

Abstract
Enzymes catalyze organic transformations with exquisite levels of selectivity, including chemoselectivity, stereoselectivity, and substrate selectivity, but the types of reactions catalyzed by enzymes are more limited than those of chemical catalysts. Thus, the convergence of chemical catalysis and biocatalysis can enable enzymatic systems to catalyze abiological reactions with high selectivity. Recently, we disclosed artificial enzymes constructed from the apo form of heme proteins and iridium porphyrins that catalyze the insertion of carbenes into a C–H bond. We postulated that the same type of Ir(Me)-PIX enzymes could catalyze the cyclopropanation of a broad range of alkenes with control of multiple modes of selectivity. Here, we report the evolution of artificial enzymes that are highly active and highly stereoselective for the addition of carbenes to a wide range of alkenes. These enzymes catalyze the cyclopropanation of terminal and internal, activated and unactivated, electron-rich and electron-deficient, conjugated and nonconjugated alkenes. In particular, Ir(Me)-PIX enzymes derived from CYP119 catalyze highly enantio- and diastereoselective cyclopropanations of styrene with ±98% ee, >70:1 dr, >75% yield, and ∼10,000 turnovers (TON), as well as 1,2-disubstituted styrenes with up to 99% ee, 35:1 dr, and 54% yield. Moreover, Ir(Me)-PIX enzymes catalyze cyclopropanation of internal, unactivated alkenes with up to 99% stereoselectivity, 76% yield, and 1300 TON. They also catalyze cyclopropanation of natural products with diastereoselectivities that are complementary to those attained with standard transition metal catalysts. Finally, Ir(Me)-PIX P450 variants react with substrate selectivity that is reminiscent of natural enzymes; they react preferentially with less reactive internal alkenes in the presence of more reactive terminal alkenes. Together, the studies reveal the suitability of Ir-containing P450s to combine the broad reactivity and substrate scope of transition metal catalysts with the exquisite selectivity of enzymes, generating catalysts that enable reactions to occur with levels and modes of activity and selectivity previously unattainable with natural enzymes or transition metal complexes alone.
Short abstract
This study describes the evolution of enzymes containing iridium porphyrins to create artificial metalloenzymes that catalyze stereoselective cyclopropanations of terminal and internal, activated and unactivated, electron-rich and electron-deficient alkenes.
The ability to evolve enzymes in the laboratory to achieve high reactivity and selectivity for unnatural substrates has motivated research toward expanding the scope of reactions catalyzed by enzymes.1,2 In some cases, this reactivity has been achieved by exploiting the promiscuity of natural active sites;3 in other cases, abiological reactivity has been achieved by incorporating synthetic transition metal complexes into natural proteins.1 Although highly enantioselective reactions catalyzed by synthetic complexes within a protein have been achieved by protein engineering and directed evolution,1,4,5 reactions catalyzed by artificial metalloenzymes that occur with modes of selectivity beyond enantioselectivity are rare.
Recently, we disclosed that the apo forms of natural heme proteins reconstituted with native-like cofactors carrying abiological metal centers catalyze abiological reactions, while retaining a native binding site that can be readily evolved to bind unnatural substrates.6 Specifically, Physeter macrocephalus myoglobin and Sulfolobus solfataricus CYP119 reconstituted with Ir(Me)-PIX catalyze the insertion of carbenes into C–H bonds with high enantioselectivities. The latter enzyme catalyzes this reaction with rates comparable to those of natural enzymes.7 The same enzyme also catalyzes the insertion of nitrenes into C–H bonds with high levels of chemo- and enantioselectivity.8
Based on these initial studies, we hypothesized that Ir(Me)-PIX enzymes could catalyze abiological reactions with the exquisite levels and range of modes of selectivity displayed by natural enzymes, and with the broad substrate scope typical of transition metal catalysts. The addition of a carbene unit to an alkene to form a cyclopropane,9 mechanistically related to the insertion of a carbene into a C–H bond,10 is a widely practiced reaction in organic synthesis. Pharmaceutically active cyclopropanes have been investigated recently with particular intensity,11−13 making this a relevant model reaction for catalysis by Ir(Me)-PIX enzymes.
Both natural and artificial metalloenzymes have been shown to catalyze cyclopropanation.14−17 Heme-containing metalloproteins catalyze the addition of monosubstituted carbenes to activated, terminal alkenes (mono- and 1,1-disubstituted styrenes) with high TON and selectivity.14,18−20 Artificial Rh(II)-oligopeptidases catalyze the addition of less reactive, disubstituted carbenes to activated, monosubstituted alkenes.15 Neither of these enzymes have been shown to catalyze the addition of carbenes to internal vinylarenes or unconjugated alkenes. We showed that myoglobins containing iridium in place of iron do catalyze the cyclopropanation of both terminal alkenes (1-octene) and internal alkenes (trans-β-methylstyrene), but the reactions occurred with low yields and TON (<10% yield and <40 TON) and moderate enantioselectivities (30–82% ee).6
Here, we report that artificial metalloenzymes created from a P450, instead of myoglobin, and Ir(Me)-PIX catalyze the addition of carbenes to a wide range of alkenes, including terminal and internal, activated and unactivated, electron-rich and electron-deficient alkenes. The cyclopropanations of vinylarenes occur with up to 99% ee, 200:1 dr, 80% yield, and 10,000 TON, and the cyclopropanations of less reactive internal and unconjugated alkenes occur with up to 99% ee, 200:1 dr, 76% yield, and 1300 TON. Mutants of Ir(Me)-PIX CYP119 also form highly diastereoselective catalysts for the functionalization of natural terpenes and their derivatives. Finally, variants of Ir(Me)-PIX P450s react preferentially with typically less reactive internal alkenes in the presence of typically more reactive terminal alkenes. Thus, Ir(Me)-PIX P450s react with a wide range of selectivity modes previously achieved by only natural enzymes.
We commenced our studies to develop artificial metalloenzymes for cyclopropanation of alkenes by evaluating reactions of ethyl diazoacetate (EDA) with four model substrates (1–4) in the presence of Ir(Me)-CYP119 (Figure 1, Table 1). Initial evaluation of approximately 100 variants of Ir(Me)-CYP119 containing mutations at positions within the active site (positions L69, A209, T213, V254, Figure 1) identified a pair of mutants that form opposite enantiomers of the cyclopropane products from reactions of olefins 1–4 and EDA with up to 40:1 dr and up to 86% ee (dr as defined in Figures 2–4; for details on the process of directed evolution, see the Supporting Information). For reactions of substrates 1–2 and 4, enzymes were identified that form the opposite diastereomer of the product than is formed from reactions catalyzed by the free Ir(Me)-PIX cofactor (Table 1). Specifically, the double mutant C317G, V254A (hereafter referred to as “CYP119(+)”) formed the (1S,2R)-enantiomer of the cis-cyclopropane 5 with 73% ee, while the second variant, the triple mutant C317G, L69F, T213V (hereafter referred as “CYP119(−)”), gave the opposite enantiomer (1R,2S) of the cis product 5 with (−)74% ee. In all cases, the analogous reactions of substrates 2–4 catalyzed by the mutant CYP119(+) formed the diastereomer of the cyclopropane products 6–8 containing the ester and the phenyl ring in a cis relationship to one another with >2:1 dr and 73–86% ee. The mutant CYP119(−) gave the opposite enantiomer of the same cyclopropane diastereomer in >3:1 dr and up to (−)86% ee. The free cofactor formed the racemic trans-isomer as the major cyclopropane product in all cases.
Figure 1.
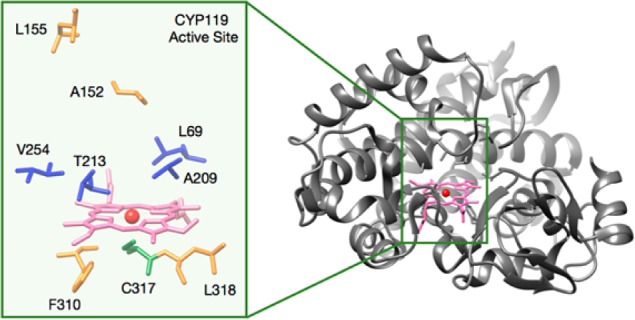
Structure of WT Fe-CYP119 (prepared in UCSF Chimera from PDB 1IO7). Right: Structure of Fe-CYP119. Left: Residues targeted in the evolution of the protein scaffold to increase activity and selectivity in C–H insertion reactions.
Table 1. Cyclopropanation of Alkenes of Differing Steric and Electronic Properties with Ethyl Diazoacetatea.
| CYP119(+)b |
CYP119(−)c |
free
Ir(Me)-PIX |
||||
|---|---|---|---|---|---|---|
| alkene | ee (%) | dr (cis:trans) | ee (%) | dr (cis:trans) | ee (%) | dr (cis:trans) |
| styrene | 73 | 2:1 | –74 | 5:1 | 0 | 1:4 |
| α-Me-styrene | 82 | 11:1 | –86 | 3:1 | 0 | 1:2 |
| cis-β-Me-styrene | 75 | 27:1* | –18 | 40:1* | 0 | 10:1* |
| trans-β-Me-styrene | 86 | 13:1** | –32 | 4:1** | 0 | 1:>25** |
| hexen-5-one | 79 | 4:1 | –88 | 8:1 | 0 | 1:4 |
| cyclopentene 12 | 2:1:t:t*** | 228:1:t:1*** | 3:1:1:3*** | |||
| 1-octene | 66 | 4:1 | –78 | 6:1 | 0 | 1:4 |
| cis-2-octene | 39 | 8:1** | –60 | 1:1** | 0 | 5:1** |
Tabulated data describes the outcome of the reactions using two mutants of CYP119 identified in the initial phase of directed evolution in comparison to the same reactions catalyzed by the free Ir(Me)-PIX cofactor. c = cis, t = trans. *Major diastereomer is 2-c,3-c:2-t,3-t, ref = COOEt; **Major diastereomer is 2-c,3-c:2-t,3-t, ref = COOEt; ***minor products not determined, major product shown as compound 13, t = trace. For details, see Table S5.
CYP119 (+): Ir(Me)-PIX CYP119 with the mutations C317G, V254A.
CYP119 (−): Ir(Me)-PIX CYP119 with the mutations C317G, L69F, T213V.
Figure 2.

Cyclopropanation of vinylarenes with different substitution patterns on the alkene. The reactions were conducted with 1 mL of alkene under the listed conditions using 20 mM alkene and 3 equiv of EDA (added over 3 h by syringe pump as a 30% (v:v) solution in DMF). c = cis, t = trans. The yields were determined by GC using dodecane as internal standard. The absolute stereochemistry of products for 1 and 2 was assigned based on prior literature (see the Supporting Information). The (1S,2R)-enantiomer of product 5 was formed in 98% ee, and the (1R,2S)-enantiomer was formed in −98% ee. The (1S,2S)-enantiomer of product 6 was formed in 82% ee, while the (1R,2R)-enantiomer was formed in −93% ee. The absolute configurations of products 7 and 8 were not assigned. All variants contained the mutations C317G in addition to those listed.
Figure 4.
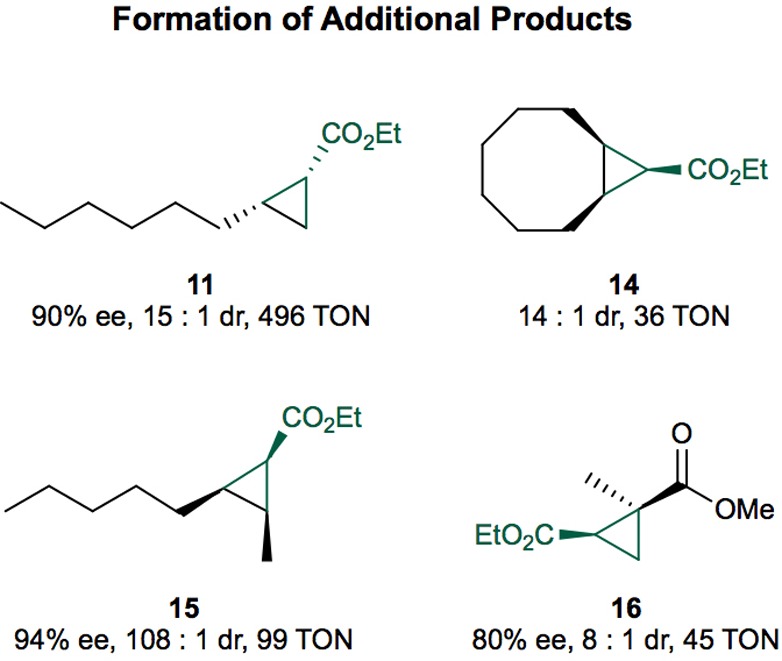
Outcomes of cyclopropanation reactions of additional aliphatic alkenes with EDA. The reactions were conducted on 1 mL scale under the listed conditions using 20 mM alkene and 3 equiv of EDA (added over 3 h by syringe). Product 11 was obtained using the variant CYP119-C317G, A209G (0.02 mol % cat.). Product 14 was obtained using the variant CYP119 C317G, T213G, V254L, L318F, L155W (0.2 mol % cat.). Product 15 was obtained using the variant CYP119(−)-V254L (0.1 mol % cat.), and product 16 was obtained using the variant CYP119(+)-L155W (0.2 mol % cat.). The TON were determined by GC using dodecane as internal standard. The stereochemistry of products was assigned based on NMR analysis and literature reports (see the Supporting Information); the absolute stereochemistry of 16 was assigned based on literature reports (see the Supporting Information); the absolute stereochemistry of products 14 and 16 was not assigned.
These results suggested that stereodivergent catalysts for the formation of cyclopropanes from diverse alkenes with high diastereoselectivity and enantioselectivity could be generated by evolving further just the two mutants CYP119(+) and CYP119(−). Consequently, we evaluated ∼100 variants of CYP119(+) and CYP119(−) that contain additional mutations at positions V254, A152, L155, F310, and L318 (Figure 1, for details on the process of directed evolution, see the Supporting Information). From this library of mutants, we identified distinct variants of Ir(Me)-CYP119 that catalyze the cyclopropanation of all four vinylarenes 1–4 with high enantioselectivity and diastereoselectivity (Figure 2). Specifically, cyclopropanation of styrene 1 with EDA in the presence of the mutant CYP119(+)-L155W occurred to form the (1S,2R)-enantiomer of the cis-cyclopropane 5 in (+)98% ee, 90:1 dr, and 80% yield (10,000 TON). The same reaction in the presence of CYP119(−)-V254L gave the opposite (1R,2S)-enantiomer of 5 with (−)98% ee, 73:1 dr, and similar yield (74%). The diastereoselectivity for the cis products formed by variants of Ir(Me)-PIX CYP119 was over 6-fold higher than that by evolved Fe-PIX enzymes,21 and variants of Ir(Me)-PIX CYP119 were identified that provide either enantiomer of the cis-cyclopropane product with high enantioselectivity. Because the solubility of EDA is greater than that of the alkene in water and because EDA competitively couples with itself to form alkene, EDA was added by syringe pump. This procedure, commonly used for the reactions of highly active iridium catalysts, leads to higher yields than does the procedure with batch addition of EDA.9,22
The cis- and trans-isomers of β-methylstyrene (3 and 4) do not react with Fe-PIX proteins or artificial metalloenzymes reported previously, but they underwent highly selective cyclopropanation reactions in the presence of variants of Ir(Me)-CYP119(+) and (−), as shown in Figure 2. In the presence of the mutant CYP119(+)-A152L, cis-alkene 3 reacted with EDA to form the all-cis diastereomer of the cyclopropane product 7 with 286 TON, 99% ee, and 36:1 dr (major vs all other diasteromers with the major diastereomer being 2-c,3-c:2-t,3-t, ref = CO2Et, c = cis, t = trans). The mutant CYP119(−)-V254L afforded the opposite enantiomer of the cyclopropane 7 with 270 TON, >100:1 dr (2-c,3-c:2-t,3-t, ref = CO2Et) and (−)88% ee. trans-Alkene 4 reacted with EDA in the presence of the mutant CYP119(+)-A152V, to form the 2-c,3-t-cyclopropane 8 with 310 TON, 30:1 dr (2-c,3-t:2-t,3-c, ref = CO2Et), and 90% ee.
By following a different evolutionary trajectory, we also identified mutants that form the opposite diastereomer of the cyclopropane formed from trans-β-methylstyrene 4 (for details on the process of directed evolution, see the Supporting Information). The reaction of trans-alkene 4 and EDA in the presence of the variant C317G, T213G, V254L, A152Y afforded the cyclopropane with a trans relationship between the ester and the phenyl group with 220 TON, 94% ee, and 6:1 dr (2-t,3-c:2-c,3-t, ref = CO2Et) (Figure 2).
Having identified highly selective Ir(Me)-PIX CYP119 variants for cyclopropanation reactions, we evaluated these artificial enzymes as catalysts for the cyclopropanation of unactivated alkenes. This class of alkene has not been reported to react in the presence of Fe-PIX enzymes. The mutants CYP119(+) and CYP119(−) catalyzed the reaction of hexen-2-one 9 and EDA to form opposite enantiomers of cis-cyclopropanes (Table 1). Highly selective variants of the enzymes were identified from the pool of 12 mutants that catalyzed the reactions of substrates 1–4 with the highest enantioselectivities (Figure 3). No further mutagenesis was needed to achieve high diastereoselectivity and enantioselectivity. In the presence of CYP119(−)-V254L-L155T, the terminal alkene 9 underwent cyclopropanation with EDA to form the cis-cyclopropane 10 in (−)99% ee, 7:1 dr (cis:trans), and 68% yield with 1006 TON. The variant CYP119(+)-L155W formed the opposite enantiomer of the cis-cyclopropane (44% yield, 440 TON, 99% ee, 19:1 dr). The related substrate 1-octene was also functionalized with high enantio- and diastereoselectivity to form cyclopropane 11 (Figure 4), although the yield of this reaction was likely limited by the poor water solubility of 1-octene.
Figure 3.
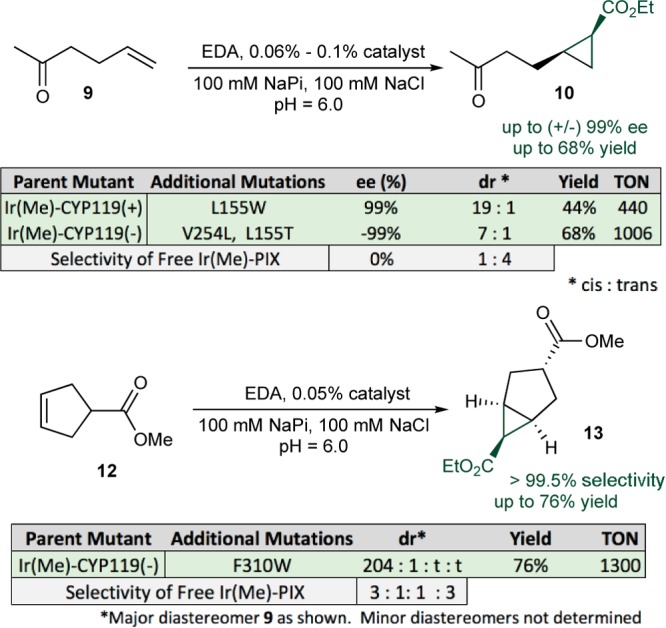
Cyclopropanation of aliphatic alkenes. The reactions were conducted on 1 mL scale under the listed conditions using 20 mM alkene and 3 equiv of EDA (added over 3 h by syringe pump as a 30% (v:v) solution in DMF). The yields were determined by GC using dodecane as internal standard. The stereochemistry of products was assigned based on NMR analysis (see the Supporting Information); the absolute stereochemistry of 10 was not assigned.
Internal, unactivated, aliphatic alkenes also underwent highly stereoselective cyclopropanation in the presence of the derivatives of Ir(Me)-PIX CYP119(+) and (−). The reaction of cyclopentene 12 and EDA formed cyclopropane 13 in 76% yield with 1300 TON in the presence of CYP119(−)-F310W (Figure 3). The stereoisomer 13 was >99.5% of the cyclopropane diastereomers. This diastereoselectivity contrasts sharply with that of the reaction in the presence of the free Ir(Me)-PIX cofactor. The free cofactor formed a mixture of four diastereomers, none of which accounted for >45% of the product. The related substrate cyclooctene reacted with EDA in the presence of the mutant T213G, V254L, L318F, L155W to form the cis-cyclopropane 14 with 14:1 dr and 36 TON (Figure 4). Like the yield of the reaction of 1-octene, the yield of this reaction was likely limited by the poor water solubility of the alkene.
The acyclic, internal alkene cis-2-octene also underwent stereoselective cyclopropanation with EDA. This reaction formed cyclopropane 15 in up to 94% ee, 108:1 dr, and with 99 TON in the presence of Ir(Me)-CYP119(−)-V254L (Figure 4). No comparable stereoselectivities have been achieved using a small molecule catalyst for the cyclopropanation of acyclic, internal, aliphatic alkenes.23
Electron poor, unactivated, 1,1-disubstituted alkenes also underwent stereoselective cyclopropanation in the presence of Ir(Me)-CYP119 enzymes, but with modest yields and turnover numbers (Figure 4). The variant CYP119(+)-155W catalyzed the reaction of ethyl methacrylate and EDA to form the product 16 in 80% ee and 8:1 dr (cis:trans) with 47 TON. The variant CYP119(−)-L155V afforded the opposite enantiomer of 16 with 88% ee, 1:1 dr, and 145 TON.
Because of this reactivity of the artificial enzymes for cyclopropanation of aliphatic alkenes, we investigated the cyclopropanation of unactivated aliphatic alkenes commonly found in natural products, such as the terpenes (Figure 5). The selective cyclopropanation of this class of molecules represents a challenging synthetic problem because of the possibility to form multiple diastereomeric products.24 In fact, we found that commonly used Rh22,25−28 catalysts form a complex mixture of all diastereomers in the reactions of chiral, nonracemic 1,1-disubstituted alkenes (Figure S3). An enzyme catalyst provides the potential to recognize the overall structure of the substrate, rather than the local orientation of substituents at the alkene or relative local size of substituents at the alkene.
Figure 5.

Cyclopropanation reactions of natural terpenes and their derivatives. Above: selectivities obtained from Ir(Me)-PIX CYP119 variants identified by directed evolution. Below: The reaction of (−)-carvone with EDA under conditions optimized to produce the product in the highest yield and diastereoselectivity. The yields were determined by GC using dodecane as internal standard. EDA was added slowly using a syringe pump.
The evaluation of the cyclopropanation reactions of seven terpenes 17–23 catalyzed by a library of mutants of Ir(Me)-PIX CYP119 revealed that specific variants of the enzyme catalyze the reactions with strong control over diastereoselectivity (Figure 5 and Table S2). For example, the reaction of β-pinene and EDA in the presence of the free Ir(Me)-PIX cofactor occurred to form an equimolar mixture of diastereomers (17), whereas the same reaction in the presence of Ir(Me)-CYP119-T213V formed predominantly one isomer of the product, with 7:1 dr. In the presence of the same variant of the enzyme, (+)-pinocarvone, a close derivative of β-pinene, underwent reaction with EDA to form one major isomer of the product 18, which is 89% of the stereoisomers formed, while the same reactions catalyzed by the free cofactor formed this isomer as only 43% of the cyclopropane product.
Cyclopropanations of the other five terpenes catalyzed by variants of Ir(Me)-CYP119 also occurred with diastereoselectivities that were distinct from those of reactions of the free cofactor. The major diastereomers from the reactions catalyzed by the enzymes were different from those of the reactions catalyzed by the free cofactor, and the reactions catalyzed by the enzymes occurred with higher diastereoselectivities. In the presence of the mutant T213G, V254L, L155W, cyclopropanes 19–21 were formed with the major diastereomer being 73–79% of the stereoisomers formed, while the same reactions catalyzed by the free Ir(Me)-cofactor produced the same diastereomers as only 9–12% of the cyclopropanation products. Similarly, in the presence of the same mutant of the enzyme, the cyclopropanation of (−)-carvone (23) occurred to form a product mixture consisting of 73% of a major diastereomer which was a minor diastereomer (11%) formed in the presence of the free cofactor. The reaction of (+)-limonene (22) with EDA occurred with higher stereoselectivity when catalyzed by a Ir(Me)-CYP119 variant containing the additional mutation L318F.
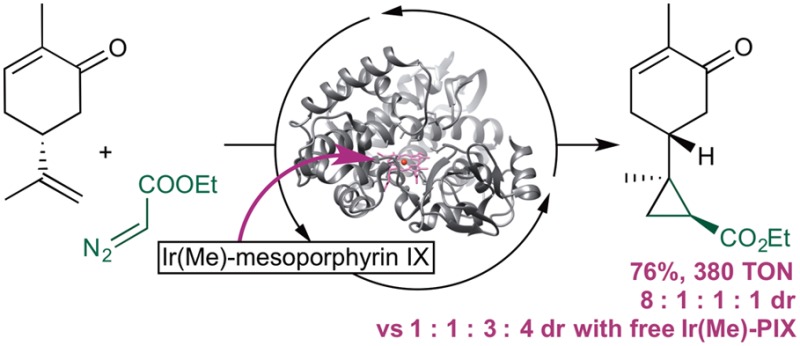
After identifying a selective enzyme for each substrate, we conducted studies to determine if modification of the reaction parameters would result in an improvement in the yield of reactions of this class of substrate. Using the reaction of carvone with EDA as a representative example, we found that the reaction of carvone 23 with 10 equiv of EDA delivered the cyclopropane products in 76% yield, with 380 TON, and 8:1:1:1 diastereoselectivity. The (1S,2R)-isomer 24 was the major product, as determined by single-crystal X-ray diffraction of the semicarbazone derivative (Figure 5). The Ir(Me)-PIX enzyme was also suitable for reactions on a preparative scale: the reaction of 1 mmol of carvone gave product 20 in 52% isolated yield with 1400 TON and with diastereoselectivity that was comparable to that observed for the reaction on an analytical scale.
To determine the relative propensity of these artificial enzymes and more conventional chiral rhodium catalysts to catalyze stereoselective cyclopropanations, we conducted the reactions of carvone with two dirhodium complexes that are well established to catalyze enantioselective cyclopropanations of certain alkenes. In contrast to the high selectivity for the reaction of carvone catalyzed by the artificial metalloenzyme, the reactions catalyzed by Rh2(5R-MEPY)4 and Rh2(4S-MEOX)4 occurred with low diastereoselectivity. The cyclopropanation of carvone catalyzed by these complexes formed the four diastereomers in 1:2:2:3 and 3:1:3:1 ratios, respectively (Table S3).29
Selective binding of one substrate in the presence of a mixture allows natural enzymes to react with substrates that are typically less reactive in the presence of those that are typically more reactive.30,31 The sizes of alkenes 25 and 26 are similar, but the position of the double bond is different. Hence, the reactivity of these two alkenes is different and the terminal alkene is more reactive toward most catalysts (Figure 6).32 The reaction of a mixture of 25 and 26 in the presence of the free Ir(Me)-PIX occurred preferentially with 1-octene 25 over 2-octene 26 to form cyclopropane 11 as 92% of the total product mixture. In sharp contrast, the reaction of EDA with a mixture of 25 and 26 catalyzed by Ir(Me)-CYP119(−)-V254L occurred predominantly with 15, producing a 65:35 ratio of 15 to 11. This result constitutes the first example of substrate-selective catalysis achieved with artificial metalloenzymes.
Figure 6.
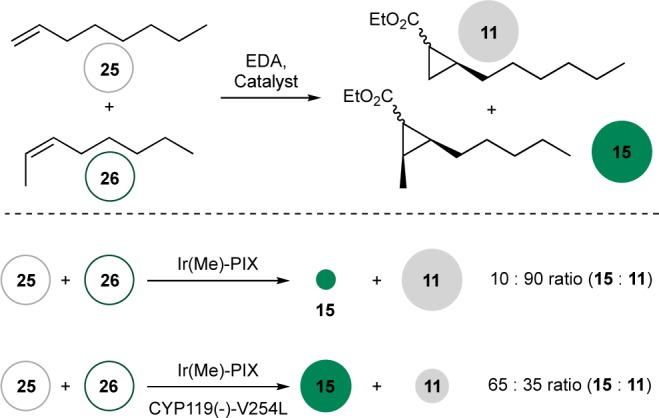
Substrate-selective cyclopropanation catalyzed by a variant of CYP119 in comparison to the same reaction catalyzed by the free cofactor. Reaction conditions: 10 mM 25, 10 mM 26, 60 mM EDA (added by syringe pump over 1 h), 0.17% catalyst, 100 mM NaPi/100 mM NaCl, pH 6.0, 3 vol % DMF. The amounts of 11 and 15 are the sum of all stereoisomers of the product. In the case of the enzyme catalyzed reaction, 11 and 15 were produced in comparable stereoselectivities to those of reactions of the individual substrates.
The body of results of this study demonstrate that artificially metalated P450 enzymes retain the properties of the active sites of native P450 enzymes. The results obtained for the substrates presented here, which have different sizes and shapes, distinct substitution patterns, and varied electronic properties, underscore the potential of the active site of CYP119 to be evolved to accommodate a wide range of substrates and the Ir(Me)-center to react with a broader scope of structures than has been achieved with Fe-PIX enzymes. Such artificial metalloenzymes perform catalytic transformations with exquisite stereoselectivity and with access to modes of selectivity of natural enzymes previously unrealized by artificial metalloenzymes. This approach creates avenues to combine, in a practical way, the abiological reactivity of transition metals with exquisite selectivity of enzymes.
Acknowledgments
The authors thank Dr. Jeff Pelton from the QB3 900 MHz NMR facility for assistance in assigning the stereochemistry of the products, Dr. Tony Iavarone for measuring the native ESI-MS of the Ir(Me)-CYP119 protein, the QB3 macrolab for competent cells, and the UC Berkeley DNA sequencing facility for sequencing the plasmids.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00391.
Author Contributions
§ H.M.K. and P.D. contributed equally.
This work was supported by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, the NSF (graduate research fellowship to H.M.K.), the NWO Netherlands Organization for Scientific Research (the Rubicon postdoctoral fellowship, No. 680-50-1306, to P.D.), and the Singapore Agency for Science, Technology and Research (National Science Scholarship to Z.L.)
The authors declare the following competing financial interest(s): J.F.H., H.M.K., P.D., and D.S.C. are inventors on PCT Application No. PCT/US2016/057032, filed October 14, 2016, by the Lawrence Berkeley National Laboratory, that covers preparation and application of the artificial metalloenzymes containing iridium porphyrins in this paper.
Supplementary Material
References
- Lewis J. C. Artificial Metalloenzymes and Metallopeptide Catalysts for Organic Synthesis. ACS Catal. 2013, 3, 2954–2975. 10.1021/cs400806a. [DOI] [Google Scholar]
- Bornscheuer U. T.; Huisman G. W.; Kazlauskas R. J.; Lutz S.; Moore J. C.; Robins K. Engineering the Third Wave of Biocatalysis. Nature 2012, 485, 185–194. 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- Reetz M. T. Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future. J. Am. Chem. Soc. 2013, 135, 12480–12496. 10.1021/ja405051f. [DOI] [PubMed] [Google Scholar]
- Reetz M. T.; Peyralans J. J.-P.; Maichele A.; Fu Y.; Maywald M. Directed Evolution of Hybrid Enzymes: Evolving Enantioselectivity of an Achiral Rh-Complex Anchored to a Protein. Chem. Commun. 2006, 4318–4320. 10.1039/b610461d. [DOI] [PubMed] [Google Scholar]
- Hyster T. K.; Ward T. R. Genetic Optimization of Metalloenzymes: Enhancing Enzymes for Non-Natural Reactions. Angew. Chem., Int. Ed. 2016, 55, 7344–7357. 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- Key H. M.; Dydio P.; Clark D. S.; Hartwig J. F. Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron. Nature 2016, 534, 534–537. 10.1038/nature17968. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Key H. M.; Nazarenko A.; Rha J. Y.-E.; Seyedkazemi V.; Clark D. S.; Hartwig J. F. An Artificial Metalloenzyme with the Kinetics of Native Enzymes. Science 2016, 354, 102–106. 10.1126/science.aah4427. [DOI] [PubMed] [Google Scholar]
- Dydio P.; Key H. M.; Hayashi H.; Clark D. S.; Hartwig J. F. Chemoselective, Enzymatic C-H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor. J. Am. Chem. Soc. 2017, 139, 1750–1753. 10.1021/jacs.6b11410. [DOI] [PubMed] [Google Scholar]
- Anding B. J.; Ellern A.; Woo L. K. Olefin Cyclopropanation Catalyzed by Iridium(III) Porphyrin Complexes. Organometallics 2012, 31, 3628–3635. 10.1021/om300135f. [DOI] [Google Scholar]
- Wang J.-C.; Xu Z.-J.; Guo Z.; Deng Q.-H.; Zhou C.-Y.; Wan X.-L.; Che C.-M. Highly Enantioselective Intermolecular Carbene Insertion to C–H and Si–H Bonds Catalyzed by a Chiral Iridium(Iii) Complex of a D 4 -Symmetric Halterman Porphyrin Ligand. Chem. Commun. 2012, 48, 4299–4301. 10.1039/c2cc30441d. [DOI] [PubMed] [Google Scholar]
- Dwivedi N.; Tewari N.; Tiwari V. K.; Chaturvedi V.; Manju Y. K.; Srivastava A.; Giakwad A.; Sinha S.; Tripathi R. P. An Efficient Synthesis of Aryloxyphenyl Cyclopropyl Methanones: a New Class of Antimycobacterial Agents. Bioorg. Med. Chem. Lett. 2005, 15, 4526–4530. 10.1016/j.bmcl.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Wang Z. J.; Renata H.; Peck N. E.; Farwell C. C.; Coelho P. S.; Arnold F. H. Improved Cyclopropanation Activity of Histidine-Ligated Cytochrome P450 Enables the Enantioselective Formal Synthesis of Levomilnacipran. Angew. Chem., Int. Ed. 2014, 53, 6810–6813. 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Tückmantel W.; Eaton J. B.; Yuen P.-W.; Yu L.-F.; Bajjuri K. M.; Fedolak A.; Wang D.; Ghavami A.; Caldarone B.; et al. Chemistry and Behavioral Studies Identify Chiral Cyclopropanes as Selective A4β2-Nicotinic Acetylcholine Receptor Partial Agonists Exhibiting an Antidepressant Profile. J. Med. Chem. 2012, 55, 717–724. 10.1021/jm201157c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyster T. K.; Arnold F. H. P450BM3-Axial Mutations: a Gateway to Non-Natural Reactivity. Isr. J. Chem. 2015, 55, 14–20. 10.1002/ijch.201400080. [DOI] [Google Scholar]
- Srivastava P.; Yang H.; Ellis-Guardiola K.; Lewis J. C. Engineering a Dirhodium Artificial Metalloenzyme for Selective Olefin Cyclopropanation. Nat. Commun. 2015, 6, 7789. 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho P. S.; Brustad E. M.; Kannan A.; Arnold F. H. Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]
- Coelho P. S.; Wang Z. J.; Ener M. E.; Baril S. A.; Kannan A.; Arnold F. H.; Brustad E. M. A Serine-Substituted P450 Catalyzes Highly Efficient Carbene Transfer to Olefins in Vivo. Nat. Chem. Biol. 2013, 9, 485–487. 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeaux M.; Tyagi V.; Fasan R. Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts. Angew. Chem., Int. Ed. 2015, 54, 1744–1748. 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gober J. G.; Rydeen A. E.; Gibson-O’Grady E. J.; Leuthaeuser J. B.; Fetrow J. S.; Brustad E. M. Mutating a Highly Conserved Residue in Diverse Cytochrome P450s Facilitates Diastereoselective Olefin Cyclopropanation. ChemBioChem 2016, 17, 394–397. 10.1002/cbic.201500624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj P.; Sreenilayam G.; Tyagi V.; Fasan R. Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem., Int. Ed. 2016, 55, 16110–16114. 10.1002/anie.201608680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fe-P450 CIS-T438S, reported previously by Arnold et al., catalyzes the same transformation producing the same stereoisomer of the product with 11.5:1 dr.
- Davies H. M. L.; Denton J. R. Application of Donor/Acceptor-Carbenoids to the Synthesis of Natural Products. Chem. Soc. Rev. 2009, 38, 3061–3071. 10.1039/b901170f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C.; Wang L.-J.; Zhu J.; Tang Y. A Chiral Cagelike Copper(I) Catalyst for the Highly Enantioselective Synthesis of 1,1-Cyclopropane Diesters. Angew. Chem., Int. Ed. 2012, 51, 11620–11623. 10.1002/anie.201206376. [DOI] [PubMed] [Google Scholar]
- Friedrich E. C.; Niyati-Shirkhodaee F. Regioselectivity and Solvent Effects in Cyclopropanation of Alkadienes. J. Org. Chem. 1991, 56, 2202–2205. 10.1021/jo00006a044. [DOI] [Google Scholar]
- Denton J. R.; Davies H. M. L. Enantioselective Reactions of Donor/ Acceptor Carbenoids Derived from α-Aryl-α-Diazoketones. Org. Lett. 2009, 11, 787–790. 10.1021/ol802614j. [DOI] [PubMed] [Google Scholar]
- Chepiga K. M.; Qin C.; Alford J. S.; Chennamadhavuni S.; Gregg T. M.; Olson J. P.; Davies H. M. L. Guide to Enantioselective Dirhodium(II)-Catalyzed Cyclopropanation with Aryldiazoacetates. Tetrahedron 2013, 69, 5765–5771. 10.1016/j.tet.2013.04.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Guptill D. M.; Varela-Alvarez A.; Musaev D. G.; Davies H. M. L. Rhodium-Catalyzed Enantioselective Cyclopropanation of Electron-Deficient Alkenes. Chemical Science 2013, 4, 2844–2850. 10.1039/c3sc50425e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanthamath S.; Iwasa S. Enantioselective Cyclopropanation of a Wide Variety of Olefins Catalyzed by Ru(II)–Pheox Complexes. Acc. Chem. Res. 2016, 49, 2080–2090. 10.1021/acs.accounts.6b00070. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Eismont M. Y.; Protopopova M. N.; Kwan M. M. Y. Enantioselective Intramolecular Cyclopropanation of N-Allylic- and N-Homoallylic Diazoacetamides Catalyzed by Chiral Dirhodium(II) Catalysts. Tetrahedron 1994, 50, 1665–1674. 10.1016/S0040-4020(01)80842-5. [DOI] [Google Scholar]
- Johnston J. B.; Ouellet H.; Podust L. M.; Ortiz de Montellano P. R. Structural Control of Cytochrome P450-Catalyzed Ω-Hydroxylation. Arch. Biochem. Biophys. 2011, 507, 86–94. 10.1016/j.abb.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse C. J. C.; Bell S. G.; Wong L.-L. P450(BM3) (CYP102A1): Connecting the Dots. Chem. Soc. Rev. 2012, 41, 1218–1260. 10.1039/C1CS15192D. [DOI] [PubMed] [Google Scholar]
- Maxwell J. L.; O’Malley S.; Brown K. C.; Kodadek T. Shape-Selective and Asymmetric Cyclopropanation of Alkenes Catalyzed by Rhodium Porphyrins. Organometallics 1992, 11, 645–652. 10.1021/om00038a023. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.