

Abstract
Objective
The objective of this study was to examine practice effects and longitudinal cognitive change in 190 clinically normal elderly classified according to a two-feature biomarker model for Alzheimer's disease.
Methods
All participants completed neuropsychological testing, MRI, FDG-PET and PiB-PET at their baseline evaluation. We divided participants into four groups based on neuroimaging measures of amyloid (A+ or A-) and neurodegeneration (N+ or N-) and reexamined cognition at 15- and 30-month intervals.
Results
The A-N- group showed significant improvements in the memory and global scores. The A+N- group also showed significant improvements in the memory and global scores as well as attention. The A-N+ group showed a significant decline in attention at 30 months. The A+N+ group showed significant improvements in memory and the global score at 15 months followed by a significant decline in the global score at 30 months.
Conclusion
Amyloidosis in the absence of neurodegeneration did not have an adverse impact on practice effects or the 30-month cognitive trajectories. In contrast, participants with neurodegeneration (either A-N+ or A+N+) had worse performance at the 30-month follow-up. Our results show that neurodegeneration has a more deleterious effect on cognition than amyloidosis in clinically normal individuals.
Keywords: Cognition, practice effects, amyloid, neurodegeneration, FDG-PET, PiB-PET, preclinical Alzheimer's disease
Introduction
The preclinical phase of Alzheimer's disease (AD) has been an area of intense study since the introduction of new criteria in 2011 which proposed a 3-stage model of a pathophysiological cascade that occurs prior to the emergence of clinical symptoms: Stage 1: asymptomatic cerebral amyloidosis; Stage 2 asymptomatic amyloidosis + “downstream” neurodegeneration; and, Stage 3: amyloidosis + neurodegeneration + subtle cognitive/behavioral decline (Sperling et al., 2011). However, there is a need to understand the relationship of cognition to the biomarker changes across the first two stages.
Emerging evidence shows that amyloidosis and neurodegeneration can occur independently prior to the onset of clinical symptoms of AD. Jack et al. describe a 2-feature biomarker system that includes the following groups based on amyloid (A-/A+) and neurodegeneration status (N-/N+): A-N- corresponds to Stage 0; A+/N- corresponds to Stage 1; A+N+ corresponds to Stages 2+3; and A-N+ denotes individuals with suspected non-Alzheimer's pathology (SNAP) (Jack et al., 2012; Jack et al., 2016; Jack, Wiste, Knopman, et al., 2014; Jack, Wiste, Weigand, et al., 2014; Knopman et al., 2013).
Several longitudinal studies have examined the impact of elevated levels of cerebral amyloid on cognition in clinically normal individuals with variable results. Some studies report that amyloid has a deleterious effect on cognition (Landau et al., 2012; Lim et al., 2012; Morris et al., 2009; Resnick et al., 2010; Snitz et al., 2013) while a meta-analysis showed that amyloid burden is associated with modestly decreased cognitive performance, with the largest associations seen in episodic memory and global cognitive function (Hedden, Oh, Younger, & Patel, 2013). Other studies show that cognitive decline is accelerated only in those who also have evidence for neuronal injury (Desikan, McEvoy, Thompson, & et al., 2012; Ewers et al., 2012; Jack et al., 2009; Okonkwo et al., 2014; Wirth, Madison, et al., 2013; Wirth, Oh, et al., 2013).
Imaging biomarker studies of clinically normal elderly support the notion that cognitive decline is accelerated in those with evidence of both amyloidosis and neurodegeneration (Mormino et al., 2014; Petersen, Wiste, Weigand, & et al., 2016; van Harten et al., 2013; Vos et al., 2013). Using neuroimaging biomarkers to classify participants, Mormino et al. found that individuals with evidence for amyloidosis were more likely to be classified as neurodegeneration positive at baseline. Further, cognitive decline, as measured by a global score, was significantly greater in this group (i.e., A+N+) than the other biomarker groups (i.e., Stage 0, Stage 1 and SNAP) (Mormino, et al., 2014). Petersen et al. (Petersen, et al., 2016) also used neuroimaging biomarkers to classify participants, and while they did not examine cognitive changes separately among A+N- and A+N+ participants, they did show that elevated amyloid was associated with worse cognition and more abnormal neurodegeneration biomarkers at baseline and with greater clinical decline and neurodegeneration.
Many studies have examined cognitive changes and practice effects associated with normal aging in older adults (Dodge, Wang, Chang, & Ganguli, 2011; Duff et al., 2010; Ivnik et al., 2000; Ivnik et al., 1999; Wilson, Beckett, Bennett, Albert, & Evans, 1999; Wilson, Li, Bienias, & Bennett, 2006). Investigators have also examined differences in the longitudinal cognitive trajectories in those that remain cognitively normal versus those who develop incident mild cognitive impairment (MCI) or dementia, with those who develop MCI or dementia showing accelerated cognitive decline prior to a clinical diagnosis (Amieva et al., 2014; Bennett et al., 2002; Grober et al., 2008; Howieson et al., 2008; Johnson, Storandt, Morris, & Galvin, 2009; Machulda et al., 2013; Small & Backman, 2007; Wilson et al., 2010; Wilson, Leurgans, Boyle, & Bennett, 2011; Yu et al., 2012).
More recently, investigators have examined the degree to which diminished or absent practice effects may reflect the earliest cognitive signal of individuals on the path to AD dementia. Jonaitis et al (Jonaitis et al., 2015) examined practice effects in a middle aged cohort of 594 individuals (mean age at baseline for individuals without a family history of AD was 56.6, those with a family history of AD was 53.30) and found that the group with a family history of AD showed attenuated practice effects on measures of working memory and speed/flexibility. Hassenstab and colleagues (Hassenstab et al., 2015) examined a cohort of 263 cognitively normal older adults (mean age at baseline was 71.74 for cognitively stable participants, 74.67 for Progressors) over a span of approximately 9½ years and found that those who progressed to a CDR > 0 with symptomatic AD showed a reduction in practice effects in episodic memory. These studies lend support for the notion that the lack of practice effects in cognitively asymptomatic individuals may be the first harbinger of future cognitive decline.
The aim of the current investigation was to expand on these previous studies by examining the cognitive trajectories in four domains (memory, attention, language, visuospatial) and a global score in a large cohort of clinically normal individuals classified according to a two-feature imaging biomarker system of amyloid and neurodegeneration. We specifically examined initial practice effects and longitudinal cognitive change to better elucidate the cognitive changes associated with different biomarkers states among clinically normal elderly. We hypothesized that individuals with evidence of neurodegeneration, irrespective of amyloid status, would not demonstrate a practice effect and would be the most likely to experience cognitive decline.
Method
Study sample
Participants were from the Mayo Clinic Study of Aging (MCSA) which is a longitudinal population-based study of cognitive aging in Olmsted County, Minnesota (Roberts et al., 2008). All participants for this study were recruited in 2008 and ≥ 70 years old at the baseline assessment. Eligibility included classification as cognitively normal (CN) based on physician and study coordinator ratings at the time of baseline imaging, which included PET and MRI (which was not known to the clinicians), and no previous exposure to neuropsychological testing at the time of the PET and MRI. Therefore, this study differs from other reports from the MCSA and the restrictive inclusion criteria accounts for the smaller number of participants. Figure 1 provides a flow chart that describes the steps involved in identifying the study sample. Thirty-six of the 190 participants (i.e., 19%) in the current study overlapped with our previous report (Machulda, et al., 2013). The Mayo Clinic and Olmsted Medical Center Institutional Review Boards approved these studies which also followed Health Insurance Portability and Accountability Act (HIPAA) guidelines. Every subject provided written informed consent.
Figure 1.
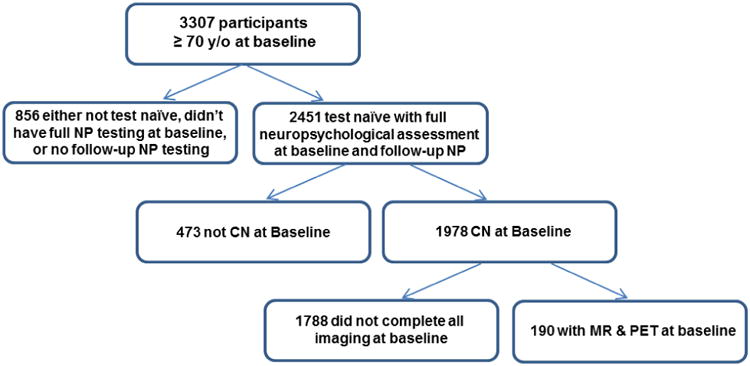
Flow chart describing the steps involved in establishing the study sample.
NP = neuropsychological; CN = clinically normal; MR = magnetic resonance imaging; FDG-PET = Fluorodeoxyglucose Positron Emission Tomography; PiB-PET = Pittsburgh Compound B Positron Emission Tomography
Evaluation
Participants were evaluated up to three times, at approximately 15 month intervals. At each visit, a study coordinator collected information regarding medical and family history. Subject interviews included questions about memory, and study partner interviews obtained information for completing the Clinical Dementia Rating (CDR) Scale, (Morris, 1993) and the Functional Activities Questionnaire (FAQ) (Pfeffer, Kurosaki, Harrah, Chance, & Filos, 1982). The neurologic evaluation included administration of the Short Test of Mental Status (STMS), (Kokmen, Smith, Petersen, Tangalos, & Ivnik, 1991), a medical history review and administration of a questionnaire developed to elicit neurologic conditions that could influence cognition. The questionnaire assessed Parkinson's disease, depression, anxiety, alcoholism, problems with balance, tremor, speech, stroke or transient ischemic attack and sleep problems. The physician also performed a complete neurologic examination.
Neuropsychological testing included the same nine measures at each evaluation. We used the delayed recall trials from three tasks [(Auditory Verbal Learning Test; (Rey, 1964), and the Wechsler Memory Scale-Revised (Wechsler, 1987) Logical Memory & Visual Reproduction subtests] to assess memory. The Boston Naming Test (Kaplan, Goodglass, & Weintraub, 1983), and a category fluency task (Strauss, Sherman, & Spreen, 2006)] assessed language. Wechsler Adult Intelligence Scale-Revised (Wechsler, 1981) Picture Completion and Block Design subtests examined visuospatial reasoning. The Trailmaking Test Part B (Reitan, 1958; Strauss, et al., 2006) and the WAIS-R Digit Symbol subtest assessed attention/executive ability. Individual test scores were first converted to z-scores using the mean and standard deviation from the MCSA 2004 enrollment cohort that consisted of non-demented participants (n=1969). The individual z-scores were averaged to create four cognitive domain scores which were then also converted to z-scores. A global cognitive score was obtained from the average of the four domain z-scores and then converted to a z-score by subtracting the mean and dividing by the standard deviation.
In the MCSA, consensus diagnoses are rendered by a committee of neurologists, study coordinators and neuropsychologists. However, to avoid circularity we used only study coordinator and physician ratings (but not neuropsychological test scores) to classify participants as clinically normal. This was a design specifically for this study. The study coordinator and physician reached a diagnostic impression of the subject's cognitive status independent of one another. We only used participants for whom the study coordinator and physician agreed on a diagnosis of cognitively normal. The study coordinator and physician evaluators were blind to any neuropsychological data.
APOE ε4 Measurement
All participants underwent a blood draw at their baseline visit. DNA extraction and apolipoprotein E (APOE) genotyping was performed for each participant using standard methods (Hixson & Vernier, 1990). The APOE carrier group included individuals with one or two copies of the ε4 allele (i.e., ε2ε4, ε3ε4, ε4ε4).
PET Methods
All participants underwent PET imaging at baseline. PET images were acquired using a GE Discovery RX PET/CT scanner. A CT image is obtained for attenuation correction. The 11C Pittsburgh Compound (Klunk et al., 2004) (PiB)-PETscan, consisting of four 5-minute dynamic frames, was acquired from 40–60 minutes after injection. Fluorodeoxyglucose (18F-FDG) PET images were obtained 1 hour after the PiB scan. Participants were injected with 18F-FDG and imaged after 30 minutes, for an 8-minute image acquisition consisting of four 2-minute dynamic frames.
Quantitative image analysis for both PiB and FDG was done with our in-house fully automated pipeline (Senjem, Gunter, Shiung, Petersen, & Jack Jr, 2005), which uses MRI to guide PET region of interest (ROI) placement and to perform partial volume correction (for PiB only) (Jack et al., 2008). A global cortical PiB-PET standardized uptake value ratio (SUVR) was formed by calculating the median uptake over voxels in the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus values for each subject and dividing this by the median uptake across voxels in the cerebellar gray matter region. For an FDG-PET SUVR, we used angular gyrus, posterior cingulate, and inferior temporal cortical regions to construct an “AD-signature meta ROI”, normalized to pons and vermis uptake (Landau et al., 2010).
MRI Methods
All participants also underwent baseline MR scanning at 3 Tesla with a standardized protocol that included a 3D-MPRAGE sequence. Scans were performed on one of 2 scanners from the same manufacturer. Hippocampal volume at baseline was measured with FreeSurfer (v5.3) and total intracranial volume (TIV) was measured using an in-house algorithm whereby a TIV mask is propagated from template space to each subject's MPRAGE image. Each subject's raw hippocampal volume was adjusted for TIV by calculating the residual from a linear regression of hippocampal volume (y) versus TIV (x) within 133 CN participants aged 30 - 59. Baseline subject classification for the neurodegeneration variable was based on this TIV-adjusted hippocampal volume (HVa).
We then divided our participants into four groups based on neuroimaging measures of amyloid (PiB-PET SUVR ≥ 1.4 [A+ or A-] and neurodegeneration (abnormal hippocampal volume [HVa < -2.40] or FDG-PET hypometabolism [FDG < 1.32] in “Alzheimer signature” regions [N+ or N-]) at baseline to create the four biomarker groups: A-N-, A-N+, A+N-, A+N+.(Jack et al., 2015). The rationale for our approach in selecting biomarker cut-points was to use the same percentile value for each biomarker which is our standard approach [i.e., 90th %tile for HVa and FDG SUVR and 10th %tile for PIB SUVR from a group of persons with AD dementia] (Jack, et al., 2015; Knopman et al., 2012; Knopman, et al., 2013).
Table 1 provides the number of participants in each biomarker group at each visit. Six participants refused to participate at visit 2 but all of these individuals agreed to participate at visit 3. At visit 3, 7 participants refused, 10 withdrew from the study and 7 were not yet eligible (i.e., were not yet scheduled or due for assessment) resulting in a group total of 166.
Table 1. Total sample sizes for each biomarker group at each visit.
| Visit | A-N- | A+N- | A-N+ | A+N+ | Total |
|---|---|---|---|---|---|
| 1 | 93 | 49 | 27 | 21 | 190 |
| 2 | 92 | 46 | 26 | 20 | 184 |
| 3 | 81 | 39 | 26 | 20 | 166 |
A-N- = amyloid negative/neurodegeneration negative
A+N- = amyloid positive/neurodegeneration negative
A-N+ = amyloid negative/neurodegeneration positive
A+N+ = amyloid positive/neurodegeneration positive
Statistics
We used generalized estimating equations (GEE) to fit marginal multilevel models (marginal models) under the SAS Mixed procedure to test for differences in z-scores between time points in four cognitive domains and a global score for each of the four imaging groups (Laird & Ware, 1982). All models were adjusted for age, sex, education, APOE and biomarker group. Participants were required to have all cognitive domains present at baseline for inclusion in the analysis. For subsequent visits we allowed participants as long as they had at least one domain score present to avoid eliminating those who might be missing domain scores. The amount of single domain data missing was very minimal (∼1%) and did not impact analyses. The marginal model's population average approach does not remove these individuals from the model because they did have baseline data present. Within group adjusted means with standard errors are presented and p-values are not adjusted for multiple comparisons. We used SAS for all statistical analyses (SAS Institute, Cary, NC, Version 9.4, 2015).
Results
Demographics
This study included 190 clinically normal individuals at baseline who completed up to two additional evaluations approximately 15 months apart [mean 15.7 ± 1.6 (range = 11.0 – 20.9)]. Table 2 provides participant demographics and clinical characteristics at enrollment. The slight differences in proportions of the A/N defined groups relative to a previous paper published by our group (Knopman, et al., 2012) is due to the strict inclusion criteria for this study which was designed to specifically examine practice effects as well as the use of different cut-points for defining A/N groups. Thirty-nine percent of this sample completed ≤ 12 years of education, 26% had some post-high school education, and 35% completed college or graduate level work. Thirty percent were APOE ε4 carriers which is largely consistent with estimates in the general population. Approximately 4% of the total sample had a Beck Depression Inventory-II (Beck & Steer, 2001) score of ≥ 13, and 15% of all participants were taking an antidepressant. None of the individuals in our study had a diagnosis of Parkinson's disease. However, 19 (10%) had a history of cerebrovascular disease and 11 (6%) had a history of transient ischemic attack. It is unlikely that a history of cerebrovascular disease or transient ischemic attack had an impact on neurodegeneration status (Knopman, et al., 2013; Vemuri & Knopman, 2015). We also examined the demographic characteristics of those with versus those without a follow-up at visit 2. The individuals without follow-up at visit 2 are similar on all demographic variables to those who reassessed. This information is provided in Table 3.
Table 2. Demographics and clinical characteristics of study participants at enrollment.
| Two feature biomarker | |||||
|---|---|---|---|---|---|
| A-N- (N=93) | A+N- (N=49) | A+N+ (N=21) | A-N+ (N=27) | Total (N=190) | |
| Age | |||||
| Mean (SD) | 74.3 (3.0) | 74.0 (2.6) | 76.4 (3.1) | 75.6 (4.1) | 74.7 (3.2) |
| Median | 73.5 | 73.7 | 76.7 | 74.6 | 73.8 |
| Range | (70.9-84.1) | (70.6-83.2) | (71.2-81.5) | (71.0-83.9) | (70.6-84.1) |
| Over 80 | |||||
| No | 88 (94.6%) | 47 (95.9%) | 17 (81.0%) | 22 (81.5%) | 174 (91.6%) |
| Yes | 5 (5.4%) | 2 (4.1%) | 4 (19.0%) | 5 (18.5%) | 16 (8.4%) |
| Gender | |||||
| Female | 51 (54.8%) | 25 (51.0%) | 8 (38.1%) | 8 (29.6%) | 92 (48.4%) |
| Male | 42 (45.2%) | 24 (49.0%) | 13 (61.9%) | 19 (70.4%) | 98 (51.6%) |
| Education (years) | |||||
| Mean (SD) | 14.3 (2.6) | 14.5 (2.7) | 14.6 (2.3) | 13.8 (2.4) | 14.3 (2.5) |
| Median | 13.0 | 14.0 | 15.0 | 13.0 | 13.0 |
| Range | (12.0-20.0) | (8.0-19.0) | (12.0-19.0) | (11.0-20.0) | (8.0-20.0) |
| Education Category | |||||
| <=12 | 38 (40.9%) | 17 (34.7%) | 7 (33.3%) | 12 (44.4%) | 74 (38.9%) |
| 13-15 | 24 (25.8%) | 12 (24.5%) | 4 (19.0%) | 9 (33.3%) | 49 (25.8%) |
| 16-20 | 31 (33.3%) | 20 (40.8%) | 10 (47.6%) | 6 (22.2%) | 67 (35.3%) |
| Marital Status | |||||
| Single, Never Married | 2 (2.2%) | 3 (6.1%) | 1 (4.8%) | 1 (3.7%) | 7 (3.7%) |
| Married | 72 (77.4%) | 40 (81.6%) | 18 (85.7%) | 18 (66.7%) | 148 (77.9%) |
| Divorced | 7 (7.5%) | 4 (8.2%) | 0 (0.0%) | 3 (11.1%) | 14 (7.4%) |
| Widowed | 12 (12.9%) | 2 (4.1%) | 2 (9.5%) | 5 (18.5%) | 21 (11.1%) |
| Abnormal PiB ratio | |||||
| No | 93 (100.0%) | 0 (0.0%) | 0 (0.0%) | 27 (100.0%) | 120 (63.2%) |
| Yes | 0 (0.0%) | 49 (100.0%) | 21 (100.0%) | 0 (0.0%) | 70 (36.8%) |
| Carrier of APOE e4 allele | |||||
| No | 74 (79.6%) | 27 (55.1%) | 10 (47.6%) | 22 (81.5%) | 133 (70.0%) |
| Yes | 19 (20.4%) | 22 (44.9%) | 11 (52.4%) | 5 (18.5%) | 57 (30.0%) |
| BDI-II Total Score | |||||
| Mean (SD) | 3.3 (3.5) | 4.0 (3.7) | 5.5 (4.6) | 4.1 (5.3) | 3.8 (4.0) |
| Median | 2.0 | 3.0 | 5.0 | 2.0 | 2.0 |
| Range | (0.0-18.0) | (0.0-15.0) | (0.0-20.0) | (0.0-26.0) | (0.0-26.0) |
| BDI depression, Total >=13 | |||||
| Missing | 0 (.%) | 1 (.%) | 0 (.%) | 0 (.%) | 1 |
| 0=No | 91 (97.8%) | 46 (95.8%) | 19 (90.5%) | 26 (96.3%) | 182 (96.3%) |
| 1=Yes | 2 (2.2%) | 2 (4.2%) | 2 (9.5%) | 1 (3.7%) | 7 (3.7%) |
| Any Antidepressant | |||||
| No | 78 (83.9%) | 42 (85.7%) | 18 (85.7%) | 24 (88.9%) | 162 (85.3%) |
| Yes | 15 (16.1%) | 7 (14.3%) | 3 (14.3%) | 3 (11.1%) | 28 (14.7%) |
A-N- = amyloid negative/neurodegeneration negative; A+N- = amyloid positive/neurodegeneration negative; A-N+ = amyloid negative/neurodegeneration positive; A+N+ = amyloid positive/neurodegeneration positive; PiB = Pittsburgh compound B; APOE = apolipoprotein E type; BDI = Beck Depression Inventory
Table 3. Demographics and clinical characteristics in participants with and without followup.
| No (N=14) | Yes (N=190) | |
|---|---|---|
| Age | ||
| N | 14 | 190 |
| Mean (SD) | 73.2 (2.3) | 74.7 (3.2) |
| Median | 72.9 | 73.8 |
| Range | (70.7-78.6) | (70.6-84.1) |
| Gender | ||
| Female | 7 (50.0%) | 92 (48.4%) |
| Male | 7 (50.0%) | 98 (51.6%) |
| Education (years) | ||
| N | 14 | 190 |
| Mean (SD) | 13.7 (2.7) | 14.3 (2.5) |
| Median | 13.0 | 13.0 |
| Range | (12.0-20.0) | (8.0-20.0) |
| Education Category | ||
| <=12 | 5 (35.7%) | 74 (38.9%) |
| 13-15 | 7 (50.0%) | 49 (25.8%) |
| 16-20 | 2 (14.3%) | 67 (35.3%) |
| Marital Status | ||
| Single, Never Married | 0 (0.0%) | 7 (3.7%) |
| Married | 9 (64.3%) | 148 (77.9%) |
| Divorced | 2 (14.3%) | 14 (7.4%) |
| Widowed | 3 (21.4%) | 21 (11.1%) |
| PiB > 1.4 | ||
| No | 10 (71.4%) | 120 (63.2%) |
| Yes | 4 (28.6%) | 70 (36.8%) |
| Carrier of APOE e4 allele | ||
| Missing | 1 | 0 |
| No | 11 (84.6%) | 133 (70.0%) |
| Yes | 2 (15.4%) | 57 (30.0%) |
| BDI-II Total Score | ||
| N | 14 | 189 |
| Mean (SD) | 3.4 (4.3) | 3.8 (4.0) |
| Median | 2.0 | 2.0 |
| Range | (0.0-12.0) | (0.0-26.0) |
| Any Antidepressant | ||
| No | 12 (85.7%) | 162 (85.3%) |
| Yes | 2 (14.3%) | 28 (14.7%) |
| Two feature biomarker | ||
| A-N- | 8 (57.1%) | 93 (48.9%) |
| A+N- | 4 (28.6%) | 49 (25.8%) |
| A+N+ | 0 (0.0%) | 21 (11.1%) |
| A-N+ | 2 (14.3%) | 27 (14.2%) |
A-N- = amyloid negative/neurodegeneration negative; A+N- = amyloid positive/neurodegeneration negative; A-N+ = amyloid negative/neurodegeneration positive; A+N+ = amyloid positive/neurodegeneration positive; PiB = Pittsburgh compound B; APOE = apolipoprotein E type; BDI = Beck Depression Inventory
Table 4 shows the descriptive statistics of the baseline z-scores for the global and cognitive domain scores. There were overall differences between the two feature biomarker groups, with the N+ groups having noticeably lower scores at baseline in the global score and all cognitive domains with the exception of visuospatial reasoning.
Table 4. Baseline z-scores by biomarker group.
| Two feature biomarker | |||||
|---|---|---|---|---|---|
| A-N- (N=93) | A+N- (N=49) | A+N+ (N=21) | A-N+ (N=27) | p value | |
| Global z-score | 0.00071 | ||||
| Mean (SD) | 0.9 (0.8) | 0.8 (0.6) | 0.3 (0.8) | 0.3 (0.9) | |
| Median | 0.9 | 0.8 | 0.5 | 0.2 | |
| Range | (-0.9-2.5) | (-0.6-2.1) | (-1.5-1.4) | (-1.2-2.6) | |
| Memory z-score | 0.00661 | ||||
| Mean (SD) | 0.7 (0.9) | 0.6 (0.8) | 0.2 (0.9) | 0.2 (0.8) | |
| Median | 0.8 | 0.6 | 0.2 | 0.2 | |
| Range | (-1.6-2.6) | (-1.0-2.0) | (-1.4-1.5) | (-0.9-2.3) | |
| Language z-score | 0.01061 | ||||
| Mean (SD) | 0.7 (0.8) | 0.5 (0.6) | 0.2 (0.8) | 0.3 (0.8) | |
| Median | 0.7 | 0.6 | 0.4 | 0.1 | |
| Range | (-1.5-2.5) | (-0.9-2.1) | (-1.2-1.7) | (-1.1-2.7) | |
| Attention z-score | 0.00421 | ||||
| Mean (SD) | 0.8 (0.7) | 0.8 (0.7) | 0.3 (0.7) | 0.2 (1.0) | |
| Median | 0.8 | 0.8 | 0.4 | 0.2 | |
| Range | (-1.1-2.4) | (-0.8-2.3) | (-1.2-1.3) | (-1.9-2.0) | |
| Visual spatial z-score | 0.06391 | ||||
| Mean (SD) | 0.6 (1.0) | 0.6 (0.8) | 0.2 (1.0) | 0.2 (1.1) | |
| Median | 0.7 | 0.4 | 0.4 | 0.0 | |
| Range | (-1.6-2.4) | (-1.0-2.5) | (-1.6-2.2) | (-2.3-2.3) | |
Kruskal Wallis
A-N- = amyloid negative/neurodegeneration negative; A+N- = amyloid positive/neurodegeneration negative; A-N+ = amyloid negative/neurodegeneration positive; A+N+ = amyloid positive/neurodegeneration positive; SD = standard deviation
Table 5 and Figure 2 provide results from the GEE for global and cognitive domain scores. A marginal model was fit for each cognitive outcome and included age, sex, education, APOE, biomarker group at baseline as predictors. The table shows the estimated mean cognitive score from the model at each visit for each biomarker group as well as the difference in means between visits for each biomarker group.
Table 5. Results from marginal models for global and cognitive domain scores.
| Visit | Two feature biomarker | LS Mean | Standard Error | P-value | |
|---|---|---|---|---|---|
| Global | 1 | A-N- | 0.8000 | 0.08555 | 0.0461 |
| 2 | A-N- | 0.9100 | 0.08479 | . | |
| 3 | A-N- | 0.9400 | 0.09480 | . | |
| Global | 2 vs 1 | A-N- | 0.1100 | 0.04470 | 0.0155 |
| 3 vs 1 | A-N- | 0.1400 | 0.07031 | 0.0436 | |
| 3 vs 2 | A-N- | 0.03000 | 0.05238 | 0.5205 | |
| Global | 1 | A+N- | 0.6900 | 0.1049 | 0.0207 |
| 2 | A+N- | 0.8600 | 0.1075 | . | |
| 3 | A+N- | 0.8500 | 0.1215 | . | |
| Global | 2 vs 1 | A+N- | 0.1700 | 0.05926 | 0.0056 |
| 3 vs 1 | A+N- | 0.1600 | 0.08829 | 0.0693 | |
| 3 vs 2 | A+N- | 0 | 0.07167 | 0.9452 | |
| Global | 1 | A+N+ | 0.2700 | 0.1533 | 0.0150 |
| 2 | A+N+ | 0.4900 | 0.1665 | . | |
| 3 | A+N+ | 0.2900 | 0.1887 | . | |
| Global | 2 vs 1 | A+N+ | 0.2200 | 0.08785 | 0.0142 |
| 3 vs 1 | A+N+ | 0.02000 | 0.1198 | 0.8803 | |
| 3 vs 2 | A+N+ | -0.2000 | 0.09947 | 0.0464 | |
| Global | 1 | A-N+ | 0.3400 | 0.1413 | 0.7677 |
| 2 | A-N+ | 0.3800 | 0.1505 | . | |
| 3 | A-N+ | 0.3200 | 0.1683 | . | |
| Global | 2 vs 1 | A-N+ | 0.04000 | 0.07822 | 0.6205 |
| 3 vs 1 | A-N+ | -0.02000 | 0.1076 | 0.8731 | |
| 3 vs 2 | A-N+ | -0.06000 | 0.09060 | 0.5372 | |
| Memory | 1 | A-N- | 0.6100 | 0.1012 | 0.0000 |
| 2 | A-N- | 0.8500 | 0.1012 | . | |
| 3 | A-N- | 1.0500 | 0.1110 | . | |
| Memory | 2 vs 1 | A-N- | 0.2400 | 0.07057 | 0.0008 |
| 3 vs 1 | A-N- | 0.4400 | 0.09042 | 0.0000 | |
| 3 vs 2 | A-N- | 0.2000 | 0.07148 | 0.0054 | |
| Memory | 1 | A+N- | 0.5100 | 0.1248 | 0.0060 |
| 2 | A+N- | 0.7600 | 0.1293 | . | |
| 3 | A+N- | 0.8700 | 0.1433 | . | |
| Memory | 2 vs 1 | A+N- | 0.2500 | 0.09551 | 0.0102 |
| 3 vs 1 | A+N- | 0.3600 | 0.1167 | 0.0024 | |
| 3 vs 2 | A+N- | 0.1100 | 0.1001 | 0.2699 | |
| Memory | 1 | A+N+ | 0.2400 | 0.1828 | 0.0883 |
| 2 | A+N+ | 0.5500 | 0.1990 | . | |
| 3 | A+N+ | 0.4400 | 0.2208 | . | |
| Memory | 2 vs 1 | A+N+ | 0.3200 | 0.1424 | 0.0278 |
| 3 vs 1 | A+N+ | 0.2100 | 0.1609 | 0.2027 | |
| 3 vs 2 | A+N+ | -0.1100 | 0.1408 | 0.4350 | |
| Memory | 1 | A-N+ | 0.2400 | 0.1680 | 0.3934 |
| 2 | A-N+ | 0.4100 | 0.1795 | . | |
| 3 | A-N+ | 0.4000 | 0.1951 | . | |
| Memory | 2 vs 1 | A-N+ | 0.1600 | 0.1258 | 0.1997 |
| 3 vs 1 | A-N+ | 0.1600 | 0.1415 | 0.2551 | |
| 3 vs 2 | A-N+ | 0 | 0.1211 | 0.9973 | |
| Language | 1 | A-N- | 0.6300 | 0.08684 | 0.8430 |
| 2 | A-N- | 0.6500 | 0.08833 | . | |
| 3 | A-N- | 0.6100 | 0.09615 | . | |
| Language | 2 vs 1 | A-N- | 0.02000 | 0.05536 | 0.7252 |
| 3 vs 1 | A-N- | -0.02000 | 0.08244 | 0.8270 | |
| 3 vs 2 | A-N- | -0.04000 | 0.07183 | 0.6020 | |
| Language | 1 | A+N- | 0.4400 | 0.1070 | 0.8709 |
| 2 | A+N- | 0.4800 | 0.1131 | . | |
| 3 | A+N- | 0.4700 | 0.1246 | . | |
| Language | 2 vs 1 | A+N- | 0.04000 | 0.07473 | 0.6056 |
| 3 vs 1 | A+N- | 0.03000 | 0.1077 | 0.7549 | |
| 3 vs 2 | A+N- | 0 | 0.1006 | 0.9607 | |
| Language | 1 | A+N+ | 0.2600 | 0.1565 | 0.7718 |
| 2 | A+N+ | 0.3000 | 0.1758 | . | |
| 3 | A+N+ | 0.2000 | 0.1884 | . | |
| Language | 2 vs 1 | A+N+ | 0.04000 | 0.1135 | 0.7029 |
| 3 vs 1 | A+N+ | -0.06000 | 0.1464 | 0.7041 | |
| 3 vs 2 | A+N+ | -0.1000 | 0.1404 | 0.4813 | |
| Language | 1 | A-N+ | 0.4000 | 0.1440 | 0.4026 |
| 2 | A-N+ | 0.4800 | 0.1568 | . | |
| 3 | A-N+ | 0.3200 | 0.1674 | . | |
| Language | 2 vs 1 | A-N+ | 0.07000 | 0.09792 | 0.4533 |
| 3 vs 1 | A-N+ | -0.09000 | 0.1301 | 0.5049 | |
| 3 vs 2 | A-N+ | -0.1600 | 0.1229 | 0.1930 | |
| Attention | 1 | A-N- | 0.6800 | 0.08475 | 0.4171 |
| 2 | A-N- | 0.7500 | 0.08494 | . | |
| 3 | A-N- | 0.7200 | 0.08662 | . | |
| Attention | 2 vs 1 | A-N- | 0.07000 | 0.05010 | 0.1913 |
| 3 vs 1 | A-N- | 0.04000 | 0.06649 | 0.5584 | |
| 3 vs 2 | A-N- | -0.03000 | 0.05613 | 0.6344 | |
| Attention | 1 | A+N- | 0.7200 | 0.1043 | 0.0531 |
| 2 | A+N- | 0.8800 | 0.1086 | . | |
| 3 | A+N- | 0.7900 | 0.1101 | . | |
| Attention | 2 vs 1 | A+N- | 0.1600 | 0.06773 | 0.0168 |
| 3 vs 1 | A+N- | 0.07000 | 0.08348 | 0.3994 | |
| 3 vs 2 | A+N- | -0.09000 | 0.07799 | 0.2346 | |
| Attention | 1 | A+N+ | 0.3700 | 0.1525 | 0.7352 |
| 2 | A+N+ | 0.3900 | 0.1666 | . | |
| 3 | A+N+ | 0.3100 | 0.1697 | . | |
| Attention | 2 vs 1 | A+N+ | 0.02000 | 0.09901 | 0.8409 |
| 3 vs 1 | A+N+ | -0.06000 | 0.1097 | 0.5658 | |
| 3 vs 2 | A+N+ | -0.08000 | 0.1078 | 0.4423 | |
| Attention | 1 | A-N+ | 0.2800 | 0.1404 | 0.1126 |
| 2 | A-N+ | 0.1500 | 0.1503 | . | |
| 3 | A-N+ | 0.08000 | 0.1523 | . | |
| Attention | 2 vs 1 | A-N+ | -0.1400 | 0.08764 | 0.1249 |
| 3 vs 1 | A-N+ | -0.2000 | 0.09988 | 0.0476 | |
| 3 vs 2 | A-N+ | -0.0600 | 0.09621 | 0.5065 | |
| Visual Spatial | 1 | A-N- | 0.6500 | 0.1059 | 0.5268 |
| 2 | A-N- | 0.7000 | 0.1002 | . | |
| 3 | A-N- | 0.7500 | 0.1066 | . | |
| Visual Spatial | 2 vs 1 | A-N- | 0.05000 | 0.06611 | 0.4833 |
| 3 vs 1 | A-N- | 0.1000 | 0.08476 | 0.2632 | |
| 3 vs 2 | A-N- | 0.05000 | 0.07718 | 0.5289 | |
| Visual Spatial | 1 | A+N- | 0.5700 | 0.1312 | 0.2537 |
| 2 | A+N- | 0.7000 | 0.1274 | . | |
| 3 | A+N- | 0.7300 | 0.1359 | . | |
| Visual Spatial | 2 vs 1 | A+N- | 0.1200 | 0.08923 | 0.1683 |
| 3 vs 1 | A+N- | 0.1500 | 0.1076 | 0.1584 | |
| 3 vs 2 | A+N- | 0.03000 | 0.1073 | 0.7875 | |
| Visual Spatial | 1 | A+N+ | 0.07000 | 0.1926 | 0.5657 |
| 2 | A+N+ | 0.2100 | 0.1961 | . | |
| 3 | A+N+ | 0.1200 | 0.2082 | . | |
| Visual Spatial | 2 vs 1 | A+N+ | 0.1400 | 0.1324 | 0.2875 |
| 3 vs 1 | A+N+ | 0.05000 | 0.1425 | 0.7155 | |
| 3 vs 2 | A+N+ | -0.09000 | 0.1503 | 0.5539 | |
| Visual Spatial | 1 | A-N+ | 0.2000 | 0.1767 | 0.6497 |
| 2 | A-N+ | 0.3000 | 0.1797 | . | |
| 3 | A-N+ | 0.3000 | 0.1870 | . | |
| Visual Spatial | 2 vs 1 | A-N+ | 0.1000 | 0.1207 | 0.4320 |
| 3 vs 1 | A-N+ | 0.1000 | 0.1297 | 0.4433 | |
| 3 vs 2 | A-N+ | 0 | 0.1374 | 0.9734 |
A-N- = amyloid negative/neurodegeneration negative; A+N- = amyloid positive/neurodegeneration negative; A-N+ = amyloid negative/neurodegeneration positive; A+N+ = amyloid positive/neurodegeneration positive; LS = adjusted least square
Figure 2.
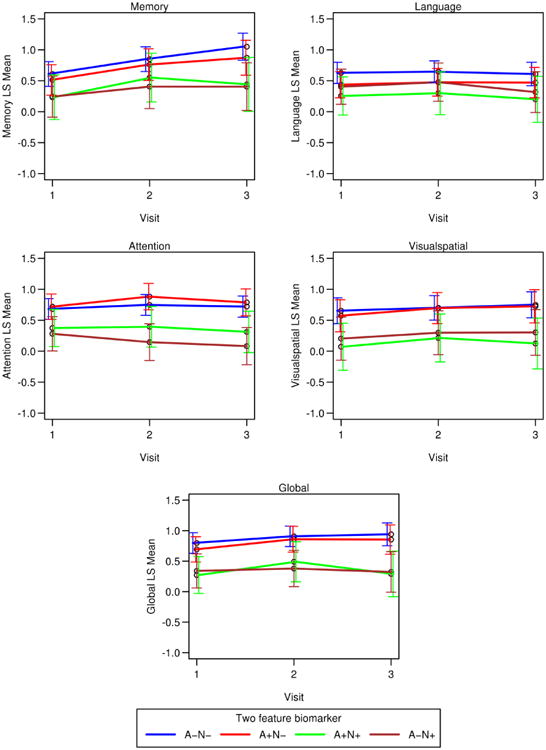
Cognitive trajectories for each biomarker group by cognitive domain. These plots represent results from marginal models and are adjusted for age, sex education, apolipoprotein E status and biomarker group.
A-N- = amyloid negative/neurodegeneration negative; A+N- = amyloid positive/neurodegeneration negative; A-N+ = amyloid negative/neurodegeneration positive; A+N+ = amyloid positive/neurodegeneration positive
Cognitive trajectories for each baseline biomarker group (See Table 5; Figure 2)
A-N-
There were 93 A-N- participants. This group showed significant improvements in the global and memory scores at visits 2 and 3 relative to baseline. Memory continued to improve from Visit 2 to Visit 3 whereas the global score plateaued. There were no significant differences in language, attention or visuospatial trajectories.
A+N-
There were 49 A+N- participants. The A+N- group showed significant improvements in memory at visits 2 and 3 relative to baseline. The A+N- group also showed an improvement in attention and the global score at visit 2 relative to baseline, followed by a plateau between visits 2 and 3. The language and visuospatial trajectories were stable.
A-N+
There were 27 A-N+ participants. This group showed a significant decline in attention at visit 3 relative to visit 1. The trajectories of the remaining domains were stable.
A+N+
There were 21 A+N+ participants. This group showed improvement in the global score at visit 2, followed by a significant decline at visit 3 relative to visit 2. The A+N+ group also showed an improvement in memory at visit 2 that was not sustained at Visit 3. Language, attention, and visuospatial trajectories were stable.
Discussion
The main findings from this study are: (1) the neurodegeneration negative groups, regardless of amyloid status, showed modest improvements or stable performances in all cognitive domains and the global score. The most noteworthy improvement was in memory, including the A+N- group which was sustained at Visit 3; (2) the neurodegeneration positive groups, regardless of amyloid status, showed stable or declining performances by Visit 3, despite the unexpected improvement in memory and global score of the A+N+ group at Visit 2; (3) we were able to measure significant cognitive change using a 2-feature biomarker classification system of preclinical AD even though a high percentage (93%) of our participants remained clinically normal at visit 3.
We found that amyloid positivity, in the absence of neurodegeneration, did not have a deleterious effect on practice effects in this population of individuals aged 70 and older. The cognitive trajectories of A+N- follow a similar pattern to A-N-, including a sustained improvement in memory. This is discrepant from previous reports of clinically normal elderly with amyloid positivity which found a decline in episodic memory (Landau, et al., 2012; Lim et al., 2014; Small, Siddarth, Kepe, & et al., 2012; Storandt, Mintun, Head, & Morris, 2009). Unexpectedly, the A+N+ group also showed a significant practice effect in memory at visit 2 but this was not sustained at visit 3. It is possible that A+N+ conferred some compensatory abilities that eventually became exhausted. In any case, the lack of a sustained practice effect may be the first signal of subtle cognitive decline described in NIA-AA Stage 3 (Sperling, et al., 2011). These findings parallel our previous paper showing that clinically normal individuals who developed incident MCI or dementia showed an initial practice effect in memory followed by a decline (Machulda, et al., 2013).
In contrast, the N+ groups started out approximately a half standard deviation below the N- groups at baseline, suggesting that the trajectories of these biomarker groups diverged at an earlier point in time. There was a dissociation of the cognitive trajectories between the N- vs N+ groups, regardless of amyloid status, with both N+ groups showing a decline in cognition over time which was most evident for the global and memory scores. Our results support the notion that cognition is more closely associated with neurodegeneration than amyloid status. Although the literature shows that there is an impact of amyloid positivity on cognition in elderly cohorts, it is subtle and not always detectable. In fact, we previously showed that the impact of amyloid elevation on cognition is mediated by neurodegeneration, and if one controls for neurodegenerative status in models of amyloidosis and cognition, the association between the latter two is abolished (Vemuri et al., 2016; Vemuri et al., 2012).
A recent paper also utilizing a 2-feature biomarker classification system to examine cognitive decline in normal elderly found that their A+ individuals were more likely classified as N+ than N-, and that the A+N+ group had accelerated cognitive decline relative to the other biomarker groups on a global index of cognition (Mormino, et al., 2014). They found a practice effect in the Stage 0 group (A-N-) group whereas the practice effect was diminished in the stage 1 (A+N-) and SNAP (A-N+) groups. These findings differ slightly from ours in that the A-N- and A+ N- groups in our study show a significant practice effect on the global score. Although the mean age of participants is comparable between the studies, the Mayo Clinic Study of Aging recruited participants who were randomly selected from a community, while the Harvard Aging Brain Study participants were highly educated and specifically recruited to participate in a memory study. The discrepant findings might also be attributed to the use of different psychometric indices to derive the global score.
The A+N+ group showed an initial improvement followed by a significant decline. The variability of the A+N+ group, particularly on the global and memory scores when examining the cognitive trajectories using a common starting point, may reflect very early cognitive changes associated with preclinical dementia. Interestingly, another report of individuals similar in age to our study found that intra-individual variability across neuropsychological test performances was associated with development of incident dementia even after adjusting for level of performance on each neuropsychological test (Holtzer, Verghese, Wang, Hall, & Lipton, 2008).
These results have implications for clinical trials in pre-clinical AD in which clinically normal individuals who are amyloid positive are the target group which includes individuals who are A+N- and A+N+ without distinguishing between the two biomarker states for inclusion purposes. The duration of these clinical trials is typically long. For example, individuals in the Anti-Amyloid Treatment in Asymptomatic Alzheimer's Disease (A4) study are followed for three years (Sperling et al., 2014). Our findings demonstrate that A+N- individuals are expected to exhibit learning effects for at least 30 months which is approximately as long as the clinical trial whereas A+N+ individuals do not have the same cognitive trajectory. This dichotomy will affect interpretation of these results and powering of trials for preclinical AD.
Serrano-Pozo: 2016 neuritic plaques and NFTs, but not Thal amyloid stages, independently associated with cognition.
Several studies show that attenuated practice effects may provide a means for identifying risk for future cognitive decline across the clinical spectrum of cognitively normal to MCI. For example, Hassenstab et al. (Hassenstab, et al., 2015) found that the magnitude of gain in episodic memory from repeated testing assessed by annual visits was inversely related to progression risk in individuals who were cognitive normal at baseline. We previously showed that individuals who developed incident MCI or dementia showed an initial practice effect in episodic memory followed by declining performance thereafter (Machulda, et al., 2013). Duff et al (Duff et al., 2011) found that individuals with MCI who showed minimal practice effects across 1 week performed significantly more poorly one year later on measures of immediate memory, delayed memory language and overall cognition than cognitive normal individuals and those with MCI who had large practice effects.
Our study has several strengths. This is among the few reports to longitudinally examine the trajectories of four cognitive domains and a global score based on a 2-feature biomarker classification system of preclinical Alzheimer's disease and addresses the often overlooked issue of practice effects (Jagust, 2015). Our participants were derived from a population-based sample which enhances the generalizability of our findings and minimizes sample biases that might be present in a volunteer sample recruited from advertisements.
Our study also has some limitations. Participants were 70- 90 years at baseline, so our results may not apply to younger and much older age groups. We divided our groups into positive vs. negative amyloid and neurodegeneration status at baseline which serves as a useful heuristic but may not fully capture the cognitive changes over time as individuals transition from one biomarker group to another (Jack, et al., 2016). The follow-up period was fairly short (i.e., 30 months). A longer period of follow-up will allow us to better characterize the cognitive trajectories of those who remain cognitively normal versus those who develop MCI and dementia. A larger sample size will also allow us the ability to examine the effect of APOE ε4 status on the cognitive trajectories of each biomarker group. It's possible that the use of a cognitive screening measure and functional scales to identify our clinically normal participants might not have identified features of subtle cognitive decline and thus created more variability in the clinically normal group. Also, inference on between group differences at any single time point isn't appropriate in the absence of overall group by time interaction significance (Nieuwenhuis, Forstmann, & Wagenmakers, 2011). This study is not able to say any two groups are statistically different from each other, rather we present within group performance in an empirical manner. Finally, although we derived our participants from a population-based sample, it is possible that individuals in our study are healthier than non-participants.
In conclusion, we show that neurodegeneration has a deleterious effect on cognition regardless of amyloid positivity in clinically normal individuals. Our study adds to the previous literature showing that the inability to sustain a practice effect and derive benefit from previous exposure to testing may represent the earliest indicator of future cognitive decline. Our results also underscore the importance of examining both amyloid and neurodegeneration when examining cognitive change during the preclinical phases of AD.
Acknowledgments
The authors wish to thank the participants and staff at the Mayo Clinic Study of Aging. This research was made possible by the Rochester Epidemiology Project (R01 AG034676) and was supported by NIH grants P50 AG016574, U01 AG006786, and R01 AG041851, by the Robert Wood Johnson Foundation, The Elsie and Marvin Dekelboum Family Foundation, and by the Mayo Foundation for Education and Research.
Footnotes
There are no conflicts of interest.
References
- Amieva H, Mokri H, Le Goff M, Meillon C, Jacqmin-Gadda H, Foubert-Samier A, et al. Dartigues JF. Compensatory mechanisms in higher-educated subjects with Alzheimer's disease: a study of 20 years of cognitive decline. Brain. 2014;137(4):1167–1175. doi: 10.1093/brain/awu035. [DOI] [PubMed] [Google Scholar]
- Beck AT, Steer RA. Manual for Beck Depression Inventory-II (BDI-II) San Antonio: TX: 2001. [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, et al. Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59(2):198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- Desikan RS, McEvoy LK, Thompson WK, et al. Amyloid-β–associated clinical decline occurs only in the presence of elevated p-tau. Archives of Neurology. 2012;69(6):709–713. doi: 10.1001/archneurol.2011.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge HH, Wang CN, Chang CCH, Ganguli M. Terminal decline and practice effects in older adults without dementia: The MoVIES project. Neurology. 2011;77(8):722–730. doi: 10.1212/WNL.0b013e31822b0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Beglinger L, Moser D, Paulsen J, Schultz S, Arndt S. Predicting cognitive change in older adults: the relative contribution of practice effects. Archives of Clinical Neuropsychology. 2010;25:81–88. doi: 10.1093/arclin/acp105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Lyketsos C, Beglinger L, Chelune G, Moser D, Arndt S, et al. McCaffrey R. Practice effects predict cognitive outcome in amnestic mild cognitive impairment. American Journal of Geriatric Psychiatry. 2011;19(11):932–939. doi: 10.1097/JGP.0b013e318209dd3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewers M, Insel P, Jagust WJ, Shaw L, Trojanowski J JQ, Aisen P, et al. Initiative, f. t. A. s. D. N. CSF Biomarker and PIB-PET–Derived Beta-Amyloid Signature Predicts Metabolic, Gray Matter, and Cognitive Changes in Nondemented Subjects. Cerebral Cortex. 2012;22(9):1993–2004. doi: 10.1093/cercor/bhr271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer's disease. Journal of the International Neuropsychological Society. 2008;14:266–278. doi: 10.1017/S1355617708080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassenstab J, Ruvolo D, Jasielec MS, Xiong C, Grant E, Morris JC. Absence of practice effects in preclinical Alzheimer's Disease. Neuropsychology. 2015;29(6):940–948. doi: 10.1037/neu0000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden T, Oh H, Younger AP, Patel TA. Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology. 2013;80(14):1341–1348. doi: 10.1212/WNL.0b013e31828ab35d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hixson J, Vernier D. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with Hhal. Journal of Lipid Research. 1990;31(3):545–548. [PubMed] [Google Scholar]
- Holtzer R, Verghese J, Wang C, Hall C, Lipton R. Within-person across-neuropsychological test variability and incident dementia. JAMA. 2008;300(7):823–830. doi: 10.1001/jama.300.7.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howieson D, Carlson N, Moore M, Wasserman D, Abendroth C, Payne_Murphy J, Kaye J. Trajectory of mild cognitive impairment onset. Journal of the International Neuropsychological Society. 2008;14:192–198. doi: 10.1017/S1355617708080375. [DOI] [PubMed] [Google Scholar]
- Ivnik RC, Smith GE, Petersen RC, Boeve BF, Kokmen E, Tangalos E. Diagnostic accuracy of four approaches to interpreting neuropsychological test data. Neuropsychology. 2000;14(2):163–177. doi: 10.1037//0894-4105.14.2.163. [DOI] [PubMed] [Google Scholar]
- Ivnik RJ, Smith GE, Lucas JA, Petersen RC, Boeve BF, Kokmen E, Tangalos EG. Testing normal older people three or four times at 1- to 2-year intervals: defining normal variance. Neuropsychology. 1999;13(1):121–127. doi: 10.1037//0894-4105.13.1.121. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, et al. Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131(Pt 3):665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, et al. Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132(Pt 5):1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Weigand SD, Wiste HJ, Vemuri P, Lowe V, et al. Petersen RC. An operational approach to National Institute on Aging–Alzheimer's Association criteria for preclinical Alzheimer disease. Annals of Neurology. 2012;71(6):765–775. doi: 10.1002/ana.22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Therneau TM, Wiste HJ, Weigand SD, Knopman DS, Lowe VJ, et al. Petersen RC. Transition rates between amyloid and neurodegeneration biomarker states and to dementia: a population-based, longitudinal cohort study. The Lancet Neurology. 2016;15:56–64. doi: 10.1016/S1474-4422(15)00323-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Knopman DS, Vemuri P, Mielke MM, Weigand SD, et al. Petersen RC. Rates of β-amyloid accumulation are independent of hippocampal neurodegeneration. Neurology. 2014;82(18):1605–1612. doi: 10.1212/WNL.0000000000000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Weigand SD, Knopman DS, Mielke MM, Vemuri P, et al. Petersen RC. Different definitions of neurodegeneration produce similar amyloid/neurodegeneration biomarker group findings. Brain. 2015 doi: 10.1093/brain/awv283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Wiste HJ, Weigand SD, Rocca WA, Knopman DS, Mielke MM, et al. Petersen RC. Age-specific population frequencies of cerebral B-amyloidosis and neurodegeneration among people with normal cognitive function aged 50-89 years: a cross-sectional study. Lancet Neurology. 2014;13:997–1005. doi: 10.1016/S1474-4422(14)70194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. Is amyloid-β harmful to the brain? Insights from human imaging studies. Brain. 2015 doi: 10.1093/brain/awv326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer Disease. Archives of Neurology. 2009;66(10):1254–1259. doi: 10.1001/archneurol.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonaitis E, Koscik R, La Rue A, Johnson SC, Hermann BP, Sager MA. Aging, practice effects, and genetic risk in the Wisconsin Registry for Alzheimer's Prevention. The Clinical Neuropsychologist. 2015;29(4):426–441. doi: 10.1080/13854046.2015.1047407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Langstrom B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Annals of neurology. 2004;55(3):306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Wiste HJ, Weigand SD, Vemuri P, Lowe V, et al. Petersen RC. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology. 2012;78(20):1576–1582. doi: 10.1212/WNL.0b013e3182563bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Wiste HJ, Weigand SD, Vemuri P, Lowe VJ, et al. Petersen RC. Brain injury biomarkers are not dependent on β-amyloid in normal elderly. Annals of Neurology. 2013;73(4):472–480. doi: 10.1002/ana.23816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics. 1982;38(4):963–974. [PubMed] [Google Scholar]
- Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, et al. Initiative, O. b. o. t. A. s. D. N. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75(3):230–238. doi: 10.1212/WNL.0b013e3181e8e8b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, et al. for the Alzheimer's Disease Neuroimaging. I. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Annals of Neurology. 2012;72(4):578–586. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YY, Ellis KA, Pietrzak RH, Ames D, Darby D, Harrington K, et al. Group, F. t. A. R. Stronger effect of amyloid load than APOE genotype on cognitive decline in healthy older adults. Neurology. 2012;79(16):1645–1652. doi: 10.1212/WNL.0b013e31826e9ae6. [DOI] [PubMed] [Google Scholar]
- Lim YY, Maruff P, Pietrzak RH, Ames D, Ellis KA, Harrington K, et al. Rowe CC. Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer's disease. Brain. 2014;137(1):221–231. doi: 10.1093/brain/awt286. [DOI] [PubMed] [Google Scholar]
- Machulda MM, Pankratz VS, Christianson TJ, Ivnik RC, Mielke MM, Roberts RO, et al. Petersen RC. Practice effects and longitudinal cognitive change in normal aging vs. incident mild cognitive impairment and dementia in the Mayo Clinic Study of Aging. The Clinical Neuropsychologist. 2013;27(8):1247–1264. doi: 10.1080/13854046.2013.836567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mormino EC, Betensky RA, Hedden T, Schultz AP, Amariglio RE, Rentz DM, et al. Sperling RA. Synergistic effect of B-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurology. 2014;2014:E1–E7. doi: 10.1001/jamaneurol.2014.2031. Published online September, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, et al. Mintun MA. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66(12):1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis S, Forstmann BU, Wagenmakers EJ. Erroneous analyses of interactions in neuroscience: a problem of significance. Nat Neurosci. 2011;14(9):1105–1107. doi: 10.1038/nn.2886. [DOI] [PubMed] [Google Scholar]
- Okonkwo OC, Oh JM, Koscik R, Jonaitis E, Cleary CA, Dowling NM, et al. Johnson SC. Amyloid Burden, Neuronal Function, and Cognitive Decline in Middle-Aged Adults at Risk for Alzheimer's Disease. Journal of the International Neuropsychological Society. 2014;20(04):422–433. doi: 10.1017/S1355617714000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Wiste HJ, Weigand SD, et al. Association of elevated amyloid levels with cognition and biomarkers in cognitively normal people from the community. JAMA Neurology. 2016;73(1) doi: 10.1001/jamaneurol.2015.3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer RI, Kurosaki TT, Harrah CH, Jr, Chance JM, Filos S. Measurement of functional activities in older adults in the community. J Gerontol. 1982;37(3):323–329. doi: 10.1093/geronj/37.3.323. [DOI] [PubMed] [Google Scholar]
- Reitan R. Validity of the Trail Making Test as an indicator of organic brain damage. Perceptual & Motor Skills. 1958;8:271–276. [Google Scholar]
- Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, et al. Wong DF. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74(10):807–815. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey A. L'examen clinique en psychologie. Paris: Presses Universitaires de France; 1964. [Google Scholar]
- Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF, et al. Rocca WA. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30(1):58–69. doi: 10.1159/000115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senjem ML, Gunter JL, Shiung MM, Petersen RC, Jack CR., Jr Comparison of different methodological implementations of voxel-based morphometry in neurodegenerative disease. Neuroimage. 2005;26(2):600–608. doi: 10.1016/j.neuroimage.2005.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small B, Backman L. Longitudinal trajectories of cognitive change in preclinical Alzheimer's Disease: a growth mixture modeling analysis. Cortex. 2007;43:826–834. doi: 10.1016/s0010-9452(08)70682-8. [DOI] [PubMed] [Google Scholar]
- Small GW, Siddarth P, Kepe V, et al. Prediction of cognitive decline by positron emission tomography of brain amyloid and tau. Archives of Neurology. 2012;69(2):215–222. doi: 10.1001/archneurol.2011.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitz BE, Weissfeld LA, Lopez OL, Kuller LH, Saxton J, Singhabahu DM, et al. DeKosky ST. Cognitive trajectories associated with β-amyloid deposition in the oldest-old without dementia. Neurology. 2013 doi: 10.1212/WNL.0b013e31828c2fc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Phelps CH. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, Aisen P. The A4 Study: Stopping AD Before Symptoms Begin? Science translational medicine. 2014;6(228):228fs213–228fs213. doi: 10.1126/scitranslmed.3007941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storandt M, Mintun MA, Head D, Morris JC. Cognitive decline and brain volume loss as signatures of cerebral amyloid-β peptide deposition identified with pittsburgh compound b: Cognitive decline associated with aβ deposition. Archives of Neurology. 2009;66(12):1476–1481. doi: 10.1001/archneurol.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss E, Sherman EMS, Spreen O. A Compendium of Neuropsychological Tests. New York: Oxford University Press; 2006. [Google Scholar]
- van Harten AC, Smits LL, Teunissen CE, Visser PJ, Koene T, Blankenstein MA, et al. van der Flier WM. Preclinical AD predicts decline in memory and executive functions in subjective complaints. Neurology. 2013;81(16):1409–1416. doi: 10.1212/WNL.0b013e3182a8418b. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Knopman DS. The role of cerebrovascular disease when there is concomitant Alzheimer disease`. Biochemica et Biophysica Acta. 2015 doi: 10.1016/j.bbadis.2015.09.013. http://dx.doi.org/10.1016/j.bbadis_2015.09.013. [DOI] [PMC free article] [PubMed]
- Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Machulda M, Lowe VJ, et al. Jack CR. Effect of intellectual enrichment on AD biomarker trajectories: Longitudinal imaging study. Neurology. 2016;86(12):1128–1135. doi: 10.1212/wnl.0000000000002490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Roberts RO, Lowe VJ, et al. Jack CR. Effect of lifestyle activities on alzheimer disease biomarkers and cognition. Annals of Neurology. 2012;72(5):730–738. doi: 10.1002/ana.23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SJB, Xiong C, Visser PJ, Jasielec MS, Hassenstab J, Grant EA, et al. Fagan AM. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. The Lancet Neurology. 2013;12(10):957–965. doi: 10.1016/S1474-4422(13)70194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Adult Intelligence Scale-Revised. San Antonio, TX: The Psychololgical Corporation; 1981. [Google Scholar]
- Wechsler D. Wechsler Memory Scale-Revised. New York: The Psychological Corporation; 1987. [Google Scholar]
- Wilson R, Aggarwal N, Barnes L, Mendes de Leon C, Hebert L, Evans D. Cognitive decline in indicent Alzheimer disease in a community population. Neurology. 2010;74:951–955. doi: 10.1212/WNL.0b013e3181d64786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Beckett L, Bennett D, Albert M, Evans D. Change in cognitive function in older persons from a community population. Archives of Neurology. 1999;56:1274–1279. doi: 10.1001/archneur.56.10.1274. [DOI] [PubMed] [Google Scholar]
- Wilson R, Leurgans S, Boyle P, Bennett B. Cognitive decline in prodromal Alzheimer Disease and mild cognitive impairment. Archives of Neurology. 2011;68(3):351–356. doi: 10.1001/archneurol.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Li Y, Bienias J, Bennett D. Cognitive decline in old age: separating retest effects from the effects of growing older. Psychology and Aging. 2006;21(4):774–789. doi: 10.1037/0882-7974.21.4.774. [DOI] [PubMed] [Google Scholar]
- Wirth M, Madison CM, Rabinovici GD, Oh HL, Landau SM, Jagust WJ. Alzheimer's Disease Neurodegenerative Biomarkers Are Associated with Decreased Cognitive Function but Not β-Amyloid in Cognitively Normal Older Individuals. The Journal of Neuroscience. 2013;33(13):5553–5563. doi: 10.1523/jneurosci.4409-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Oh H, Mormino EC, Markley C, Landau SM, Jagust WJ. The effect of amyloid B on cognitive decline is modulated by neural integrity in cognitively normal elderly. Alzheimer's & Dementia. 2013;9:687–698. doi: 10.1016/j.jalz.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Boyle P, Wilson R, Segawa E, Leurgans S, De Jager P, Bennett D. A random change point model for cognitive decline in Alzheimer's Disease and mild cognitive impairment. Neuroepidemiology. 2012;39:73–83. doi: 10.1159/000339365. [DOI] [PMC free article] [PubMed] [Google Scholar]