic site and numerous other locations, while a peritoneal tumor in the mesentery of the small bowel continued to grow, albeit at a slower rate. After 9 months of treatment, the resistant lesion and responding sites were resected. Pathology of the subhepatic, pembrolizumab-responsive tumor site demonstrated dense and collagenous tissue with foreign body giant cell reaction, granuloma, and hemosiderin-laden macrophages. The treatment-resistant mesenteric tumor demonstrated a 9.0cm myxoid leiomyosarcoma involving the bowel wall, which was highly mitotic (117 cells/10 high-power fields) with necrosis (up to 20% tumor volume) and tissue hemorrhage. No tumor tissue remained for sampling in the treatment-responsive lesion, but a sample from the treatment-resistant tumor was taken for genomic comparison with the primary uterine tumor and germline blood. The clinical trial of pembrolizumab was sponsored by Merck Sharp & Dohme Corp. (NCT02054806) and approved by the institutional review board of the Dana-Farber/Harvard Cancer Center. The patient provided written informed consent for molecular profiling (DFCI Protocol #11-104).
Whole-exome and whole-transcriptome sequencing
Whole exome and transcriptome sequencing and quality control of tumor and germline samples were performed as previously described (Van Allen et al., 2015; Van Allen et al., 2014). Tumor-specific somatic point mutations, small insertions and deletions (indels), copy-number alterations, and neoantigens were identified by standard genomic analysis pipelines (Van Allen et al., 2015). Mutational clonality (presence of a mutation in all sampled tumor cells versus a subclonal tumor population), tumor purity, and tumor ploidy were assessed using ABSOLUTE (Carter et al., 2012), and used as inputs for phylogenetic analyses (Stachler et al., 2015). For purposes of neoantigen prediction, all six 4-digit patient HLA alleles were inferred from germline WES using Polysolver (Shukla et al., 2015). All tumor-specific 9- and 10-amino-acid peptides generated by nonsynonymous tumor-specific nonsynonymous somatic point mutations and small insertions and deletions were stored in a FASTA file and then assessed for strength of binding to each of the patient’s 6 HLA alleles using NetMHCpan-2.4 (Hoof et al., 2009; Nielsen et al., 2007). Any 9- or 10-amino-acid peptide with predicted binding affinity to any HLA allele with IC50 ≤ 500nM was considered a neoantigen, such that one nonsynonymous mutation could theoretically generate many neoantigens. HLA binding affinity to tumor-specific neoantigens was further confirmed using NetMHCpan-3.0 (Karosiene et al., 2013). A neoantigen was considered expressed if gene-level TPM exceeded 1 by RSEM (Li and Dewey, 2011). All sequencing data is available at dbGap accession number phs000694.
TCGA analyses
Whole exome mutation annotation files and whole transcriptome mRNASeq gene expression files for the TCGA sarcoma cohort were downloaded from Firebrowse (data version 2016_01_28). RNA-seq from matched normal tissue was excluded. Copy number loss, gain, and amplification and oncogenic alterations in MYC and PTEN were defined in accordance with TCGA standards (http://firebrowse.org, http://www.cbioportal.org) (Cerami et al., 2012; Gao et al., 2013).
Neoantigen-reactivity experiments
PBMCs were obtained with the patient’s consent following resection of the treatment-resistant metastatic lesion and evaluated for reactivity to 7 synthetic in silico predicted neoantigens and their wildtype analogs using standard protocols (Lin et al., 2009; Maeda et al., 2014; Rizvi et al., 2015). Tumor-specific and wild-type peptides were synthesized by GenScript USA Inc. Briefly, healthy donor or patient-derived PBMCs were stimulated with 10 μg/mL of mutant peptides for 10 or 12 days in the presence of IL-2 and IL-15. Cells were washed and then re-stimulated with monocyte-derived dendritic cells pulsed with the same concentration of either mutant or wildtype peptides for 24 hours or PMA and Ionomycin for 6 hours. Protein transport inhibitor cocktail (Golgi plug/stop) was added in the last 5 hours of culture. Cells were acquired using fluorescence-activated cell sorting (FACS) on BD LSRFortessa (BD Biosciences) prior to staining for interferon-γ. Data were analyzed with Flowjo software (Tree Star).
Statistical analysis
All statistical analyses were done using R version 3.2.3. Student’s t-tests with unequal variances were used for all comparisons of continuous variables between groups. All tests were two-tailed with an alpha level of 0.05.
Supplementary Material
Highlights.
Anti-PD-1 monotherapy can induce complete tumor remission in uterine leiomyosarcoma
Longitudinal profiling of patient tumors reveals response and resistance mechanisms
Tumor neoantigens are patient-specific and can induce long-term immunoreactivity
Biallelic PTEN loss is associated with resistance to PD-1 blockade in sarcoma
Acknowledgments
This work was supported by BroadIgnite (E.M.V.), BroadNext10 (E.M.V., D.M., P.H.), the Catherine England Leiomyosarcoma Research Fund (S.G.), the Ludwig Center at Harvard (G.D.), the Erica Kaitz LMS RESEARCH NOW Fund (G.D.), NIH K08CA188615 (E.M.V.), NIH P50CA101942 (G.F.), the Alexandra J. Miliotis Pediatric Oncology Research Fund (D.M.), and the Howard Hughes Medical Institute Medical Research Fellowship (D.M). The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Information
Supplemental information includes four figures, two tables and Supplemental Experimental Procedures, and can be found at (link to be added by production).
Author contributions:
S.G., G.D., and C.P.R contributed to clinical care and sample procurement. S.G., K.K.W. and E.M.V. supervised the project. S.J.R. and M.L. performed immunohistochemistry. A.A., S.S., D.M., and E.M.V. performed genomic and statistical analyses. D.A. and K.K.W. performed the in vitro neoantigen experiments. P.H. G.J.F, C.J.W., and P.A.O. contributed immunology assay development. D.M., S.G. and E.M.V. wrote the manuscript with contributions from all authors.
The authors declare no conflicts of interest.
References
- Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discovery. 2017 doi: 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Taylor BS, Banerji S, Ramos AH, Lagos-Quintana M, Decarolis PL, Shah K, Socci ND, Weir BA, Ho A, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42:715–721. doi: 10.1038/ng.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5:1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res. 2014;2:522–529. doi: 10.1158/2326-6066.CIR-13-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Chang MT, Johnsen HC, Gao SP, Sylvester BE, Sumer SO, Zhang H, Solit DB, Taylor BS, Schultz N, Sander C. 3D clusters of somatic mutations in cancer reveal numerous rare mutations as functional targets. Genome Medicine. 2017;9:4. doi: 10.1186/s13073-016-0393-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397–404.e399. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, Bahl S, Cao Y, Amin-Mansour A, Yamauchi M, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016;15:857–865. doi: 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics. 2013;65:711–724. doi: 10.1007/s00251-013-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Gallardo HF, Ku GY, Li H, Manukian G, Rasalan TS, Xu Y, Terzulli SL, Old LJ, Allison JP, et al. Optimization and validation of a robust human T-cell culture method for monitoring phenotypic and polyfunctional antigen-specific CD4 and CD8 T-cell responses. Cytotherapy. 2009;11:912–922. doi: 10.3109/14653240903136987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Nishikawa H, Sugiyama D, Ha D, Hamaguchi M, Saito T, Nishioka M, Wing JB, Adeegbe D, Katayama I, Sakaguchi S. Detection of self-reactive CD8(+) T cells with an anergic phenotype in healthy individuals. Science. 2014;346:1536–1540. doi: 10.1126/science.aaa1292. [DOI] [PubMed] [Google Scholar]
- Maki RG, Jungbluth AA, Gnjatic S, Schwartz GK, D’Adamo DR, Keohan ML, Wagner MJ, Scheu K, Chiu R, Ritter E, et al. A pilot study of anti-CTLA4 antibody ipilimumab in patients with synovial sarcoma. Sarcoma. 2013;2013:168145. doi: 10.1155/2013/168145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips GK, Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol. 2015;27:39–46. doi: 10.1093/intimm/dxu095. [DOI] [PubMed] [Google Scholar]
- Rizvi NA, Chan TA. Immunotherapy and oncogenic pathways: The PTEN connection. Cancer Discov. 2016;6:128–129. doi: 10.1158/2159-8290.CD-15-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19:747–752. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, JL D, Steelman S, et al. Polysolver for the detection of somatic mutations in MHC class I alleles across cancers. Nat Biotechnol. 2015;33:1152–1158. doi: 10.1038/nbt.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachler MD, Taylor-Weiner A, Peng S, McKenna A, Agoston AT, Odze RD, Davison JM, Nason KS, Loda M, Leshchiner I, et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat Genet. 2015;47:1047–1055. doi: 10.1038/ng.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, Pinton S, Zhang J, Kalathur M, Civenni G, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014;9:75–89. doi: 10.1016/j.celrep.2014.08.044. [DOI] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. T-cell transfer therapy targeting mutant KRAS in cancer. New England Journal of Medicine. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MH, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, Jane-Valbuena J, Friedrich DC, Kryukov G, Carter SL, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20:682–688. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31:e439–442. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536:91–95. doi: 10.1038/nature18945. [DOI] [PubMed] [Google Scholar]
- Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med. 2012;18:1254–1261. doi: 10.1038/nm.2883. [DOI] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.