

Abstract
Background
Platelet Endothelial Aggregation Receptor 1 (PEAR1), a membrane protein highly expressed in platelets and endothelial cells, plays a role in platelet contact-induced activation, sustained platelet aggregation and endothelial function. Previous reports implicate PEAR1 rs12041331 as a variant influencing risk in patients with coronary heart disease. We investigated whether genetic variation in PEAR1 predicts cardiovascular outcome in a white population.
Methods
In 1938 participants enrolled in the Flemish Study on Environment, Genes and Health Outcomes (51.3% women; mean age 43.6 years), we genotyped 9 tagging SNPs in PEAR1, measured baseline cardiovascular risk factors, and recorded Cardiovascular disease incidence. For SNPs, we contrasted cardiovascular disease incidence of minor-allele heterozygotes and homozygotes (variant) vs. major-allele homozygotes (reference) and for haplotypes carriers vs. non-carriers. In adjusted analyses, we accounted for family clusters and baseline covariables, including sex, age, body mass index, mean arterial pressure, the total-to-HDL cholesterol ratio, smoking and drinking, antihypertensive drug treatment, and history of cardiovascular disease and diabetes mellitus.
Results
Over a median follow-up of 15.3 years, 238 died and 181 experienced a major cardiovascular endpoint. The multivariable-adjusted hazard ratios of eight PEAR1 SNPs, including rs12566888, ranged from 0.87 to 1.07 (P ≥0.35) and from 0.78 to 1.30 (P ≥0.15), respectively. The hazard ratios of three haplotypes with frequency ≥10% ranged from 0.93 to 1.11 (P ≥0.49) for mortality and from 0.84 to 1.03 (P ≥0.29) for a cardiovascular complications. These results were not influenced by intake of antiplatelet drugs, nonsteroidal anti-inflammatory drugs, or both (P-values for interaction ≥ 0.056).
Conclusions
In a White population, we could not replicate previous reports from experimental studies or obtained in patients suggesting that PEAR1 might be a susceptibility gene for cardiovascular complications.
Electronic supplementary material
The online version of this article (doi:10.1186/s12881-017-0411-x) contains supplementary material, which is available to authorized users.
Keywords: Clinical genetics, Cardiovascular risk, PEAR1, Population science, Replication research
Background
Platelet Endothelial Aggregation Receptor 1 (PEAR1) is a membrane tyrosine kinase receptor highly expressed in platelets and endothelial cells. PEAR1 mediates platelet contact-induced activation [1] and sustains aggregation by supporting activation of the platelet specific integrin αIIbβ3 [2, 3]. Variation in the PEAR1 gene, including SNP rs12041331, is associated with increased platelet responses to agonists [4] and with the inter-individual variability in the response to antiplatelet drugs [5, 6]. Among patients with coronary heart disease taking antiplatelet agents [5], rs12041331 A-allele carriers experienced more adverse cardiovascular outcomes and had higher death rates than GG homozygotes. Moreover, in experimental studies [7], we observed an inverse correlation between endothelial PEAR1 expression and vascular assembly. In the Heredity and Phenotype Intervention Heart Study [8], there was significant association between flow-mediated dilation and rs12041331. A meta-analysis of 75,000 publicly available microarrays [8] revealed that expression of PEAR1 is highly correlated with genes, such as ANG2, ACVRL1 and ENG, and phenotypes, such as endothelial cell migration and angiogenesis, which play a pivotal role in endothelial function.
Platelet [5] and endothelial [9] dysfunction precede adverse cardiovascular outcomes, but the impact of genetic variability in PEAR1 on cardiovascular outcome remains poorly understood and requires further clarification, in particular in unbiased population samples. To this end, we analysed the database of the Flemish Study on Environment, Genes and Health Outcomes (FLEMENGHO [10–12]) to search for association between the incidence of cardiovascular complications and genetic variation in PEAR1.
Methods
Study population
The recruitment and follow-up of FLEMENGHO participants are fully described in previous publications [10–12]. In this study, of 3343 FLEMENGHO participants, we excluded 1405 from analysis, because blood stored in the biobank was exhausted with no material left for genotyping (n = 521), because of DNA degradation (n = 314), because one or more PEAR1 SNPs were missing (n = 16), because participants were less than 20 years old at enrolment (n = 372) with no contribution to incident cardiovascular disease, or because follow-up data were lacking (n = 182). Of the 16 participants with missing information on SNPs, none experienced a cardiovascular event during follow-up and one men died from prostate carcinoma. Finally, the number of participants carried through in all statistical analyses totalled 1938.
Measurements at baseline
The information of blood pressure, anthropometric characteristics, medical history, and smoking and drinking habits were obtained by trained nurses as described elsewhere previously [12]. Blood pressure was the average of five consecutive auscultatory readings obtained with a standard mercury sphygmomanometer after participants had rested in the sitting position for at least 5 min. Mean arterial blood pressure was diastolic blood pressure plus one third of the difference of systolic minus diastolic blood pressure. The nurses also administered a standardised questionnaire inquiring about each participant’s medical history, smoking and drinking habits, and intake of medications. Antiplatelet agents included aspirin, dipyridamole, ticlopidine and clopidogrel. Plasma glucose and serum total and high-density lipoprotein (HDL) cholesterol and serum creatinine were measured by automated methods in certified laboratories.
Follow-up of mortality and morbidity
We ascertained the vital status of participants at annual intervals until 31 December 2014 via the Belgian Population Registry. In addition, we obtained the International Classification of Disease codes for the immediate and underlying causes of death from the Flemish Registry of Death Certificates. For 1838 participants, we collected information on the incidence of non-fatal cardiovascular events either via face-to-face follow-up visits with repeated administration of the same standardised questionnaire as used at baseline (n = 1660) or via a structured telephone interview (n = 178). Follow-up data involving face-to-face contact were available from one visit in 501 participants, from two in 352, from three in 388, and from four or more in 419 participants.
Trained nurses used the International Classification of Diseases to code incident adverse health outcomes. Investigators blinded with regard to the genotypic results adjudicated the cause of death and non-fatal cardiovascular events against the medical records of general practitioners or the hospitals in the catchment area of the study. Coronary events included sudden death, fatal and non-fatal myocardial infarction, and surgical or percutaneous coronary revascularisation. Cerebrovascular disease included ischaemic stroke and transient ischaemic attack. Cardiovascular events included all of the foregoing coronary and cerebrovascular events plus pulmonary embolism, deep vein thrombosis, aortic dissection or aneurysm, and thrombosis or revascularisation of visceral or peripheral arteries. In the outcome analyses, we only considered the first event within each category.
Genotyping
PEAR1 (22704 base-pairs) maps to a genomic area characterised by high linkage disequilibrium (Fig. 1) on chromosome 1. We selected nine tagging SNPs (rs2768762, rs2644620, rs12566888, rs2768744, rs6671392, rs822441, rs11264581, rs12137505 and rs749256; Additional file 1: Table S1) that are in high linkage disequilibrium (R 2 > 0.80) with 53 SNPs, covering the entire PEAR1 gene and have a minor allele frequency of at least 1%. After extraction of genomic DNA from peripheral blood cells [13], the SNPs were genotyped, using the TaqMan® OpenArray™ Genotyping System (Life Technologies, Foster City, CA). All DNA samples were loaded at 50 ng per microliter and amplified on customised arrays following the manufacturer’s instructions. For analysis of the genotypes, we used autocalling methods implemented in the TaqMan Genotyper software version 1.3 (Life Technologies). Next, genotype clusters were evaluated manually with the sample call rate set above 0.90. Sixteen duplicate samples gave 100% reproducibility for all SNPs on the custom made array [12]. The participants were genotyped for these SNPs by using the strategy as described previously [12, 13]. In our study population, rs12041331 was in complete linkage with genotyped rs12566888 (R 2 = 0.99; D’ = 1.000). We used Minimac software [14] to impute rs12041331, which in previous reports [5, 15] was associated with platelet function and cardiovascular outcomes.
Fig. 1.
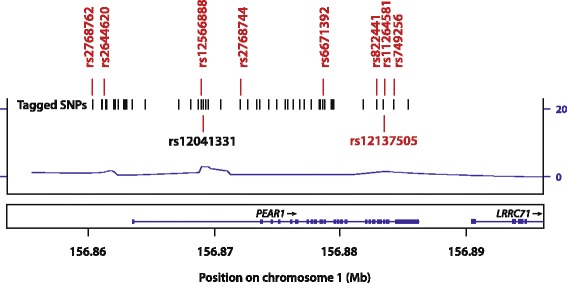
Plot of the PEAR1 gene (p13.1–12.3). The x-axis represents the physical position on chromosome 1 (build 37, hg19). The y-axis and the line indicate the recombination rate. The nine selected tagging SNPs (rs number and position given) were in high linkage disequilibrium (R 2 > 0.80) with ~50 tagged SNPs denoted by vertical lines. Imputed rs12041331 was in complete linkage with genotyped rs12566888 (R 2 = 0.99; D’ = 1.000)
Statistical analysis
For database management and statistical analysis, we used SAS software, version 9.4 (SAS Institute, Cary, NC). For comparison of means and proportions, we applied the large sample z test or ANOVA and Fisher’s exact, respectively. We tested Hardy-Weinberg equilibrium in unrelated founders, using the exact statistics available in the PROC ALLELE procedure of the SAS package. For analysis of single SNPs, we compared minor allele carriers with major allele homozygotes. We tested linkage disequilibrium, using the SAS procedures PROC ALLELE. Using the expectation-maximisation algorithm as implemented in the PROC HAPLOTYPE procedure of the SAS software, we used all SNPs included in the statistical analysis to reconstruct haplotypes. In the context of this article, haplotype refers to a set of statistically associated PEAR1 SNPs.
We compared the cumulative incidence of adverse health outcomes in relation to genetic variants, using Cox proportional hazards models adjusted for sex and age. Next, we assessed the prognostic value of the genetic variants in multivariable-adjusted Cox regression. We checked the proportional hazard assumption by applying the Kolmogorov-type supremum test as implemented in the ASSESS statement of the PROC PHREG procedure. To account for family clusters, we used the PROC SURVIVAL procedure of the SAS-callable SUDAAN 11.0.1 software (Research Triangle Institute, NC)[12]. In this procedure, clustering within pedigrees was accounted for by including family as a random effect in the Cox models along with other covariables modelled as fixed effects. We analysed genotypes and haplotypes using major allele homozygotes and non-carriers as reference groups, respectively. Throughout our manuscript, statistical significance refers to a 2-sided P-value of 0.05 or less.
Results
Baseline characteristics
As described in a previous publication [11], all 1938 participants were White Europeans, of whom 994 (51.3%) were women. The study population consisted of 335 singletons and 1603 related subjects, belonging to 45 single-generation families and 176 multi-generation pedigrees. Age averaged (±SD) 43.6 ± 14.3 years, body mass index 25.7 ± 4.3 kg/m2, blood pressure 125.2 ± 15.7 mm Hg systolic and 76.2 ± 9.5 mm Hg diastolic, and total cholesterol 5.50 ± 1.15 mmol/L. Among all participants, 478 (24.7%) had hypertension, of whom 208 (44.0%) were on antihypertensive drug treatment, 32 (1.7%) had diabetes mellitus, and 52 (2.7%) reported a history of cardiovascular disease. Previous cardiovascular complications included coronary heart disease, ischaemic cerebrovascular disease, peripheral arterial disease and pulmonary embolism in 41 (2.1%), 9 (0.46%), 2 (0.10%) and 1 (0.052%) patients, respectively.
Of 994 women and 944 men, 275 (27.7%) women and 329 (34.8%) men were smokers, and 166 (16.7%) women and 394 (41.7%) men reported intake of alcohol. In smokers, median tobacco use was 15 cigarettes per day (interquartile range, 10 to 20 cigarettes per day). In drinkers, the median alcohol consumption was 14 g per day (8 to 28 g per day). Table 1 lists the baseline characteristics of participants according to the rs12566888 genotype. None of the risk factors differed between minor allele carriers and major allele homozygotes (0.30 ≤ P ≤ 0.97).
Table 1.
Baseline characteristics of participants by rs12566888 genotypes
| Characteristic | T allele carriers | GG homozygotes | All |
|---|---|---|---|
| N° | 363 | 1575 | 1938 |
| N° with characteristics (%) | |||
| Women | 195 (53.7) | 799 (50.7) | 994 (51.3) |
| Current smoker | 101 (27.8) | 503 (31.9) | 604 (31.2) |
| Drinking alcohol | 112 (30.9) | 448 (28.4) | 560 (28.9) |
| Diabetes mellitus | 3 (0.8) | 29 (1.8) | 32 (1.7) |
| Hypertension | 84 (23.1) | 394 (25.0) | 478 (24.7) |
| Treated hypertension | 39 (10.7) | 169 (10.7) | 208 (10.7) |
| History of CVD | 8 (2.2) | 44 (2.8) | 52 (2.7) |
| Antiplatelet drugs | 70 (19.3) | 305 (19.4) | 375 (19.3) |
| Mean of characteristic (±SD) | |||
| Age, years | 42.8 ± 14.1 | 43.6 ± 14.3 | 43.6 ± 14.3 |
| Body mass index, kg/m2 | 25.6 ± 4.3 | 25.7 ± 4.3 | 25.7 ± 4.3 |
| Waist-to-hip ratio | 0.84 ± 0.09 | 0.85 ± 0.09 | 0.84 ± 0.09 |
| Systolic blood pressure, mm Hg | 124.5 ± 15.2 | 125.3 ± 15.8 | 125.2 ± 15.7 |
| Diastolic blood pressure, mm Hg | 76.0 ± 9.4 | 76.2 ± 9.6 | 76.2 ± 9.5 |
| Mean arterial pressure, mm Hg | 92.2 ± 10.2 | 92.6 ± 10.6 | 92.5 ± 10.5 |
| Heart rate, beats per minute | 69.2 ± 9.5 | 69.4 ± 9.8 | 69.3 ± 9.6 |
| Total cholesterol, mmol/L | 5.47 ± 1.18 | 5.51 ± 1.15 | 5.50 ± 1.15 |
| HDL cholesterol, mmol/L | 1.39 ± 0.41 | 1.39 ± 0.39 | 1.39 ± 0.38 |
| Total-to-HDL cholesterol ratio | 4.31 ± 1.77 | 4.27 ± 1.65 | 4.27 ± 1.67 |
| Serum creatinine, μmol/L | 90.2 ± 16.6 | 91.0 ± 17.3 | 90.9 ± 17.1 |
| Plasma glucose, mmol/L | 5.12 ± 1.55 | 5.03 ± 1.32 | 5.04 ± 1.37 |
Abbreviations: HDL, high-density lipoprotein cholesterol. Mean arterial pressure was diastolic pressure plus one third of the difference of systolic minus diastolic pressure. Diabetes mellitus was a fasting or random plasma glucose level of ≥ 7.0 or ≥11.1 mmol/L (≥126 mg/dL or ≥200 mg/dL), or use of antidiabetic agents. Hypertension was a blood pressure of ≥140 mm Hg systolic or ≥90 mm Hg diastolic or use of antihypertensive drugs. There were no differences between minor allele carriers and major allele homozygotes (0.30 ≤ P ≤ 0.97)
Incidence of events
Over a median follow-up of 15.3 years (5th to 95th percentile interval, 7.4 to 27.2 years), 238 participants died. Table 2 lists incident events by fatality and disease endpoint. Coronary events (n = 107) comprised 7 sudden deaths, 17 fatal and 35 non-fatal myocardial infarctions, 78 cases of surgical (n = 29) or percutaneous (n = 56) coronary revascularisation. Ischaemic cerebrovascular events (n = 61) included 15 fatal and 38 non-fatal cases of ischaemic stroke and 14 cases of transient ischaemic attack. The composite cardiovascular endpoint, consisting of 53 fatal and 128 non-fatal events, included the aforementioned coronary and cerebrovascular events plus 4 fatal and 14 non-fatal cases of pulmonary embolism or deep vein thrombosis, 7 fatal cases of aortic dissection or aneurysm, and 3 fatal and 13 non-fatal cases of thrombosis of visceral or peripheral arteries. Non-fatal events do not sum up, as only the first event in each category was analysed.
Table 2.
Fatal and Nonfatal Cardiovascular Events
| Endpoint | Type | Number of events |
|---|---|---|
| Sudden death | Fatal | 7 |
| Myocardial infarction | Fatal | 17 |
| Non-fatal | 35 | |
| Coronary revascularisation | Non-fatal | 78 |
| Ischaemic cardiomyopathy | Non-fatal | 22 |
| Ischaemic stroke | Fatal | 15 |
| Non-fatal | 38 | |
| Transient ischaemic attack | Non-fatal | 14 |
| Pulmonary embolism | Fatal | 4 |
| Pulmonary embolism or deep venous thrombosis | Non-fatal | 14 |
| Aortic aneurysm or dissection | Fatal | 7 |
| Peripheral arterial diseases | Fatal | 2 |
| Non-fatal | 13 | |
| Visceral arterial thrombosis | Fatal | 1 |
| Total number | 181 |
Follow-up of the 1938 participants spanned a median of 15.3 years (5th to 95th percentile interval, 7.4–27.2 years). Participants could experience multiple non-fatal events, so that number do not add up. In the outcome analyses, only the first event within each category was considered
Use of antiplatelet agents
Of 1938 participants, 375 (19.3%), 374 (19.3%) and 84 (4.3%) were on antiplatelet therapy, non-steroidal anti-inflammatory drugs and both at any time point, while 250 (12.9%), 240 (12.4%) and 53 (2.7%) took these agents for at least 25% of their follow-up. Of 375 patients on antiplatelet agents at any time, 369 (19.0%) took aspirin, 9 (0.46%) dipyridamole, 11 (0.57%) ticlopidine and 43 (2.2%) clopidogrel, either in monotherapy (n = 318 [16.4%]) or in combination (n = 57 [2.9%]). Among the 250 participants on antiplatelet agents during at least 25% of their follow-up, 249 (12.8%) took aspirin, 1 (0.052%) dipyridamole, 2 (0.10%) ticlopidine and 15 (7.7%) clopidogrel prescribed in monotherapy (n = 233 [12.0%]) or in combination (n = 17 [0.88%]).
Analyses of SNPs
Additional file 1: Table S2 describes the position of the 9 SNPs on chromosome 1. In 787 unrelated founders (first generation participants), all SNPs complied with Hardy-Weinberg equilibrium (0.08 ≤ P ≤0.88) with the exception of rs749256 (P = 0.01), which was therefore excluded for further analysis. In the whole study population (Additional file 1: Table S3), excluding rs749256, the frequencies of the minor alleles in all of the SNPs ranged from 9.9 to 42.9%. The frequencies of minor allele homozygotes in all of the analysed 8 SNPs were less than 5% (from 1.1 to 4.2%), except for rs12137505 (18.5%).
As illustrated for total mortality and the composite cardiovascular endpoint in relation to rs12566888 in Fig. 2, for all SNPs (0.23 ≤ P ≤0.98), the sex- and age-adjusted cumulative incidence of all endpoints under study did not differ between minor allele carriers and major allele homozygotes. There were also no differences in these estimates between homozygous and heterozygous minor allele carriers (0.38 ≤ P ≤0.92). Next, we accounted for family clusters and adjusted the hazard ratios for baseline characteristics, including sex, age, body mass index, mean arterial pressure, the total-to-HDL cholesterol ratio, smoking and drinking, antihypertensive drug treatment and history of cardiovascular disease and diabetes mellitus. As illustrated for all endpoints under study in relation to rs12566888, cardiovascular risk was similar among minor allele carriers and major allele homozygotes (0.25 ≤ P ≤0.95; Table 3). Analysis of the seven other PEAR1 SNPs similarly produced non-significant findings (Additional file 1: Table S4). Among the 1938 participants, 250 and 240 were taking antiplatelet agents or nonsteroidal anti-inflammatory drugs for at least 25% of their follow-up. The aforementioned results with respect to rs12566888 and the seven other SNPs were consistent, irrespective of the intake of antiplatelet drugs, nonsteroidal anti-inflammatory drugs, or both (Additional file 1: Table S5; P-values for interaction ≥ 0.27).
Fig. 2.
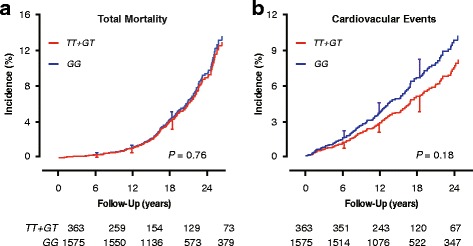
Incidence of total mortality (a) and cardiovascular disease (b) by rs12566888 genotype. Estimates were standardised to the mean of the distributions of sex and age in the whole study population. Vertical bars denote the standard error. Cumulative incidence did not differ between minor allele homozygotes and heterozygotes (0.38 ≤ P ≤ 0.92). Median follow-up was 15.3 years. Tabulated data are the number of participants at risk by genotype at 6-year intervals
Table 3.
Multivariable-adjusted hazard ratios by rs12566888 genotype
| Event | N° of events (rate per 1000 person-years) | Hazard ratio (95% CI) | P | |
|---|---|---|---|---|
| T allele carriers (N = 363) | GG homozygotes (N = 1575) | |||
| Total mortality | 40 (6.80) | 198 (7.58) | 0.99 (0.72–1.36) | 0.95 |
| Cardiovascular mortality | 7 (1.19) | 46 (1.79) | 0.79 (0.36–1.73) | 0.55 |
| Cardiovascular events | 27 (4.76) | 154 (6.15) | 0.78 (0.50–1.20) | 0.25 |
| Coronary events | 18 (3.13) | 89 (3.50) | 0.93 (0.55–1.58) | 0.79 |
| Ischaemic cerebrovascular events | 8 (1.37) | 53 (2.05) | 0.72 (0.35–1.46) | 0.36 |
Numbers of events do not add up, because only the first event in each category was analysed. Hazard ratios (95% confidence interval) express the risk of minor allele carriers vs. major allele homozygotes. Hazard ratios account for family clusters, and were adjusted for baseline characteristics including sex, age, body mass index, mean arterial pressure, total-to-HDL cholesterol ratio, smoking and drinking, antihypertensive drug treatment, and history of cardiovascular disease and diabetes mellitus
Analysis of haplotypes
Using the expectation-maximisation algorithm as implemented in the PROC HAPLOTYPE procedure of the SAS software, three haplotypes had a frequency of over 10% (Additional file 1: Table S6) and were carried through in the analysis. With letters referring to the rs2768762, rs2644620, rs12566888, rs2768744, rs6671392, rs822441, rs11264581, and rs12137505 alleles, respectively (Additional file 1: Tables S1 and S2), these haplotypes (Additional file 1: Table S6) were TTGATGGA (37.1%), TTGATGGG (22.8%), and TTGATGAG (12.0%). As illustrated for total mortality and the composite cardiovascular endpoint in relation to the most frequent haplotype (TTGATGGA) in Fig. 3, the sex-and age-adjusted cumulative incidence of any endpoint did not differ between carriers and non-carriers of the three haplotypes (0.24 ≤ P ≤0.86). Similarly, the multivariable-adjusted hazard ratios did not reveal increased risk associated with any haplotype (0.11 ≤ P ≤0.97; Table 4). As shown for TTGATGGA in Additional file 1, Table S7, the haplotype results were independent of the use of antiplatelet drugs, non-steroidal anti-inflammatory drugs, or both (P-values for interaction ≥ 0.056).
Fig. 3.
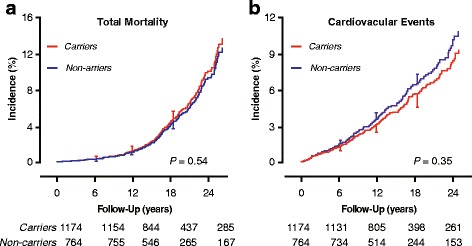
Incidence of total mortality (a) and cardiovascular disease (b) in PEAR1 TTGATGGA haplotype carriers and non-carriers. Letters coding the haplotype refer to the rs2768762, rs2644620, rs12566888, rs2768744, rs6671392, rs822441, rs11264581, and rs12137505 alleles respectively (Additional file 1: Tables S1 and S2). Vertical bars denote the standard error. P-values refer to the difference between non-carriers and carriers. Median follow-up was 15.3 years. Tabulated data are the number of participants at risk by TTGATGGA carrying status at 6 year intervals
Table 4.
Multivariable-adjusted hazard ratios by PEAR1 haplotypes
| Haplotypes Event | N° of events (rate per 1000 person-years) | Hazard ratio (95% CI) | P | |
|---|---|---|---|---|
| Carriers | Non carriers | |||
| TTGATGGA (N° at risk) | 1174 | 764 | ||
| Total mortality | 146 (7.46) | 92 (7.37) | 1.05 (0.81–1.35) | 0.73 |
| Cardiovascular mortality | 29 (1.48) | 24 (1.92) | 0.79 (0.43–1.45) | 0.45 |
| Cardiovascular events | 102 (5.43) | 79 (6.62) | 0.84 (0.60–1.17) | 0.29 |
| Coronary events | 64 (3.37) | 43 (3.53) | 0.98 (0.62–1.53) | 0.91 |
| Ischaemic cerebrovascular events | 31 (1.60) | 30 (2.43) | 0.67 (0.41–1.09) | 0.11 |
| TTGATGGG (N° at risk) | 788 | 1150 | ||
| Total mortality | 97 (7.48) | 141 (7.39) | 0.93 (0.72–1.20) | 0.58 |
| Cardiovascular mortality | 21 (1.62) | 32 (1.68) | 0.90 (0.51–1.58) | 0.72 |
| Cardiovascular events | 78 (6.25) | 103 (5.65) | 1.03 (0.76–1.39) | 0.87 |
| Coronary events | 43 (3.39) | 64 (3.46) | 0.93 (0.63–1.37) | 0.71 |
| Ischaemic cerebrovascular events | 28 (2.18) | 33 (1.75) | 1.17 (0.68–2.01) | 0.58 |
| TTGATGAG (N° at risk) | 435 | 1503 | ||
| Total mortality | 65 (8.87) | 173 (7.00) | 1.11 (0.83–1.48) | 0.49 |
| Cardiovascular mortality | 17 (2.32) | 36 (1.46) | 1.33 (0.73–2.45) | 0.35 |
| Cardiovascular events | 44 (6.26) | 137 (5.78) | 1.00 (0.70–1.43) | 0.98 |
| Coronary events | 26 (3.66) | 81 (3.36) | 1.01 (0.63–1.63) | 0.97 |
| Ischaemic cerebrovascular events | 15 (2.07) | 46 (1.88) | 0.96 (0.54–1.70) | 0.89 |
Numbers of events do not add up, because only the first event in each category was analysed. Letters coding the haplotypes refer to the rs2768762, rs2644620, rs12566888, rs2768744, rs6671392, rs822441, rs11264581, and rs12137505 alleles, respectively (see Additional file 1: Tables S1 and S2). Haplotypes were reconstructed using the expectation-maximisation algorithm as implemented in the PROC HAPLOTYPE procedure of the SAS software version 9.4. Haplotypes with a frequency of ≥10% were carried forward in the analysis. Hazard ratios (95% confidence interval) express the risk of haplotype carriers vs. non carriers. Hazard ratios account for family clusters, and were adjusted for baseline characteristics including sex, age, body mass index, mean arterial pressure, total-to-HDL cholesterol ratio, smoking and drinking, antihypertensive drug treatment, and history of cardiovascular disease and diabetes mellitus
Discussion
We could not confirm our working hypothesis that incidence of cardiovascular disease is associated with genetic variation in PEAR1. These results were independent of the use of antiplatelet agents or anti-inflammatory drugs. Our hypothesis originated from studies showing association of variability in the responses to antiplatelet drugs [4, 6] or of the incidence of cardiovascular complications [5] with rs12041331. A recent genome-wide association study [15] also demonstrated association of platelet aggregation with rs12566888. In our current study, we covered the entire PEAR1 gene by analysis of eight tagging SNPs, which are in high linkage disequilibrium with 53 other SNPs in PEAR1. For analysis of single SNPs, we contrasted minor allele carriers with major allele homozygotes, because of the low frequency of the minor alleles and minor allele homozygotes (Additional file 1: Table S3) and because the sex- and age-adjusted cumulative incidence of all endpoints under study was similar in minor allele homozygotes and heterozygotes. rs12566888 genotyped in the present study is a proxy for rs12041331. In line with the reported R 2 of 0.97 [15], in the present study population this measure of linkage disequilibrium was 0.99.
Experimental data [1, 2, 4, 15, 16] and studies of platelet aggregation in humans [17] further underpinned our hypothesis that genetic variation in PEAR1 might predict cardiovascular risk. Indeed, recent studies [1, 2] generated the molecular and functional evidence that PEAR1 is a platelet transmembrane protein that is activated by signalling molecules or platelet contact. Agonists, such as ADP and epinephrine, enhance the membrane expression of PEAR1 and its activation by tyrosine phosphorylation, which can be blocked by eptifibatide, an αIIbβ3 antagonist [2]. Platelet proximity induced by centrifugation also increases PEAR1 tyrosine phosphorylation, independent of αIIbβ3 [2]. In a functional genomics approach, several PEAR1 SNPs (rs3737224, rs41299597, rs41273215, rs82242 and rs11264579) were associated with increased platelet responses to collagen-related peptide and enhanced PEAR1 protein expression [16]. Similarly, rs12041331 [15] and rs12566888 [4] were associated with PEAR1 expression [15] or functionality [4]. C-allele carriers of rs2768759 recruited among families with premature coronary artery disease [17] showed increased native platelet aggregation to agonists in vitro before and after two weeks of aspirin treatment. Relevant for our current report, two studies [4, 15] demonstrated that platelet aggregation in response to aspirin [15] or agonists [4], including adenosine diphosphate, epinephrine and collagen, is related to intronic variation at rs12566888, but proposed that additional studies would be needed to clarify the importance of genetic variation in PEAR1 to cardiovascular disease progression and response to antiplatelet therapy.
Our current null findings obtained in a general population are at variance with observations in selected patients with coronary heart disease on treatment with aspirin. Lewis and coworkers [5] addressed the association of cardiovascular outcomes with genetic variation in PEAR1 in two independent aspirin-treated cohorts: 227 percutaneous coronary intervention patients and 1000 patients of the International Verapamil SR/Trandolapril Study Genetic Substudy (INVEST-GENES). In 144 white and 83 black patients undergoing percutaneous coronary intervention, A-allele carriers of rs12041331 were more likely to experience a cardiovascular event or death compared with GG homozygotes. The hazard ratios were 2.62 (95% confidence interval, 0.96–7.10; P = 0.059) and 3.97 (1.10–14.3; P = 0.035), respectively). In aspirin-treated INVEST-GENES patients [5], rs12041331 A-allele carriers had a significantly twofold increased risk of myocardial infarction compared with GG homozygotes. This is in apparent contradiction with several reports showing association of the rs12041331 A-allele with lower platelet function or PEAR1 expression [5, 15, 18, 19] or with endothelial dysfunction [8]. This contradictory results highlight the necessity of further investigations, in particular in unbiased population samples.
The absence of association between the risk of cardiovascular complications and genetic variation in PEAR1 demonstrates that the results from experimental studies are difficult to translate in to human health outcomes. In the Duke Databank for Cardiovascular Disease patients [20], candidate SNPs associated with in vitro aspirin resistance, including PEAR1 rs2768759, were not associated with clinical outcomes in aspirin-treated patients with coronary artery disease. Several issues might underlie the divergence between the experimental input and the epidemiological observations for the research question we addressed. First, the platelet function tests as used in the aforementioned genetic studies are conducted in an artificial environment, which might distort the responses to various agonists [21] and ignore the complexity of arterial thrombus formation [22]. Indeed, in clinical trials [22–24] platelet function tests set up to guide dosage of antiplatelet treatment failed to reduce cardiovascular risk. Second, as evidenced by genome-wide association studies [5], the statistical power to detect association of genetic variation with platelet function depends on other genetic variants close to the marker SNP and the frequency of the minor allele carriers, which may differ across populations and ethnicities. However, the PEAR1 SNP frequencies in our current study are in agreement with those reported in 1000 Genomes (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) or HapMap (ftp://ftp.ncbi.nlm.nih.gov/hapmap/), as showed in Additional file 1: Table S8. Finally, the problem of publication bias cannot be ignored, certainly not in the field of smaller size studies with focus on a single gene. Ginsel and coworkers [25] demonstrated a biased distribution of P-values in abstracts listed in Medline 2012 with an apparent increase in significance levels immediately below 0.05 relative to the frequency immediately above 0.05. This finding is likely to be evidence of P-hacking (biased analysis and reporting) or publication bias.
Conclusion
Based on a predefined hypothesis originating from experimental studies and observations in patients, we could not confirm that PEAR1 is a major susceptibility gene for cardiovascular disease in the population at large. Our study illustrates that experimental studies and observations in selected patients cannot be readily extrapolated to the general population. The divergence between experimental findings and the current study result might be related to the complex network of molecular pathways, in which PEAR1 is involved over and beyond its physiological role in platelet and endothelial function. Furthermore, epigenetic regulation of gene function might be another factor to be accounted for in future research of the functional significance of PEAR1.
Acknowledgements
The authors gratefully acknowledge the clerical assistance of Vera De Leebeeck and Renilde Wolfs and the contribution of Linda Custers, Marie-Jeanne Jehoul, Daisy Thijs and Hanne Truyens in data collection at the field centre.
Funding
The European Union (HEALTH-2011.2.4.2-2-EU-MASCARA, HEALTH-F7-305507 HOMAGE), the European Research Council (Advanced Researcher Grant 2011-294713-EPLORE and Proof-of-Concept Grant 713601-uPROPHET) and the Fonds voor Wetenschappelijk Onderzoek Vlaanderen, Ministry of the Flemish Community, Brussels, Belgium (G.0881.13, G.0880.13, 11Z0916N and 12M2715N) currently support the Studies Coordinating Centre in Leuven.
Availability of data and materials
Scientifically motivated request for data sharing will be considered. If justified, all or part of the data can be made available in anonymized format.
Authors’ contributions
JAS coordinated the Flemish Study on Environment, Genes and Health Outcomes and managed funding. LT and LJ coordinated the construction and updates of the master database and the management of the biobank. LC, SDC, and ES did the genotyping. CB, LC and DC supervised DNA extraction and amplification, set up the genotyping procedures and managed quality control of the genotypes. W-YY, TP, NC, Z-YZ, C-SS, F-FW and Y-MG collected phenotypic data and participated in checking the consistency of the database. W-YY, TK, NC and JAS collected outcome data. W-YY and JAS did the statistical analysis with guidance provided by LT. W-YY and JAS wrote the first draft of the manuscript. BI, CV, MFH and PV assisted in translating and interpreting the basic science data. All authors commented on successive drafts of the manuscript and approved the final version.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The FLEMENGHO population study complies with the Helsinki declaration for research in human subjects and the Belgian legislation for the protection of privacy (http://www.privacycommission.be). The Ethics Committee of the University of Leuven approved the study. At each contact, participants gave or renewed informed written consent.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ACVRL1
Activin receptor-like kinase 1
- ADP
Adenosine diphosphate
- ANG2
Angiopoietin 2
- CpG
-C-phosphate-G-
- DNA
Deoxyribonucleic acid
- ENG
Endoglin
- FLEMENGHO
Flemish study on environment, genes and health outcomes
- GWAS
Genome-wide association study
- HDL
High-density lipoprotein
- INVEST-GENES
International verapamil SR/trandolapril study genetic substudy
- PEAR1
Platelet endothelial aggregation receptor 1
- SNP
Single nucleotide polymorphism.
Additional file
Common tagging SNPs in PEAR1. Table S2. PEAR1 allele and genotype frequencies by SNP in unrelated founders. Table S3. PEAR1 allele and genotype frequencies by SNP in 1938 analysed participants. Table S4. Hazard ratios for all-cause mortality and the composite cardiovascular endpoint by PEAR1 SNPs. Table S5. Multivariable-adjusted hazard ratios associated with rs12566888 by antiplatelet treatment status. Table S6. Frequencies of haplotypes. Table S7. Multivariable-adjusted hazard ratios associated with the TTGATGGA haplotype by antiplatelet treatment status. Table S8. Minor allele frequencies in current study population and in European subjects in 1000 Genomes and HapMap. (DOC 211 kb)
Contributor Information
Wen-Yi Yang, Email: wenyi.yang@med.kuleuven.be.
Thibault Petit, Email: thibault.petit@uzleuven.be.
Nicholas Cauwenberghs, Email: nicholas.cauwenberghs@med.kuleuven.be.
Zhen-Yu Zhang, Email: zhenyu.yang@med.kuleuven.be.
Chang-Sheng Sheng, Email: scsheng2004@163.com.
Lutgarde Thijs, Email: lutgarde.thijs@med.kuleuven.be.
Erika Salvi, Email: erika.salvi@unimi.it.
Benedetta Izzi, Email: benedetta.izzi@med.kuleuven.be.
Christophe Vandenbriele, Email: christophe.vandenbriele@med.kuleuven.be.
Fang-Fei Wei, Email: fangfei.wei@med.kuleuven.be.
Yu-Mei Gu, Email: yumei.gu@med.kuleuven.be.
Lotte Jacobs, Email: lotte.jacobs@med.kuleuven.be.
Lorena Citterio, Email: citterio.lorena@hsr.it.
Simona Delli Carpini, Email: dellicarpini.simona@hsr.it.
Cristina Barlassina, Email: cristina.barlassina@unimi.it.
Daniele Cusi, Email: daniele.cusi@unimi.it.
Marc F. Hoylaerts, Email: marc.hoylaerts@med.kuleuven.be
Peter Verhamme, Email: peter.verhamme@uzleuven.be.
Tatiana Kuznetsova, Email: tatiana.kouznetsova@med.kuleuven.be.
Jan A. Staessen, Phone: +32-16-34-7104, Email: jan.staessen@med.kuleuven.be, Email: ja.staessen@maastrichtuniversity.nl
References
- 1.Nanda N, Bao M, Lin H, Clauser K, Komuves L, Quertermous T, et al. Platelet endothelial aggregation receptor 1 (PEAR1), a novel epidermal growth factor repeat-containing transmembrane receptor, participates in platelet contact-induced activation. J Biol Chem. 2005;280:24680–24689. doi: 10.1074/jbc.M413411200. [DOI] [PubMed] [Google Scholar]
- 2.Kauskot A, Di Michele M, Loyen S, Freson K, Verhamme P, Hoylaerts MF. A novel mechanism of sustained platelet αIIbβ3 activation via PEAR1. Blood. 2012;119:4056–4065. doi: 10.1182/blood-2011-11-392787. [DOI] [PubMed] [Google Scholar]
- 3.Qayyum R, Becher LC, Becher DM, Faraday N, Yanek LR, Leal SM, et al. Genome-wide association study of platelet aggregation in African Americans. BMC Genet. 2015;16:58–68. doi: 10.1186/s12863-015-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson AD, Yanek LR, Chen MH, Faraday N, Larson MG, Tofler G, et al. Genome-wide meta-analyses identifies seven loci associated with platelet aggregation in response to agonists. Nat Genet. 2010;42:608–613. doi: 10.1038/ng.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis JP, Ryan K, O’Connell JR, Horenstein RB, Damcott CM, Gibson Q, et al. Genetic variation in PEAR1 is associated with platelet aggregation and cardiovascular outcomes. Circ Cardiovasc Genet. 2013;6:184–192. doi: 10.1161/CIRCGENETICS.111.964627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiang Q, Cui Y, Zhao X, Zhao N. Identification of PEAR1 SNPs and their influences on the variation in prasugrel pharmacodynamics. Pharmacogenomics. 2013;14:1179–1189. doi: 10.2217/pgs.13.108. [DOI] [PubMed] [Google Scholar]
- 7.Vandenbriele C, Kauskot A, Vandersmissen I, Criel M, Geenens R, Craps S, et al. Platelet endothelial aggregation receptor-1: A novel modifier of neoangiogenesis. Cardiovasc Res. 2015;108:124–138. doi: 10.1093/cvr/cvv193. [DOI] [PubMed] [Google Scholar]
- 8.Fisch AS, Yerges-Armstrong LM, Bachman JD, Wang H, Donnelly P, Ryan KA, et al. Genetic variation in the platelet endothelial aggregation receptor 1 gene results in endothelial dysfunction. PLoS ONE. 2015;10:e0138795. doi: 10.1371/journal.pone.0138795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shechter M, Shechter A, Koren-Morag N, Feinberg MS, Hiersch L. Usefulness of brachial artery flow-mediated dilatation to predict long-term cardiovascular events in subjects without heart disease. Am J Cardiol. 2014;113:162–167. doi: 10.1016/j.amjcard.2013.08.051. [DOI] [PubMed] [Google Scholar]
- 10.Staessen JA, Wang JG, Brand E, Barlassina C, Birkenhäger WH, Herrmann SM, et al. Effects of three candidate genes on prevalence and incidence of hypertension in a Caucasian population. J Hypertens. 2001;19:1349–1358. doi: 10.1097/00004872-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Zagato L, Kuznetsova T, Tripodi G, Zerbini G, Richart T, et al. Angiotensin-converting enzyme I/D and α-adducin Gly460Trp polymorphisms. From angiotensin-converting enzyme activity to cardiovascular outcome. Hypertension. 2007;49:1291–1297. doi: 10.1161/HYPERTENSIONAHA.106.085498. [DOI] [PubMed] [Google Scholar]
- 12.Yang WY, Petit T, Thijs L, Zhang ZY, Jacobs L, Hara A, et al. Coronary risk in relation to genetic variation in MEOX2 and TCF15 in a Flemish population. BMC Genet. 2015;16:116. doi: 10.1186/s12863-015-0272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Citterio L, Simonini M, Zagato L, Salvi E, Delli Carpini S, Lanzani C, et al. Genes involved in vasoconstriction and vasodilation affect salt-sensitive hypertension. PLoS One. 2011;6:e19620. doi: 10.1371/journal.pone.0019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faraday N, Yanek LR, Yang XP, Mathias RA, Herrera-Galeano JE, Suktitipat B, et al. Identification of a specific intronic PEAR1 gene variant associated with greater platelet aggregability and protein expression. Blood. 2011;118:3367–3375. doi: 10.1182/blood-2010-11-320788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones CI, Bray S, Garner SF, Stephens J, de Bono B, Angenent WGJ, et al. A functional genomics approach reveals novel quantitative trait loci associated with platelet signaling pathways. Blood. 2009;114:1405–1416. doi: 10.1182/blood-2009-02-202614. [DOI] [PubMed] [Google Scholar]
- 17.Herrera-Galeano JE, Becher DM, Wilson AF, Yanek LR, Bray P, Vaidya D, et al. A novel variant in the platelet endothelial aggregation receptor-1 gene is associated with increased platelet aggregability. Arterioscler Thromb Vasc Biol. 2008;28:1484–1490. doi: 10.1161/ATVBAHA.108.168971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wurtz M, Nissen PH, Grove EL, Kristensen SD, Hvas AM. Genetic determinants of on-aspirin platelet reactivity: focus on the influence of PEAR1. PLoS ONE. 2014;9:e111816. doi: 10.1371/journal.pone.0111816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim Y, Suktitipat B, Yanek LR, Faraday N, Wilson AF, Becker DM, et al. Targeted deep resequencing idetifies coding variants in the PEAR1 gene that play a role in platelet aggregation. PLoS ONE. 2013;8:1–8. doi: 10.1371/annotation/c8b2e360-b78a-4c2f-a1a3-c53325f18211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voora D, Horton J, Shah SH, Shaw LK, Newby LK. Polymorphisms associated with in vitro aspirin resistance are not associated with clinical outcomes in patients with coronary artery disease who report regular aspirin use. Am Heart J. 2011;162:166–172. doi: 10.1016/j.ahj.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 21.Gorog DA, Sweenly JM, Fuster V. Antiplatelet drug ‘resistance’. Part 2: laboratory resistance to antiplatelet drugs-fact or artifact? Nat Rev Cardiol. 2009;6:365–373. doi: 10.1038/nrcardio.2009.13. [DOI] [PubMed] [Google Scholar]
- 22.Gorog DA, Fuster V. Platelet function tests in clinical cardiology: unfulfilled expectations. J Am Coll Cardiol. 2013;61:2115–2129. doi: 10.1016/j.jacc.2012.11.080. [DOI] [PubMed] [Google Scholar]
- 23.Current-Oasis 7 Investigators. Mehta SR, Bassand JP, Chrolavicius S, Diaz R, Eikelboom JW, et al. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med. 2010;363:930–942. doi: 10.1056/NEJMoa0909475. [DOI] [PubMed] [Google Scholar]
- 24.Price MJ, Berger PB, Teirstein PS, Tanguay JF, Angiolillo DJ, Spriggs D, et al. Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. J Am Med Assoc. 2011;305:1097–1105. doi: 10.1001/jama.2011.290. [DOI] [PubMed] [Google Scholar]
- 25.Ginsel B, Aggarwal A, Xuan W, Harris I. The distribution of probability values in medical abstracts: an obvervational study. BMC Res Notes. 2015;8:721. doi: 10.1186/s13104-015-1691-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Scientifically motivated request for data sharing will be considered. If justified, all or part of the data can be made available in anonymized format.