

Abstract
Chiral pentose sugars mediate the enantioselective synthesis of amino acid precursors, with the magnitude of the chiral induction dictated by a subtle cooperativity between sugar hydroxyl groups. Ribose and lyxose give opposite chiral preferences, and theoretical calculations reveal the pseudoenantiomeric nature of transition state structures from the two sugars. Prebiotically plausible mixtures of natural d-sugars lead to enantioenrichment of natural l-amino acid precursors. Temporal monitoring and kinetic modeling of the reaction reveal an unusual dynamic kinetic resolution that shifts toward an enantioselective pathway over time, providing an amplification mechanism for the transfer of chiral information. This work adds to growing evidence for synergy in the etiology of the single chirality of the two most important classes of biological molecules, the sugars that make up DNA and RNA and the amino acids that form proteins.
Short abstract
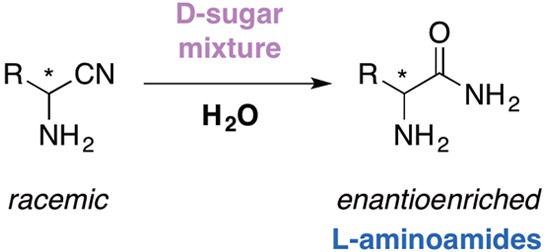
d-Aldopentose sugar mixtures mediate the hydration of racemic aminonitriles under prebiotically plausible conditions to yield the natural l-aminoamide products. The stereochemical outcome is dictated by the sugar C2 and C3 hydroxyl stereochemistry.
The single chirality of biological molecules is a signature of life, and its origin remains an unanswered fundamental question. Theoretical and experimental proposals for the emergence of single chirality in amino acids and sugars have been considered for more than half a century. Models for enantioenrichment have been proposed1−4 based on both chemical reactions5−7 and physical phase behavior,8−16 with limitations, however, in that either the systems under study involve chemistry that is not prebiotically plausible (as in the Soai autocatalytic formation of pyrimidyl alcohols6) or they apply only to a specific and narrow range of chiral molecules (as in attrition-enhanced deracemization of chiral conglomerate crystals14−16). Thus, a general rationalization for the emergence of the single chirality of sugars and amino acids in the context of plausible prebiotic chemistry remains a challenging goal.
The formose reaction17 and the Strecker reaction18 represent prebiotically plausible routes to sugar and amino acid building blocks, respectively (Scheme 1). While recent advances have expanded our chemistry toolbox leading to biological building blocks, the issue of chirality has often been left to the side.19−29
Scheme 1. Prebiotic Reactions Implicated in the Emergence of Biological Homochirality.

A number of studies3,30−39 have begun to postulate routes to enantioenrichment of sugars or amino acids via either asymmetric catalysis or kinetic resolution, where the chirality of one class of molecules induces enantioenrichment in the other. For example, several pathways toward enantioenrichment of glyceraldehyde mediated by amino acids have been explored. Breslow and co-workers30 showed that amino acids catalyze the formose reaction to produce glyceraldehyde in small enantiomeric excesses that could be amplified further by physical processes (Scheme 2a). While proline gave the largest absolute ee values in the catalytic reaction, the major enantiomer proved to be the unnatural d-amino acid. However, Hein and Blackmond3 showed that the natural hand of glyceraldehyde is the major product when a prolinate salt rather than proline was employed as the catalyst in the formose reaction (Scheme 2b). Further work by Breslow offered detailed mechanistic proposals to rationalize these preferences.31 Blackmond and co-workers also showed that amino acid additives could induce enantioenrichment in the Powner/Sutherland ribonucleotide synthesis both to amplify glyceraldehyde enantiomeric excess (Scheme 2c) and to produce enantiopure activated pyrimidine nucleotide precursors, again aided by physical amplification processes.32 Interestingly, as seen by comparing Schemes 2c and 2d, this kinetic resolution may be configured so that either the amino acid resolves the sugar or, conversely, so that the sugar resolves the amino acid.
Scheme 2. Examples of Enantioenrichment of Sugars and Amino Acids in Prebiotically Relevant Reactions.

(a, b) Formose reaction; (c, d) kinetic resolution accompanying synthesis of ribonucleotide precursors; (e) kinetic resolution in Strecker hydration of aminonitriles.
Amino acid enantioenrichment via Strecker chemistry was also studied more than a quarter century ago by the groups of Taillades and Commeyras36−39 who probed enantioselective carbonyl-mediated hydration of amino acid precursors (Scheme 2e). All of these studies hint at a synergy between sugars and amino acids in processes for enantioenrichment in both classes of molecules.
Our current study of sugar-mediated enantioenrichment in amino acid synthesis was inspired by the Commeyras studies.36 The prebiotic relevance of that work was limited by the use of alcohol reaction media and complex natural products as chiral catalysts. While later studies40−44 have implicated carbonyl catalysis in amino acid formation, including Eschenmoser’s examination of the etiological relevance of the acetaldehyde addition to the HCN-tetramer—a “classic of prebiotic chemistry”40—these subsequent investigations either did not address chirality40−42 or did not employ prebiotically relevant conditions.43,44
Our work sought to probe the viability of chiral aldopentose sugars to mediate enantioenrichment in the Strecker synthesis of amino acid precursors under prebiotically relevant conditions (Scheme 3).45 Derivatives of these sugars recently identified on meteoritic samples were shown to exhibit significant enantiomeric excesses toward the natural (d) enantiomer,46 and their formation in interstellar ice analogues, albeit without indication of their stereochemistry, has recently been reported.47
Scheme 3. Enantioselective Synthesis of Amino Acids Mediated by Chiral Sugars.

For the reaction of Scheme 3, we envisioned that equilibria favoring the pyranose and furanose cyclic forms of aldopentoses may shift toward the less abundant open chain form as the carbonyl group of the sugar reacts with aminonitriles, drawing more of the sugar into the catalytic cycle. Table 1 confirms that reactions using chiral pentose sugars gave aminoamide products in 9–29% isolated yield,48 with modest to significant product enantiomeric excess (11–83% ee) leading to the aminoamides of alanine (Ala-II), phenylalanine (Phe-II), and tryptophan (Trp-II). Table 2 shows that l-sugars give results opposite to their d-enantiomers as expected. In the absence of sugar, the reaction gives racemic product.48
Table 1. Enantioenrichment of Amino Acid Precursors Driven by d-Sugars (Scheme 3)a.
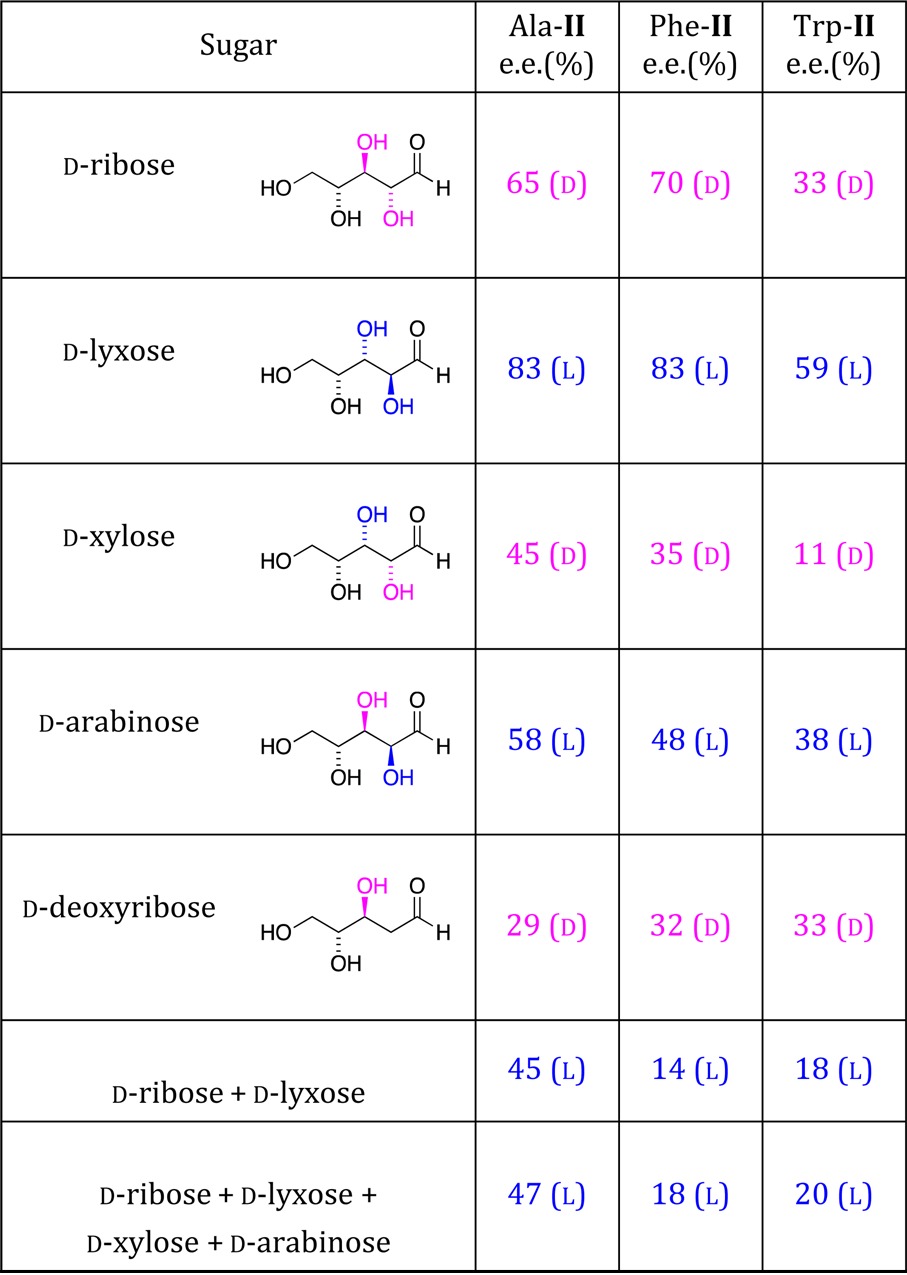
Reaction conditions: 0.25 M AM-I with 0.50 M sugar in H2O with 0.25 M NaOH at 22–24 °C; Ala-I (7 d), Phe-I (7 d), Trp-I (5 d). For reactions employing mixtures of sugars: 0.25 M in each sugar; Ala-I (8 d), Phe-I (7 d), Trp (7 d). Enantiomeric excess measured using chiral HPLC after derivatization of AM-II.
Table 2. Opposite Sense of Enantioenrichment of Phe-II for l-Sugarsa.
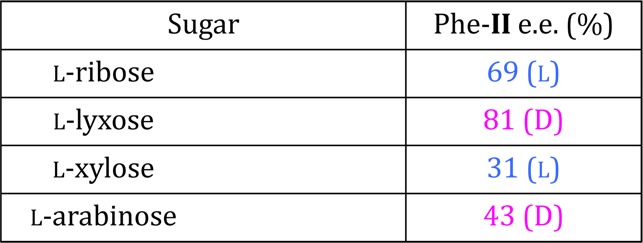
Reaction conditions: 0.25 M Phe-I with 0.50 M sugar in H2O with 0.25 M NaOH at 22–24 °C; (7 d). Enantiomeric excess measured using chiral HPLC after derivatization of AM-II.
Preliminary studies also show similar trends using chiral C4 and C6 aldoses.48 Hydrolysis of enantioenriched aminoamides to amino acids occurs with fidelity in the chiral center.49 Similar trends are observed in reactions using catalytic quantities of sugar as low as 0.025 M (Table 3) and at pH values as low as 7 (Table 4), with both conversion and enantioenrichment developing more slowly at lower pH and sugar concentrations. These results demonstrate sugar-driven enantioenrichment under prebiotically relevant reaction conditions.
Table 3. Effect of Sugar Concentration on Phe-II ee (%) for Reaction Mediated by d-Ribosea.
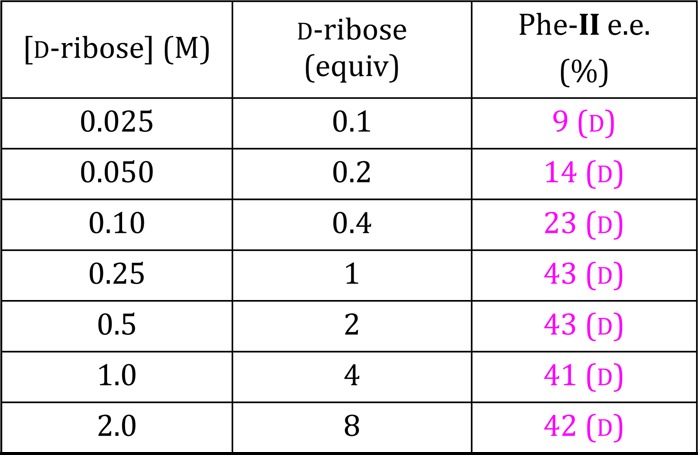
Reaction conditions: Phe-I concentrations as shown with 0.25 M NaOH in H2O at 22–24 °C; (1 d). Enantiomeric excess measured using chiral HPLC after derivatization of AM-II.
Table 4. Effect of Solution pH on Phe-II ee (%) for Reaction Mediated by d-Ribosea.
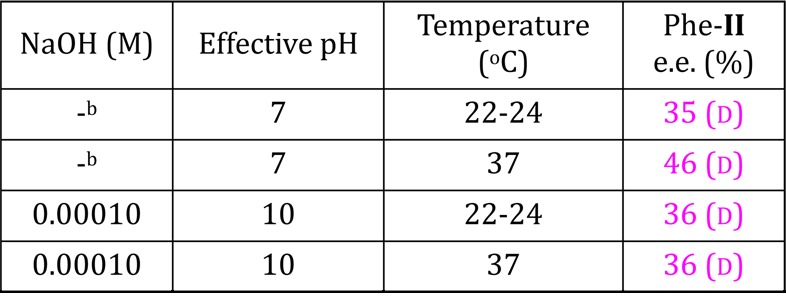
Reaction conditions: 0.25 M Phe-I with 0.50 M sugar in H2O with NaOH concentration and temperature as listed; (35 d). Enantiomeric excess measured using chiral HPLC after derivatization of AM-II.
Deionized H2O employed with no added NaOH.
When the reaction of Scheme 2 is mediated by a mixture of equal parts d-ribose and d-lyxose, or equal mixtures of four d-pentoses, the observed enantiomeric excess favors the natural l-amino acid precursor (Table 1, final two entries), implying that lyxose and arabinose compete better than ribose and xylose in the mixture. It is important to note that sugars synthesized under prebiotically plausible conditions50 or extracted from meteoritic samples46 yield derivatives of a mixture of the pentoses shown in Table 1, even though in modern biology these pentoses do not play equal roles and are not formed in similar relative concentrations. For example, enantioenriched derivatives of the biologically rare lyxose were found in similar abundance to derivatives of ribose on two separate meteorites.46 While d-ribose and d-deoxyribose ultimately came to serve as key building blocks of biological molecules, our results suggest a key role for other prebiotically common d-pentoses such as d-lyxose in mediating the emergence of l-amino acid homochirality.
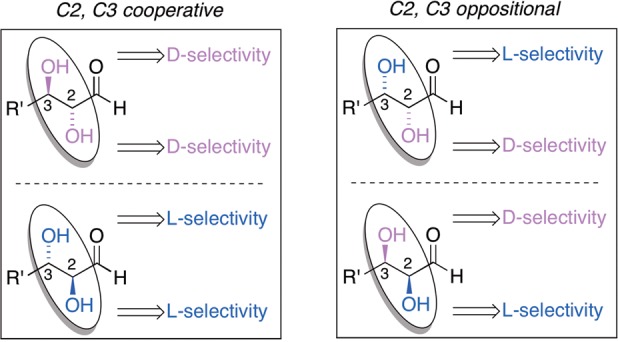
The sense of enantioenrichment exhibits a striking correlation with the chirality of the hydroxyl group of the C2 carbon alpha to the carbonyl group of the sugar (Scheme 4). Thus, d-ribose and d-lyxose favor opposite enantiomers of AM-II, as do d-xylose and d-arabinose. Interestingly, d-deoxyribose, which lacks the chiral center at C2 but retains d-ribose’s R chiral center at the beta C3 carbon, gives the same sense of the aminoamide product as d-ribose, albeit with lower enantioselectivity for Ala-II and Phe-II. This suggests that the magnitude of chiral induction is a subtle function of both C2 and C3 stereochemistry of the sugar: the (R,R) and (S,S) configurations respectively at (C2, C3) for d-ribose and d-lyxose show higher absolute enantiomeric excesses than do d-xylose (R,S) and d-arabinose (S,R), because the C3 selectivity works in concert with C2 for the former two and in weak opposition for the latter two (Scheme 4).
Scheme 4. Stereochemical Rationalization of Enantioenrichment by Chiral Sugars.
Monitoring the entire reaction progress allows a full range of reactant concentrations to be probed over the course of the reaction, including low concentrations of prebiotic relevance that naturally prevail at high conversion. Strikingly, the enantiomeric excess of Phe-II increases from nearly racemic at the outset of the reaction (Figure 1, symbols) and continues to rise for nearly 1 week, long after full consumption of the aminonitrile, which disappears in under 4 h. This behavior is unusual both in a conventional kinetic resolution, where product ee is predicted to be at its maximum at the reaction outset, and in a dynamic kinetic resolution, where product ee should remain constant over time. Temporal reaction profiling by NMR spectroscopy confirmed that not all of the reacted aminonitrile is immediately captured as aminoamide product, suggesting the buildup of a reservoir of intermediates. Two-dimensional NMR studies implicate the development of covalently bound aminonitrile-sugar species over time.29 Further investigations to confirm the nature of these intermediates are ongoing.
Figure 1.
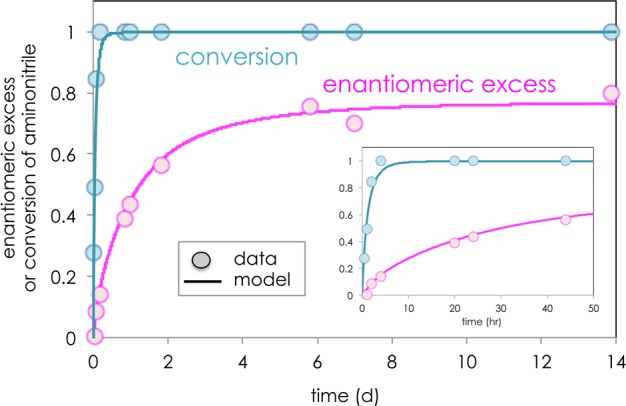
Temporal evolution of product enantiomeric excess in the reaction of Phe-I to form Phe-II mediated by d-ribose (Scheme 3). Experimental data (symbols) and kinetic modeling48 (solid lines) of ee and conversion as a function of time. Inset expands early stages of the reaction in hours.
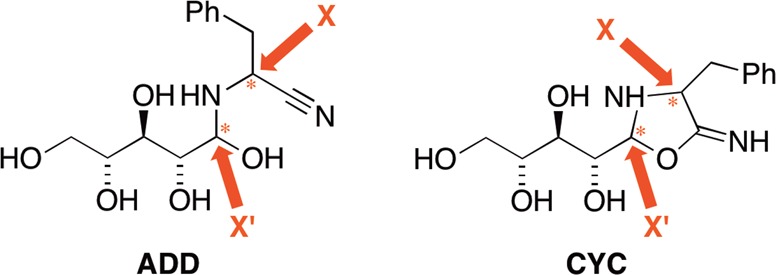
A general network for the reaction of racemic aminonitriles mediated by aldopentoses that is consistent with these observations is proposed in Figure 2. The mechanism is illustrated here for a generic aminonitrile AM-I and d-lyxose, the sugar that in all cases afforded the highest enantiomeric excess toward the natural l-aminoamides. The mechanism proposes that diastereomeric linear hemiaminals ADD are formed that may cyclize to form imine intermediates CYC, in analogy to the Bucherer–Bergs hydantoin synthesis.51ADD species may hydrate directly and unselectively to AM-II, but enantioenrichment emerges over time as the reaction is increasingly channeled through the major cyclic intermediate species CYCmaj.
Figure 2.

Proposed reaction network for the synthesis of amino acid precursors via the dynamic kinetic resolution of racemic aminonitriles. The network is shown for reaction of a generic aminonitrile AM-I mediated by d-lyxose. Shaded area highlights the enantioselective pathway forming a reservoir of the major cyclic intermediate CYCmaj that effectively channels both enantiomers of the aminonitrile toward the aminoamide l-AM-II, the natural enantiomer of its corresponding amino acid.
Figure 1 (solid lines) shows that a simple kinetic model48,52 based on the mechanism of Figure 2 captures both the temporal increase in enantiomeric excess of aminoamide and the initial rapid consumption of aminonitrile. The buildup of a reservoir of CYCmaj intermediates becomes the source of selectivity in the sugar-mediated reaction, channeling both enantiomers of the aminonitrile substrate into the major pathway, as indicated by the shaded portion of Figure 2. This network provides a unique mechanism for the transmission and amplification of chiral information from the sugars to the amino acids.
Calculations were performed to evaluate the relative stability and reactivity of hemiaminal conformers ADD, the viability of the deprotonated form of these conformers to undergo cyclization forming cyclic imine intermediates CYC, and the relative stability of the conformers CYC.48 Four families of conformers of the Phe-I-d-ribose hemiaminal system are defined by (X, X′) for the configurations of both the chiral center in the aminonitrile (X = R or S), and therefore of the aminoamide product, and that of the hemiaminal formed at the sugar carbonyl (X′ = R or S). The relative energies of the conformers are calculated along with the dihedral angle Dhcyc between the C–CN and C–O bonds that are involved in hemiaminal cyclization. The smaller the dihedral angle Dhcyc, the higher the probability of effective cyclization from ADD to CYC.
Table 5 shows that there is little difference in relative energy between the most stable ADDR and ADDS conformers formed from Phe-I and d-ribose. Significant selectivity is not expected in direct hydration of ADD to Phe-II because this barrier should not be strongly influenced by the configuration and intramolecular interactions for different diastereomeric open-chain hemiaminal ADD conformers. This calculation supports the assumption of the kinetic model that kADDmaj ≈ kADDmin. However, in the case of Phe-I reaction mediated by d-ribose, the free energy barrier for cyclization of the deprotonated form to the imine intermediate CYCR (specifically the R,S conformer) leading to the major R aminoamide product of Phe-II was found to be lower by 2.1 kcal/mol than for cyclization of CYCS. Thus, the major intermediate species CYCR (both R,R and R,S) are more stable than the minor species CYCS. Taken together, these calculations for ADD and CYC support the kinetic model’s assumption of Keq,CYCmaj > Keq,CYCmin, as well as the experimental observation of an initially unselective pathway giving way over time to enantioselective reaction.
Table 5. Calculations for Intermediates in Reaction Network of Figure 2 for the Case of Phe-I and d-Ribose.
| energy (kcal/mol) for (X,X′) |
||||
|---|---|---|---|---|
| conformer | (S,R) | (S,S) | (R,R) | (R,S) |
| most stable ADD | 1.2 | 0.1 | 0.7 | 0 |
| most stable CYC | –6.0 | –7.0 | –8.4 | –7.8 |
| free energy barrier for cyclization of deprotonated ADD to CYC | 3.9 | 3.2 | 3.9 | 1.1 |
Calculations also help to rationalize the opposite stereoselectivity observed in reactions mediated by ribose and lyxose. Figure 3 reveals the pseudoenantiomeric nature of the transition state structures for cyclization of ADD to CYC for Phe-I with (R,S)-d-ribose and (S,R)-d-lyxose. A series of symmetry operations—reflection followed by C4 inversion (since the stereochemistry at C4 is identical for the two sugars) and C4, C5 rotations—maps the transition state of the cyclic imine d-ribose-CYCR into d-lyxose-CYCS. The relevant interactions enabling cyclization are mirrored in the ribose and lyxose transition states, the only difference being that the C4 hydroxyl groups exhibit d stereochemistry in both cases, which affects the positioning of the distal C5 hydroxyl group.
Figure 3.
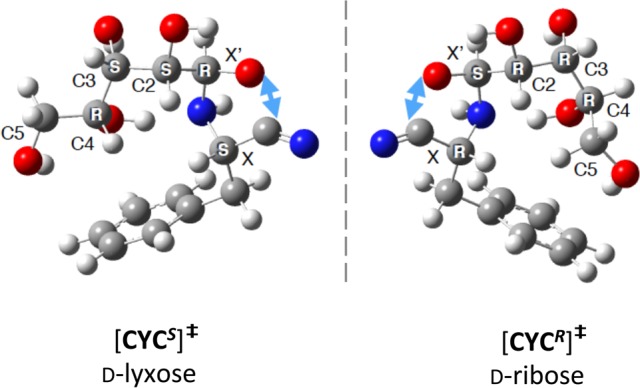
Pseudoenantiomeric character of the lowest energy transition states for cyclization from the corresponding anionic hemiaminals of Phe-I for (R,S) d-ribose and (S,R) d-lyxose. Light blue arrow shows O–C distance for attack of carbonyl oxygen on nitrile carbon.
These results demonstrate the viability of prebiotically important chiral aldopentose sugars in mediating enantioenrichment to over 80% enantiomeric excess in amino acid precursors. Enantioselectivity is in the opposite sense for reactions mediated by ribose and lyxose, and by xylose and arabinose, with product stereochemistry informed by a subtle synergy between the sugar’s chiral hydroxyl groups such that they may act either cooperatively (for the former two sugars) or in opposition (for the latter two). The pseudoenantiomeric nature of the hemiaminal transition states for ribose and lyxose rationalizes the opposite enantioselectivity observed for the two sugars. Remarkably, mixtures of equal amounts of the four natural d-aldopentose sugars yield enantioenriched natural l-amino acid precursors. While d-ribose and d-deoxyribose ultimately became critical building blocks for biological molecules, our work suggests an important role for biologically rare but prebiotically plausible mixtures of d-aldopentose sugars including d-lyxose in l-amino acid enantioenrichment. These findings highlight the complementary nature of these two classes of molecules in the emergence of biological homochirality
Acknowledgments
The authors gratefully acknowledge funding from the Simons Foundation Simons Collaboration on the Origins of Life (D.G.B. and A.J.W., SCOL 287625; D.Y.Z., SCOL 291937). D.Y.Z. acknowledges the Research Computing Group at Harvard University. D.G.B. acknowledges the TSRI High Performance Computing Facility and technical aid from R. E. Plata and P. Jain. Valuable discussions with G. F. Joyce, R. Krishnamurthy, and R. Ghadiri are acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00085.
Details of the experimental work (reaction procedures, compound characterization, analytical procedures) and computational studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Blackmond D. G. The origin of biological homochirality. Cold Spring Harbor Perspect. Biol. 2010, 2, a002147. 10.1101/cshperspect.a002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow R. A likely possible origin of homochirality in amino acids and sugars on prebiotic earth. Tetrahedron Lett. 2011, 52, 2028–2032. 10.1016/j.tetlet.2010.08.094. [DOI] [Google Scholar]
- Hein J. E.; Blackmond D. G. On the origin of single chirality of amino acids and sugars in biogenesis. Acc. Chem. Res. 2012, 45, 2045–2054. 10.1021/ar200316n. [DOI] [PubMed] [Google Scholar]
- Elsila J. E.; Aponte J. C.; Blackmond D. G.; Burton A. S.; Dworkin J. P.; Glavin D. P. Meteoritic amino acids: diversity in compositions reflects parent body histories. ACS Cent. Sci. 2016, 2, 370–379. 10.1021/acscentsci.6b00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank F. C. On spontaneous asymmetric synthesis. Biochim. Biophys. Acta 1953, 11, 459–463. 10.1016/0006-3002(53)90082-1. [DOI] [PubMed] [Google Scholar]
- Soai K.; Shibata T.; Morioka H.; Choji K. Asymmetric autocatalysis and amplification of enatiomeric excess of a chiral molecule. Nature 1995, 378, 767–768. 10.1038/378767a0. [DOI] [Google Scholar]
- Blackmond D. G.; McMillan C. R.; Ramdeehul S.; Schorm A.; Brown J. M. Origins of asymmetric amplification in autocatalytic alkylzinc additions. J. Am. Chem. Soc. 2001, 123, 10103–10104. 10.1021/ja0165133. [DOI] [PubMed] [Google Scholar]
- Morowitz H. J. A mechanism for the amplification of fluctuations in racemic mixtures. J. Theor. Biol. 1969, 25, 491–494. 10.1016/S0022-5193(69)80035-4. [DOI] [PubMed] [Google Scholar]
- Klussmann M.; Iwamura H.; Mathew S. P.; Wells D. H. Jr.; Pandya U.; Armstrong A.; Blackmond D. G. Thermodynamic control of asymmetric amplification in amino acid catalysis. Nature 2006, 441, 621–623. 10.1038/nature04780. [DOI] [PubMed] [Google Scholar]
- Klussmann M.; White A. J. P.; Armstrong A.; Blackmond D. G. Rationalization and prediction of solution enantiomeric excess in ternary phase systems. Angew. Chem., Int. Ed. 2006, 45 (47), 7985–7989. 10.1002/anie.200602520. [DOI] [PubMed] [Google Scholar]
- Klussmann M.; Izumi T.; White A. J. P.; Armstrong A.; Blackmond D. G. Emergence of solution-phase homochirality via crystal engineering of amino acids. J. Am. Chem. Soc. 2007, 129, 7657–7660. 10.1021/ja0708870. [DOI] [PubMed] [Google Scholar]
- Breslow R.; Levine M. S. Amplification of enantiomeric concentrations under credible prebiotic conditions. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 12979–12980. 10.1073/pnas.0605863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow R.; Cheng Z.-L. On the origin of terrestrial homochirality for nucleosides and amino acids. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 9144–9146. 10.1073/pnas.0904350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorduin W. L.; Izumi T.; Millemaggi A.; Leeman M.; Meekes H.; Van Enckevort W. J. P.; Kellogg R. M.; Kaptein B.; Vlieg E.; Blackmond D. G. Emergence of a single solid chiral state from a nearly racemic amino acid derivative. J. Am. Chem. Soc. 2008, 130, 1158–1159. 10.1021/ja7106349. [DOI] [PubMed] [Google Scholar]
- Viedma C.; Ortiz J. E.; Torres T. d.; Izumi T.; Blackmond D. G. Evolution of solid-phase homochirality for a proteinogenic amino acid. J. Am. Chem. Soc. 2008, 130, 15274–15275. 10.1021/ja8074506. [DOI] [PubMed] [Google Scholar]
- Hein J. E.; Huynh Cao B.; Viedma C.; Kellogg R. M.; Blackmond D. G. Pasteur’s tweezers revisited: on the mechanism of attrition-enhanced deracemization and resolution of chiral conglomerate solids. J. Am. Chem. Soc. 2012, 134, 12629–12636. 10.1021/ja303566g. [DOI] [PubMed] [Google Scholar]
- Butlerow A. Bildung einer zuckerartigen Substanz durch Synthese. Justus Liebigs Annalen der Chemie 1861, 120, 295–298. 10.1002/jlac.18611200308. [DOI] [Google Scholar]
- Strecker A. Ueber die künstliche Bildung der Milchsäure und einen neuen, dem Glycocoll homologen Körper. Annalen der Chemie und Pharmacie 1850, 75, 27–45. 10.1002/jlac.18500750103. [DOI] [Google Scholar]
- a Powner M. W.; Gerland B.; Sutherland J. D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. 10.1038/nature08013. [DOI] [PubMed] [Google Scholar]
- Patel B. H.; Percivalle C.; Ritson D. J.; Duffy C. D.; Sutherland J. D. Common origins of RNA, protein, and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 2015, 7, 301–307. 10.1038/nchem.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasi C.; Crowe M. A.; Powner M. W.; Sutherland J. D. Direct assembly of nucleoside precursors from two- and three-carbon units. Angew. Chem., Int. Ed. 2006, 45, 6176–6179. 10.1002/anie.200601267. [DOI] [PubMed] [Google Scholar]
- Xu J.; Tsanakopoulou M.; Magnani C. J.; Szabla R.; Sponer J. E.; Sponer J.; Gora R. W.; Sutherland J. D. A prebiotically plausible synthesis of pyrimidine β-ribonucleosides and their phosphate derivatives involving photoanomerization. Nat. Chem. 2016, 10.1038/nchem.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powner M. W.; Sutherland J. D.; Szostak J. W. Chemoselective multicomponent one-pot assembly of purine precursors in water. J. Am. Chem. Soc. 2010, 132, 16677–16688. 10.1021/ja108197s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggins A. J.; Powner M. W. Prebiotic synthesis of phosphoenol pyruvate by α-phosphorylation-controlled triose glycolysis. Nat. Chem. 2016, 10.1038/nchem.2624. [DOI] [PubMed] [Google Scholar]
- Islam S.; Bucar D.-K.; Powner M. W. Prebiotic selection and assembly of proteinogenic amino acids and natural nucleotides from complex mixtures. Nat. Chem. 2017, 10.1038/nchem.2703. [DOI] [Google Scholar]
- Becker S.; Thoma I.; Deutsch A.; Gehrke T.; Mayer P.; Zipse H.; Carell T. A high-yielding, strictly regioselective prebiotic purine nucleoside formation pathway. Science 2016, 352, 833–836. 10.1126/science.aad2808. [DOI] [PubMed] [Google Scholar]
- Kim H.-J.; Ricardo A.; Illangkoon H. I.; Kim M. J.; Carrigan M. A.; Frye F.; Benner S. A. Synthesis of carbohydrates in mineral-guided prebiotic cycles. J. Am. Chem. Soc. 2011, 133, 9457–9468. 10.1021/ja201769f. [DOI] [PubMed] [Google Scholar]
- Chen M. C.; Cafferty B. J.; Mamajanov I.; Gállego I.; Khanam J.; Krishnamurthy R.; Hud N. V. Spontaneous prebiotic formation of a β-ribofuranoside that self-assembles with a complementary heterocycle. J. Am. Chem. Soc. 2014, 136, 5640–5646. 10.1021/ja410124v. [DOI] [PubMed] [Google Scholar]
- Eschenmoser A. Etiology of potentially primordial biomolecular structures: from vitamin B12 to the nucleic acids and an inquiry into the chemistry of life’s origin: a retrospective. Angew. Chem., Int. Ed. 2011, 50, 12412–12472. 10.1002/anie.201103672. [DOI] [PubMed] [Google Scholar]
- Breslow R.; Cheng Z.-L. L-Amino acids catalyze the formation of an excess of D-glyceraldehyde, and thus of other D sugars, under credible prebiotic conditions. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5723–5725. 10.1073/pnas.1001639107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow R.; Ramalingam V.; Appayee C. Catalysis of glyceraldehyde synthesis by primary or secondary amino acids under prebiotic conditions as a function of pH. Origins Life Evol. Biospheres 2013, 43, 323–329. 10.1007/s11084-013-9347-0. [DOI] [PubMed] [Google Scholar]
- Hein J. E.; Tse E.; Blackmond D. G. A route to enantiopure RNA precursors from nearly racemic starting materials. Nat. Chem. 2011, 3, 704–706. 10.1038/nchem.1108. [DOI] [PubMed] [Google Scholar]
- Levine M.; Kenesky C. S.; Mazori D.; Breslow R. Enantioselective synthesis and enantiomeric amplification of amino acids under prebiotic conditions. Org. Lett. 2008, 10, 2433–2436. 10.1021/ol8007099. [DOI] [PubMed] [Google Scholar]
- Breslow R.; Levine M.; Cheng Z.-L. Imitating prebiotic homochirality on earth. Origins Life Evol. Biospheres 2010, 40, 11–26. 10.1007/s11084-009-9179-0. [DOI] [PubMed] [Google Scholar]
- Breslow R.; Appayee C. Transketolase reaction under credible prebiotic conditions. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4184–4187. 10.1073/pnas.1301522110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagriffoul P.-H.; Tadros Z.; Taillades J.; Commeyras A. Influence of a hydroalcoholic solvent on the enantioselectivity of α-aminonitrile hydration catalysed by chiral ketones. J. Chem. Soc., Perkin Trans. 2 1992, 2, 1279–1285. 10.1039/P29920001279. [DOI] [Google Scholar]
- Taillades J.; Commeyras A. Systemes de Strecker et apparentes—I: Etude de la decomposition en solution aqueuse des α-alcoyl-aminonitriles tertiaires. Mécanisme d’élimination du groupement nitrile. Tetrahedron 1974, 30, 127–132. 10.1016/S0040-4020(01)97226-6. [DOI] [Google Scholar]
- Taillades J.; Commeyras A. Systemes de Strecker et apparentes—II: Mécanisme de formation en solution aqueuse des α-alcoylaminoisobutyronitrile à partir d’acétone, d’acide cyanhydrique et d’ammoniaque, methyl ou diméthylamine. Tetrahedron 1974, 30, 2493–2501. 10.1016/S0040-4020(01)97121-2. [DOI] [Google Scholar]
- Taillades J.; Beuzelin I.; Garrel L.; Tabacik V.; Bied C.; Commeyras A. N-Carbamoyl-α-amino acids rather than free α-amino acids formation in the primitive hydrosphere: a novel proposal for the emergence of prebiotic peptides. Origins Life Evol. Biospheres 1998, 28, 61–77. 10.1023/A:1006566810636. [DOI] [PubMed] [Google Scholar]
- Koch K.; Schweizer W. B.; Eschenmoser A. Reactions of the HCN-tetramer with aldehydes. Chem. Biodiversity 2007, 4, 541–551. 10.1002/cbdv.200790049. [DOI] [PubMed] [Google Scholar]
- Edward J. T.; Chubb F. L. Pseudo-enzymatic hydrolyses of amino-nitriles to amino-amides. Proc. R. Ir. Acad. 1983, 83B, 57–64. [Google Scholar]
- Chitale S.; Derasp J. S.; Hussain B.; Tanveer K.; Beauchemin A. M. Carbohydrates as efficient catalysts for the hydration of α-amino nitriles. Chem. Commun. 2016, 52, 13147–13150. 10.1039/C6CC07530D. [DOI] [PubMed] [Google Scholar]
- Kunz H.; Sager W. Diastereoselective Strecker synthesis of α-aminonitriles on carbohydrate templates. Angew. Chem., Int. Ed. Engl. 1987, 26, 557–559. 10.1002/anie.198705571. [DOI] [Google Scholar]
- Kunz H.; Pfrengle W. Asymmetric synthesis on carbohydrate templates: stereoselective Ugi synthesis of α-amino acid derivatives. J. Am. Chem. Soc. 1988, 110, 651–652. 10.1021/ja00210a084. [DOI] [Google Scholar]
- While conditions on prebiotic earth can only be postulated, and plausible prebiotic environments remain hotly debated, we have carried out our work in aqueous media under sugar and aminonitrile concentrations similar to those of reactant and catalyst concentrations employed in other recent studies of prebiotically relevant reactions (such as Refs. (19−25) and (30−35)). Although most of our reactions employed pH values higher than those considered prebiotically relevant, we have demonstrated (Table 4) that the same trends are observed at neutral pH with extended reaction times required at lower pH.
- Cooper G.; Rios A. C. Enantiomer excesses of rare and common sugar derivatives in carbonaceous meteorites. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E3322–E3331. 10.1073/pnas.1603030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinert C.; Myrgorodska I.; de Marcellus P.; Buhse T.; Nahon L.; Hoffmann S. V.; le Sergeant d’Hendecourt L. L.; Meierhenrich U. J. Ribose and related sugars from ultraviolet irradiation of interstellar ice analogs. Science 2016, 352, 208–212. 10.1126/science.aad8137. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for details.
- Wang J.; Liu X.; Feng X. Asymmetric Strecker reactions. Chem. Rev. 2011, 111, 6947–6983. 10.1021/cr200057t. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy R.; Pitsch S.; Arrhenius G. Mineral induced formation of pentose-2,4-bisphosphates. Origins Life Evol. Biospheres 1999, 29, 139–152. 10.1023/A:1006540518676. [DOI] [PubMed] [Google Scholar]
- Ware E. The chemistry of the hydantoins. Chem. Rev. 1950, 46, 403–470. 10.1021/cr60145a001. [DOI] [PubMed] [Google Scholar]
- Hoops S.; Sahle S.; Gauges R.; Lee C.; Pahle J.; Simus N.; Singhal M.; Xu L.; Mendes P.; Kummer U. COPASI--a COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.