

Abstract
Cellular binding-proteins (BP), including CRBP1, CRBP2, CRABP1, CRABP2, and FABP5, shepherd the poorly aqueous soluble retinoids during uptake, metabolism and function. Holo-BP promote efficient use of retinol, a scarce but essential nutrient throughout evolution, by sheltering it and its major metabolite all-trans-retinoic acid from adventitious interactions with the cellular milieu, and by imposing specificity of delivery to enzymes, nuclear receptors and other partners. Apo-BP reflect cellular retinoid status and modify activities of retinoid metabolon enzymes, or exert non-canonical actions. High ligand binding affinities and the nature of ligand sequestration necessitate external factors to prompt retinoid release from holo-BP. One or more of cross-linking, kinetics, and colocalization have identified these factors as RDH, RALDH, CYP26, LRAT, RAR and PPARβ/δ. Michaelis-Menten and other kinetic approaches verify that BP channel retinoids to select enzymes and receptors by protein-protein interactions. Function of the BP and enzymes that constitute the retinoid metabolon depends in part on retinoid exchanges unique to specific pairings. The complexity of these exchanges configure retinol metabolism to meet the diverse functions of all-trans-retinoic acid and its ability to foster contrary outcomes in different cell types, such as inducing apoptosis, differentiation or proliferation. Altered BP expression affects retinoid function, for example, by impairing pancreas development resulting in abnormal glucose and energy metabolism, promoting predisposition to breast cancer, and fostering more severe outcomes in prostate cancer, ovarian adenocarcinoma, and glioblastoma. Yet, the extent of BP interactions with retinoid metabolon enzymes and their impact on retinoid physiology remains incompletely understood.
Keywords: cellular retinol binding-protein, cellular retinoic acid binding-protein, retinol dehydrogenase, retinal dehydrogenase, retinoic acid, retinoids, retinol
1. Introduction
Retinol (vitamin A) and its major metabolites retinal and all-trans-retinoic acid (atRA) are known collectively as naturally-occurring retinoids (Napoli, 1999; Das et al., 2014). Retinal serves as the cofactor for the visual cycle, whereas atRA supports systemic effects of vitamin A by sustaining spermatogenesis, embryonic development, epithelial differentiation, and nervous and immune system functions (Ulbricht et al., 2012; Hogarth & Griswold, 2013). atRA also controls energy balance and adiposity and suppresses inflammation (Chen & Chen, 2014; Brown & Noelle, 2015).
Vitamin A deficiency remains one of the three most prevalent nutrient deficiencies worldwide, with highest incidence in developing countries, but also occurring in pockets of developed countries http://www.who.int/vmnis/vitamina/data/database/countries/en/). Severe vitamin A/atRA depletion primarily affects children, because of their need related to rapid growth (Sommer & Vyas, 2012; Akhtar et al., 2013). The most deleterious consequences of severe depletion include xerophthalmia, a degeneration and ulceration of the corneal epithelium that can cause permanent blindness (as opposed to nyctalopia or night blindness, a fairly benign condition that manifests early during suboptimum vitamin A status), and immune suppression, associated with a higher incidence of infection and increased mortality (Wiseman et al. 2016).
Vitamin A has limited solubility in aqueous media (~60 nM), which occurs below its concentrations in serum and most tissues (Szuts & Harosi, 1991). Its poor aqueous solubility and often scarce availability induced mechanisms to enhance its efficiency of absorption, distribution, metabolism and function. These challenges have been addressed by evolution of cellular lipid binding-proteins (BP) that sequester and chaperone retinoids and other lipids to protect them from the cellular milieu, and direct them to dedicated enzymes and receptors (Napoli, 1993; Ross, 1993; Ong, 1994; Napoli, 2012).
The conserved gene family of cellular lipid BP, FABP, includes a subset that binds retinoids exclusively (Bass, 1993). Four cellular retinol BP (CRBP) have been identified that bind retinol and retinal specifically, along with two cellular BP (CRABP) that bind all-trans-retinoic acid (atRA) with high affinity. In addition, the “non-retinoid” FABP5 also has high affinity for atRA, as well as for long-chain fatty acids (LCFA) (Shaw et al., 2003; Levi et al., 2013) (Table 1). The most thoroughly studied of the retinoid binding-proteins, CRBP1, CRBP2, CRABP1, CRABP2, and FABP5, appear essential to efficient retinoid homeostasis and function. Although the function(s) of CRBP3 and CRBP4 remain to be studied as comprehensively as the others, they also facilitate retinoid metabolism and action. This article will focus on functions of the first five BP as integral to retinoid homeostasis under physiological conditions, will emphasize kinetic data that support BP-target interactions, and will discuss the health-related contributions of retinoid BP.
Table 1. Characteristics of retinoid BP.
Specified tissues represent sites of most intense expression.
| BP | Specificity | kd (nM)d | Tissue distribution | References to kd values |
|---|---|---|---|---|
|
| ||||
| CRBP1 (Rbp1) | retinol retinal |
0.1 to 3 9 |
extensive | Li et al., 1991; Malpeli et al., 1995; Kane et al., 2011 |
| CRBP2 (Rbp2) | retinol retinal |
10 11 |
limited (intestinal enterocytes) | Li et al., 1991; Kane et al., 2011 |
| hCRBP3a (RBP5) | retinol retinal |
60 | <CRBP1 & >CRBP2 (liver and kidney) | Folli et al., 2001 |
| mCRBP4b,c (Rbp7) | retinol | 109 | limited (heart, muscle, adipose, mammary) | Vogel et al., 2001; Folli et al., 2002 |
| hCRBP4 (RBP7) | retinol | 200 | extensive | Folli et al., 2001 |
| CRABP1 | atRA = metabolites ≫ isomers | 0.4–16 | widespread | Ong & Chytil, 1978; Fiorella et al., 1993; Norris et al., 1994; Wang & Yan,1997 |
| CRABP2 | atRA ≫ RA isomers | 2–14 | limited (skin, uterus, ovary) | Giguère et al., 1990; Fiorella et al., 1993; Norris et al., 1994; Wang & Yan, 1997 |
| FABP5 | atRA LCFA |
35 15–20 |
many (liver) | Shaw et al., 2003a; Schug, et al., 2007b; Levi et al., 2015b |
The mouse does not express an ortholog of human CRBP3. Many other species express all retinoid BP.
Mouse CRBP4 is the only CRBP that binds 13-cis-retinol and 9-cis-retinol with kd values similar to all-trans-retinol.
Originally also named CRBP3, but encoded by a homologous gene.
For technical reasons, where a range of values are given, the lower values are probably more accurate.
2. Diverse metabolic functions of cellular retinol binding-proteins
Where assessed, CRBP1 and 2 occur in concentrations that exceed their ligands and sequester most, if not all cellular retinoids, despite the ability of membranes to absorb much greater amounts of retinoids than occur in cells (Ong et al., 1976; Harrison et al., 1987; Ong et al., 1994). For example, testes of vitamin A-sufficient rats express 200 pmol CRBP1/g tissue, whereas human testes have 445 pmol/g tissue (Ong et al., 1976; Arnold et al., 2015). Thirty to 43% of the CRBP1 in rat testes occurs occupied by retinol. Retinol concentrations in mouse testes occur at ~80 pmol/g tissue (Kane et al., 2008a), which agrees well with the earlier conclusion about the degree of saturation in rats, ~40%. This outcome of seemingly total sequestration of retinol should not amaze, based on the high affinity of CRBP1 and 2 for their ligands and their concentrations in excess relative to their ligands. Ratios of ligand to BP still need to be determined for CRBP3 and 4.
2.1. CRBP2 channels diet-derived retinoids into RE biosynthesis
Sequestration of retinoids within BP with a negligible dissociation rate suggests that the holo-BP itself would promote uptake of retinoids and chaperone them selectively to enzymes that interact with the BP “cassette”, while preventing metabolism by enzymes that cannot interact with the BP. CRBP2 function supports this model. CRBP2 comprises ~1% of the cytosolic protein in the intestinal enterocyte, where its expression exceeds other cells by 500-fold and CRBP1 abundance by 1000-fold (Ong & Page, 1987). Ablation of CRBP2 impairs efficient uptake of retinol in mice fed a diet copious in vitamin A and causes embryonic lethality in mice fed a vitamin A-marginal diet (E et al., 2002). The process, however, is much more complex than simply increasing retinoid uptake. Carotenoids, especially β-carotene, usually provide the major dietary sources of vitamin A. Carotenoids undergo central cleavage by the cytosolic dioxygenase BCO1 to generate retinal, the immediate precursor of atRA (Olson & Hayaishi, 1965; Goodman & Huang, 1965; Redmond et al., 2001; von Lintig & Vogt, 2000; dela Seña et al., 2014). The irreversible conversion of retinal into atRA occurs beyond the rate-limiting reaction of atRA biosynthesis, catalyzed by retinol dehydrogenases (RDH) converting retinol into retinal (Napoli, 1999). Thus, consumption of pro-vitamin A carotenoids in large amounts ought to cause illness, because excess atRA produces both toxicity and teratogenicity. Carotenoids, however, are neither toxic nor teratogenic (Biesalski, 1989; Collins & Mao, 1999). The abundant and high-affinity CRBP2 affords protection from excess atRA by chaperoning retinal into reduction (Fig. 1a) (Kakkad & Ong, 1988). The rate of reduction of CRBP2-delivered retinal exceeds its conversion into atRA by at least 300-fold.
Fig. 1.

Impact of CRBP1 and 2 on retinal reduction and RE biosynthesis. a) Model of CRBP2 actions in the intestinal enterocyte that direct retinoids into RE formation and arrests atRA biosynthesis. A brush border REH converts dietary vitamin A esters into retinol for absorption (Rigtrup & Ong, 1992; Rigtrup et al., 1994a; Rigtrup et al., 1994b). Alternatively, metabolism of carotenoids produces retinal. CRBP2 directs retinoids into RE formation and prevents atRA biosynthesis (Kakkad & Ong, 1988). b) Michaelis-Menten relationships of CRBP2-mediated retinal reduction and retinol esterification (Ong et al., 1987; MacDonald & Ong, 1988b). c) Kinetic constants demonstrating holo-CRBP2 channeling retinol through protein-protein interactions into RE biosynthesis. d) Model showing CRBP1 and 2 actions in liver directing retinol to LRAT for RE formation (Ong et al., 1988; MacDonald & Ong, 1988a; Yost et al., 1988; Randolph et al., 1991; Herr & Ong, 1992). e) Kinetic constants that illustrate ability of holo-CRBP1 to deliver retinol through protein-protein interaction to LRAT for RE biosynthesis in liver.
Retinol esters (RE) provide another source of dietary vitamin A. A brush border retinyl ester hydrolase (REH), similar to/identical with phospholipase B, catalyzes RE hydrolysis into retinol (Rigtrup & Ong, 1992; Rigtrup et al., 1994a; Rigtrup et al., 1994b). Regardless of the source, CRBP2 delivers retinol to lecithin:retinol acyltransferase (LRAT) for conversion into RE (Ong et al., 1987). Esterification of CRBP2-bound retinol specifically requires phosphatidyl choline (lecithin) as co-substrate, and does not proceed with other phospholipids or acyl-CoA as acyl group donor, which accounts for LRAT producing RE in vitro with a composition that reflects the composition of endogenous RE (MacDonald & Ong, 1988b; Ong et al., 1987).
Kinetic data demonstrate that the reductase and LRAT recognize the CRBP2-retinoid complexes and access the retinoids and incorporate them into their active sites. Low “free” retinoid concentrations, if any, occur in cells that express BP, as predicted by the kd values and demonstrated by the intracellular association of retinoids with BP despite alternative acceptors with much higher capacity, albeit with lower affinity, such as membranes and non-specific proteins. Michaelis-Menten relationships reveal BP concentration-dependent, saturable reactions that would occur only if the holo-BPs deliver substrates directly to enzymes and by-pass diffusion (Fig. 1b). Michaelis constants (Km values) identical with the values generated with “free” retinol with rates similar to those generated with “free” retinol (within experimental error) also demonstrate a substrate-delivery relationship between BP and reductase and BP and LRAT that does not rely on diffusion (Fig. 1c). These rates far exceed rates that would be supported by the calculated hypothetical concentrations of “free” retinoids in the presence of BP. The selectivity of both reactions further supports unique relationships between CRBP2 and these enzymes. CRBP2-bound retinal does not undergo dehydrogenation efficiently. Nor does CRBP2-bound retinol undergo esterification efficiently by acyl-CoA:retinol acyltransferase (ARAT) activity, which relies on long-chain acyl-CoA derivatives and “free” retinol as co-substrates.
Thus, CRBP2 in the intestinal enterocyte enhances dietary carotene and retinol absorption by effecting irreversible uptake through fostering efficient retinal reduction and RE biosynthesis, while preventing toxicity from atRA. Not incidentally, the selectivity of CRBP2-retinol for the co-substrate lecithin led to the discovery of LRAT.
2.2. CRBP1 chaperones retinol into RE biosynthesis in cells other than intestinal enterocytes
CRBP1 functions as a master regulator of retinol homeostasis in numerous human and rodent tissues and cell types, including the developing embryo, liver, kidney, lung, sex organs and various areas of the brain (Porter, et al., 1983; Ong & Page, 1986; Levin, et al., 1987; Rajan, et al., 1990; Folli et al., 2001). Liver serves as the major site of vitamin A storage as RE, and LRAT catalyzes RE biosynthesis in liver, recognizing CRBP1-retinol as delivery vehicle (Fig. 1d). The criteria discussed above for CRBP2-retinoid also apply to the reaction between CRBP1-retinol and LRAT (Ong et al., 1988; MacDonald & Ong, 1988a; Yost et al., 1988; Randolph et al., 1991; Herr & Ong, 1992). Namely, the Michaelis constant (Km) and maximum reaction rate (Vm) do not support retinol having to dissociate from CRBP1 and diffuse through the aqueous medium to LRAT, because the values for both “free” retinol and CRBP1-retinol do not differ within experimental error (Fig. 1e). This would not occur if LRAT did not recognize CRBP1-retinol and have ability to tease retinol from the BP. If only “free” retinol were substrate, adding the BP would greatly diminish the rate, because of its effect on the concentration of “free” retinol.
CRBP1, like CRBP2, imposes selectively on the esterification reaction. Only LRAT catalyzes RE formation in liver and in most other tissues under physiological conditions. Two exceptions are mammary gland and skin—organs with lower concentrations of CRBP1. In these organs, the activity known as ARAT, now identified as diacylglycerol acyltransferase 1 (DGAT1), contributes to RE formation (Randolph et al., 1991; Orland et al., 2005; Yen et al., 2005; Shih et al., 2009). DGAT1-ablation does not reduce the amount of hepatic RE (Wongsiriroj et al., 2008), whereas LRAT-ablation greatly diminishes the amount of RE in tissues (Batten et al., 2004; Liu & Gudas, 2005; O’Byrne et al., 2005). This indicates that LRAT generates the predominant amount of RE under physiological conditions in tissues that express a form of CRBP. During LRAT ablation, however, DGAT1 can partially compensate for LRAT loss by generating a limited amount of RE, ~10% of normal. DGAT1 also generates RE from pharmacological exposure to retinol (Wongsiriroj et al., 2008). This implies a mechanism for retinol toxicity, i.e. dietary amounts of retinol that exceed BP concentrations can contribute to retinoid pools. This also illustrates the point that the retinoid BP facilitate retinoid homeostasis, but are not obligatory, especially if diets have copious vitamin A.
2.3. The ratio apo-CRBP1/holo-CRBP1 signals cellular retinol status
apo-CRBP1 inhibits LRAT competitively, with ~1 μM of the former causing ~60% inhibition (Herr & Ong, 1992) (Fig. 2a). Inhibition is inherent to the CRBP1 structure and is specific, because chemically disabled apo-CRBP1 that cannot bind retinol still inhibits LRAT, and apo-CRBP2 does not have a similar effect. apo-CRBP1 also stimulates hydrolysis of endogenous RE (Boerman & Napoli, 1991). Other proteins that bind retinol non-specifically do not duplicate this effect, such as albumin, which has lower affinity but higher capacity, and therefore might have affected the reaction by removing product. Both effects of apo-CRBP1 display saturable kinetics, which implies a direct relationship of apo-BP with LRAT/REH. Sensing of the intracellular quantity of retinol by the ratio apo-CRBP1/holo-CRBP1 affords a simple but elegant mechanism to direct retinol traffic into and out of RE, to balance storage vis-à-vis substrate availability for atRA biosynthesis.
Fig. 2.

Apo-CRBP1 enables mobilization of RE. a) Kinetics of apo-CRBP1 inhibiting LRAT and stimulating endogenous RE hydrolysis (Herr & Ong, 1992; Boerman & Napoli, 1991). b) Model relating actions of apo-CRBP1 to cellular retinoid homeostasis. Apo-CRBP1 acts as a sink to draw retinol into cells from the serum BP, RBP4, and also signals cell retinoid status. In the absence of apo-CRBP1, CRBP1 directs retinol into RE formation, while maintaining atRA biosynthesis. As apo-CRBP1 increases, inhibition of LRAT preserves remaining holo-CRBP1 to support atRA biosynthesis.
2.4. CRBP1 directs retinol to RDH for retinal biosynthesis
Crosslinking CRBP1-retinol with microsomal RDH indicated that the two approach each other closely (Fig. 3a). Crosslinking was achieved by derivatizing holo-CRBP1 with a cleavable, radio-iodinated reagent, activated upon light exposure. Incubating derivatized holo-CRBP1 with microsomes, exposing the reaction to light and then cleaving the reagent, produced only two radio-iodinated bands on SDS-PAGE, one at ~37 kDa and the other at ~25 kDa (Boerman & Napoli, 1995). The 37 kDa band appeared only in presence of pyridine nucleotide co-factor (NADP+ or NAD+). Dehydrogenases function through an ordered bi-substrate mechanism, i.e. they first must bind cofactor before they can bind co-substrate. Crosslinking therefore, occurred as expected if CRBP1 delivers retinol to RDH via channeling. The 25 kDa band was identified as LRAT (Ruiz et al., 1999; Zolfaghari & Ross, 2000). Cross-linking of only two bands, both retinoid metabolizing enzymes, reveals the selective nature of holo-CRBP1’s delivery of retinol in an environment of numerous enzymes.
Fig. 3.

Contribution of CRBP1 to atRA homeostasis. a) Crosslinking with holo-CRBP1 identified RDH as members of the short-chain dehydrogenase/reductase gene family (SDR) by radio-iodinating this CRBP1 target enzyme (Boerman & Napoli, 1995). b) Michaelis-Menten kinetics confirmed ability of holo-CRBP1 to deliver retinol to RDH. The lack of change in Km or Vm with differences in the ratio CRBP1/retinol further confirmed direct interaction between holo-CRBP1 and RDH (Posch et al., 1991). c) Illustration of CRBP1’s impact on the amount of “free” retinol, calculated using a kd value of 2 nM. d) Kinetics of the reductase DHRS4 with free retinal or CRBP1-retinal (Lei et al., 2003). e) Inhibition of the DHRS4 reaction by apo-CRBP1, illustrating the mechanism of a decreased Vm with CRBP1-retinal as substrate shown in d. The filled blue symbols show that partial sequestration of retinal does not lower the reaction rate, indicating that both free and bound retinal are available as substrate. The red symbol shows the impact of equimolar amounts of BP and retinal, indicating ability of apo-CRBP1 to inhibit DHRS4.
Kinetics also showed that holo-CRBP1 provides substrate for retinal biosynthesis directly to microsomal RDH (Posch et al., 1991). The rate of retinal production from holo-CRBP1 proceeds via a Michaelis-Menten relationship, and exceeds the rate observed experimentally from the calculated concentrations of unbound retinol that would maximally occur in the presence of BP (Fig. 3b, c). Holo-CRBP1 also supports a lower Km value (~1.6 μM) than free retinol (4.7 μM) (Song et al., 2003). The Vm with BP is lower than the Vm supported by free retinol, but the rate with BP generates more than sufficient retinal to satisfy cellular demands for atRA, which occurs in low nM in tissues (Obrochta et al., 2014). The lower rate with BP might reflect transfer of retinol from BP to RDH becoming the rate-limiting event, rather than release of retinal from the RDH. Varying the ratio total CRBP1/retinol had no effect on the reaction rate or the Km value. Changing the ratio retinol/CRBP1 from 1/1.1 to 1/1.5 would decrease any unbound retinol 5-fold, which would have a major impact on the reaction rate, if only “free” retinol were substrate.
Compared to cytosol, microsomes harbor 80–94% of the enzyme units that convert CRBP1-bound retinol (5:2, holo-CRBP1:apo-CRBP1) into retinal (Boerman & Napoli, 1996). Apo-CRBP1 and ethanol inhibit cytosolic activity with IC50 values of 1 and 20 μM, respectively, but have no effect on microsomal RDH activities. These data indicate that most RDH activity occurs in microsomes, and corroborate inability of chronic alcohol exposure to inhibit atRA biosynthesis in vivo (Kane et al., 2010).
Amino acid sequence obtained from the cross-linked 37 kDa band enabled cloning the cDNA of rat RDH2 and RDH7 (initially RoDHI and RoDHII) (Chai et al., 1995a; Chai et al., 1995b; Zhang et al., 2001) and confirmed identity of RDH as short-chain dehydrogenase/reductase gene family members (Krozowski, 1994). These data supported identification and cDNA cloning of multiple additional RDH, including RDH1 (mouse ortholog of RDH2, aka hRDH16), RDH10 (mouse, rat, human), and DHRS9 (mouse, rat, human) and the reductases DHRS3 and DHRS4 (Napoli, 2012; Kedishvili, 2013).
2.5. CRBP1 presents retinal to DHRS4 for reduction
Several reductases of the short-chain dehydrogenase/reductase gene family convert retinal into retinol, including DHRS3, DHRS4, and RDH11. Of these, only DHRS4 (formerly RRD) has been tested for ability to recognize CRBP1-retinal as substrate (Lei et al., 2003). DHRS4 accesses retinal bound to CRBP1 without requiring dissociation of retinal prior to uptake, as indicated by saturable kinetics, generated with a 2:1 ratio of total CRBP1/retinal at each concentration (Fig. 3d). The presence of BP decreases the Vm by ~75%. This could result because the rate of retinal transfer from the BP to DHRS4 behaves as the rate-limiting event in the overall reaction, rather than release of reduced co-factor from the dehydrogenase. Alternatively, apo-CRBP1 might inhibit the reaction. To determine whether inhibition by apo-CRBP1 caused the decreased rate, the effect of adding graded doses to 4 μM retinal was assayed. Adding 1 or 2 μM total apo-CRBP1 to 4 μM retinal decreases the free retinal concentration by ~1 and 2 μM, respectively, but did not decrease the rate of retinal reduction (two blue points in Fig. 3e). An equal molar amount of CRBP1/retinal (red point in Fig. 3e) decreased the rate by 57%, but also left ~0.18 μM apo-CRBP1 in the reaction (calculated from a kd of 9 nM, Kane et al., 2011). Increasing the total CRBP1 beyond the concentration of retinal decreased the rate proportionately. These data are consistent with both free retinal or CRBP1-retinal delivering substrate, but also with apo-CRBP1 inhibiting. This indicates apo-CRBP1 as a signal of cellular retinoid status that prioritizes atRA biosynthesis over retinal reduction, when retinoid concentrations are too low to saturate BP.
2.6. CRBP1 conducts retinal into atRA biosynthesis
Microsomes are not efficient at producing atRA from retinal—cytosolic aldehyde dehydrogenases (ALDH) catalyze this reaction. At least 19 genes contribute to the ALDH gene family (Vasiliou et al., 2013). Of these, RALDH1 (ALDH1A1) was the first unequivocally identified to contribute physiologically to atRA biosynthesis (Posch et al., 1992). The activity was identified, distinguished from other cytosolic ALDH (especially the phenobarbital-inducible ALDH, which had been postulated as identical) and purified based on ability to produce atRA from retinal generated in situ by microsomes with CRBP1-retinol as substrate (Fig. 4a). These data are consistent with the observation that retinal generated from CRBP1-retinol occurs bound with CRBP1 (Ottonello, et al., 1993). Recombinant RALDH1 generated Michaelis-Menten kinetics with CRBP1-retinal produced from a 2:1 ratio of total CRBP1/retinal, to minimize the amount of “free” retinoid, albeit with a Vm ~20% of the maximum rate with free retinal (Fig. 4b) (Penzes et al., 1997). Competition between apo-CRBP1 and CRBP1-retinal caused the lower rate, as shown by plotting 1/v vs the amount of apo-CRBP1 at each substrate concentration. This graph allowed calculating the rate as if only CRBP1-retinal were present (theoretical zero apo-CRBP1), which verified that the rate in the absence of apo-CRBP1 was 91% of the rate with free retinal (red point in Fig. 4c). The Km value with CRBP1-retinal was 0.8 μM vs. 1.4 μM with free retinal, indicating the reaction was more efficient (kcat/Km) with the former delivering substrate. The reaction was run at 25°C, which decreases the rate by 8-fold, to avoid excess substrate consumption that occurred at 37°C for the lower substrate concentrations, i.e. to maintain the steady-state assumption, crucial to enzyme kinetics.
Fig. 4.
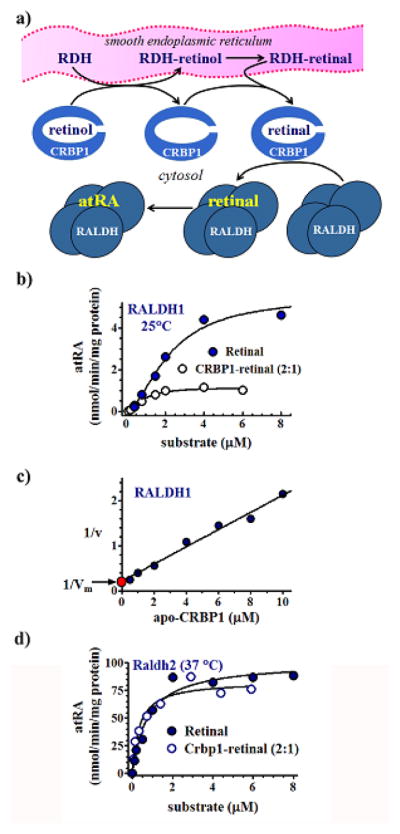
Interactions of RALDH1 and 2 with CRBP1. A) Model depicting ability of cytosolic RALDH to access retinal generated in microsomes by RDH. CRBP1 acts as intermediate delivering retinal to the tetrameric RALDH for conversion into atRA. b) Kinetics of retinal generation from free retinal or CRBP1-bound retinal by RALDH1 (Penzes et al., 1997). A 2/1 ratio of CRBP1/retinal ensured total binding of retinal. The Vm from this ratio was lower than from retinal because apo-CRBP1 inhibits RALDH1. c) A plot allowing calculation of the Vm in the presence of an exact 1/1 ratio of CRBP1/retinal (red point). This approach showed that the Vm with no apo-CRBP1 present was 91% of the value using free retinal, which could occur only with direct interaction between CRBP1 and RALDH1. d) Michaelis-Menten kinetics illustrating channeling between CRBP1 and RALDH2, and lack of inhibition of RALDH2 by apo-CRBP1 (Wang et al., 1996).
A second RALDH, RALDH2 (ALDH1A2), was identified, cloned, expressed in E. coli and characterized (Wang et al., 1996), and subsequently cloned from embryonic cells (Zhao et al., 1996). The purified recombinant enzyme produces atRA from retinal generated in situ by microsomes using CRBP1 to deliver retinol. RALDH2 recognizes CRBP1-retinal with enzymatic efficiency similar to its kinetics with free retinal (Fig. 4d). Apo-CRBP1 does not inhibit RALDH2, as disclosed by equivalent rates with free retinal and a 2:1 ratio of CRBP1/retinal. The lower Km value with CRBP1-retinal (0.2 μM) than the 0.7 μM value with free retinal, suggests a possibility of a more efficient reaction when the BP delivers substrate.
The differences between RALDH1 and RALDH2 with respect to apo-CRBP1 suggest that during reduced vitamin A status, RALDH2 would continue to generate atRA, whereas RALDH1 would have a reduced contribution. Different subcellular locations also suggest different participation in generating atRA. In primary astrocytes, RALDH1 localizes to both cytosol and nuclei, whereas RALDH2 localizes most intensely in the perinuclear regions. These differences suggest that multiple RALDH exist to generate atRA in specific regions of the cell, perhaps to support different vitamin A functions.
RALDH3 (ALDH1A3, aka ALDH6) and RALDH4 (ALDH8A1) complete the complement of known ALDH that catalyze retinoid metabolism (Grün et al., 2000; Lin et al., 2003). RALDH3 has a Km value of 0.3 μM with free all-trans-retinal (Grün et al., 2000) and has the same Vm with CRBP1-retinal as substrate—indicating that it too interacts with CRBP1 (Arnold et al., 2015). RALDH4 has highest activity with 9-cis-retinal and low activity with all-trans-retinal. It has not been tested with CRBP1.
2.7. CRBP1 colocalizes with LRAT, RDH and RALDH
Since its discovery in 1973, CRBP1 has been viewed as a soluble protein, because it localizes in the cytosolic fraction upon differential centrifugation (Bashor et al., 1973). Later work concluded that CRBP1 localizes to lipid droplets (LD), and was mistaken as cytosolic (Kuppumbatti, et al., 2001). Subsequent work also relying on confocal microscopy, however, showed CRBP1 colocalizing precisely with the mitochondria marker MitoTracker, when expressed with a C-terminal GFP conjugate in COS7 cells (Jiang & Napoli, 2012). Incubation of the cells with oleate or retinol to stimulate LD biosynthesis induced CRBP1 to migrate to the surfaces of LD and colocalize with LRAT, which originates in microsomes (Fig. 5). A major RDH, RDH10, also colocalizes with MitoTracker in the absence of LD biosynthesis (Jiang & Napoli, 2013). Like CRBP1 and LRAT, RDH10 migrates to the surfaces of LD during triacylglycerol or RE biosynthesis. Co-localizations of CRBP1, LRAT and RDH10 on the surfaces of LD corroborate the kinetic data indicating BP-enzyme interactions, and place the three proteins at a major source of retinoids for substrate storage and access.
Fig. 5.

LRAT, RDH10 and CRBP1 co-localize around LD during RE biosynthesis. Confocal microscopy of LRAT, CRBP1 and RDH10 with fluorescent reporters (Jiang & Napoli, 2012; Jiang & Napoli, 2013). LRAT localizes around LD containing RE, as does CRBP1. White arrows designate LD. RDH10 also concentrates on LD surfaces. This places LRAT and RDH10 at a major source of substrate for their reactions and also places the enzymes with the substrate delivery vehicle CRBP1. CRBP1 does not associate as tightly with LD as the two enzymes, consistent with its function of conducting retinol from the cell membrane to LD. These data suggest that LD act as organelles of hormone generation.
Additional data consistent with interactions between CRBP1 and other elements of the retinoid metabolon include in situ hybridization, which revealed mRNAs of CRBP1, RDH2 and RDH7 co-localizing in multiple cell types of liver, kidney, lung and testis (Zhai et al., 1997). Also, CRBP1 and DHRS9 proteins express concurrently during estrus in the rat uterine lining (Rexer & Ong, 2002), and co-localize in multiple cells of the rat respiratory and urogenital tracts and pancreas, although there are mismatches, likely due to presence of RDH other than DHRS9 (Everts et al., 2005). The observation that CRBP1 co-localizes with either RALDH2 or 3 supports this conclusion. Co-localization of retinoid metabolon constituents occurs in a complex array during various stages of the hair cycle, consistent with spatial-temporal regulation of atRA biosynthesis (Everts et al, 2007).
2.8. CRBP1 modulates retinoid homeostasis, regulates intermediary metabolism, and suppresses tumorigenesis
Mice with an Rbp1 knockout (encodes CRBP1), when fed a diet containing copious vitamin A (25,000 IU retinyl palmitate/kg), are fertile, grow normally, seem healthy, and present no obvious morphological abnormalities (Ghyselinck et al., 1999). These mice, however, have a metabolic phenotype. Hepatic stellate cells normally store up to 80% of the body’s vitamin A as RE and express CRBP1 and retinoid metabolizing enzymes (Blaner et al., 2009; Senoo et al., 2007). Stellate cells in Rbp1-null mice had fewer and smaller cytoplasmic lipid droplets than WT mice. RE concentrations in knockout mice were ~50% of WT at 4 weeks old, due in part to a 6-fold faster RE elimination rate. The impact of ablating CRBP1 is especially noteworthy considering that dietary vitamin A exceeded the 4000 IU/kg recommended for rodents by the National Research Council (Reeves, 1997). Thus, CRBP1 enhances efficiency of retinol uptake and storage and protects retinol from metabolism by enzymes that cannot access CRBP1-bound retinol (Napoli, 2000).
The Rbp1-null mouse has disrupted retinoid homeostasis in multiple tissues, which impairs function. This includes abnormal pancreas development, illustrated by α-cell infiltration into the islet β-cell field and an 80-fold increase in islet Rbp2 mRNA (encodes CRBP2) with reduced Pdx-1, Glut2, and GK (glucokinase) expression (Kane et al., 2011). The four-fold increase in fasting glucagon in the null mouse produces an abnormally high rate of gluconeogenesis after re-feeding causing hyperglycemia (glucose intolerance). Null mice, moreover, have an increased rate of fatty acid oxidation and resist obesity when fed a high-fat diet. Thus, CRBP1 contributes to post-natal glucose homeostasis and lipid metabolism.
The totality of studies in mammary cell models verifies that retinoid signaling through atRA promotes differentiation and suppress tumorigenesis, which relies on CRBP1-mediated retinol uptake. About 24% of human breast carcinomas express relatively low CRBP1 concentrations, suggesting its modulation of retinoid homeostasis prevents disease (Kuppumbatti et al., 2000). Indeed, CRBP1 expression suppresses foci and colony formation by decreasing cell survival through inhibiting the PI3K/AKT survival pathway (Kuppumbatti et al., 2001). The SV40-immortalized human mammary epithelial cell line MTSV1-7 does not express Rbp1. Retinol uptake relies on transfection with CRBP1 and/or LRAT, which prompts cells to activate RAR, consistent with biosynthesis of atRA (Farias et al., 2005). MTSV1-7 cells transfected with Rbp1 point mutants with lower affinities for retinol (L29A and R58E) or with an LRAT mutant (C161A) that eliminates catalytic activity have impaired RAR activation (Mondal, et al., 2000).
Tumorigenic epithelial cell lines that lack CRBP1 are ~50% as active as non-tumorigenic cells generating atRA from retinol, and Rbp1-null mice have 40% less atRA in mammary tissue compared to WT (Pierzchalski et al., 2013). atRA-depleted mammary tissue suffers epithelial hyperplasia and stromal hypercellularity.
Concurrent loss of CRBP1 expression and decreased atRA biosynthesis also occurs early during onset of human ovarian cancer (Roberts et al., 2002; Williams et al., 2009). CRBP1 loss also may contribute to the onset of nasopharyngel and esophageal squamous cell carcinoma (Kwong et al., 2005; Mizuiri et al., 2005). Hypermethylation of Rbp1 seems to underlie the loss of expression that precedes the onset of human cancers (Esteller et al., 2002).
2.9. CRBP4 provides retinol to LRAT
LRAT accesses CRBP4-retinol to form RE, but kinetic values were not determined, so efficiency relative to CRBP1 has not been determined (Piantedosi et al., 2005). Unlike apo-CRBP1, apo-CRBP4 (encoded by Rbp7) does not inhibit LRAT. The 30% fewer RE in milk from Rbp7-null mice compared to WT illustrate the physiological importance of this association. Rbp7-null mice fed a high-fat diet reduce their food intake by ~25% and experience ~50% less hepatic steatosis, a decreased rate of adipose lipolysis, an 11% decrease in fat mass, increased expression of fatty acid oxidation genes, a slightly increased rate of carbohydrate use (respiratory exchange ratio), and increased cold tolerance. The phenotype does not result from a direct effect on the hypothalamus, because the brain does not express Rbp7. atRA concentrations were not determined, leaving open the question of how CRBP4 impacts retinoid activation.
3. CRABPs perform multiple tasks fundamental to atRA metabolism and function
As indicated in Table 1, retinoic acid BP very much favor the trans forms of RA and its metabolites. For example, 13-cis-retinoic acid (13cRA) binds CRABP1 and CRABP2 with affinities an order of magnitude or more lower than atRA, and has little if any affinity for RAR (Fiorella et al., 1993; Kim et al., 1994). These observations indicate that 13cRA, which converts into atRA in vivo, serves as a slow-release reservoir of atRA (i.e. a prodrug), thereby accounting for its more favorable therapeutic index. This does not preclude 13cRA functioning independently of retinoid BP or RAR, but such remains to be determined.
CRABP1 concentrations exceed atRA where tested. Testes of vitamin A-sufficient rats, for example, express 117 pmol CRABP1/g tissue (Ong et al., 1976), whereas mouse testes have atRA concentrations ~9 pmol/g tissue (Kane et al., 2008a; Kane et al., 2008b). In contrast to CRABP1, CRABP2 concentrations are low outside of select tissues. Unfortunately, comprehensive data relating concentrations of either BP to atRA concentrations in multiple tissues and species have not been generated.
As to sites of atRA biosynthesis, CRABP1 does not correlate strictly with cells that biosynthesize atRA, but CRABP2 correlates with many areas of atRA biosynthesis. Oddly, two tissues of fairly robust atRA biosynthesis, liver and kidney, do not express CRABP2, but express CRABP1. In one of a few detailed studies, CRABP2 co-localized in multiple cells of the rat respiratory and urogenital tract that express atRA biosynthesis enzymes, although there are mismatches (Everts et al., 2005). Detailed studies also have been done of CRABP1 and 2 localization in the rat uterus (Bucco et al., 1997). Prepubertal and proliferative stromal cells and smooth muscle cells express CRABP1 at all stages of the estrus cycle, whereas surface epithelial cells express CRABP2. CRABP2 expression in cultured uterine epithelial cells occurs only during atRA biosynthesis. Uterine cells that express CRABP1 do not biosynthesize atRA. These and other studies have prompted the hypothesis that CRABP2 enhances, whereas CRABP1 limits, atRA function. This seems true to a large extent, but the situation can be more complex.
3.1. CRABPs deliver atRA to catabolic enzymes
Cytochrome P450 (CYP)-catalyzed oxidation of atRA at multiple carbon atoms in the β-ionone ring initiates its catabolism (Fig. 6a). The microsomal CYP26A1, B1 and C1, which are induced by atRA and its catabolites, function as the most dominant oxidases (Ross & Zolfaghari, 2011; Topletz et al., 2012, 2015). Each has unique tissue-specific expression patterns, which often overlap, and each seems to have effects on specific atRA-functions.
Fig. 6.

Functions of atRA BP. a) Structure of atRA designating two of the carbons oxidized by CYP26, C4 and C18. b) Michaelis-Menten kinetics revealing the differences between catabolism of free atRA vs. CRABP1-bound atRA. The reaction with BP was done with a 3/1 ratio of total CRABP1 to atRA to ensure lack of free atRA, yet enhanced the reaction rate at low concentrations of substrate. The reaction with BP-bound atRA followed typical Michaelis-Menten kinetics, whereas that of unbound atRA seemed unsaturable at concentrations that exceed the concentrations of atRA in tissues. The table shows the effects of binding atRA and two of its metabolites on their rates of catabolism, suggesting high concentrations of catabolites bound to CRABP contribute to atRA toxicity by blocking further catabolism of atRA (Fiorella & Napoli, 1991, 1994). c) Rate of 4-OH-atRA biosynthesis from free atRA or albumin bound atRA showing that not all protein-bound atRA behaves similar to CRABP-bound atRA. Michaelis-Menten kinetics illustrating protein-protein interactions between recombinant CYP26B1 and CRABP1 and 2. Impact of CRABP1 and 2 on the ability of recombinant CYP26B1 to catalyze atRA catabolism, indicating inhibition of CRP26B1 by the two atRA BP (Nelson et al., 2016). d) Effect of increasing the RAR concentration on the rate of transfer of atRA from BP. The decrease in the transfer rate indicates that only CRABP2 directly interacts with RAR (Dong et al., 1999). e) Summary of CRABP and FABP5 functions.
Holo-CRABP1 delivers substrate to microsome-catalyzed atRA catabolism in multiple tissues through direct interactions with enzymes (Fiorella & Napoli, 1991, 1994). Several observations support this conclusion, including: 1) high affinity of CRABP1 for atRA, which reduces free atRA to negligible amounts; 2) a Michaelis-Menten relationship between BP-bound atRA and catabolic enzymes; 3) a lower Km value for CRABP1-atRA (1.8 nM) compared to free atRA (49 nM) (Fig. 6b); 4) a 7-fold higher reaction efficiency (Vm/Km) for CRABP1-atRA than with free atRA; 5) a marginal decrease in the reaction rate (~35%) in the presence of a 3-fold excess of total CRABP1 over atRA, which could not occur if diffusion were required from the BP; 6) an elimination t1/2 for atRA that does not differ between free and BP bound atRA. These data indicate no need for catabolic enzymes to rely only on atRA that diffuses from CRABP1.
CRABP1 binding has tissue-specific effects on catabolite compositions through binding major catabolites, such as 4-OH-RA, 4-oxo-RA, and 18-OH-RA. Rates of 4-OH-RA and 4-oxo-RA catabolism decrease 2- and 70-fold, respectively, when CRABP1 bound, despite affinities close to atRA, suggesting decreased ability of enzymes to tease them from the BP (Fiorella et al., 1993). The latter data suggest that 4-oxo-RA and 4-OH-RA contribute to atRA toxicity. Normally, 4-oxo-RA and 4-OH-RA tissue concentrations are very low relative to atRA. They increase markedly during atRA excess and have high affinities for RAR. In addition, they may inhibit atRA catabolism through their BP complexes blocking CYP.
The aforementioned work used microsomes as source of CYP, because specific CYP that catalyze atRA metabolism had not been identified. Studies of interactions between CRABP1 and CRABP2 with several recombinant CYP, which recognize free atRA as substrate, revealed that only CYP2B1 recognizes BP-bound atRA: CYP3A4 and CYP2C8 do not (Nelson et al., 2016). CYP26A1 and C1 have not been tested. Binding atRA with albumin greatly arrests catabolism, in contrast to CRABP binding (Fig. 6c). Isotope dilution studies that evaluated competition between unlabeled atRA vs deuterium labeled atRA, with either one bound to BP and the other “free”, corroborate BP-enzyme interactions. Km values were lower for holo-CRABP1 (~22 nM) and holo-CRABP2 (~24 nM) than for free atRA (~65 nM). Overall reaction efficiencies (kcat/Km) were somewhat higher with holo-CRABP1 and 2 (relative values of 8 and 12 respectively vs 7 for free atRA). These data are consistent with the earlier results. This study, however, found that both apo-BPs compete with holo-CRABP. With CYP26B1, both apo-BP titrated in excess of atRA had a substantial impact on rates of catabolism (Fig. 6c, third panel). The differences may reflect examining multiple CYP in their native environment (microsomes) vs a specific recombinant CYP in a reconstituted environment, or may reflect differences among CYP26 isomers. Nevertheless, the cumulative data indicate that both CRABP1 and CRABP2 deliver atRA to CYP for catabolism via BP-enzyme interactions.
In cells that express CRABP, non-CRABP-recognizing CYP probably would not have access to atRA. This suggests a mechanism for discriminating metabolically between endogenous atRA and pharmacologically-dosed retinoids that exceed CRABP capacity or have low affinity for CRABP.
The principle that CRABP1 delivers atRA to catabolism has been verified in several tumorigenic cell lines. Transfection of CRABP1 into tumorigenic cells lines that either do not express the BP or to enhance expression confers or increases resistance to the anti-proliferative effects of atRA. These cell lines include the mouse teratocarcinoma F9, the human head and neck squamous cell carcinoma line AMC-HN-7, and the human renal cell carcinoma line A-498 (Boylan & Gudas, 1991; Won et al., 2004; Pfoertner et al., 2005). CRABP1, however, may have more complex, tumor specific effects (see below).
3.2. The CRABP1-atRA complex regulates the cell cycle
Upon atRA binding, CRABP1 activates ERK1/2 independently of RAR (Persaud, et al., 2013). Activated ERK1/2 translocates to the nucleus and induces p27 phosphorylation and stabilization, thereby inhibiting G1-S progression in an embryonic stem cell model. Holo-CRABP1 also activates protein phosphatase 2A, which similarly delays the cell cycle and sensitizes the CRABP1-positive breast cancer cell line MCF7 to apoptosis. This does not occur in CRABP1-null A2780 (ovarian) and KPC (pancreatic duct) cancer cell lines (Persaud et al., 2016).
3.3. CRABP2 delivers atRA to nuclear RAR
CRABP1 and 2 have been detected in both cytosol and nucleus in several cell lines and mouse tissues, but the function(s) of nuclear CRABP1 remain(s) unclear (Takase et al., 1986; Venepally et al., 1996; Gustafson et al., 1996; Gaub et al., 1998). Diffusion controls transfer of atRA from CRABP1 to atRA nuclear receptors (RAR), whereas transfer from CRABP2 occurs via protein-protein interaction between the BP and RAR, as revealed by kinetics (Dong et al., 1999). In a diffusion controlled process, the rate of ligand transfer from a BP will not increase with increases in acceptor concentration, whereas ligand transfer through protein-protein interactions will accelerate with increases in acceptor concentration, by increasing the collision rate between donor and acceptor. Kinetic differences between transfer from CRABP1 or 2 to RAR illustrate this phenomenon (Fig. 6d). Direct interaction of CRABP2 with RAR denotes evolution of a specific process, which implies physiological function. In support, expression of CRABP1 does not increase the transcription rate from atRA response elements that drive reporters in COS-7 or CV-1 cells, but CRABP2 expression does (Venepally et al., 1996; Dong et al., 1999). In further support of CRABP2 delivery to RAR modifying atRA action, the level of CRABP2 in the MCF-7 mammary carcinoma cell line correlates directly with sensitivity to atRA-induced growth inhibition (Budhu & Noy, 2002).
3.4. A CRABP2 Interaction with HuR promotes mRNA stabilization and controls cytosolic/nuclear cycling
The mRNA BP HuR shuttles between the cytosolic face of the endoplasmic reticulum and the nucleus. When in cytosol, HuR attaches to AU-rich sections in 3′-untranslated regions of mRNAs involved in cell proliferation, apoptosis and differentiation. HuR binding protects transcripts from degradation and regulates translation (Meisner & Filipowicz, 2011; Grammatikakis et al., 2016). HuR binding with apo-CRABP2 enhances its affinity for select mRNAs >3 orders of magnitude, i.e. lowers kd values from ~400 nM to <0.1 nM (Vreeland et al., 2014; Zhang et al., 2016). atRA binding causes CRABP2 to change its conformation, releasing it from HuR and forming a nuclear localization signal, not present in apo-CRABP2 (Sessler & Noy, 2005). This conformational change also results in SUMOylation at K102, an event critical for nuclear transfer (Majumdar et al., 2011). Transfer of CRABP2 to the nucleus concludes within 30 min. Once CRABP2 delivers atRA to nuclear RAR, it returns to cytosol. Short residence time in the nucleus explains how CRABP2 shuttling has only a minor effect on its ability to stabilize mRNA. CRABP2 entrance into the nucleus requires multiple components of the nuclear import apparatus regulated by HuR. Thus, the HuR-CRABP2 interaction augments functions of both proteins.
4. The FABP5/PPARδ/β partnership enables atRA to stimulate proliferation and survival
Three intracellular lipid BPs, CRABP2, FABP4 and FABP5, partner selectively with the nuclear receptors, RARα, PPARγ, and PPARδ/β, respectively (Tan et al., 2002). These partnerships result in each BP transferring a select few of their ligands from cytosol to their cognate nuclear receptors. This implies that binding select ligands imparts conformational changes to BP that expose nuclear localization signals or prompt post-translational modifications to induce nuclear uptake. This BP “bridge” would instill selectivity by controlling ligand access to receptors, which in turn controls idiosyncratic gene expression. BP expression, therefore, contributes to regulating cell-specific gene expression.
4.1. FABP5 transfers atRA to the nucleus to transactivate PPARδ/β target genes
atRA and the PPARδ/β agonist GW0742 bind FABP5 with kd values ~42 and ~35 nM, respectively, i.e. with affinities similar to pan-PPAR ligand parinaric acid (~42 nM) and the synthetic PPAR ligand L165041 (~46 nM) (Schug et al., 2007a). The former two bind FABP5 selectively, whereas the latter two bind FABP4 and 5 with similar affinities. Although atRA has much lower affinity for FABP5 than for CRABP1 or 2, atRA and GW0742 induce transfer of FABP5 from cytosol to the nuclei of COS7, NaF and HaCaT (keratinocytes) cells, in contrast to parinaric acid, L165041, and another pan-PPAR ligand, the saturated long-chain fatty acid (LCFA) stearate.
FABP5-mediated transfer to the nucleus delivers atRA specifically to PPARδ/β and activates target genes, but does not activate PPARα or PPARγ-responsive genes (Shaw et al., 2003b; Schug et al., 2007a). Binding affinities of atRA (~0.1–3 nM) for the three RAR isoforms exceed affinity for PPARδ/β (17 nM) by 3-orders of magnitude (Allegretto et al., 1993; Allenby et al., 1993). Likely, the kd values of atRA for both FABP5 and PPARδ/β have been underestimated because of technical limitations of determining kd values by fluorescence titration. Nevertheless, the much higher affinity of atRA for the CRABP2/RAR pair vs the FABP5/PPARδ/β pair indicates that the former pair will prevail over the latter in most cells, and the later pair will effect atRA function only in cells that have an especially low CRABP2/FABP5 ratio. This has been demonstrated to occur in the breast cancer cell lines MCF-7 and NaF, and in the keratinocyte cell line HaCaT. Actions of CRABP1 and 2 are summarized in Fig. 6e.
4.2. Physiological consequences of atRA activating PPARδ/β vs RAR
atRA has cell-specific functions. In some cells, atRA inhibits cell proliferation and induces differentiation or apoptosis; in other cell types atRA promotes proliferation and enhances cell survival. These dueling effects have been attributed to the intracellular ratio CRABP2/FABP5, because CRABP2 delivers atRA to RAR, whereas FABP5 delivers atRA to PPARδ/β (Schug et al., 2007a). RARs mediate atRA inhibition of proliferation, but PPARδ/β mediates atRA stimulation of proliferation. The sensitivity of breast cancer cell lines to atRA growth inhibition seems directly proportional to the degree of RAR expression, especially RARα (Centritto et al., 2015; Carrier et al., 2016). It follows that breast cancer cell sensitivity should depend on the CRABP2/FABP5 ratio. To test this premise, CRABP2 concentrations were manipulated in a model of atRA-resistant breast cancer, the MMTV-her2/neu transgenic mouse (Tari et al., 2002), by overexpressing CRABP2 or cross breeding these mice with CRABP2-null mice (Schug et al., 2008b). The resulting strains did not express RAR, PPARδ/β, or FABP5 differently, and had either no or enhanced CRABP2 expression. In tumors of the unmodified MMTV model, which have a relatively low CRABP2/FABP5 ratio, atRA upregulated PPARδ/β-target genes. In mice lacking CRABP2 totally, atRA up-regulated PPARδ/β-target genes to a greater extent. In high CRABP2/FABP5 ratio mice, atRA up-regulated some RAR-target genes to a much greater extent than in the unmodified mice, decreased tumor growth, increased apoptosis and increased differentiation. Ablation of FABP5 has effects on mammary tumor growth similar to over-expression of CRABP2 (Levi et al., 2013). FABP5-null mice cross-bred with MMTV-ErbB2/HER2 oncomice have decreased PPARδ-target gene expression and mammary tumor development.
To demonstrate the importance in vivo of PPARδ/β activation by atRA, mice with a global reporter driven by a RAR response element were dosed with atRA, and mice with a global reporter activated by PPARδ/β were dosed with atRA or the PPARδ/β-specific ligand GW1516. In the former, atRA (1 mg) activated RAR response elements. In the latter, atRA (4 mg or ~140 μg/kg) activated PPAR response elements similar to GW1616. Therefore, exogenously-dosed atRA affects both response elements, and does so at reasonably low pharmacological doses, but in a dose-dependent manner (Levi et al., 2015). This suggests exercising caution in choosing pharmacological doses of atRA.
Ligand interactions with FABP5 have complex consequences. Both saturated and unsaturated LCFA bind FABP5 with kd values about twice the affinity of atRA, but do not prompt the same outcome (Levi et al., 2015). Saturated LCFA do not induce FABP5 to migrate to the nucleus and do not activate PPARδ/β. Competition between saturated LCFA, which occur in higher concentrations than atRA, displace atRA from FABP5. This increases the amount of atRA available to bind with RAR. Both actions of saturated LCFA promote the anti-cancer actions of atRA. Unsaturated LCFA induce FABP5 migration to the nucleus to activate PPARδ/β. This still increases the amount of atRA available to activate RAR, which competes with activation of PPARδ/β, but the outcome would not occur to the same degree of clarity as with saturated LCFA. These data suggest that saturated LCFA may function as anti-cancer agents.
5. Impact of CRABP1, CRABP2 and FABP5 on human cancer in vivo
Teasing out the relationships between CRABP1, CRABP2 and FABP5 in human cancer samples vs. non-tumorigenic tissues remains in its infancy. Although the preponderance of data from primary tumors show that CRABP1 and FABP5 correlate with deleterious cancer outcomes, and CRABP2 has an ameliorating effect, contrary observations have been reported for Wilms tumors (nephroblastomas) (Takahashi et al., 2002). In Wilms tumors, CRABP2 expresses 11-fold more intensely than in non-cancerous adult kidney tissue, and has especially high expression in tumors associated with poor prognoses. It is not clear whether this reflects the tumor type or confounding factors.
5.1. Prostate cancer
The transcription repressor capicua impedes prostate cancer progression by suppressing proliferation, invasion, and migration of tumorigenic cells. Human prostate intra-epithelial tumors express relatively low capicua, and advanced prostate adenocarcinomas do not express capicua (Choi et al., 2015). Loss of capicua increases expression of its target gene CRABP1. CRABP1 increases proliferation and invasiveness of PC-3 cells, an aggressive grade IV human prostate adenocarcinoma cell line. Because PC-3 cells respond to retinoids (Hammond et al., 2001), the deleterious action of CRABP1 may reflect sequestration of atRA followed by delivery to catabolism. This is the opposite of shifting the atRA dose-response curve to the left (lower IC50 value) by inhibiting catabolism (Williams & Napoli, 1985). In contrast, carcinoma cells express CRABP2 mRNA and protein less intensely relative to non-tumor glandular cells (Okuducu et al., 2005), and CRABP2 cytobands co-localize with loci of prostate cancer predisposition (Thompson et al., 2008).
5.2. Breast cancer
Increased cytoplasmic CRABP1 expression levels correlate with tumor virulence and lower patient survival in multiple classes of human breast cancers, but higher nuclear CRABP2 levels associate with improved prognoses (Liu et al., 2015). These actions were attributed to CRABP1 sequestering atRA in cytosol, inhibiting its nuclear action, and ability of CRABP2 to activate RAR. In these cells, knocking down CRABP1 up-regulated CYP26A1 at high atRA concentrations, prompting the argument that this negative association shows that CRABP1 does not contribute to catabolism. In the absence of CRABP1 and high doses of atRA, however, the efficiency of atRA catabolism would decrease, shunting atRA to CRABP2, which activates RAR to induce CYP, as well as atRA-induced anti-cancer genes. Thus, negative association of CRABP1 with tumor prognosis in the presence of high atRA doses likely reflects increased ligand available for CRABP2 and activation of RAR target genes, such as CYP. Non-genomic actions of cytosolic CRABP1 also must be considered.
5.3. Other cancers
A recent study concluded that reduced CRABP1 correlates with serious cases of human ovarian adenocarcinoma and denotes poorer overall survival (Miyake et al., 2011). This study, however, did not evaluate atRA effects on the tumors or catabolic rates.
The median ratio FABP5/CRABP2 mRNA expression of 3.6 in short-term survivors (≤ 6 months) with glioblastomas significantly surpassed the ratio of 1.4 in long-term survivors (≥ 36 months) (Barbus et al., 2011). Differences in ratios were contributed solely by increased FABP5 mRNA. Consistent with the functions credited to FABP5 and CRABP2, RAR target genes were expressed to a much lesser extent in the short-term survivors, compared to the long-term survivors, and PPARδ/β-target genes were expressed robustly in tumors from short-term survivors. A second study observed no decrease in CRABP2 mRNA in 282 astrocytic gliomas, but found decreased protein expression that correlated with tumor malignancy and shorter patient survival (Campos et al., 2011).
5.4. Clinical uses of retinoids for chemoprevention or cancer treatment
A review of the vast literature that summarizes successes and failures of retinoid therapy for cancer chemoprevention and treatment lies well beyond the scope of this review. Briefly, notable successes have been achieved with use of retinoids to treat skin cancers and other skin maladies, neuroblastomas, breast cancer, and some leukemias, and in select instances of chemoprevention. Retinoids, however, have not realized the wide-ranging potential in clinical trials that was anticipated from their mechanisms of action regarding cell proliferation and differentiation (Tang and Gudas, 2011; Uray et al., 2016). Multiple factors have stymied systemic use of retinoids, including toxicity, differential effects on different cell types, pharmacology (absorption, distribution, metabolism and excretion) and epi-genetic changes induced onto retinoid signaling apparatus, such as silencing CRBP1 and the tumor suppressor RARβ. Despite not providing a panacea, retinoids continue to undergo basic and clinical research that ventures beyond the initial straightforward assumptions about their promise. Emerging data about BP, for example, recurrently provide new insight that should direct therapy, or at least aid diagnosis, and new retinoids are consistently evaluated to improve therapeutic utility, as well use as a component of combination therapy. Alternative approaches to increasing intracellular atRA by inhibiting catabolism also undergo consideration to treat retinoid-related diseases (Stevison et al., 2015).
6. Structures of retinoid binding-proteins
Members of the lipid BP gene family fold into similar three dimensional structures despite low primary amino acid sequence similarities (Newcomer, 1995; Li & Norris, 1996). CRBP1 (135 residues, 15.9 kDa) and CRBP2 (134 residues, 15.7 kDa), however, share ~56% primary amino acid identify. CRABP1 and 2 share ~78% identity. Different species share high amino acid identities for each retinoid BP. Between human and mouse, as an example, CRABP1 differs in only one amino acid residue (15.6 kDa) and CRABP2 differs in 9 of 138 residues (15.7 kDa). FABP5 (135 residues, 15.1 kDa) does not have the same degree of conservation between human and mouse, differing by 23 residues, but with more than half conservative substitutions.
6.1. CRBP1 structure
Among the retinoid BP, the structure of CRBP1 has been investigated most thoroughly by X-ray crystallography, NMR, and proteolytic digestion followed by mass spectrometric analysis of the fragments. X-ray crystallography revealed that the residues of CRBP1 arrange into a flattened β-clam formed by two orthogonal sheets, each consisting of five anti-parallel β-strands (Cowan et al., 1993). Loops connect the strands, except for strands βA and βB, which a helix-loop-helix connects. Retinol occupies the binding-cavity in a planar conformation, hydroxyl group deep within, isolated totally from the cellular milieu (Fig. 7a–d). The N-terminus blocks the end of the ligand-binding pocket. Eight water molecules in the binding cavity surround the isoprene side-chain. The helix-loop-helix covers an entrance portal along with turns βC/βD and βE/βF. The β-ionone ring occurs in a hydrophobic environment formed by close contacts with residues around the entrance portal. Side-chains L29, I32, and L36 project from the αII helix inward. L29 associates with the β-ionone ring and with residues F57 and R58. I32, also in αII helix, associates with F57. L36 associates with the β-ionone ring and F57. The side-chains of F57 and R58, which occur in the turn between βC and βD, or in βD, respectively, project inward to associate with each other, the β-ionone ring, and the aforementioned residues. Residue I77 projects inward from the turn between βE and βF to associate with the isoprene side chain. These associations stabilize and impart rigidity to holo-CRBP1.
Fig. 7.

Structure-activity relationships of retinoid BP. a) Ribbon diagram of CRBP1 illustrating the entrance portal, key residues, orientation of retinol in the binding pocket, and the structure of the β-clam (Cowan et al., 1993; Franzoni et al., 2002; Lu et al., 2003; Careri et al., 2006; Mittag et al., 2006; Franzoni et al., 2010; 2016 Silvaroli et al., 2016). Red shows the hydroxyl group of retinol. Structure PDB ID 5HBS, NCBI. b) Partial structure focusing on interactions of entrance portal residues with each other and with the β-ionone ring. Red depicts the retinol hydroxyl group. c) Partial space-filling structure illustrating close contacts of residues in the entrance portal. Red illustrates the β-ionone ring. d) Total space-filling structure of CRBP1 showing sequestration of retinol from the cellular milieu, and residues that project from the αII helix out from the BP, which serve as putative “handles” that allow enzymes to tease retinol from the BP. Red indicates the β-ionone ring. e) Differences in proteolysis rates between apo- and holo-BP. BP were exposed to the endopeptidase Arg-C (clostripain). Degrees of proteolysis were analyzed by SDS-PAGE: scale: 0, no proteolysis; 1, low (<50%); 2, moderate (>50%); 3, complete. These data were obtained after 1 hour incubation, but holo-forms remained protease resistant up to 20 hours (Jamison et al., 1994). f) Effects of point mutations on ability of CRBP1 to deliver retinol to RDH, ligand affinity and rigidity (proteolysis rate) (Penzes and Napoli, 1999). g) Structure of CRABP1, which folds similarly to CRABP2, showing the carboxyl group (red) of atRA and the location of the three residues that enable RAR binding in CRABP2. Structure PDB ID 1CBR, NCBI.
The hydrophilic environment of the cavity would exert pressure on the isoprene ring to push the β-ionone ring against the cap (Jamison et al., 1994). This pressure would oppose the stability of the cap. Ligand egress would be prompted by external forces, such as enzymes interacting with external residues. Significant differences between apo- and holo-BP supported these notions, revealed by a difference in chromatographic mobility, and further by limited proteolysis (Herr & Ong, 1992b; Jamison et al., 1994). One hour treatment with the endopeptidase Arg-C (clostripain) cleaved each apo-BP at a conserved R residue (R30 in CRBP1) in helix αII (Fig. 7e). Notably, this area includes the residues in the αII helix that contact the β-ionone ring and each other. The data in Fig. 7e show results of 1 hr incubation with Arg-C, but holo-BP resisted proteolysis at least 20 hours. These data confirm biochemically the less ordered structure of apo-BP and the flexible nature of the entrance portal. Single mutants of portal residues decreased the rigidity of the holo-BP, as evaluated by proteolysis rates and decreased ligand affinities (Penzes and Napoli, 1999). No consistent relationship occurred between ability of mutant holo-BP to deliver substrates for retinal biosynthesis (Vm values) and rigidity or ligand affinities, dissociating these internal residues from consistently affecting the substrate delivering properties of CRBP1 (Fig. 7f).
High conservation of external residues among species of individual retinoid BP, despite no requirement for such conservation to allow family members to fold in the same secondary and tertiary structures, implies function for exterior residues other than protein shape. This was confirmed by site-directed mutagenesis (Penzes & Napoli, 1999). Residues V27, K31, L35 and K37 also occur in helix αII, but point outward into the cellular milieu (Fig. 7d). Mutation of L35 decreased ability of CRBP1 to deliver retinol to RDH by 50% (Vm), but did not affect ligand binding affinity and affected rigidity modestly. Contrast this with L36, the inward projecting neighbor, which associates with the β-ionone ring and F57, and had 2-fold increased efficiency as substrate, accompanied by a 5-fold decrease in ligand affinity and 4.5-fold decrease in rigidity. These data suggest that L36 enhances ligand binding and holo-BP stability, and L35 provides a “handle” for enzyme interaction, but does not affect ligand binding.
Data generated since the first X-ray crystallography and the proteolysis and mutagenesis experiments support the results and deductions of these early studies, concerning differences in structure between apo and holo CRBP1, a conformation change upon ligand binding, the contribution of the helix-turn-helix and other portal residues to the conformation change and/or to ligand binding, and ligand egress relying on “external” partners, rather than by diffusion (Li et al., 1991; Franzoni et al., 2002; Lu et al., 2003; Careri et al., 2006; Mittag et al., 2006; Franzoni et al., 2010; Silvaroli et al., 2016). These studies have refined and added additional detailed insight into the nature of the apo- and holo-CRBP1 structures. Kinetics and cross-linking, discussed above, have identified external partners as the intuitive candidates: LRAT, RDH, and RALDH.
6.2. CRBP2 structure
The CRBP2 structure resembles closely that of CRBP1, but is not identical. Multidimensional NMR indicated that major differences between the structures localize to the αII helix, and likely account for differences in ligand binding and interactions with enzymes (Lu et al., 2003).
6.3. CRABP structures
The crystal structures of CRABP1 (Fig. 7g) and CRABP2 show a more open ligand access portal, with the β-ionone ring readily accessible to interacting enzymes that derivatize on the 4 and 18-carbons (Kleywegt et al., 1994; Thompson et al., 1995). The precise residues essential to ligand binding have not been verified through site-directed mutagenesis, but the RAR interaction domain has been identified. Three spatially close residues in the entrance of the portal have been identified as necessary and sufficient for the difference between CRABP1 and 2 with respect to RAR binding (Budhu et al., 2001). In CRABP2, these include Q75, P81 and K102. In CRABP1, E, K and E, respectively, provide the similarly situated residues. Mutating CRABP1 and 2 to effect an exchange of residues, enables CRABP1 binding of RAR and prevents CRABP2 from binding RAR. Other than this, the only other information available notes the absolute need for R132 to enable ligand binding (Chen et al., 1995).
7. Summary
Cellular retinoid BPs contribute to cell uptake of retinoids (CRBP1 & 2), protect retinoids from non-specific interactions with enzymes, while delivering retinoids for metabolism by enzymes capable of teasing ligands from the BP (CRBP1 & 2, CRABP1 & 2), regulate intracellular retinol flux (apo-CRBP1), deliver atRA to nuclear receptors (CRABP2, FABP5), and contribute non-canonical functions in mediating atRA action (CRABP1 & 2). These actions foster efficient use of retinoids, scarce throughout evolution. Low nM binding affinities and the nature of ligand sequestration require “external factors” to prompt release of BP ligands. These external factors include RDH, RALDH, CYP26, LRAT, RAR and PPARβ/δ. Although ablation of BP does not cause extensive impairment of morphological developmental, loss of CRBP1 results in aberrant pancreas development, abnormal glucose and energy metabolism, and a predisposition to breast cancer. Loss of CRBP2 impairs retinol absorption during low or normal vitamin A dietary intake, resulting in pup death during low intake, but is tolerated when diets contain copious vitamin A. Variations in CRABP1 & 2 and FABP5 expression affect severity of multiple human cancers. Despite advances in knowledge concerning retinoid BP functions in the last 40 years, much still remains to learn about enzymatic and other protein partners, and the physiological consequences of diminished BP expression. Although cancer has been a focus of diseases related to aberrant BP expression, given the functions of retinol through its major metabolite atRA, it is reasonable to expect that retinoid BP will affect the immune system and the nervous system, to cite just a few atRA dependent processes.
Abbreviations
- 1 RA
13-cis-retinoic acid
- atRA
all-trans-retinoic acid
- BP
binding-protein(s)
- CRABP
cellular retinoic acid BP
- CRBP
cellular retinol BP
- CYP
cytochrome P-450
- FABP
fatty acid BP
- LCFA
long-chain fatty acids
- LD
lipid droplet(s)
- LRAT
lecithin:retinol acyltransferase
- PPAR
peroxisome proliferator activated receptor
- RAR
retinoic acid receptor(s)
- RALDH
retinal dehydrogenase(s)
- RDH
retinol dehydrogenase(s)
- RE
retinyl ester(s)
Footnotes
Conflict of Interest
The author declares that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhtar S, Ahmed A, Randhawa MA, Atukorala S, Arlappa N, Ismail T, Ali Z. Prevalence of vitamin A deficiency in South Asia: causes, outcomes, and possible remedies. Journal of Health, Population, and Nutrition. 2013;31(4):413–423. doi: 10.3329/jhpn.v31i4.19975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretto EA, McClurg MR, Lazarchik SB, Clemm DL, Kerner SA, Elgort MG, … Heyman RA. Transactivation properties of retinoic acid and retinoid X receptors in mammalian cells and yeast. Correlation with hormone binding and effects of metabolism. The Journal of Biological Chemistry. 1993;268(35):26625–26633. [PubMed] [Google Scholar]
- Allenby G, Bocquel MT, Saunders M, Kazmer S, Speck J, Rosenberger M, … Chambon P. Retinoic acid receptors and retinoid X receptors: interactions with endogenous retinoic acids. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(1):30–34. doi: 10.1073/pnas.90.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Kent T, Hogarth CA, Schlatt S, Prasad B, Haenisch M, Walsh T, Muller CH, Griswold MD, Amory JK, Isoherranen N. Importance of ALDH1A enzymes in determining human testicular retinoic acid concentrations. The Journal of Lipid Research. 2015;56:342–357. doi: 10.1194/jlr.M054718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbus S, Tews B, Karra D, Hahn M, Radlwimmer B, Delhomme N, … Lichter P. Differential retinoic acid signaling in tumors of long- and short-term glioblastoma survivors. Journal of the National Cancer Institute. 2011;103(7):598–606. doi: 10.1093/jnci/djr036. [DOI] [PubMed] [Google Scholar]
- Bashor MM, Toft DO, Chytil F. In vitro binding of retinol to rat-tissue components. Proceedings of the National Academy of Sciences of the United States of America. 1973;70(12):3483–3487. doi: 10.1073/pnas.70.12.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass NM. Cellular binding proteins for fatty acids and retinoids: similar or specialized functions? Molecular and Cellular Biochemistry. 1993;123(1–2):191–202. doi: 10.1007/BF01076492. [DOI] [PubMed] [Google Scholar]
- Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, … Palczewski K. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. The Journal of Biological Chemistry. 2004;279(11):10422. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesalski HK. Comparative assessment of the toxicology of vitamin A and retinoids in man. Toxicology. 1989;57(2):117–161. doi: 10.1016/0300-483x(89)90161-3. [DOI] [PubMed] [Google Scholar]
- Blaner WS, O’Byrne SM, Wongsiriroj N, Kluwe J, D’Ambrosio DM, Jiang H, … Libien J. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochimica Et Biophysica Acta. 2009;1791(6):467–473. doi: 10.1016/j.bbalip.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerman MH, Napoli JL. Cholate-independent retinyl ester hydrolysis. Stimulation by Apo-cellular retinol-binding protein. The Journal of Biological Chemistry. 1991;266(33):22273–22278. [PubMed] [Google Scholar]
- Boerman MH, Napoli JL. Characterization of a microsomal retinol dehydrogenase: a short-chain alcohol dehydrogenase with integral and peripheral membrane forms that interacts with holo-CRBP (type I) Biochemistry. 1995;34(21):7027–7037. doi: 10.1021/bi00021a014. [DOI] [PubMed] [Google Scholar]
- Boerman MH, Napoli JL. Cellular retinol-binding protein-supported retinoic acid synthesis. Relative roles of microsomes and cytosol. The Journal of Biological Chemistry. 1996;271(10):5610–5616. doi: 10.1074/jbc.271.10.5610. [DOI] [PubMed] [Google Scholar]
- Boylan JF, Gudas LJ. Overexpression of the cellular retinoic acid binding protein-I (CRABP-I) results in a reduction in differentiation-specific gene expression in F9 teratocarcinoma cells. The Journal of Cell Biology. 1991;112(5):965–979. doi: 10.1083/jcb.112.5.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CC, Noelle RJ. Seeing through the dark: New insights into the immune regulatory functions of vitamin A. European Journal of Immunology. 2015;45(5):1287–1295. doi: 10.1002/eji.201344398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucco RA, Zheng WL, Davis JT, Sierra-Rivera E, Osteen KG, Chaudhary AK, Ong DE. Cellular retinoic acid-binding protein(II) presence in rat uterine epithelial cells correlates with their synthesis of retinoic acid. Biochemistry. 1997;36(13) doi: 10.1021/bi962094o. [DOI] [PubMed] [Google Scholar]
- Budhu A, Gillilan R, Noy N. Localization of the RAR interaction domain of cellular retinoic acid binding protein-II. Journal of Molecular Biology. 2001;305(4):939–949. doi: 10.1006/jmbi.2000.4340. [DOI] [PubMed] [Google Scholar]
- Budhu AS, Noy N. Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Molecular and Cellular Biology. 2002;22(8):2632–2641. doi: 10.1128/MCB.22.8.2632-2641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos B, Centner FS, Bermejo JL, Ali R, Dorsch K, Wan F, … Herold-Mende C. Aberrant expression of retinoic acid signaling molecules influences patient survival in astrocytic gliomas. The American Journal of Pathology. 2011;178(5):1953–1964. doi: 10.1016/j.ajpath.2011.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careri M, Elviri L, Mangia A, Zagnoni I, Torta F, Cavazzini D, Rossi GL. Mass spectrometry techniques for detection of ligand-dependent changes in the conformational flexibility of cellular retinol-binding protein type I localized by hydrogen/deuterium exchange. Rapid Communications in Mass Spectrometry: RCM. 2006;20(13):1973–1980. doi: 10.1002/rcm.2547. [DOI] [PubMed] [Google Scholar]
- Carrier M, Joint M, Lutzing R, Page A, Rochette-Egly C. Phosphoproteome and Transcriptome of RA-Responsive and RA-Resistant Breast Cancer Cell Lines. PloS One. 2016;11(6):e0157290. doi: 10.1371/journal.pone.0157290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centritto F, Paroni G, Bolis M, Garattini SK, Kurosaki M, Barzago MM, … Garattini E. Cellular and molecular determinants of all-trans retinoic acid sensitivity in breast cancer: Luminal phenotype and RARα expression. EMBO Molecular Medicine. 2015;7(7):950–972. doi: 10.15252/emmm.201404670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai X, Boerman MH, Zhai Y, Napoli JL. Cloning of a cDNA for liver microsomal retinol dehydrogenase. A tissue-specific, short-chain alcohol dehydrogenase. The Journal of Biological Chemistry. 1995a;270(8):3900–3904. doi: 10.1074/jbc.270.8.3900. [DOI] [PubMed] [Google Scholar]
- Chai X, Zhai Y, Popescu G, Napoli JL. Cloning of a cDNA for a second retinol dehydrogenase type II. Expression of its mRNA relative to type I. The Journal of Biological Chemistry. 1995b;270(47):28408–28412. doi: 10.1074/jbc.270.47.28408. [DOI] [PubMed] [Google Scholar]
- Chen LX, Zhang ZP, Scafonas A, Cavalli RC, Gabriel JL, Soprano KJ, Soprano DR. Arginine 132 of cellular retinoic acid-binding protein (type II) is important for binding of retinoic acid. The Journal of Biological Chemistry. 1995;270(9):4518–4525. doi: 10.1074/jbc.270.9.4518. [DOI] [PubMed] [Google Scholar]
- Chen W, Chen G. The Roles of Vitamin A in the Regulation of Carbohydrate, Lipid, and Protein Metabolism. Journal of Clinical Medicine. 2014;3(2):453–479. doi: 10.3390/jcm3020453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi N, Park J, Lee JS, Yoe J, Park GY, Kim E, … Lee Y. miR-93/miR-106b/miR-375-CIC-CRABP1: a novel regulatory axis in prostate cancer progression. Oncotarget. 2015;6(27):23533–23547. doi: 10.18632/oncotarget.4372. https://doi.org/10.18632/oncotarget.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MD, Mao GE. Teratology of retinoids. Annual Review of Pharmacology and Toxicology. 1999;39:399–430. doi: 10.1146/annurev.pharmtox.39.1.399. [DOI] [PubMed] [Google Scholar]
- Cowan SW, Newcomer ME, Jones TA. Crystallographic studies on a family of cellular lipophilic transport proteins. Refinement of P2 myelin protein and the structure determination and refinement of cellular retinol-binding protein in complex with all-trans-retinol. Journal of Molecular Biology. 1993;230(4):1225–1246. doi: 10.1006/jmbi.1993.1238. [DOI] [PubMed] [Google Scholar]
- Das BC, Thapa P, Karki R, Das S, Mahapatra S, Liu TC, … Evans T. Retinoic acid signaling pathways in development and diseases. Bioorganic & Medicinal Chemistry. 2014;22(2):673–683. doi: 10.1016/j.bmc.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dela Seña C, Riedl KM, Narayanasamy S, Curley RW, Schwartz SJ, Harrison EH. The human enzyme that converts dietary provitamin A carotenoids to vitamin A is a dioxygenase. The Journal of Biological Chemistry. 2014;289(19):13661–13666. doi: 10.1074/jbc.M114.557710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Ruuska SE, Levinthal DJ, Noy N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. The Journal of Biological Chemistry. 1999;274(34):23695–23698. doi: 10.1074/jbc.274.34.23695. [DOI] [PubMed] [Google Scholar]
- EX, Zhang L, Lu J, Tso P, Blaner WS, Levin MS, Li E. Increased neonatal mortality in mice lacking cellular retinol-binding protein II. The Journal of Biological Chemistry. 2002;277(39):36617–36623. doi: 10.1074/jbc.M205519200. [DOI] [PubMed] [Google Scholar]
- Esteller M, Guo M, Moreno V, Peinado MA, Capella G, Galm O, … Herman JG. Hypermethylation-associated Inactivation of the Cellular Retinol-Binding-Protein 1 Gene in Human Cancer. Cancer Research. 2002;62(20):5902–5905. [PubMed] [Google Scholar]
- Everts HB, Sundberg JP, King LE, Jr, Ong DE. Immunolocalization of enzymes, binding proteins, and receptors sufficient for retinoic acid synthesis and signaling during the hair cycle. The Journal of Investigative Dermatology. 2007;127(7):1593–1604. doi: 10.1038/sj.jid.5700753. [DOI] [PubMed] [Google Scholar]
- Everts HB, Sundberg JP, Ong DE. Immunolocalization of retinoic acid biosynthesis systems in selected sites in rat. Experimental Cell Research. 2005;308(2):309–319. doi: 10.1016/j.yexcr.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Farias EF, Ong DE, Ghyselinck NB, Nakajo S, Kuppumbatti YS, Mira y Lopez R. Cellular retinol-binding protein I, a regulator of breast epithelial retinoic acid receptor activity, cell differentiation, and tumorigenicity. Journal of the National Cancer Institute. 2005;97(1):21–29. doi: 10.1093/jnci/dji004. [DOI] [PubMed] [Google Scholar]
- Fiorella PD, Giguère V, Napoli JL. Expression of cellular retinoic acid-binding protein (type II) in Escherichia coli. Characterization and comparison to cellular retinoic acid-binding protein (type I) The Journal of Biological Chemistry. 1993;268(29):21545–21552. [PubMed] [Google Scholar]
- Fiorella PD, Napoli JL. Expression of cellular retinoic acid binding protein (CRABP) in Escherichia coli. Characterization and evidence that holo-CRABP is a substrate in retinoic acid metabolism. The Journal of Biological Chemistry. 1991;266(25):16572–16579. [PubMed] [Google Scholar]
- Fiorella PD, Napoli JL. Microsomal retinoic acid metabolism. Effects of cellular retinoic acid-binding protein (type I) and C18-hydroxylation as an initial step. The Journal of Biological Chemistry. 1994;269(14):10538–10544. [PubMed] [Google Scholar]
- Folli C, Calderone V, Ottonello S, Bolchi A, Zanotti G, Stoppini M, Berni R. Identification, retinoid binding, and x-ray analysis of a human retinol-binding protein. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(7):3710–3715. doi: 10.1073/pnas.061455898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folli C, Calderone V, Ramazzina I, Zanotti G, Berni R. Ligand binding and structural analysis of a human putative cellular retinol-binding protein. The Journal of Biological Chemistry. 2002;277(44):41970–41977. doi: 10.1074/jbc.M207124200. [DOI] [PubMed] [Google Scholar]
- Franzoni L, Cavazzini D, Rossi GL, Lücke C. New insights on the protein-ligand interaction differences between the two primary cellular retinol carriers. Journal of Lipid Research. 2010;51(6):1332–1343. doi: 10.1194/jlr.M002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzoni L, Lücke C, Pérez C, Cavazzini D, Rademacher M, Ludwig C, … Ruterjans H. Structure and backbone dynamics of Apo- and holo-cellular retinol-binding protein in solution. The Journal of Biological Chemistry. 2002;277(24):21983–21997. doi: 10.1074/jbc.M201994200. [DOI] [PubMed] [Google Scholar]
- Gaub MP, Lutz Y, Ghyselinck NB, Scheuer I, Pfister V, Chambon P, Rochette-Egly C. Nuclear detection of cellular retinoic acid binding proteins I and II with new antibodies. The Journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society. 1998;46(10):1103–1111. doi: 10.1177/002215549804601002. [DOI] [PubMed] [Google Scholar]
- Ghyselinck NB, Bavik C, Sapin V, Mark M, Bonnier D, Hindelang C, … Chambon P. Cellular retinol-binding protein I is essential for vitamin A homeostasis. The EMBO Journal. 1999;18(18):4903–4914. doi: 10.1093/emboj/18.18.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguère V, Lyn S, Yip P, Siu CH, Amin S. Molecular cloning of cDNA encoding a second cellular retinoic acid-binding protein. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(16):6233–6237. doi: 10.1073/pnas.87.16.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman DS, Huang HS. Biosynthesis of vitamin A with rat intestinal enzymes. Science. 1965;149(3686):879–880. doi: 10.1126/science.149.3686.879. [DOI] [PubMed] [Google Scholar]
- Grammatikakis I, Abdelmohsen K, Gorospe M. Wiley Interdisciplinary Reviews. 2016. Posttranslational control of HuR function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün F, Hirose Y, Kawauchi S, Ogura T, Umesono K. Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominently expressed in sensory neuroepithelia during development. The Journal of Biological Chemistry. 2000;275(52):41210–41218. doi: 10.1074/jbc.M007376200. [DOI] [PubMed] [Google Scholar]
- Gustafson AL, Donovan M, Annerwall E, Dencker L, Eriksson U. Nuclear import of cellular retinoic acid-binding protein type I in mouse embryonic cells. Mechanisms of Development. 1996;58(1–2):27–38. doi: 10.1016/s0925-4773(96)00554-0. [DOI] [PubMed] [Google Scholar]
- Hammond LA, Van Krinks CH, Durham J, Tomkins SE, Burnett RD, Jones EL, … Brown G. Antagonists of retinoic acid receptors (RARs) are potent growth inhibitors of prostate carcinoma cells. British Journal of Cancer. 2001;85(3):453–462. doi: 10.1054/bjoc.2001.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison EH, Blaner WS, Goodman DS, Ross AC. Subcellular localization of retinoids, retinoid-binding proteins, and acyl-CoA:retinol acyltransferase in rat liver. Journal of Lipid Research. 1987;28(8):973–981. [PubMed] [Google Scholar]
- Herr FM, Ong DE. Differential interaction of lecithin-retinol acyltransferase with cellular retinol binding proteins. Biochemistry. 1992;31(29):6748–6755. doi: 10.1021/bi00144a014. [DOI] [PubMed] [Google Scholar]
- Hogarth CA, Griswold MD. Retinoic acid regulation of male meiosis. Current Opinion in Endocrinology, Diabetes, and Obesity. 2013;20(3):217–223. doi: 10.1097/MED.0b013e32836067cf. [DOI] [PubMed] [Google Scholar]
- Jamison RS, Newcomer ME, Ong DE. Cellular retinoid-binding proteins: limited proteolysis reveals a conformational change upon ligand binding. Biochemistry. 1994;33(10):2873–2879. doi: 10.1021/bi00176a017. [DOI] [PubMed] [Google Scholar]
- Jiang W, Napoli JL. Reorganization of cellular retinol-binding protein type 1 and lecithin:retinol acyltransferase during retinyl ester biosynthesis. Biochimica et Biophysica Acta. 2012;1820(7):859–869. doi: 10.1016/j.bbagen.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Napoli JL. The retinol dehydrogenase Rdh10 localizes to lipid droplets during acyl ester biosynthesis. The Journal of Biological Chemistry. 2013;288(1):589–597. doi: 10.1074/jbc.M112.402883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakkad BP, Ong DE. Reduction of retinaldehyde bound to cellular retinol-binding protein (type II) by microsomes from rat small intestine. The Journal of Biological Chemistry. 1988;263(26):12916–12919. [PubMed] [Google Scholar]
- Kane MA, Bright FV, Napoli JL. Binding affinities of CRBPI and CRBPII for 9-cis-retinoids. Biochimica et Biophysica Acta. 2011;1810(5):514–518. doi: 10.1016/j.bbagen.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MA, Folias AE, Napoli JL. HPLC/UV quantitation of retinal, retinol, and retinyl esters in serum and tissues. Analytical Biochemistry. 2008a;378(1):71–79. doi: 10.1016/j.ab.2008.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MA, Folias AE, Pingitore A, Perri M, Krois CR, Ryu JY, … Napoli JL. CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Molecular and Cellular Biology. 2011;31(16):3277–3285. doi: 10.1128/MCB.05516-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MA, Folias AE, Wang C, Napoli JL. Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Analytical Chemistry. 2008b;80(5):1702–1708. doi: 10.1021/ac702030f. https://doi.org/10.1021/ac702030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MA, Folias AE, Wang C, Napoli JL. Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: a potential mechanism of ethanol toxicity. FASEB Journal. 2010;24(3):823–832. doi: 10.1096/fj.09-141572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedishvili NY. Enzymology of Retinoic Acid Biosynthesis and Degradation. Journal of Lipid Research. 2013 doi: 10.1194/jlr.R037028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIm YW, Sharma RP, Li RK. Characterization of heterologously expressed recombinant retinoic acid receptors with natural or synthetic retiniods. J Biochem Toxicol. 1994;9(5):225–234. doi: 10.1002/jbt.2570090502. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Bergfors T, Senn H, Le Motte P, Gsell B, Shudo K, Jones TA. Crystal structures of cellular retinoic acid binding proteins I and II in complex with all-trans-retinoic acid and a synthetic retinoid. Structure. 1994;2(12):1241–1258. doi: 10.1016/s0969-2126(94)00125-1. [DOI] [PubMed] [Google Scholar]
- Krozowski Z. The short-chain alcohol dehydrogenase superfamily: variations on a common theme. The Journal of Steroid Biochemistry and Molecular Biology. 1994;51(3–4):125–130. doi: 10.1016/0960-0760(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Kuppumbatti YS, Bleiweiss IJ, Mandeli JP, Waxman S, Mira-Y-Lopez R. Cellular retinol-binding protein expression and breast cancer. Journal of the National Cancer Institute. 2000;92(6):475–480. doi: 10.1093/jnci/92.6.475. [DOI] [PubMed] [Google Scholar]
- Kuppumbatti YS, Rexer B, Nakajo S, Nakaya K, Mira-y-Lopez R. CRBP suppresses breast cancer cell survival and anchorage-independent growth. Oncogene. 2001;20(50):7413–7419. doi: 10.1038/sj.onc.1204749. [DOI] [PubMed] [Google Scholar]
- Kwong J, Lo KW, Chow LSN, To KF, Choy KW, Chan FL, … Huang DP. Epigenetic silencing of cellular retinol-binding proteins in nasopharyngeal carcinoma. Neoplasia (New York, NY) 2005;7(1):67–74. doi: 10.1593/neo.04370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Chen W, Zhang M, Napoli JL. Reduction of all-trans-retinal in the mouse liver peroxisome fraction by the short-chain dehydrogenase/reductase RRD: induction by the PPAR alpha ligand clofibrate. Biochemistry. 2003;42(14):4190–4196. doi: 10.1021/bi026948i. [DOI] [PubMed] [Google Scholar]
- Levi L, Lobo G, Doud MK, von Lintig J, Seachrist D, Tochtrop GP, Noy N. Genetic ablation of the fatty acid-binding protein FABP5 suppresses HER2-induced mammary tumorigenesis. Cancer Research. 2013;73(15):4770–4780. doi: 10.1158/0008-5472.CAN-13-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi L, Wang Z, Doud MK, Hazen SL, Noy N. Saturated fatty acids regulate retinoic acid signalling and suppress tumorigenesis by targeting fatty acid-binding protein 5. Nature Communications. 2015;6:8794. doi: 10.1038/ncomms9794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin MS, Li E, Ong DE, Gordon JI. Comparison of the tissue-specific expression and developmental regulation of two closely linked rodent genes encoding cytosolic retinol-binding proteins. The Journal of Biological Chemistry. 1987;262(15):7118–7124. [PubMed] [Google Scholar]
- Li E, Norris AW. Structure/function of cytoplasmic vitamin A-binding proteins. Annual Review of Nutrition. 1996;16:205–234. doi: 10.1146/annurev.nu.16.070196.001225. [DOI] [PubMed] [Google Scholar]
- Li E, Qian SJ, Winter NS, d’Avignon A, Levin MS, Gordon JI. Fluorine nuclear magnetic resonance analysis of the ligand binding properties of two homologous rat cellular retinol-binding proteins expressed in Escherichia coli. The Journal of Biological Chemistry. 1991;266(6):3622–3629. [PubMed] [Google Scholar]
- Lin M, Zhang M, Abraham M, Smith SM, Napoli JL. Mouse retinal dehydrogenase 4 (RALDH4), molecular cloning, cellular expression, and activity in 9-cis-retinoic acid biosynthesis in intact cells. The Journal of Biological Chemistry. 2003;278(11):9856–9861. doi: 10.1074/jbc.M211417200. [DOI] [PubMed] [Google Scholar]
- Liu L, Gudas LJ. Disruption of the lecithin:retinol acyltransferase gene makes mice more susceptible to vitamin A deficiency. The Journal of Biological Chemistry. 2005;280(48):40226–40234. doi: 10.1074/jbc.M509643200. [DOI] [PubMed] [Google Scholar]
- Liu RZ, Garcia E, Glubrecht DD, Poon HY, Mackey JR, Godbout R. CRABP1 is associated with a poor prognosis in breast cancer: adding to the complexity of breast cancer cell response to retinoic acid. Molecular Cancer. 2015;14:129. doi: 10.1186/s12943-015-0380-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Cistola DP, Li E. Two homologous rat cellular retinol-binding proteins differ in local conformational flexibility. Journal of Molecular Biology. 2003;330(4):799–812. doi: 10.1016/s0022-2836(03)00629-6. [DOI] [PubMed] [Google Scholar]
- MacDonald PN, Ong DE. A lecithin:retinol acyltransferase activity in human and rat liver. Biochemical and Biophysical Research Communications. 1988a;156(1):157–163. doi: 10.1016/s0006-291x(88)80818-0. [DOI] [PubMed] [Google Scholar]
- MacDonald PN, Ong DE. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. The Journal of Biological Chemistry. 1988b;263(25):12478–12482. [PubMed] [Google Scholar]
- Majumdar A, Petrescu AD, Xiong Y, Noy N. Nuclear translocation of cellular retinoic acid-binding protein II is regulated by retinoic acid-controlled SUMOylation. The Journal of Biological Chemistry. 2011;286(49):42749–42757. doi: 10.1074/jbc.M111.293464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpeli G, Stoppini M, Zapponi MC, Folli C, Berni R. Interactions with retinol and retinoids of bovine cellular retinol-binding protein. European Journal of Biochemistry/FEBS. 1995;229(2):486–493. [PubMed] [Google Scholar]
- Meisner NC, Filipowicz W. Properties of the Regulatory RNA-Binding Protein HuR and its Role in Controlling miRNA Repression. Advances in Experimental Medicine and Biology. 2011;700:106–123. doi: 10.1007/978-1-4419-7823-3_10. [DOI] [PubMed] [Google Scholar]
- Mittag T, Franzoni L, Cavazzini D, Schaffhausen B, Rossi GL, Günther UL. Retinol modulates site-specific mobility of apo-cellular retinol-binding protein to promote ligand binding. Journal of the American Chemical Society. 2006;128(30):9844–9848. doi: 10.1021/ja0616128. [DOI] [PubMed] [Google Scholar]
- Miyake T, Ueda Y, Matsuzaki S, Miyatake T, Yoshino K, Fujita M, … Kimura T. CRABP1-reduced expression is associated with poorer prognosis in serous and clear cell ovarian adenocarcinoma. Journal of Cancer Research and Clinical Oncology. 2011;137(4):715–722. doi: 10.1007/s00432-010-0930-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuiri H, Yoshida K, Toge T, Oue N, Aung PP, Noguchi T, Yasui W. DNA methylation of genes linked to retinoid signaling in squamous cell carcinoma of the esophagus: DNA methylation of CRBP1 and TIG1 is associated with tumor stage. Cancer Science. 2005;96(9):571–577. doi: 10.1111/j.1349-7006.2005.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal MS, Ruiz A, Bok D, Rando RR. Lecithin retinol acyltransferase contains cysteine residues essential for catalysis. Biochemistry. 2000;39(17):5215–5220. doi: 10.1021/bi9929554. [DOI] [PubMed] [Google Scholar]
- Napoli JL. Biosynthesis and metabolism of retinoic acid: roles of CRBP and CRABP in retinoic acid: roles of CRBP and CRABP in retinoic acid homeostasis. The Journal of Nutrition. 1993;123(2 Suppl):362–366. doi: 10.1093/jn/123.suppl_2.362. [DOI] [PubMed] [Google Scholar]
- Napoli JL. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochimica et Biophysica Acta. 1999;1440(2–3):139–162. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- Napoli JL. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochimica Et Biophysica Acta. 1999;1440(2–3):139–162. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- Napoli JL. A gene knockout corroborates the integral function of cellular retinol-binding protein in retinoid metabolism. Nutrition Reviews. 2000;58(8):230–236. doi: 10.1111/j.1753-4887.2000.tb01870.x. [DOI] [PubMed] [Google Scholar]
- Napoli JL. Physiological insights into all-trans-retinoic acid biosynthesis. Biochimica Et Biophysica Acta. 2012;1821(1):152–167. doi: 10.1016/j.bbalip.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CH, Peng CC, Lutz JD, Yeung CK, Zelter A, Isoherranen N. Direct protein-protein interactions and substrate channeling between cellular retinoic acid binding proteins and CYP26B1. FEBS Letters. 2016;590(16):2527–2535. doi: 10.1002/1873-3468.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomer ME. Retinoid-binding proteins: structural determinants important for function. FASEB Journal. 1995;9(2):229–239. doi: 10.1096/fasebj.9.2.7781925. [DOI] [PubMed] [Google Scholar]
- Norris AW, Cheng L, Giguère V, Rosenberger M, Li E. Measurement of subnanomolar retinoic acid binding affinities for cellular retinoic acid binding proteins by fluorometric titration. Biochimica Et Biophysica Acta. 1994;1209(1):10–18. doi: 10.1016/0167-4838(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Obrochta KM, Kane MA, Napoli JL. Effects of diet and strain on mouse serum and tissue retinoid concentrations. PloS One. 2014;9(6):e99435. doi: 10.1371/journal.pone.0099435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Byrne SM, Wongsiriroj N, Libien J, Vogel S, Goldberg IJ, Baehr W, … Blaner WS. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT) The Journal of Biological Chemistry. 2005;280(42):35647–35657. doi: 10.1074/jbc.M507924200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuducu AF, Janzen V, Ko Y, Hahne JC, Lu H, Ma ZL, … Wernert N. Cellular retinoic acid-binding protein 2 is down-regulated in prostate cancer. International Journal of Oncology. 2005;27(5):1273–1282. [PubMed] [Google Scholar]
- Olson JA, Hayaishi O. The enzymatic cleavage of beta-carotene into vitamin A by soluble enzymes of rat liver and intestine. Proceedings of the National Academy of Sciences of the United States of America. 1965;54(5):1364–1370. doi: 10.1073/pnas.54.5.1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong DE. Cellular transport and metabolism of vitamin A: roles of the cellular retinoid-binding proteins. Nutrition Reviews. 1994;52(2 Pt 2):S24–31. doi: 10.1111/j.1753-4887.1994.tb01383.x. [DOI] [PubMed] [Google Scholar]
- Ong DE, Chytil F. Cellular retinoic acid-binding protein from rat testis. Purification and characterization. The Journal of Biological Chemistry. 1978;253(13):4551–4554. [PubMed] [Google Scholar]
- Ong DE, Kakkad B, MacDonald PN. Acyl-CoA-independent esterification of retinol bound to cellular retinol-binding protein (type II) by microsomes from rat small intestine. The Journal of Biological Chemistry. 1987;262(6):2729–2736. [PubMed] [Google Scholar]
- Ong DE, MacDonald PN, Gubitosi AM. Esterification of retinol in rat liver. Possible participation by cellular retinol-binding protein and cellular retinol-binding protein II. The Journal of Biological Chemistry. 1988;263(12):5789–5796. [PubMed] [Google Scholar]
- Ong DE, Page DL. Quantitation of cellular retinol-binding protein in human organs. The American Journal of Clinical Nutrition. 1986;44(3):425–430. doi: 10.1093/ajcn/44.3.425. [DOI] [PubMed] [Google Scholar]
- Ong DE, Page DL. Cellular retinol-binding protein (type two) is abundant in human small intestine. Journal of Lipid Research. 1987;28(6):739–745. [PubMed] [Google Scholar]
- Ong DE, Tsai CH, Chytil F. Cellular retinol-binding protein and retinoic acid-binding protein in rat testes: effect of retinol depletion. The Journal of Nutrition. 1976;106(2):204–211. doi: 10.1093/jn/106.2.204. [DOI] [PubMed] [Google Scholar]
- Orland MD, Anwar K, Cromley D, Chu CH, Chen L, Billheimer JT, … Cheng D. Acyl coenzyme A dependent retinol esterification by acyl coenzyme A: diacylglycerol acyltransferase 1. Biochimica Et Biophysica Acta. 2005;1737(1):76–82. doi: 10.1016/j.bbalip.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Ottonello S, Scita G, Mantovani G, Cavazzini D, Rossi GL. Retinol bound to cellular retinol-binding protein is a substrate for cytosolic retinoic acid synthesis. The Journal of Biological Chemistry. 1993;268(36):27133–27142. [PubMed] [Google Scholar]
- Penzes P, Napoli JL. Holo-cellular retinol-binding protein: distinction of ligand-binding affinity from efficiency as substrate in retinal biosynthesis. Biochemistry. 1999;38(7):2088–2093. doi: 10.1021/bi982228t. [DOI] [PubMed] [Google Scholar]
- Penzes P, Wang X, Napoli JL. Enzymatic characteristics of retinal dehydrogenase type I expressed in Escherichia coli. Biochimica et Biophysica Acta. 1997;1342(2):175–181. doi: 10.1016/s0167-4838(97)00102-7. [DOI] [PubMed] [Google Scholar]
- Persaud SD, Lin YW, Wu CY, Kagechika H, Wei LN. Cellular retinoic acid binding protein I mediates rapid non-canonical activation of ERK1/2 by all-trans retinoic acid. Cellular Signalling. 2013;25(1):19–25. doi: 10.1016/j.cellsig.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persaud SD, Park SW, Ishigami-Yuasa M, Koyano-Nakagawa N, Kagechika H, Wei LN. All trans-retinoic acid analogs promote cancer cell apoptosis through non-genomic Crabp1 mediating ERK1/2 phosphorylation. Scientific Reports. 2016;6:22396. doi: 10.1038/srep22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfoertner S, Goelden U, Hansen W, Toepfer T, Geffers R, Ukena SN, … Schrader AJ. Cellular retinoic acid binding protein I: expression and functional influence in renal cell carcinoma. Tumour Biology: The Journal of the International Society for Oncodevelopmental Biology and Medicine. 2005;26(6):313–323. doi: 10.1159/000089262. [DOI] [PubMed] [Google Scholar]
- Piantedosi R, Ghyselinck N, Blaner WS, Vogel S. Cellular retinol-binding protein type III is needed for retinoid incorporation into milk. The Journal of Biological Chemistry. 2005;280(25):24286–24292. doi: 10.1074/jbc.M503906200. [DOI] [PubMed] [Google Scholar]
- Pierzchalski K, Yu J, Norman V, Kane MA. CrbpI regulates mammary retinoic acid homeostasis and the mammary microenvironment. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2013;27(5):1904–1916. doi: 10.1096/fj.12-219410. [DOI] [PubMed] [Google Scholar]
- Porter SB, Fraker LD, Chytil F, Ong DE. Localization of cellular retinol-binding protein in several rat tissues. Proceedings of the National Academy of Sciences of the United States of America. 1983;80(21):6586–6590. doi: 10.1073/pnas.80.21.6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posch KC, Boerman MH, Burns RD, Napoli JL. Holocellular retinol binding protein as a substrate for microsomal retinal synthesis. Biochemistry. 1991;30(25):6224–6230. doi: 10.1021/bi00239a021. [DOI] [PubMed] [Google Scholar]
- Posch KC, Burns RD, Napoli JL. Biosynthesis of all-trans-retinoic acid from retinal. Recognition of retinal bound to cellular retinol binding protein (type I) as substrate by a purified cytosolic dehydrogenase. The Journal of Biological Chemistry. 1992;267(27):19676–19682. [PubMed] [Google Scholar]
- Rajan N, Blaner WS, Soprano DR, Suhara A, Goodman DS. Cellular retinol-binding protein messenger RNA levels in normal and retinoid-deficient rats. Journal of Lipid Research. 1990;31(5):821–829. [PubMed] [Google Scholar]
- Randolph RK, Winkler KE, Ross AC. Fatty acyl CoA-dependent and -independent retinol esterification by rat liver and lactating mammary gland microsomes. Archives of Biochemistry and Biophysics. 1991;288(2):500–508. doi: 10.1016/0003-9861(91)90227-a. [DOI] [PubMed] [Google Scholar]
- Redmond TM, Gentleman S, Duncan T, Yu S, Wiggert B, Gantt E, Cunningham FX. Identification, expression, and substrate specificity of a mammalian beta-carotene 15,15’-dioxygenase. The Journal of Biological Chemistry. 2001;276(9):6560–6565. doi: 10.1074/jbc.M009030200. [DOI] [PubMed] [Google Scholar]
- Reeves PG. Components of the AIN-93 diets as improvements in the AIN-76A diet. The Journal of Nutrition. 1997;127(5 Suppl):838S–841S. doi: 10.1093/jn/127.5.838S. [DOI] [PubMed] [Google Scholar]
- Rexer BN, Ong DE. A novel short-chain alcohol dehydrogenase from rats with retinol dehydrogenase activity, cyclically expressed in uterine epithelium. Biology of Reproduction. 2002;67(5):1555–1564. doi: 10.1095/biolreprod.102.007021. [DOI] [PubMed] [Google Scholar]
- Rigtrup KM, Kakkad B, Ong DE. Purification and partial characterization of a retinyl ester hydrolase from the brush border of rat small intestine mucosa: probable identity with brush border phospholipase B. Biochemistry. 1994a;33(9):2661–2666. doi: 10.1021/bi00175a039. [DOI] [PubMed] [Google Scholar]
- Rigtrup KM, McEwen LR, Said HM, Ong DE. Retinyl ester hydrolytic activity associated with human intestinal brush border membranes. The American Journal of Clinical Nutrition. 1994b;60(1):111–116. doi: 10.1093/ajcn/60.1.111. [DOI] [PubMed] [Google Scholar]
- Rigtrup KM, Ong DE. A retinyl ester hydrolase activity intrinsic to the brush border membrane of rat small intestine. Biochemistry. 1992;31(11):2920–2926. doi: 10.1021/bi00126a011. [DOI] [PubMed] [Google Scholar]
- Roberts D, Williams SJ, Cvetkovic D, Weinstein JK, Godwin AK, Johnson SW, Hamilton TC. Decreased expression of retinol-binding proteins is associated with malignant transformation of the ovarian surface epithelium. DNA and Cell Biology. 2002;21(1):11–19. doi: 10.1089/10445490252810276. [DOI] [PubMed] [Google Scholar]
- Ross AC. Cellular metabolism and activation of retinoids: roles of cellular retinoid-binding proteins. FASEB Journal. 1993;7(2):317–327. doi: 10.1096/fasebj.7.2.8440409. [DOI] [PubMed] [Google Scholar]
- Ross AC, Zolfaghari R. Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annual Review of Nutrition. 2011;31:65–87. doi: 10.1146/annurev-nutr-072610-145127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Winston A, Lim YH, Gilbert BA, Rando RR, Bok D. Molecular and biochemical characterization of lecithin retinol acyltransferase. The Journal of Biological Chemistry. 1999;274(6):3834–3841. doi: 10.1074/jbc.274.6.3834. [DOI] [PubMed] [Google Scholar]
- Schug TT, Berry DC, Shaw NS, Travis SN, Noy N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell. 2007a;129(4):723–733. doi: 10.1016/j.cell.2007.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug TT, Berry DC, Toshkov IA, Cheng L, Nikitin AY, Noy N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARbeta/delta to RAR. Proceedings of the National Academy of Sciences of the United States of America. 2008b;105(21):7546–7551. doi: 10.1073/pnas.0709981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo H, Kojima N, Sato M. Vitamin A-storing cells (stellate cells) Vitamins and Hormones. 2007;75:131–159. doi: 10.1016/S0083-6729(06)75006-3. [DOI] [PubMed] [Google Scholar]
- Sessler RJ, Noy N. A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Molecular Cell. 2005;18(3):343–353. doi: 10.1016/j.molcel.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Shaw N, Elholm M, Noy N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor beta/delta. The Journal of Biological Chemistry. 2003;278(43):41589–41592. doi: 10.1074/jbc.C300368200. [DOI] [PubMed] [Google Scholar]
- Shih MYS, Kane MA, Zhou P, Yen CLE, Streeper RS, Napoli JL, Farese RV., Jr Retinol Esterification by DGAT1 Is Essential for Retinoid Homeostasis in Murine Skin. The Journal of Biological Chemistry. 2009;284(7):4292–4299. doi: 10.1074/jbc.M807503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvaroli JA, Arne JM, Chelstowska S, Kiser PD, Banerjee S, Golczak M. Ligand Binding Induces Conformational Changes in Human Cellular Retinol-binding Protein 1 (CRBP1) Revealed by Atomic Resolution Crystal Structures. The Journal of Biological Chemistry. 2016;291(16):8528–8540. doi: 10.1074/jbc.M116.714535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer A, Vyas KS. A global clinical view on vitamin A and carotenoids. The American Journal of Clinical Nutrition. 2012;96(5):1204S–6S. doi: 10.3945/ajcn.112.034868. [DOI] [PubMed] [Google Scholar]
- Song MS, Chen W, Zhang M, Napoli JL. Identification of a mouse short-chain dehydrogenase/reductase gene, retinol dehydrogenase-similar. Function of non-catalytic amino acid residues in enzyme activity. The Journal of Biological Chemistry. 2003;278(41):40079–40087. doi: 10.1074/jbc.M304910200. [DOI] [PubMed] [Google Scholar]
- Sporn MB, Roberts AB, Goodman DS, editors. The Retinoids: biology, chemistry, and medicine. 2. New York: Raven Press; 1994. [Google Scholar]
- Stevison F, Jing J, Tripathy S, Isoherranen N. Role of retinoic acid-metabolizing cytochrome P450s, CYP26, in inflammation and cancer. Adv Pharmacol. 2015;74:373–412. doi: 10.1016/bs.apha.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuts EZ, Harosi FI. Solubility of retinoids in water. Archives of Biochemistry and Biophysics. 1991;287(2):297–304. doi: 10.1016/0003-9861(91)90482-x. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Yang XJ, Lavery TT, Furge KA, Williams BO, Tretiakova M, … Teh BT. Gene expression profiling of favorable histology Wilms tumors and its correlation with clinical features. Cancer Research. 2002;62(22):6598–6605. [PubMed] [Google Scholar]
- Takase S, Ong DE, Chytil F. Transfer of retinoic acid from its complex with cellular retinoic acid-binding protein to the nucleus. Archives of Biochemistry and Biophysics. 1986;247(2):328–334. doi: 10.1016/0003-9861(86)90591-6. [DOI] [PubMed] [Google Scholar]
- Tan NS, Shaw NS, Vinckenbosch N, Liu P, Yasmin R, Desvergne B, … Noy N. Selective cooperation between fatty acid binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Molecular and Cellular Biology. 2002;22(14):5114–5127. doi: 10.1128/MCB.22.14.5114-5127.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XH, Gudas LJ. Retinoids, retinoic acid receptors and cancer. Annu Rev Pathol. 2011;6:345–364. doi: 10.1146/annurev-pathol-011110-130303. [DOI] [PubMed] [Google Scholar]
- Tari AM, Lim S-J, Hung M-C, Esteva FJ, Lopez-Berestein G. Her2/neu induces all-trans retinoic acid (ATRA) resistance in breast cancer cells. Oncogene. 2002;21(34):5224–5232. doi: 10.1038/sj.onc.1205660. [DOI] [PubMed] [Google Scholar]
- Thompson JR, Bratt JM, Banaszak LJ. Crystal structure of cellular retinoic acid binding protein I shows increased access to the binding cavity due to formation of an intermolecular beta-sheet. Journal of Molecular Biology. 1995;252(4):433–446. doi: 10.1006/jmbi.1995.0509. [DOI] [PubMed] [Google Scholar]
- Thompson M, Lapointe J, Choi YL, Ong DE, Higgins JP, Brooks JD, Pollack JR. Identification of candidate prostate cancer genes through comparative expression-profiling of seminal vesicle. The Prostate. 2008;68(11):1248–1256. doi: 10.1002/pros.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topletz AR, Thatcher JE, Zelter A, Lutz JD, Tay S, Nelson WL, Isoherranen N. Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochemical Pharmacology. 2012;83(1):149–163. doi: 10.1016/j.bcp.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topletz AR, Tripathy S, Foti RS, Shimshoni JA, Nelson WL, Isoherranen N. Induction of CYP26A1 by metabolites of retinoic acid: evidence that CYP26A1 is an important enzyme in the elimination of active retinoids. Molecular Pharmacology. 2015;87(3):430–441. doi: 10.1124/mol.114.096784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht C, Basch E, Chao W, Conquer J, Costa D, Culwell S, … Zhou S. An evidence-based systematic review of vitamin A by the natural standard research collaboration. Journal of Dietary Supplements. 2012;9(4):299–416. doi: 10.3109/19390211.2012.736721. [DOI] [PubMed] [Google Scholar]
- Uray IP, Dmitrovsky E, Brown PH. Retinoids and rexinoids in cancer prevention: from laboratory to clinic. Seminars in Oncology. 2016;43:49–64. doi: 10.1053/j.seminoncol.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V, Thompson DC, Smith C, Fujita M, Chen Y. Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells. Chemico-Biological Interactions. 2013;202(1–3):2–10. doi: 10.1016/j.cbi.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venepally P, Reddy LG, Sani BP. Analysis of the effects of CRABP I expression on the RA-induced transcription mediated by retinoid receptors. Biochemistry. 1996;35(31):9974–9982. doi: 10.1021/bi9603276. https://doi.org/10.1021/bi9603276. [DOI] [PubMed] [Google Scholar]
- Vogel S, Mendelsohn CL, Mertz JR, Piantedosi R, Waldburger C, Gottesman ME, Blaner WS. Characterization of a new member of the fatty acid-binding protein family that binds all-trans-retinol. The Journal of Biological Chemistry. 2001;276(2):1353–1360. doi: 10.1074/jbc.M005118200. [DOI] [PubMed] [Google Scholar]
- von Lintig J, Vogt K. Filling the gap in vitamin A research. Molecular identification of an enzyme cleaving beta-carotene to retinal. The Journal of Biological Chemistry. 2000;275(16):11915–11920. doi: 10.1074/jbc.275.16.11915. [DOI] [PubMed] [Google Scholar]
- Vreeland AC, Yu S, Levi L, de Barros Rossetto D, Noy N. Transcript stabilization by the RNA-binding protein HuR is regulated by cellular retinoic acid-binding protein 2. Molecular and Cellular Biology. 2014;34(12):2135–2146. doi: 10.1128/MCB.00281-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li Y, Yan H. Structure-function relationships of cellular retinoic acid-binding proteins. Quantitative analysis of the ligand binding properties of the wild-type proteins and site-directed mutants. The Journal of Biological Chemistry. 1997;272(3):1541–1547. doi: 10.1074/jbc.272.3.1541. [DOI] [PubMed] [Google Scholar]
- Wang X, Penzes P, Napoli JL. Cloning of a cDNA encoding an aldehyde dehydrogenase and its expression in Escherichia coli. Recognition of retinal as substrate. The Journal of Biological Chemistry. 1996;271(27):16288–16293. doi: 10.1074/jbc.271.27.16288. [DOI] [PubMed] [Google Scholar]
- Williams JB, Napoli JL. Metabolism of retinoic acid and retinol durng differentiation of F9 embryonal carcinoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:4658–4662. doi: 10.1073/pnas.82.14.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SJ, Cvetkovic D, Hamilton TC. Vitamin A metabolism is impaired in human ovarian cancer. Gynecologic Oncology. 2009;112(3):637–645. doi: 10.1016/j.ygyno.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman EM, Bar-El Dadon S, Reifen R. The vicious cycle of vitamin A deficiency: A review. Critical Reviews in Food Science and Nutrition. 2016 doi: 10.1080/10408398.2016.1160362. https://doi.org/10.1080/10408398.2016.1160362. [DOI] [PubMed]
- Won JY, Nam EC, Yoo SJ, Kwon HJ, Um SJ, Han HS, … Kim SY. The effect of cellular retinoic acid binding protein-I expression on the CYP26-mediated catabolism of all-trans retinoic acid and cell proliferation in head and neck squamous cell carcinoma. Metabolism: Clinical and Experimental. 2004;53(8):1007–1012. doi: 10.1016/j.metabol.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Wongsiriroj N, Piantedosi R, Palczewski K, Goldberg IJ, Johnston TP, Li E, Blaner WS. The molecular basis of retinoid absorption: a genetic dissection. The Journal of Biological Chemistry. 2008;283(20):13510–13519. doi: 10.1074/jbc.M800777200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen CLE, Monetti M, Burri BJ, Farese RV. The triacylglycerol synthesis enzyme DGAT1 also catalyzes the synthesis of diacylglycerols, waxes, and retinyl esters. Journal of Lipid Research. 2005;46(7):1502–1511. doi: 10.1194/jlr.M500036-JLR200. [DOI] [PubMed] [Google Scholar]
- Yost RW, Harrison EH, Ross AC. Esterification by rat liver microsomes of retinol bound to cellular retinol-binding protein. The Journal of Biological Chemistry. 1988;263(35):18693–18701. [PubMed] [Google Scholar]
- Zhai Y, Higgins D, Napoli JL. Coexpression of the mRNAs encoding retinol dehydrogenase isozymes and cellular retinol-binding protein. Journal of Cellular Physiology. 1997;173(1):36–43. doi: 10.1002/(SICI)1097-4652(199710)173:1<36::AID-JCP5>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Zhang M, Chen W, Smith SM, Napoli JL. Molecular characterization of a mouse short chain dehydrogenase/reductase active with all-trans-retinol in intact cells, mRDH1. The Journal of Biological Chemistry. 2001;276(47):44083–44090. doi: 10.1074/jbc.M105748200. [DOI] [PubMed] [Google Scholar]
- Zhang W, Vreeland AC, Noy N. The RNA-binding protein HuR regulates protein nuclear import. Journal of Cell Science. 2016 doi: 10.1242/jcs.192096. https://doi.org/10.1242/jcs.192096. [DOI] [PMC free article] [PubMed]
- Zhao D, McCaffery P, Ivins KJ, Neve RL, Hogan P, Chin WW, Dräger UC. Molecular identification of a major retinoic-acid-synthesizing enzyme, a retinaldehyde-specific dehydrogenase. European Journal of Biochemistry/FEBS. 1996;240(1):15–22. doi: 10.1111/j.1432-1033.1996.0015h.x. [DOI] [PubMed] [Google Scholar]
- Zolfaghari R, Ross AC. Lecithin:retinol acyltransferase from mouse and rat liver. CDNA cloning and liver-specific regulation by dietary vitamin a and retinoic acid. Journal of Lipid Research. 2000;41(12):2024–2034. [PubMed] [Google Scholar]