

Abstract
Background & Objectives
Chromatin structure is the single most important feature that distinguishes a cancer cell from a normal cell histologically. Chromatin remodeling proteins regulate chromatin structure and high mobility group A (HMGA1) proteins are among the most abundant, nonhistone chromatin remodeling proteins found in cancer cells. These proteins include HMGA1a/HMGA1b isoforms, which result from alternatively spliced mRNA. The HMGA1 gene is overexpressed in cancer and high levels portend a poor prognosis in diverse tumors. HMGA1 is also highly expressed during embryogenesis and postnatally in adult stem cells. Overexpression of HMGA1 drives neoplastic transformation in cultured cells, while inhibiting HMGA1 blocks oncogenic and cancer stem cell properties. Hmga1 transgenic mice succumb to aggressive tumors, demonstrating that dysregulated expression of HMGA1 causes cancer in vivo. HMGA1 is also required for reprogramming somatic cells into induced pluripotent stem cells. HMGA1 proteins function as ancillary transcription factors that bend chromatin and recruit other transcription factors to DNA. They induce oncogenic transformation by activating or repressing specific genes involved in this process and an HMGA1 “transcriptome” is emerging. Although prior studies reveal potent oncogenic properties of HMGA1, we are only beginning to understand the molecular mechanisms through which HMGA1 functions. In this review, we summarize the list of putative downstream transcriptional targets regulated by HMGA1. We also briefly discuss studies linking HMGA1 to Alzheimer’s disease and type-2 diabetes.
Conclusion
Further elucidation of HMGA1 function should lead to novel therapeutic strategies for cancer and possibly for other diseases associated with aberrant HMGA1 expression.
Keywords: High mobility group A1, HMGA, chromatin, cancer, tumor progression, metastasis, genes, cancer stem cells, embryonic stem cells
1. INTRODUCTION
The structural organization of the nucleus and its chromatin is the single most important histologic feature that distinguishes a cancer cell from a normal cell. Chromatin consists of nuclear DNA and proteins, and chromatin structure dictates whether genes will be actively transcribed or silent. Not surprisingly, chromatin structure is central to normal cell function, both in undifferentiated embryonic stem cells during development and in differentiated, mature cells. Eukaryotic chromatin exists in a condensed form as depicted in the “beads-on-a-string model” in which repeating units of DNA tightly wound around small basic histone proteins, called nucleosomes (the bead), are joined by linker DNA sequences (the string) to form chromatin fibers. This compact, yet malleable structure, not only provides an intricately organized framework for DNA, but also endows eukaryotes with the ability to repress or activate gene expression. As such, induction or repression of gene expression requires transient reorganization of the highly-ordered chromatin structure in which that gene resides, a process known as chromatin remodeling. Increasing evidence underscores the importance of aberrant chromatin remodeling in cancer and many other diseases.
Chromatin structure is maintained largely by chromatin binding proteins, which include both histone and nonhistone proteins. While many groups have investigated histones and chromatin binding proteins that enzymatically modify histones or DNA, few have focused on the high mobility group A (HMGA) chromatin remodeling proteins. In fact, HMGA proteins are among the most abundant, nonhistone chromatin binding proteins found in the nucleus of cancer cells [1–3]. They are members of the high mobility group (HMG) superfamily of proteins comprised of diverse, low molecular weight proteins named by their rapid migration or high mobility in polyacrylamide gel. HMGA1 proteins are primarily located in the nucleus and function in chromatin remodeling, although they lack an identifiable enzymatic activity. This superfamily of proteins was first discovered over 30 years ago in highly proliferative human cervical HeLa cancer cells [1–3]. Within the next two decades, members of the HMGA family (HMGA1a, HMGA1b, and HMGA2) were isolated from cancer cells and their genes were cloned [3–30]. All HMG proteins share an acidic, carboxyl terminus and associate with chromatin, but are distinguished by unique functional domains that confer distinct DNA binding motifs and biologic activities [21]. The HMGA family is defined by the AT-hook DNA binding domains [13–30], which mediate binding to nuclear chromatin at AT-rich regions in the minor groove, where HMGA1 proteins bend DNA to allow other transcription factors to bind [13–31]. Prior studies also show that HMGA1 proteins bind to mitochondrial DNA at AT-rich regions [32, 33; reviewed in 34]. The HMGA1 subfamily – the subject of this review - consists of both the HMGA1a and HMGA1b protein isoforms (formerly HMG-I and HMG-Y), which result from alternative splicing of the HMGA1 mRNA [6–8]; HMGA1a differs from HMGA1b by an additional 11 internal amino acids upstream of the second AT hook [6–8] (Fig. 1). The biological significance of two distinct isoforms is not yet clear, as functional studies indicate many overlapping roles [28, 29].
Fig. (1).
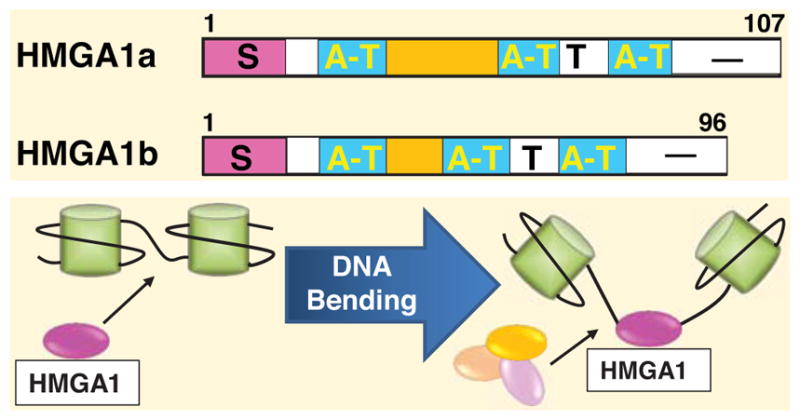
HMGA1a and HMGA1b protein isoforms are depicted with the serine (S) and threonine (T)-rich regions, AT-hook DNA binding domains (AT), and the acidic carboxyl terminal (−) region (top). HMGA1 functions as an architectural transcription factor that bends chromatin to enable binding of transcriptional complexes (bottom).
To date, HMGA1 proteins are known to participate in a myriad of cellular processes [1–119] including transcriptional regulation [17–24], neoplastic transformation [22–25, 28–30, 50, 54, 60, 68, 70–73, 75, 77, 86, 88, 90, 91, 100, 117–119], embryogenesis [98], anoikis [71, 77], metastatic progression [28, 29, 53, 54, 68, 70, 117], cell cycle regulation [100–108], repair of DNA damage [109–112], cellular senescence [113–115], mitochondrial function [32–34], and retroviral integration [116]. Most of these varied biological activities of HMGA1 are thought to result from its ability to alter chromatin structure and modulate gene expression, leading to different molecular pathways depending upon the cellular context. Promoter analyses and gene expression profile studies have uncovered downstream gene targets and an HMGA1 “transcriptome” is emerging (Fig. 2). In this review, we outline prior studies that reveal a central role of HMGA1 in diverse, aggressive cancers and normal development. We focus on the transcriptional targets regulated by HMGA1 in cancer and stem cells. In addition, we briefly consider studies that implicate HMGA1 in the pathogenesis of diabetes [35–38] and Alzheimer’s disease [39–42].
Fig. (2).

HMGA1 transcriptional networks involve all hallmarks of cancer.
2. HMGA1 IS UP-REGULATED IN RAPIDLY PROLIFERATING CELLS & CANCER
The first evidence linking HMGA1 proteins to cancer was their discovery as abundant chromatin binding proteins in HeLa cells, the aggressive human cervical carcinoma cells with a remarkable proliferative capacity [3]. Subsequent studies showed high levels of HMGA1 proteins in rat and mouse cells after oncogenic transformation by retroviral transduction [4, 5]. HMGA1 proteins are also elevated in spontaneous mouse tumors and tumors induced by either carcinogens or viral oncogenes compared to normal tissue [4, 5, 8]. High levels of HMGA1 proteins are found in rapidly proliferating tissues and neoplastic cells, with absent or low levels in normal, differentiated, adult tissues [43–46]. The Hmga1 gene was identified early on as a gene induced by serum or individual growth factors in quiescent murine fibroblasts, an experimental model that facilitated the discovery of several key oncogenic transcription factors [44]. In this model, Hmga1 is a “delayed-early” gene whose expression follows the initial wave of “immediate-early” genes [44]. Many immediate- and delayed-early genes are required by cells to traverse the G1/S boundary of the cell cycle and function as oncogenes when aberrantly expressed. Further studies uncovered high levels of HMGA1 expression at the mRNA or protein level in human cancer cells or primary tumors from diverse tissues, including thyroid [45–48], lung [49–51], breast [52–59, 117], bladder [58], prostate [60–62], colon [63–68], pancreas [69–74], uterine corpus [75], uterine cervix [76], kidney [77], head and neck [78], nervous system [58, 79–84], stomach [85, 86], liver [87], and hematopoietic system [88–93, 118, 119]. HMGA1 expression is low or undetectable in normal tissue counterparts. Taken together, these findings suggest a central role for HMGA1 in neoplastic transformation.
Subsequent studies found that high levels of HMGA1 expression correlate with adverse clinical outcomes and more advanced disease in cancer [reviewed in 24]. For example, high expression of HMGA1 at the mRNA or protein level was found in cultured cells derived from metastatic tumors compared to localized tumors, including breast [29, 54, 117], colon [63–65, 68], prostate [60], and pancreatic [69, 70, 74, 94] cancers. Further evidence that HMGA1 overexpression portends a poor prognosis in diverse cancers came with the advent of global gene and protein microarray technology. The first such study found that HMGA1 gene expression correlates with poor prognosis in primary medulloblastomas [79]. In squamous cell carcinoma and adenocarcinoma lung, an inverse correlation was observed for HMGA1 protein staining by immunohistochemical analysis and survival [49]. Higher HMGA1a mRNA and protein levels were discovered in hepatocellular carcinoma with intrahepatic metastases compared to those without intrahepatic metastases [87]. In breast cancer, HMGA1 protein levels correlate with high-grade/poor differentiation [55] and recurrent disease [56]. HMGA1 gene expression also associates with high-grade in uterine cancers [75]. In pancreatic cancer, HMGA1 protein levels are positively correlated with both poor differentiation status and decreased survival [74]. In pediatric B-lineage acute lymphoblastic leukemia, HMGA1 expression is higher in patients at relapse [119]. Moreover, a study comparing global gene expression profiles from many independent studies found that HMGA1 is among a core “signature” comprised of 9 transcription factor genes enriched in embryonic stem cells and high-grade/poorly differentiated cancers (breast, bladder and brain) [58]. Importantly, overexpression of this signature was associated with poor survival in patients with these cancers. Collectively, these studies suggest that HMGA1 promotes tumor progression and is a biomarker and potential target for more advanced disease.
3. HMGA1 IS HIGHLY EXPRESSED IN PLURI-POTENT STEM CELLS
Normally, HMGA1 genes are expressed at high levels during embryogenesis, with low or absent levels in most adult, differentiated murine tissues [23, 95, 98]. Similarly, HMGA1 is enriched in human and murine embryonic stem cells as well as induced pluripotent stem cells and adult stem cells, such as CD34+ hematopoietic stem cells and intestinal stem cells [58, 92, 93, 96–98, 120]. As noted above, HMGA1 is a core transcription factor in embryonic stem cells [58]. Because pluripotency factors (NANOG, OCT4, SOX2, KLF4, or cMYC) were not identified in this stem cell signature, it was postulated that HMGA1 and other core factors regulate the same cellular pathways induced by stem cell pluripotency factors [58]. Consistent with this hypothesis, HMGA1 is a transcripional target of cMYC, an oncogenic protein and pluripotency factor (see below) [88]. In embryonic stem cells, HMGA1 expression falls with differentiation and its expression parallels that of pluripotency genes, NANOG, OCT4 and SOX2 [98]. A global gene expression study in a murine model of leukemia identifed HMGA1 as a member of the core leukemic stem cell signature [99]. Studies in human embryonic stem cells indicate that HMGA1 regulates key stem cell and pluripotency genes [98]. In fact, a recent landmark paper showed that HMGA1 is required for cellular reprogramming of somatic cells to induced pluripotent stem cells by the Yamanaka factors [98]. This study also showed that HMGA1 enhances reprogramming to induced pluripotent stem cells by the Yamanaka factors. These findings suggest HMGA1 orchestrates transcriptional networks that maintain a primitive, poorly differentiated, stem-like state, both in cancer and normal development.
4. HMGA1 PROTEINS FUNCTION IN REGULATING GENE EXPRESSION
Once it became known that HMGA1 expression is closely linked to aggressive malignancy, several groups began to study its functional role in cancer to uncover novel mechanisms that could be blocked in therapy. Early studies focused on chromatin structure and transcription [121–140]. HMGA1 proteins were found to alter gene expression through at least two separate mechanisms. First, HMGA1 proteins bind to the matrix- and scaffold-associated regions (MARs or SARs) of chromatin, which contain AT-rich sequences and high affinity binding to the nuclear matrix [121–125]. The nuclear matrix is a dynamic structure that maintains nuclear organization and provides sites for transcription, replication, and alternative splicing of mRNA. After binding to MARs/SARs, HMGA1 proteins anchor chromatin to the nuclear scaffold. This interaction serves to topologically organize independent DNA domains that function in both replication and transcription, although a detailed understanding of HMGA1’s role in chromatin domains and the organization of the nuclear matrix is lacking. HMGA1 proteins also displace histone H1 proteins, which repress transcription by maintaining a tightly wound, heterochromatic chromatin configuration in experimental models [123]. Thus, HMGA1 proteins could facilitate global activation of gene expression by relieving histone H1-mediated repression of transcription. Interestingly, the homology of HMGA1 proteins to histone H1 in plants and lower organisms suggest that they evolved from the same protein [141], although their transcriptional regulatory functions have since diverged.
In addition to a role in global chromatin structure, HMGA1 proteins also regulate specific target genes. The repertoire of genes induced by HMGA1 is extensive, and likely only beginning to emerge. Interferon-β (IFN-β) gene is among the most studied HMGA1 targets, which was identified by screening a cDNA expression library with a regulatory region of the IFN-β promoter for DNA binding proteins [126–140]. HMGA1 binds to two of four positive regulatory regions (PRD II and IV) in an enhancer region located in the 5′UTR gene upstream of the transcription start site for IFN-β [126–140]. After binding to DNA, HMGA1 promotes cooperative DNA binding of additional essential transcription factors through two distinct mechanisms. First, HMGA1 binds to an AT-rich site in the minor groove at the PRDII element, which “unbends” DNA to enable NF-κB (p50/p65) to gain access and bind to the opposite side of the DNA helix. Second, at the PRD IV element, HMGA1 interacts with the ATF-2/c-Jun heterodimer to enhance its affinity to DNA. These protein-protein and protein-DNA interactions result in the assembly of a prototypical transcriptional enhancer complex or “enhanceosome” that includes ATF-2/c-Jun and IRF proteins in a highly cooperative fashion [126–140]. The enhanceosome is required for synergy between the involved transcription factors and results in reversing the intrinsic bend in DNA at this enhancer site. Once bound, the enhanceosome recruits histone acetyltransferase proteins (p300/CBP-associated factor PCAF in mammals or GCN5 in yeast), which acetylate HMGA1 at a critical lysine amino acid (amino acid 71) in addition to modifying histone proteins by acetylation. Acetylation at amino acid 71 stabilizes the enhanceosome and leads to recruitment of the CBP-Pol II (CREB-binding protein-Polymerases II) enzyme complex that replaces PCAF/GCN5. Next, the SWI/SNF holoenzyme/nucleosome remodeling complex [130, 131] is recruited and induces a conformational change in a nucleosome position that results in binding of TFIID transcription factor to activate transcription. HMGA1-mediated enhanceo-some formation is necessary for induction of IFN-β following viral infection of cells. Conversely, acetylation at the HMGA1 amino acid 65 by CBP leads to disruption of the enhanceosome and cessation of transcription [130, 131]. Recent studies suggest that HMGA1 may not be present in the final enhanceosome structure, but rather acts as a molecular chaperone during different stages in the assembly process of the pre-initiation complex, and ultimately dissociates from the final enhancer complex [138–140]. Studies of additional downstream transcriptional targets indicate that an HMGA1 enhanceosome may regulate many other genes and their pathways (discussed in this review). Although HMGA1 proteins do not function like transcription factors alone, they organize the framework of nuclear protein-DNA transcriptional complexes to modulate transcription. Because they alter the conformation of DNA, they have been termed “architectural transcription factors”. How these transcriptional activities relate to HMGA1 function in cancer and other diseases is an area of active investigation, which we consider in this review.
5. HMGA1 PROTEINS INDUCE ONCOGENIC AND METASTATIC PROPERTIES IN CULTURED CELLS
Cloning and characterization of the promoter for murine Hmga1 led to the discovery that Hmga1 is a direct transcriptional target of the cMyc oncoprotein [88] and provided additional clues that HMGA1 functions in cancer. HMGA1 proteins also induce potent oncogenic properties in cultured mammalian cells, similar to those of cMYC [22–24, 28–30, 50, 54, 68, 70, 75, 86, 117, 142]. To illustrate, ectopic expression of the murine Hmga1a or Hmga1b isoform results in colony formation/anchorage-independent cell growth in rat fibroblasts (Rat1a) or human lympho-blastoid cells (CB33) [28, 88]. Similarly, fibroblasts overexpressing Hmga1a or Hmga1b are tumorigenic in nude mice and metastasize to the lungs. In addition, overexpression of human HMGA1a or HMGA1b induces anchorage-independent cell growth in a noninvasive, human breast cancer cell line (MCF-7) in vitro [29]. The breast cells engineered to overexpress HMGA1b metastasize locally following injection into mammary fat pads [29]. Interestingly, the tumors have histopathologic features and express proteins characteristic of an epithelial-mesenchymal transition (EMT), a molecular program whereby cells lose their epithelial characteristics (planar, apical-basal polarity, lack of motility) and acquire mesenchymal features, including motility, invasiveness, and resistance to apoptosis. Although first described during embryonic development, and later in tissue culture models, EMT-like reprogramming is now known to drive tumor progression [29, 68, 117]. These findings demonstrate that HMGA1 could play a causal role in both tumor initiation and metastatic progression.
Studies that interfered with HMGA1 function or expression provide further insight into HMGA1’s role in tumorigenesis [28, 29 50, 68, 70, 75, 88, 117, 143–146]. Inhibiting HMGA1 expression dramatically blocks both cellular proliferation and anchorage-independent cell growth (colony formation) in soft agar in Burkitts lymphoma cells [88], which are derived from an aggressive childhood lymphoid malignancy characterized by a translocation event that leads to deregulated cMYC expression. HMGA1 is also up-regulated in cell lines derived from Burkitts lymphoma or leukemia [88]. Adenovirus-mediated antisense knock-down of HMGA1 results in apoptotic cell death in cultured human thyroid, colon, lung, and breast cancer cells, but not in cells derived from normal tissue [143]. Interfering with HMGA1 function through an antisense or dominant-negative approach in human breast cancer cells also blocks proliferation and colony formation in soft agar [29, 54, 117]. In an orthotopic xenograft model of pancreatic cancer, knock-down of HMGA1 blocks metastatic progression [70]. This group also found that HMGA1 confers protection from anoikis, the process whereby cells undergo apoptosis when they are deprived of attachment to a matrix or placed in anchorage-independent culture conditions [71]. Inhibition of anoikis is thought to promote metastatic progression since viable, unattached cells are needed to migrate from the primary tumor to a metastatic site. Knock-down of HMGA1 confers sensitivity to gemcitabine, a nucleoside analogue and first line therapy in pancreatic adenocarcinoma [73]. Another group found that silencing HMGA1 also blocks anoikis in renal cell carcinoma cells [77]. In colon cancer cells, knock-down of HMGA1 interferes with metastatic progression in a murine model [68], as well as anchorage-independent cell growth, migration, and invasion. Moreover, the stem cell property, growth in three-dimensional spheres, was blocked [68, 117]. Silencing HMGA1 also prevents tumorigenesis when limited numbers of cancer cells were injected, indicating that silencing HMGA1 depletes cancer stem cells. This work further highlights the functional role for HMGA1 in driving tumor progression.
More recently, studies in poorly differentiated, triple-negative breast cancer cells showed that HMGA1 functions as a key molecular switch required by cancer cells for oncogenic and stem cell properties. Silencing HMGA1 using a potent lentivirus to deliver short hairpin to HMGA1 abruptly halts cell growth [117]. Silencing HMGA1 also dramatically changed the cancer cell morphology from mesenchymal, spindle-shaped cells to more differentiated-appearing, epithelial, cuboidal cells [117]. In addition, silencing HMGA1 impaired oncogenic (colony-formation, migration/invasion, xenograft tumorigenesis) and cancer stem cell (growth as spheres, limiting dilution tumorigenesis) properties. Switching-off HMGA1 also prevented seeding and metastatic progression to the lungs, both from mammary fat pads or tail vein injections. Together, these findings suggest that HMGA1 is a master regulator of tumor progression, stem cell properties, and resistance to therapy.
6. HMGA1 ANIMAL MODELS FOR MALIGNANCY & OTHER DISEASES
Genetically engineered mouse models provide the most convincing evidence that HMGA1 causes cancer in vivo [68, 75, 90, 100]. Transgenic mice misexpressing murine Hmga1a develop aggressive lymphoid malignancy by 2–10 months with complete penetrance [90]. In this model, the murine Hmga1a transgene is driven by the H-2K promoter and immunoglobin μ enhancer, which directs transgene expression in the T and B cell lymphoid compartments. As expected, the transgene is expressed in B and T lymphoid cells, with highest levels in T-cells. All founders develop lymphoid malignancy, with an aggressive T-cell leukemia/lymphoma phenotype that recapitulates salient histopathologic and molecular features of human T-cell leukemia and lymphoma. The transgene is expressed at levels ranging from 2 to 10-fold above that observed in normal murine lymphocytes. This mouse model mimics human B- and T-cell lymphoblastic leukemia where HMGA1a expression is increased by 2 to 10-fold over that observed in normal human T and B cells [90]. When crossed onto an Ink4a/Arf null background, leukemogenesis is markedly accelerated [118]. The transgene is also misexpressed in the uterus and all females develop uterine sarcomas by 8–10 months of age [75]. In the uterus, the transgene is expressed at levels of 5 to 15-fold above that found in control uteri [75], which is similar to high-grade human uterine tumors where HMGA1a mRNA is 2 to 20-fold higher than levels observed in normal uterine tissue [75]. These Hmga1 mice also develop hyperproliferative changes in the small and large intestines [68], with polyposis similar to that observed in APC mutated mouse models [147]. In the gut tissue, Hmga1a expression is increased by about 3 to 5-fold above that observed in controls [68]. Preliminary studies in the transgenics indicate that there is enhanced intestinal stem cell function and number in the transgenics, further linking HMGA1 to diverse stem cells (Xian and Resar, unpublished data).
Another transgenic mouse model was engineered which expresses the human HMGA1b isoform under the control of a CMV promoter, which drives expression in most tissues [100]. These mice develop natural killer T-cell (NK) lymphomas, although with a lower penetrance and at a later age than the Hmga1a mouse model [90]. Females also develop pituitary adenomas with a penetrance of 80% by 16 months; 15% of males develop pituitary adenomas by 22 months. The differences in tumor incidence between male and female mice is intriguing and suggests that gender-specific hormones may influence tumor development. The basis for the different phenotypes in the two transgenic models is not known, but could result from differences in murine or human gene function, levels of expression, or the specific isoforms expressed by the transgene. Nonetheless, these independent mouse models demonstrate that HMGA1 causes cancer in mammals.
Studies describing embryonic stem cell lines [144] and mice [35, 145] deficient in Hmga1 were also reported. When cultured under conditions to differentiate into hematopoietic cells, murine embryonic stem cells lacking Hmga1 were reported to exhibit skewed hematopoietic differentiation, with a decrease in T-cell precursors [145]. Similarly, the same group reported that Hmga1a knock-out mice have decreased numbers of T-cells and develop skewed hematopoietic differentiation with leukemic transformation in some cases, which led to the speculation that Hmga1 could also have tumor-suppressor effects [145]. The lympho-proliferative disease also resembles lymphoproliferative disease in patients with T cell immunosuppression and could reflect a decrease in T cell number and/or function. Although additional studies are needed to elucidate the mechanisms involved in the aberrant hematopoiesis with Hmga1 deficiency, these studies further support an important role for Hmga1 in T-cell development and leukemic transformation. Interestingly, a subsequent study reported that mice deficient in Hmga1 are less susceptible to chemically induced skin cancers, highlighting Hmga1’s role in oncogenic transformation [146].
7. HMGA1, MITOCHONDRIAL FUNCTION, & THE WARBURG EFFECT
In addition to binding to nuclear chromatin to regulate gene expression, HMGA1 proteins also translocate to the mitochondria where they bind to mitochondrial DNA at AT-rich sequences in the D-loop control region [32–34]. Studies in MCF-7 breast cancer cells found that forced overexpression of HMGA1 results in decreased mitochondrial DNA and mitochondrial mass, while simultaneously causing increased reactive oxygen species and sensitivity to glycolytic inhibitors [32–34]. These findings suggest that HMGA1 promotes aerobic glycolysis, at least in part, by interfering with mitochondrial function while increasing oxidative phosphorylation and the formation of reactive oxygen species in the remaining mitochondria. The dependence of cancer cells on aerobic glycolysis was first described in the 1920s by Otto Warburg when he discovered that cancer cells avidly take up glucose and convert it almost exclusively to lactate in the presence of adequate oxygen (aerobic glycolysis) [148, 149]. Later dubbed the “Warburg Effect”, this metabolic alteration and others associated with tumor metabolism have emerged as hallmarks of cancer [150]. More recent studies of global metabolic alterations in intestines of Hmga1 transgenic mice indicate that aerobic glycolysis is enhanced [151–153]. Indeed, most major tumor suppressors and oncogenes alter cellular metabolism. cMYC, for example, promotes aerobic glycolysis in cancer by inducing genes that enhance glucose uptake, while inhibiting oxidative phosphorylation in the mitochondria [154–158]. In MCF-7 cells, HMGA1 also induces expression of a subset of mitochondrial genes, including NADH dehydrogenase subunit 2, NADH dehydrogenase subunit 6, cytochrome c oxidase subunit 1, and ATP synthase 6 [32–34]. Importantly, some of these mitochondrial genes are up-regulated in cancer, although their functional roles in malignant transformation are incompletely understood. In addition to increasing sensitivity to glycolytic inhibitors and reactive oxygen species, this study also found that the efficiency of DNA repair decreases with increasing HMGA1 expression. These studies suggest that HMGA1 overexpression in cancer cells could produce mutations in genomic and mitochondrial DNA, while disrupting DNA repair. Further studies will be needed to elucidate the roles of HMGA1 in mitochondrial function and cellular metabolism because recent evidence indicates that targeting cancer metabolism could be an effective therapeutic approach [150–158].
8. TYPE-2 DIABETES MELLITUS
Decreased expression of the insulin receptor (Ir) gene (discussed below), decreased insulin signaling, hyperglycemia, and a type-2 diabetes-like phenotype [35–38] were also reported in the Hmga1 deficient mice. This group also described increases in the glucose transporter 3 (Glut3) protein, possibly as a compensatory mechanism in response to decreased Ir expression and signaling. Four patients with mutations that decrease expression of HMGA1 were also described from a screen of 148 patients with type-2 diabetes [35]. Two cases were from the same family and both individuals have a hemizygous deletion of the HMGA1 gene locus. The remaining two cases have a single nucleotide deletion in the 3′ untranslated regions, which was reported to decrease HMGA1 gene expression [35]. More recent studies (outlined below) identified additional genes involved in glucose metabolism that are regulated by HMGA1. The human correlates suggest that decreased HMGA1 expression could contribute to the development of type-2 diabetes in a subset of patients [35]. This group also reported cardiac hypertrophy in the knock-out mice, although there are no studies in humans that link genetic alterations affecting HMGA1 with cardiac hypertrophy [145].
9. ALZHEIMER’S DISEASE
In addition to cancer and diabetes, recent studies implicate HMGA1 in the pathogenesis of Alzheimer’s disease, a neurodegenerative disorder that currently afflicts over 5 million Americans [39–41, 159–162]. Alzheimer’s disease is characterized by neuronal loss, glial cell proliferation, aberrant angiogenesis, and accumulation of “senile plaques” composed of amyloid-β [159–161]. The Presenilin-1 (PS1) and Presenilin-2 (PS2) genes generate proteins (PS1 and PS2) involved in the production of amyloid-β [39–42]. An aberrantly spliced form of PS2 was demonstrated in the brains of sporadic Alzheimer’s disease, the most common form of this disease. The aberrant splicing results from exon 5 skipping, which causes a frameshift with a premature termination codon and could lead to an accumulation of a deleterious protein (called PSV2) in the brain and the development of Alzheimer’s disease [39–42, 159–161]. In vitro studies show that HMGA1 binds to a site within exon 5 and inactivates normal splicing, resulting in aberrantly spliced, truncated PSV2 protein [42]. In addition, HMGA1 proteins are elevated in the hippocampus of patients with sporadic Altzheimer’s disease and HMGA1 is induced with hypoxia in neuronal cells [39–42]. Moreover, inhibitory oligonucleotides to HMGA1 block the abnormal splicing event. Although further work is needed, these studies uncover a potential role for HMGA1 in Altzheimer’s disease and possibly other neurodegenerative diseases associated with abnormal PS2 accumulation.
10. HMGA1 TRANSCRIPTIONAL TARGETS
Despite progress in our understanding of HMGA1 in development, cancer, and other diseases, there remain important questions about the molecular programs that are regulated by HMGA1 as well as the pathways that govern its function and expression. Global genomic studies are beginning to uncover HMGA1 pathways and define an HMGA1 transcriptome (Figs. 2, 3, Table 1).
Fig. (3). The HMGA1 Transcriptome.

Multiple factors induce HMGA1 expression, after which the protein isoforms become up-regulated and translocate to the nucleus to induce expression of genes involved in fundamental pathways required for tumor initiation and progression, normal development, and adult stem cell function.
Table 1.
Putative gene targets of HMGA1.
| Gene | Function | Induce/Repressa | Organismb | Cell Typec | Approach/Rationaled | Validation Studiese | HMGA1-Dependent Expression in Tissues/Cell Linesf | Required for HMGA1 Phenotypesg | NF-κB Sitesh | Primary Refs. |
|---|---|---|---|---|---|---|---|---|---|---|
| Cytokine and cytokine receptor genes with promoters with similar (IFN-β– like) enhancer regions | ||||||||||
| Interferon-β (IFN-β) | Antiviral defense, inflammatory response | Induce | H, F, M | HeLa cervical cancer cells, Osteosarcoma, Jurkat T-leukemia cells, MG63 osteosarcoma cells | Expression library screen for IFN-β promoter binding proteins | EMSA, Methylation interference Transfection transcription assay 1, 2, 3 | ND | ND | + | Falvo [126], Thanos [127], Du [128] |
| IFN-γ | Induce T helper cell development | Induce | H, M | Transgenic mouse T cells, Human EL4 T cells | Homology to IL2RA | RNase protection assay, qRT-PCR and ELISA in Transgenic mice, DNase footprinting Assay, EMSA, Transfection transcription assay | Yes | ND | + | Chau [166] |
| Interleukin-4 (IL-4) | B-cell proliferation/differentiation | Repress | H, M | Mouse E14 T lymphoma cells, human peripheral blood T-cells, Jurkat T-leukemia cells | Promoter analysis | EMSA, Transfection transcription assay DNAase Footprinting | ND | ND | + | Chuvpilo [169] |
| Interleukin-2 (IL-2) | T-cell proliferation/differentiation, antigen-induced T-cell activation | Induce | H, M | Jurkat T-leukemia cells, human peripheral blood lymphocytes | Promoter analysis | EMSA, Transfection transcription assay DNAase footprinting | + | Y | + | Chuvpilo [169], Himes [189], Fedele [100] |
| E-Selectin (ELAM-1) | Cell adhesion molecule (CAM), leukocyte rolling and recruitment, inflammatory response | Induce | H, B | Human umbilical vein endothelial cells, bovine aortic endothelial cells (BAEC) | Promoter analysis | EMSA, Methylation interference Transfection transcription assay | ND | ND | + | Whitley [173] |
| P-Selectin | Cell adhesion molecule (CAM), leukocyte rolling and recruitment, inflammatory response | Induce | M, B | Bovine aortic endothelial cells (BAEC), primary murine lung enothelial cells (MLEC), murine endothelial cells | Promoter analysis | EMSA, Methylation interference ChIP | + | ND | + | Baron [176] |
| Interleukin-2 Receptor α(IL-2Rα) | T cell proliferation/differentiation, antigen-induced T-cell immune function | Induce | H, M, Mo | Jurkat T-leukemia cells, MT-2 T-lymphocytes, Kit 225 T cells | Promoter analysis | EMSA, Methylation interference Transfection transcription assay DNAase footprinting ChIP Ethylation interference | + | ND | + | John [177, 178], Fedele [100] |
| Tumor Necrosis Factor-β (Tnf-β) | Inflammation, maintenance of lymphoid tissue & Peyer’s patches | Induce | M | PD pre-B murine cells | Promoter analysis | EMSA Transfection transcription assay | ND | ND | + | Fashena [181] |
| Melanoma Growth Stimulatory Activity/Growth Regulate α (MGSA/GROα) | Chemokine, inflammation, neutrophil chemo-attractant, induced as melanocytes progress to melanoma | Induce | H | Hs294T melanoma cells, human retinal pigment epithelial cells | Promoter analysis | EMSA, Transfection transcription assay DNAase footprinting | ND | ND | + | Wood [184] |
| Additional cytokine and chemokine genes | ||||||||||
| Interleukin-15 (IL-15) | T-cell proliferation, Generation of cytotoxic and NK T cells, B cell immuneglobulin synthesis | Induce | M | HMGA1b transgenic mouse splenocytes | Defined as HMGA2 target in HMGA2/T transgenic mouse model | Transfection transcription assay | + | ND | + | Fedele [100] |
| Interleukin-15 Receptor alpha (Il-15Rα) | T-cell proliferation, Generation of cytotoxic and NK T cells, B cell immuneglobulin synthesis | Induce | M | HMGA1b transgenic mouse splenocytes | HMGA2 target in HMGA2/T transgenic mouse model | Transfection transcription assay | + | ND | + | Fedele [100] |
| Interleukin-10 (IL-10) | Promotes B cell proliferation, maintains TH1 cells in an undifferentiated state | Induce | H | Raji Burrkitt cells, U937 and K562 acute myeloid leukemia cells, Jurkat T-leukemia cells | Promoter analysis | EMSA | ND | ND | None | Lin [187] |
| Granulocyte Macrophage (GM-CSF) | Stimulates hematopoietic stem cells to produce granulocytes & monocytes | Induce | H | Jurkat T-leukemia cells | Promoter analysis | EMSA, Transfection transcription assay | + | ND | + | Himes [189] |
| Interleukin 2 Receptor β (IL-2Rβ) | IL-2 signaling, T-cell proliferation, inflammatory response | Induce | H, M | HMGA1a transgenic mouse splenocytes, Jurkat T-leukemia cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Schuldenfrei [108] |
| Interleukin 18 receptor (IL-18r1) | Member of IL-1 super-family involved in cell-mediated immunity and inflammation following microbial infection | Induce | H, M | HMGA1a transgenic mouse splenocytes | Microarray gene expression profile analysis | qRT-PCR | + | ND | + | Schuldenfrei [108] |
| CXCR3 | Leukocyte trafficking, inflammatory cell recruitment, inflammation | Induce | H, M | HMGA1a transgenic mouse splenocytes | Microarray gene expression profile analysis | qRT-PCR | + | ND | + | Schuldenfrei [108] |
| Other genes involved in leukocyte function and inflammation | ||||||||||
| T-Cell Receptor α (TCR-α) | Tissue and stage-specific expression and V(D)J recombination in T-& B-lymphocytes | Repress | H | Jurkat T-leukemia cells HeLa cervical cancer cells | Promoter analysis | In vitro transcription | + | ND | + | Bagga [191] |
| Immuneglobulin heavy chain ε enhancer (ε enhancer) (Ighe) | V(D)J recombination in B-lymphocytes; IgE production | Repress | M | BALB/c – derived mouse B lymphoma cells M12.4.1, Abelson virus transformed pre-B cells 18.81, mouse splenocytes | Promoter analysis | EMSA, Transfection transcription assay, DNAase footprinting | + | ND | + | Kim [192] |
| Immuneglobulin heavy chain mu promoter (μ enhancer) (Ighm) | V(D)J recombination in B-lymphocytes; IgM production | Induce | M | Ag8.653 murine mature plasmacytoma B cells, 2017 pro-T cells, 38B9 preB cells, BAL-17 mature B cells | Promoter analysis | EMSA, Methylation interference ChIP Pull-down | + | ND | + | Lewis [193] |
| GP91-PHOX | ROS production in phagocytosis (encodes the heme-binding beta subunit of NADPH oxidase or cytochrome b) | Not known | H | Human endothelial cell library | cDNA expression library screen for CCAAT-box binding proteins in gp91-phox promoter | Southwester blot | ND | ND | + | Skalnif [196] |
| CD8β1 | Cytotoxic T-cell receptor, antigen-specific T-cell activation | Induce | H, M | HMGA1a transgenic mouse splenocytes, Jurkat T-leukemia cells | Microarray gene expression profile analysis | qRT-PCR | + | ND | + | Schuldenfrei [108] |
| Granzyme M (GZMM) | Protein stored in granzymes and involved in apoptosis | Induce | H, M | HMGA1a transgenic mouse splenocytes | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Schuldenfrei [108] |
| Spleen tyrosine kinase (syk) | Signal transduction in T and B-cells, inflammation, angiogenesis, transformation | Induce | M | Mouse embryonic stem cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Martinez-Hoyos [198] |
| Transcription factors | ||||||||||
| Gata-1 | Zinc finger transcription factor involved in hematopoiesis | Repress | M | Mouse embryonic stem cells | Gata1 was tested based on phenotypes found in hematopoietic cells derived from hmga1null ES cells | EMSA, Methylation interference ChIP qRT-PCR | + | ND | + | Battista [144] |
| Gata-4 | Zinc finger transcription factor involved in embryogenesis (meso and endoderm-derived tissues), cardiogenesis | Induce | M | Mouse embryonic stem cells and mouse embryonic fibroblasts | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Martinez-Hoyos [198] |
| Gata-6 | Zinc finger transcription factor involved in embryogenesis (meso- and endoderm-derived tissues), cardiogenesis | Induce | M | Mouse embryonic stem cells and mouse embryonic fibroblasts | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Martinez-Hoyos [198] |
| Lung Krupple-like factor (Lklf) | Zinc finger transcription factor involved in hematopoiesis and an anti-inflammatory response | Induce | M | Mouse embryonic stem cells and mouse embryonic fibroblasts | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Martinez-Hoyos [198] |
| Zif268 | Immediate-early gene involved in mitogenesis and differentiation | Induce | M | Mouse embryonic stem cells, embryonic fibroblasts and liver | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Martinez-Hoyos [198] |
| JunB | Immediate-early/proto-oncogene | Induce | M | Mouse embryonic stem cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Martinez-Hoyos [198] |
| c-Fos | Immediate-early/proto-oncogene | Induce | M, F | Murine embryonic stem cells, SL2 droso-phila cells | Microarray gene expression profile analysis, Promoter analysis | RT-PCR, EMSA, Transfection transcription assay | + | ND | + | Martinez-Hoyos [198], Chin [204] |
| cMYC | Immediate-early/proto-oncogene, pluripotency factor | Induce | M, H | Murine embryonic stem cells, human embryonic stem cells | Microarray gene expression profile analysis; analysis of pluripotency gene expression | RT-PCR, ChIP | + | ND | + | Martinez-Hoyos [198], Shah [98] |
| NANOG | Pluripotency gene important in stem cells | Induce | H | Human embryonic stem cells, induced pluripotent stem cells | Stem cell gene expression profile analysis | RT-PCR | + | ND | + | Shah [98] |
| OCT4 | Pluripotency gene important in stem cells | Induce | H | Human embryonic stem cells, induced pluripotent stem cells | Stem cell gene expression profile analysis | RT-PCR, ChiP | + | ND | + | Shah [98] |
| SOX2 | Pluripotency gene important in stem cells | Induce | H | Human embryonic stem cells, induced pluripotent stem cells | Stem cell gene expression profile analysis | RT-PCR, ChiP, Protein IF | + | ND | + | Shah [98] |
| Signal Transducer and Activator of Transcription (STAT-3) | Transcription factor, Intracellular Signaling; oncogene | Induce | H, M, R | HMGA1a transgenic mouse splenocytes and lymphoid tumors, HMGA1a- and 1b Rat1a fibroblast cells, HEL erythroleukemia cells | Microarray gene expression profile analysis | EMSA, Transfection transcription assay, ChIP, qRT-PCR, western blot, IH | + | Y | + | Hillon [91] |
| Forkhead Box Transcription Factor p1 (FOXP1) | Neuronal development | Repress | H, M | HMGA1a transgenic mouse splenocytes | Microarray gene expression profile analysis | qRT-PCR | ND | ND | None | Schuldenfrei [108] |
| Eomesodemin (EOMES) | Embryonic development, nueronal and trophoblast development | Induce | H, M | HMGA1a transgenic mouse splenocytes, Jurkat T-leukemia cells | Microarray gene expression profile analysis | qRT-PCR | + | ND | + | Schuldenfrei [108] |
| TWIST | Embryonic development, epithelial to mesenchymal transition | Induce | H, M | HCT116 & SW480 human colorectal cancer cells | Defined as HMGA2 target | qRT-PCR | + | ND | + | Belton [68] |
| HAND1 | Placental formation, cardiac morphogenesis | Repress | H, M, R | Mouse embryonic stem cells, human thyroid primary and carcinoma cells*, rat thyroid carcinoma cells | Microarray gene expression profile analysis | EMSA, Transfection transcription assay, ChIP, RT-PCR, IH | + | Y | + | Matinez-Hoyos [198] |
| Tfeb | Lysosomal biogenesis & autophagy | Induce | M | Mouse embryonic stem cells | Microarray gene expression profile analysis | RT-PCR, | + | ND | + | Martinez-Hoyos [198] |
| DNA damage tolerance, genomic instability | ||||||||||
| Xeroderma Pigmentosa A (XPA) | DNA Repair (Nuclear excision repair) | Repress | H | MCF-7 breast adeno-carcinoma cells expressing HMGA1a | Microarray gene expression profile analysis | Methylation interference ChIP RT-PCR Western blot | + | Y | + | Adair [112] |
| Breast Cancer Type 1 Susceptibility (BRCA1) | Cell cycle regulation, DNA repair (Double-stranded break repair) | Repress | H, M | MCF-7 breast adeno-carcinoma, HMEC human mammary epithelial cells | Promoter analysis | EMSA, Methylation interference Transfection transcription assay, ChIP Western blot Northern blot | + | ND | + | Baldassarre [110] |
| Genes involved in cell cycle regulation, pro-survival and/or anti-apoptotic pathways | ||||||||||
| B-Cell Lymphoma Gene 2 (BCL-2) | Apoptosis Inhibitor | Induce | H, M | ND7 mouse neuroblastoma cells, MCF-7 breast adeno-carcinoma cells, mouse embryo fibrobroblasts, SAOS-2 osteosarcoma cells | HMGA1-interacting protein array identified p53; p53-regulated genes were therefore studied | EMSA, Transfection transcription assay ChIP Pull-down qRT-PCR Western blot Co-IP | + | ND | + | Esposito [221] |
| c-KIT Ligand | Hematopoiesis, hematopoietic stem cell niche, RAS/ERK signaling | Induce | H | MCF-7 breast adeno-carcinoma cells, OCC1 human ovarian cancer cells | Microarray gene expression profile analysis | EMSA, Transfection transcription assay ChIP qRT-PCR | + | ND | + | Treff [222, 223] |
| Caveolin 1 CAV1 | Cellular Signaling | Repress | H | MCF-7 breast adeno-carcinoma cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Treff [222] |
| Caveolin 2 CAV2 | Cellular Signaling | Repress | H | MCF-7 breast adeno-carcinoma cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Treff [222] |
| p96 Differentially expressed in ovarian cancer 2 (DOC2) | Negative regulator of mitogenic signaling, induces of c-Fos | Repress | M | Mouse embryonic stem cells & embryonic fibroblasts | Microarray gene expression profile analysis | EMSA, ChIP qRT-PCR | + | ND | + | Martinez-Hoyos [198] |
| Gly96, immediate-early response gene (IEX-1) | Immediate early gene that regulates proliferation (positively or negatively) | Repress | M | Mouse embryonic stem cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Martinez-Hoyos [198] |
| Vacular smooth muscle proliferation and function | ||||||||||
| Inducible Nitric Oxide Synthase (iNOS) | Nitric oxide biosynthesis, blood vessel dilation, angiogenesis | Induce | R, F, H | RASMCs (rat aortic smooth muscle cells), SL2 droso-phila cells, RINm5F insulinoma cells, A549 human alveolar Type II alveolar l epithelial cells | Defined as an NF-κB gene target | EMSA, Transfection transcription assay | ND | ND | + | Perrella [230], Takamiya [231], Darville [233] |
| Smooth Muscle specific gene 22 (SM22) | Endothelial cell proliferation | Induce | F | SL2 droso-phila cells | Promoter Analysis | EMSA, Transfection transcription assay | ND | ND | + | Chin [237] |
| Cd44 | Cellular adhesion, stem cell marker | Induce | R | RASMCs (rat aortic smooth muscle cells), F9 mouse embryonal carcinoma cells, | Defined as a cytokine-regulated gene & Promoter analysis | EMSA, Transfection transcription assay | ND | ND | + | Foster [239] |
| Vascular smooth muscle actin (Vsm-actin) | Cytoskeleton | Induce | M | Mouse embryonic stem cells | Promoter Analysis | EMSA, Transfection transcription assay | ND | ND | + | Martinez-Hoyos [198] |
| Helix-Loop-Helix factors | ||||||||||
| DNA-binding protein inhibitor Id-3 (Id3) | Cell growth, differentiation, angiogenesis | Induce | M, R | Mouse embryonic stem cells, mouse splenocytes, FRTL-5 rat epithelial cells | Microarray gene expression profile analysis | EMSA, ChIP, RT-PCR, Western | + | ND | + | Martinez-Hoyos [198] |
| Adipogenesis | ||||||||||
| Obese | Adipogenesis & energy intake | Induce | H, M | Human embryonic kidney HEK) 293 cells3T3-L1 mouse fibroblasts | HMGA interacts with RB & C/EBPβ, C-EBPβ regulated genes studied | Transfection transcription assay | + | ND | + | Melillo [244] |
| Migration/Invasion/Metastasis | ||||||||||
| Matrix Metallo-Proteinase 2 (MMP-2) | Extracellular matrix degradation | Induce | R, H | G-Dunning rat prostate cancer, H1299 lung cancer cells | Microarray gene expression profile analysis in rat prostate cells | Western blot (prostate cells) RT-PCR, ChiP (H1299 lung cancer cells) | + | Y | + | Takaha [60], Hillion [50] |
| Matrix Metallo-Proteinase 9 (MMP-9) | Extracellular Matrix degradation | Induce | H | MiaPaCa2 and PANC1 huamn pancreatic adenocarcinoma cells | HMGA1 regulates other MMPs and functions in invasion | MMP-9 activity (fluorometric assay), RT-PCR, Transfection transcription assay | + | Y | + | Liau [70] |
| Matrix Metallo-Proteinase 13 (MMP-13) | Extracellular Matrix degradation | Induce | H | MCF-7 breast cancer cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Reeves [29] |
| Integrin-β1 | Cellular Adhesion, migration | Induce | H | MCF-7 breast cancer cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Reeves [29] |
| Regulators of Multiple Transformation Pathways | ||||||||||
| Cyclooxygenase 2 (Prostaglandin G/H Synthase 2) | Lipid Biosynthesis, inflammatory response | Induce | H, M | Vascular endothelium, HMGA1a transgenic mouse uterine neoplasms, MES-SA uterine sarcoma cells | Promoter analysis | EMSA, Transfection transcription assay, ChIP, qRT-PCR, western blot | + | Y | + | Ji [255], Tesfaye [75], DiCello [256] |
| Ocular Function | ||||||||||
| Rhodopsin (Rho) | Retinal function, photoreceptor differentiation | Induce | H, M | C57B6 and BALBc mouse ocular tissues, WERI-Rb1 & Y79 retinoblastoma cells | Expression of HMGA1a in photorecep tor cells led to exploring a proposed role in gene turnover | EMSA, Transfection transcription assay, DNAse Footprinting, GST-pulldown, qRT-PCR, Western blot, Northern blot, IH | + | ND | + | Chau [258] |
| Alpha crystalline B chain (CRYAB) | Heat shock protein | Induce | H | Human Lens Epithelial Cells (HLE-B3), SW-13 cells | Differential analysis | EMSA, Transfection transcription assay, ChIP, Nucleosome Mapping | ND | ND | + | Duncan [260] |
| Diabetes and Insulin Resistance | ||||||||||
| Insulin Receptor (IR) | Cell signaling | Repress | H, M, R | IM-9 Lymphocytes, HepG2 and HTC rat Hepatoma cells, 3T3-L1 mouse fibroblasts, pancreatic cells, MCF-7 breast carcinoma | Promoter analysis | EMSA, Transfection transcription assay, ChIP, western, co-IP Southwestern | + | Y | + | Brunetti [261], Foti [262] |
| RBP4 | Insulin Resistance | Induce | H, M | HMGA1-deficient mouse adipocytes, Hepa1 & HepG2 Hepatoma cells, kidney | Defined as gene involved in glucose homeostasis | Transient transfection transcription assays, ChIP, qRT-PCR, western blot, northern blot | + | Y | + | Chiefari [263] |
| Insulin-Like Growth Factor Binding Protein-1 (IGFBP-1) | Glucose Homeostasis | Induce | H, M | HMGA1-deficient mouse hepatocytes, HepG2 hepatoma cells, MCF-7 breast cancer cells, NIH 3T3 mouse fibroblasts | Promoter analysis | EMSA, Transient transfection transcription Assays, ChIP, qRT-PCR, western, co-IP | + | ND | + | Allander [265], Iiritano [266] |
| Insulin-Like Growth Factor Binding Protein-3 (IGFBP-3) | Glucose Homeostasis | Induce | H, M | HMGA1-deficient mouse hepatocytes, HepG2 hepatoma cells, MCF-7 breast cancer cells, NIH 3T3 mouse fibroblasts | Promoter Analysis | EMSA, Transient transfection trancription assays, ChIP, RT-PCR, western, co-IP | + | ND | + | Allander [265], Iiritano [266] |
| Developmental, Stem Cell and EMT | ||||||||||
| Jagged-1 (JAG1) | Notch1 ligand involved in cell fate decisions in hematopoiesis & cardiac development | Induce | H | MCF-7 breast cancer cells | Microarray gene expression profile analysis | RT-PCR | + | ND | + | Reeves [29] Reeves |
| Vimentin (VIM) | Embryonic development, epithelial to mesenchymal transition | Induce | H, M | MCF-7-breast cancer cells, HCT116 & SW480 colon cancer cell lines | Microarray gene expression profile analysis (MCF-7 cells) | RT-PCR IH | + | ND | + | [29], Belton [68] |
| E-Cadherin | Cellular Adhesion, epithelial to mesenchymal transition | Repress | H | SW480 colon cancer cell line | Expression screen for genes involved in EMT | qRT-PCR | + | ND | + | Belton [68] |
| Spindle Activating Checkpoint Genes | ||||||||||
| Budding Uninhibited by Benzimidazoles 1 Homolog β (Bub1β) | Mitosis, cell cycle regulation | Induce | H, M | HMGA1a transgenic mouse splenocytes (ref 108), Jurkat T-ALL cells, HCT116, NIH3T3, HeLa cells (ref 277) | Microarray gene expression profile analysis | qRT-PCR | + | ND | + | Schuldenfrei [108], Pierantoni [277] |
| Budding Uninhibited by Benzimidazoles (Bub1) | Mitosis, cell cycle regulation | Induce | H | HCT116, NIH3T3, HeLa cells | Promoter analysis, Function in phenotypes associated with HMGA1 | qRT-PCR, Western | + | ND | + | Pierantoni [277] |
| TTK | Mitosis, cell cycle regulation | Induce | H | HCT116, NIH3T3, HeLa cells | Promoter analysis, Function in phenotypes associated with HMGA1 | qRT-PCR | + | ND | + | Pierantoni [277] |
| MAD2/1 | Mitosis, cell cycle regulation | Induce | H | HCT116, NIH3T3, HeLa cells | Promoter analysis, Function in phenotypes associated with HMGA1 | qRT-PCR, Western | + | ND | + | Pierantoni [277] |
Legend:
- Induce or Repress: Refers to whether the gene expression is induced (up-regulated) or repressed (down-regulated) by HMGA1/Hmga1.
- Organism: Refers to the organism from which the cell lines or other reagents were used. H=Human, F=Fly, M=Mouse, R=Rat, Mo=Monkey, B=Bovine.
- Cell Type: Refers to the specific cell line(s) used for the studies.
- Approach/Rationale: Refers to the initial approach or model used to identify the gene target.
- Validation Studies: Refers to the experimental approaches used to validate the original observation, including: Gel Shift or Electrophoretic Mobility Shift Assay (EMSA); Methylation Interference, Transcription Transfection Assays (an in vitro transfection experiments in cells with a reporter construct), DNAse Footprinting, Chromatin immunoprecipitation (ChIP), Ethylation Interference, in vitro GST or biotinylated-pull-down; Southwestern blot analysis, qRT-PCR, Western blot, Northern blot, Immunohistochemical analysis, co-immunoprecipitation.
HMGA1-dependent expression in tissues/cell lines: Refers to whether expression of the target gene depends upon HMGA1 levels.
- Required for HMGA1 Phenotypes: Refers to whether knock-down or silencing of the target gene also impairs HMGA1 phenotypes (transformation, proliferation, others).
- NF-κB binding sites: Refers to the presence of NF-κB binding sites in the promoters. These were determined either experimentally or using an in silico prediction algorithm (MatInspector) [297].
ND = Not done.
Of the more than 75 HMGA1 transcriptional targets that have been reported thus far, most include Nuclear Factor-κB (NF-κB) regulatory elements in their promoter regions and many participate in mediating inflammatory pathways. Given the increasing evidence that inflammation is a precursor lesion in diverse cancers, this link between HMGA1 and NF-κB suggests a model whereby HMGA1 and NF-κB proteins cooperate to induce inflammatory signals and drive transformation [23]. Indeed, HMGA1 regulates many genes with roles in both inflammation and cancer. Like HMGA1, NF-κB is also known to play an important role in cancer progression, resistance to therapy, and a stem-like state. HMGA1 could therefore enhance NF-κB function in these processes. Proteomic studies have identified additional proteins that interact with HMGA1 and modify its function [163, 164]. In the following section, we review what is known about HMGA1 transcriptional targets and how their associated pathways could mediate HMGA1 function. We limited our review to reports of putative targets in which HMGA1-dependent function or expression has been validated by more than one experimental approach.
10.1. Inflammatory Cytokine and Cytokine Receptor Genes with Similar Enhancer Regions
10.1.1. IFN-β
IFN-β is among the most studied HMGA1 target genes and was first discovered just over two decades ago [126–140]. IFN-β expression is up-regulated following viral infection of cells, and IFN-β, in turn, induces expression of a large set of genes encoding antiviral gene products. These include chemokines and cytokines that stimulate an inflammatory response to counter viral infections. IFN-β plays a role in mediating cell-cell interactions involved in innate and acquired immune function. In addition, some chemokines play a role in metastatic progression and HMGA1-IFN-β-chemokine pathways could contribute to tumor progression. IFN-β also functions in osteogenesis by inhibiting osteoclast differentiation [165]. Similar pathways in cancer could promote inflammatory pathways and a poorly differentiated state. Moreover, the elegant studies that defined the IFN-β enhanceosome uncovered a unique role for HMGA1 as an architectural transcription factor that orchestrates the assembly of transcription factor complexes in enhancer regions to modulate gene expression [125–140].
10.1.2. IFN-γ
Following the identification of IFN-β as an HMGA1 transcriptional target, other putative target genes were discovered based on their homology to the IFN-β enhancer-promoter regions. The IFN-γ gene was also reported to be a downstream gene target regulated by HMGA1 [166]. IFN-γ is produced primarily by CD8+ T cells, and to a lesser extent by CD4+ T cells and NK cells. IFN-γ functions as a key inducer of T helper cells whose expression is increased following TCR stimulation in the setting of specific cytokines, such as IL-2, IL-12, and IL18. Like IFN-β, IFN-γ functions in intercellular communication during innate and acquired immune responses and in tumor surveillance. A prior study reported that transgenic mice expressing HMGA1b driven by the lck promoter have increased expression of IFN-γ mRNA and protein in mature T cells as compared to controls. This group also found that HMGA1 binds directly to 2 AT rich regulatory regions within the promoter of the IFN-γ gene using electrophoretic mobility or gel shift experiments. In addition, they reported that HMGA1 induced expression of the promoter in transfection experiments, while a dominant-negative HMGA1 repressed promoter expression. Together, these data are consistent with direct regulation of IFN-γ expression by HMGA1.
10.1.3. IL-4 and IL-2
The murine IL-4 and IL-2 genes harbor similar AT-rich, transcriptional enhancer sites to IFN-β in their upstream regulatory regions [167–171]. In vitro studies show that HMGA1 binds to AT-rich sites in the IL-4 promoter to repress transcription [169], while it enhances IL-2 transcription [167–169]. IL-2 stimulates T-cell proliferation and differentiation into effector cells [172]. For example, IL-2 promotes the development of T cell immunologic memory by stimulating the growth, differentiation and survival of antigen-specific CD4+ and CD8+ T cells. IL-2 also induces division of T regulatory cells, proliferation and differentiation of NK cells, and immunoglobulin production by B cells. Thus, IL-2 plays an important role in immunologic memory and adaptive immunity. IL-2 is also expressed in T- and B-cell malignancies [167–170]. In contrast, IL-4 is involved in B cell proliferation and differentiation [171]. These observations are consistent with the existing mouse models that misexpress HMGA1 genes, which develop T-cell lymphoid malignancies [90, 100]. In addition to IL-2, the IL-2 receptor α, IL-15, and the IL-15 receptor α, were reported to be up-regulated in HMGA1b transgenic mice [100] (discussed in more detail below). In human T-cell ALL, HMGA1 is also overexpressed [90, 108, 118]. Thus, the T-cell specific transcriptional targets further support a role for HMGA1 in T-cell development and malignancy.
10.1.4. E-Selectin (ESEL, ELAM-1)
ESEL is up-regulated by HMGA1. The organization and expression of the ESEL promoter has similarities to the IFN-β enhancer, including AT-rich regions in positive regulatory domains (PRD) I-IV regions [173]. This gene encodes cell adhesion molecules (CAMs) induced by inflammatory cytokines, such as interleukin-1-β and tumor necrosis factor alpha (TNF-α). Similar to the IFN-β promoter, HMGA1 binds to an enhancer site and promotes the binding of ATF-2 and NF-κB [173]. Like other selectins, ESEL plays an important role in inflammation, although ESEL is expressed only in endothelial cells (therefore designated E) [174]. It serves to recruit white cells to sites of inflammation by mediating the adhesion of white cells to the vascular lining and is associated with inflammatory processes and disease states [175].
10.1.5. P-Selectin (PSEL)
HMGA1 also induces expression of PSEL, another CAM gene that is expressed in platelets (thus designated PSEL) as well as in endothelial cells [176]. HMGA1 binds to the PSEL promoter, as demonstrated in vitro by gel shift experiments and in vivo by chromatin immunoprecipitation in bovine aortic endothelial cells (BAECs) at an AT-rich site in a complex that includes the p50/p65 members of the NF-κB family. In transfection experiments in BAECs, HMGA1 cooperates with p50/p65 to induce expression of PSEL. Distamycin, a drug that binds to the minor groove, blocks HMGA1 binding to the PSEL promoter in gel shift experiments and decreases PSEL expression in transfection experiments without affecting ESEL expression. Distamycin also blocks PSEL expression in a murine model for inflammation during endotoxemia, with no decrease in ESEL expression [176]. The basis for the differential effects on P- and ESEL are not clear and further studies are needed to determine under what cellular contexts HMGA1 regulates P- and ESEL. In cancer, P-Selectin has a similar functional role to E-Selectin, recruiting CAMs that mediate the interaction of activated platelets with leukocytes. P-Selectin also facilitates cancer cell invasion into the bloodstream and metastatic progression.
10.1.6. Interleukin 2 Receptor α (IL-2Rα, IL-2RA)
The IL-2Rα gene is rapidly and potently induced in T-cells following mitogenic stimuli and it is also overexpressed in Jurkat T-cell leukemia cells. The promoter includes an enhancer region with positive regulatory regions (PRR) I and II, which are essential to activate IL-2Rα expression following mitogenic stimulation [177, 178]. The Elf-1 transcription factor binds at PRRII, and HMGA1 interacts with Elf-1 in vitro. PRRI contains an NF-κB binding motif and biochemical studies showed that Elf-1 interacts with the NF-κB family members, p50 and c-Rel, bound to PRRI. In this setting, it was proposed that HMGA1 serves as the molecular “glue” bridging the interactions between Elf-1 and NF-κB. Alternatively, HMGA1 could maintain Elf-1 in a transcriptionally active confirmation. In contrast to the IFN-β gene, HMGA1 does not appear to enhance Elf-1 binding to PRR II. Like IL-2, activation of IL-2Rα gene facilitates T-cell proliferation and antigen-induced T-cell immune function. Subsequent studies showed that HMGA1 binds to the IL-2Rα promoter following T-cell activation, and remodels an inhibitory nucleosome, thereby inducing IL-2Rα gene expression [178–180]. IL-2 and IL-2Rα were reported to be up-regulated in the HMGA1b transgenic model and could contribute to the development of the T-cell leukemias [100].
10.1.7. Tumor necrosis factor-β (Tnf-β, lymphotoxin α)
HMGA1 up-regulates expression of the gene encoding the pro-inflammatory cytokine, tumor necrosis factor-β (TNF-β; also known as lymphotoxin α) [181]. The murine Tnf-β promoter includes an AT-rich region to which HMGA1 binds by gel shift experiments. In transfection experiments, HMGA1 activates expression of a Tnf-β promoter construct with the AT-rich region in murine leukemia cells. TNF-β is a cytokine produced by mitogenic-activated macrophages and monocytes and plays a key role in inflammation. Knock-out mouse models demonstrate that Tnf-β is essential for the formation and maintenance of lymphoid tissues, including lymph nodes, the spleen, and Peyer’s patches in the gut [182]. Tnf-β/Lymphotoxin-α is a member of the Tnf family which forms heterotetramers with lymphotoxin-β and serves to anchor Tnf-β to the cell surface. The primary function of Tnf-β is to mediate diverse inflammatory, immunostimulatory, and antiviral processes. It is also involved in NF-κB signaling. Although originally discovered as an anti-tumor factor, increasing evidence suggests that it is involved in inflammatory pathways that can also promote oncogenesis, particularly in settings of prolonged inflammation [183]. TNF-β is also constitutively expressed in some human leukemia cell lines [183].
10.1.8. MGSA/GROα/Chemokine (C-X-C Motif) Ligand CXCL1
HMGA1 directly activates expression of the gene encoding the chemokine, MGSA/GROα/CXCL1, which is up-regulated as melanocytes progress to melanoma [184]. The MGSA/GROα/CXCL1 promoter has similar regulatory elements as ESEL and HMGA1 was found to bind to an AT-rich region nested within the NF-κB element similar to its binding to the ESEL promoter. Transfection experiments with promoter constructs containing mutations in the regulatory elements showed that HMGA1, NF-κB, and SP1 are required for transactivation of this gene. These findings suggest that HMGA1 could mediate progression to melanoma by up-regulating the MGSA/GROα/CXCL1 cytokine gene. This protein serves as a chemoattractant for neutrophils and thereby has a role in inflammation. Aberrant expression of MGSA/GROα/CXCL1 is also associated with growth and progression in a subset of tumors [175].
10.2. Additional Cytokine and Chemokine Genes
10.2.1. Interleukin 15 (Il-15 and Il-15Rα)
Il-15 and Il-15Rα were also reported to be up-regulated in the HMGA1b transgenic model [100]. Like IL-2, IL-15 is a proinflammatory cytokine that is secreted by macrophages following viral infection [185, 186]. Moreover, both IL-2 and IL-15 bind to common erythropoietin receptor subunits and they may compete for receptor binding, thereby negatively regulating each other. IL-15 also stimulates T-cell proliferation, the generation of cytotoxic T cells, stimulation of immunoglobulin synthesis by B cells, and the generation and persistence of NK cells. IL-15 induces expression of TNF-α, IL-1β, and other inflammatory cytokines [185]. In contrast to IL-2, IL-15 is not required for T regulatory cells [185]. Proliferation of NK cells induced by IL-15 results in downstream JAK-STAT signaling. In murine studies, Il-15 also has anti-apoptotic effects by inducing expression of apoptosis inhibitors Bcl2/Bcl1/Bcl-x(L) [175, 185, 186]. Abnormalities in IL-15 have been observed in immunologic diseases, such as rheumatoid arthritis, inflammatory bowel disease and colitis, and lymphoid malignancies associated with human T-cell lymphotropic virus 1 infection [185, 186]. More recently, both IL-15 and soluble IL-15Rα were found to be elevated in the serum of patients with large granular lymphocyte (LGL) leukemia, a T-cell lymphoproliferative disorder characterized by clonal expansion of mature T or NK cells [185]. IL-15Rα mRNA was also increased in the peripheral blood monocytes of patients LGL leukemia and it was proposed that enhanced signaling of IL-15 pathways contribute to the pathogenesis of the disease. Although it is not known if HMGA1 is involved in LGL leukemia, these findings suggest that IL-15 induces inflammatory pathways in this lymphoproliferative disorder [185].
10.2.2. IL-10
IL-10 is produced by monocytes, and to a lesser extent lymphocytes, and involved in inflammation and immune-regulation. It is commonly known to be an anti-inflammatory cytokine because it inhibits the synthesis of a pro-inflammatory cytokines, including IFN-gamma, IL-2, IL-3, TNF and GM-CSF produced by activated macrophages and by helper T-cells. IL-10 is also associated with signaling through the JAK-STAT pathway [175]. Gel shift experiments show that HMGA1 enhances binding of the nuclear factory Y (NF-Y) to the IL-10 promoter in a Burkitts lymphoma cell line [187]. In contrast, antibodies to HMGA1 diminish the intensity of this band, suggesting that HMGA1 is part of the NF-Y complex binding to the IL-10 promoter. Although promoter expression studies were not performed, it was postulated that HMGA1 and NF-Y cooperate to enhance IL-10 expression. IL-10 has the ability to suppress TH1 cell differentiation, which could maintain T cells in an undifferentiated state. IL-10 also promotes B cell proliferation, survival and antibody production which could contribute to malignant transformation in B cells [175, 187]. Studies demonstrating HMGA1 overexpression in both T and B lymphoid malignancies provide additional evidence for a link between HMGA1, IL-10, and lymphoid tumors [90]. Another study found that IL-10 stimulates self-renewal of hematopoietic stem cells ex vivo, suggesting that HMGA1 could contribute to the maintenance of hematopoietic stem cells through IL-10 [188]. Studies in mice indicate that it is an essential immunoregulatory molecule in the intestines [175].
10.2.3. Granulocyte Macrophase Colony Stimulating Factor (GM-CSF) and IL-2
In Jurkat T-cell acute lymphoblastic leukemic cells, HMGA1 induces expression of the GM-CSF [189] in addition to IL-2 cytokine growth factors. HMGA1 binds to the AT-rich sequence within a CD28 response element of the GM-CSF and IL-2 promoters to enhance binding of the NF-κB family member, c-Rel. Antisense RNA targeting HMGA1 or c-Rel abolishes activation of these promoters, suggesting that HMGA1, together with c-Rel, induce expression of GM-CSF and IL-2. GM-CSF stimulates hematopoietic stem cells to produce granulocytes and monocytes as part of an immune and inflammatory cascade, and could promote inflammatory pathways mediated by HMGA1 [190]. As detailed above, IL-2 drives T-cell proliferation and is overexpressed in some T-cell malignancies [109, 189]. Targeted disruption of Il-2 is also associated with colitis in mice [175].
10.2.4. lL-2rb and Il-18r1
Genome-wide analysis of lymphoid tumors from Hmga1a transgenic mice identified additional genes that are regulated by HMGA1 during tumorigenesis and involved in inflammation and white blood cell signaling [108]. Genes expressed in lymphoid cells from Hmga1a transgenic mice early in tumorigenesis (2 months) and in established tumors (12 months) were compared to control lymphoid cells to identify genes induced by Hmga1 during leukemic transformation [108]. Microarray analysis and quantitative RT-PCR show that the Il-2 receptor β (Il-2rb) and Il-18 receptor 1 (Il-18r1) are up-regulated in early and late tumorigenesis. The beta subunit of Il-2 receptor (Il2rb) forms a heterodimer with Il-2 receptor α (Il2ra) or heterotrimer with Il2ra and Il-2 receptor γ (Il2rg) to form an Il-2 receptor, which mediates mitogenic stimulation or receptor-mediated endocytosis induced by Il-2. The Il2ra-Il2rb heterodimer is a high affinity receptor, while the heterotrimer (Il2ra, Il2rb and Il2rg) is an intermediate affinity receptor. As noted above, IL-2 signaling is involved in T-cell proliferation, further linking HMGA1 to T cell function. This study also found that IL-2RB is repressed in human Jurkat T-cell leukemia cell lines following knockdown of HMGA1 expression, suggesting that this pathway is functional in human T-cell acute lymphoblastic leukemia in addition to the T-cell acute lymphoblastic leukemia in the Hmga1 transgenics. The Il-18 receptor 1 (Il-18r1) gene encodes the Il-18 receptor which binds to the Il-18 cytokine. IL-18 belongs to the IL-1 superfamily that induces cell-mediated immunity following infection with microbial products. Following stimulation by IL-18, natural killer and T cells release IFN-γ to activate macrophages and other cells involved in cell-mediated immunity and inflammation [175]. Further studies are needed to determine if the IL-2 receptor β (IL2rb) and IL-18 receptor 1 are directly regulated by HMGA1.
10.2.5. Chemokine (CXC-Motif) Receptor 3 (CXCR3)
Early in lymphoid tumorigenesis, the gene encoding the Cxcr3 receptor chemokine is up-regulated in lymphoid cells from the Hmga1a transgenics compared to controls, as shown by microarray gene expression profile analysis and quantitative RT-PCR in mouse and human cells [108]. Cxcr3 is a chemokine receptor protein in the CXC chemokine receptor family [175]. Normally expressed in activated T cells and NK T cells, it functions in leukocyte trafficking and recruitment of inflammatory cells. The Cxcr3 could mediate inflammatory pathways that contribute to tumorigenesis. It is not yet known if Cxcr3 is directly regulated by HMGA1 [108].
10.3. Additional Genes Involved Leukocyte Function and Inflammation
10.3.1. T Cell Receptor α-chain (TCRα, TRA)
HMGA1 represses the TCRα gene, which directs tissue- and stage-specific expression and V(D)J recombination of this gene locus [191]. Using an in vitro transcription assay system in Jurkat T-cell leukemia cells, investigators found that HMGA1 is part of a T cell-specific repressor complex that is sensitive to DNA topology. These studies suggest that HMGA1 binds to the repressor and inactivates the TCRα promoter when DNA is in a supercoiled state. In the absence of the repressor complex, TCRα gene expression is not dependent upon the 3′ enhancer and DNA topology. This pathway could also serve to maintain T-cells in a less differentiated state prior to expression of the TCR complex. The overexpression of HMGA1 in T-cell acute lymphoblastic leukemia is also consistent with a role for HMGA1 in maintaining an aberrantly differentiated, leukemogenic phenotype in T cells [90, 100]. Other known pathways associated with TCRα are Class I MHC mediated antigen processing and presentation. TCRα is also associated with T-cell acute lymphoblastic leukemia and colitis [175].
10.3.2. Immunoglobulin heavy chain ε (Ighe)
Transcription of the germ-line immunoglobulin heavy chain ε enhancer is required for the immuno-globulin isotype switch from the μ heavy chain constant region that encodes IgM to the ε heavy chain constant region that encodes IgE. Transcription from the ε enhancer in B-cells is repressed by Hmga1 and induced by IL-4 and lipopolysaccharide [192]. Gel shift and DNAase footprint experiments demonstrate that Hmga1 binds directly to an AT-rich region at the ε enhancer and transient transfection experiments in mouse B lymphoma cells show that Hmga1 represses immunoglobulin heavy chain ε transcription. This suggests that Hmga1 could interfere with immunoglobulin isotype switch from IgM to IgE in B cells.
10.3.3. Immunoglobulin Heavy Chain μ, Immunoglobulin Heavy Constant μ, (IGHM)
Subsequent studies showed that HMGA1 co-activates expression of the IGHM enhancer in human B cells, although in vitro studies suggest that HMGA1 indirectly associates with DNA at this enhancer [192–195]. HMGA1 interacts with the PU.1 transcription factor to enhance PU.1 binding to the μ enhancer and potentiation of the PU.1/ETS-1 interaction at the enhancer. While HMGA1 is not associated with the μ enhancer by chromatin immunoprecipitation, silencing HMGA1 expression results in repression of the μ enhancer. These findings suggest that HMGA1 orchestrates the transcriptional complex, but is not present when transcription commences. The μ enhancer mediates V(D)J rearrangement of the immunoglobulin heavy chain required for IgM production during B cell development [192]. Repression of Ighe enhancer and activation of the IGHM by HMGA1 could help to maintain IgM production and repress IgE production in B cells. IgM antibodies are critical for primary defense mechanisms and recognizing external invaders into the body [175].
10.3.4. GP91-PHOX (Cytochrome B-245, CYBB)
HMGA1 also binds upstream of the GP91-PHOX gene in vitro, which encodes the heme-binding beta subunit of NADPH oxidase (cytochrome b) [195]. This interaction was discovered by screening an expression library with the GP91-PHOX promoter CCAAT-box region that includes an AT-rich motif [196]. HMGA1 binds specifically to this AT-rich region; studies were not done to show if gene expression was altered by HMGA1 binding. NADPH oxidase is a mitochondrial enzyme that produces reactive oxygen species (ROS) required by neutrophils to kill bacteria in phagocytic vacuoles. ROS is involved in diverse processes, including cancer, artherosclerosis, and Alzheimer’s disease.
10.3.5. CD8β1 (CD8b1)
Gene expression studies in lymphoid tumors from the Hmga1a transgenics also show that the CD8β1 (CD8b1) gene is up-regulated in the Hmga1a-driven lymphoid tumors [108]. CD8β1 forms a dimer with CD8α (CD8a) to form the CD8 T cell receptor (TCR), which is normally expressed on the surface of cytotoxic T cells. It is also found on natural killer cells, cortical thymocytes, and dendritic cells, where it is involved in antigen-specific T cell activation [175]. CD8 is also expressed in T-cell acute lymphoblastic leukemia. Knocking-down HMGA1 in Jurkat T-cell acute lymphoblastic leukemia cells represses CD8b1 expression, further implicating the HMGA1-CD8Rb1 pathway in T cell leukemia.
10.3.6. Granzyme M (GZMM)
The Gzmm gene, which encodes granzyme M, is also up-regulated in the Hmga1a transgenic lymphoid cells, both early and late in tumorigenesis [108]. This was confirmed by qRT-PCR in human and mouse cells. This gene is expressed in natural killer and activated T cells and the protein product is stored in large cytoplasmic vacuoles (called granzymes) together with granzyme A, B, and H. Granzyme M is a serine protease involved in apoptosis through caspase activation [175]. Whether HMGA1 directly regulates GZMM remains to be determined.
10.3.7. Spleen Tyrosine Kinase (Syk)
Syk encodes a non-receptor tyrosine kinase which transduces signals downstream of diverse trans- membrane receptors involved in innate and adaptive immunity, proliferation, and survival [197]. Although it was originally identified as an essential factor downstream of B-cell receptor (BCR) signaling, Syk also functions in T-cell receptor signaling. Syk transduces signals involved in cellular adhesion, osteoclast maturation, platelet activation, and angiogenesis [197]. It also induces inflammatory pathways and, together with NF-κB, Syk activates expression of chemokine and cytokines following infection by pathogens. Moreover, Syk is constitutively active in hematologic malignancies and efforts are underway to target its activity in lymphoid malignancies [197]. Syk was reported as a gene induced by Hmga1 in mouse embryo stem cells (mESCs), both in gene expression profile analysis and quantitative RT-PCR studies [198].
10.4. Transcription Factor Genes
10.4.1. Gata-1, Gata-4 and Gata-6
A comparison of mouse embryonic stem cells (mESCs) deficient in Hmga1 to wildtype mESCs led to the identification of target genes involved in endoderm and mesoderm-drived tissues, including blood cell commitment and [144, 198]. When mESC-derived embryoid bodies (EBs) were cultured in methylcellulose under conditions that enable hematopoietic differentiation, the Hmga1 deficient mESCs were reported to form fewer “hematopoietic EB bodies”, defined as EBs that differentiate into colonies with macrophages, granulocytes, and hemoglobin-producing erythroid precursors. This group also reported a decrease in cells expressing the T-cell marker, Thy1.2, with an increase in B cells expressing the pan-B-cell marker, B220. Conversely, these investigators reported a decrease in myeloid and monocytic differentiation, while both erythroid and megakaryocyte differentiation were enhanced. Following treatment of embryoid bodies with retinoic acid to induce mesodermal differentiation, this group reported an increase in globin (ζ, α, β) gene expression in the Hmga1 deficient cells. Because the Gata-1 zinc finger transcription factor regulates globin gene expression, Gata-1 expression was assessed and both mRNA and protein were reported to be increased in the Hmga1 deficient cells after culture under conditions that induce hematopoietic differentiation. The Gata family of zinc finger transcription factors regulate differentiation and fate specification in diverse tissues; the first member to be identified, Gata-1, was discovered in hematopoiesis [175]. A Gata-1 promoter regulatory region (−3.9 to −2.6 kilobases upstream of the first exon) linked to the luciferase reporter was expressed at higher levels in the Hmga1 null ES cells compared to the wildtype ES cells. Chromatin immunoprecipitation show that HMGA1 binds to this region as well as a second downstream regulatory region. Electrophoretic mobility shift assays (EMSA) confirmed HMGA1 binding to a 46 base pair AT-rich site in the downstream regulatory region; the upstream regions were not studied. Together, these studies suggest that Hmga1 directly represses Gata-1 during hematopoietic differentiation.
Both Gata-4 and Gata-6 were also reported as genes repressed by Hmga1 in a similar microarray study comparing genes expressed in wildtype mouse embryonic stem cells (mESCs) to mESCs deficient in Hmga1 [198]. The differential regulation of Gata-4 was confirmed by RT-PCR in mESCs and mouse embryo fibroblasts (MEFs), respectively. Gata-4 and Gata-6 are expressed in diverse mesoderm- and endoderm-derived tissues, including the heart, lung, liver, gut, and gonad [199, 200]. Gata-4 regulates genes involved in embryogenesis, including cardiac muscle differentiation and function, while Gata-6 is thought to regulate terminal differentiation and proliferation. Mutations in GATA-4 and GATA-6 are associated with septal defects and other forms of congenital heart disease in humans.
10.4.2. Lung Krupple-like Factor (Lklf)
Lklf encodes a member of the Kruppel-like family of zinc finger transcription factors that is highly related to the erythroid kruppel-like factor (EKLF) and recognizes a similar DNA binding motif [201, 202]. Deletion of Lklf in mice results in embryonic lethality with retarded growth, craniofacial abnormalities, abnormal bleeding and anemia in utero [201]. Although yolk sac erythropoiesis is normal, fetal liver cultures from these embryos fail to generate erythroid cells. Studies in lung epithelia reveal that LKLF is an important anti-inflammatory factor that blocks pro-inflammatory cytokine production [202]. In mESCs, Hmga1 was reported to up-regulate Lklf expression in gene expression profile analysis and RT-PCR, although studies in more differentiated MEFs suggest that Hmga1 represses Lklf [201].
10.4.3. Zif268, Early Growth Response Protein 1 (Egfr1), Nerve Growth Factor-Induced Protein A (Ngfr-1), or Krox-24
Zif268 [also known as early growth response protein 1 (Egfr1), nerve growth factor-induced protein A (Ngfr-1), or Krox-24] is an immediate-early gene induced by growth factors, cytokines, and stress signals that encodes a zinc finger transcription factor [203]. In the absence of stimulation, Zif268 expression is low in most tissues, except the brain. In the brain, it is associated with neuronal activity and it may function in neuronal plasticity [175]. Zif268 also regulates genes involved in mitogenesis and differentiation. Although some studies suggest that it has tumor suppressor function, recent work in prostate cancer indicates that ZIF268 promotes proliferation [203]. A report of gene expression profile analyses and quantitative, RT-PCR in mESCs, MEFs, and liver tissue deficient in Hmga1 compared to wildtype fibroblasts showed that Zif268 is induced by Hmga1 [198].
10.4.4. JunB and cFos
JunB, cFos, and cMyc are additional immediate-early genes that encode proto-oncogenic transcription factors and are up-regulated by Hmga1 [198]. Gene expression profile analysis and qRT-PCR studies reported induction of these genes in wildtype mESCs compared to mESCs deficient in Hmga1 [198]. JunB and cFos are members of the AP1 family with oncogenic properties. Human cFOS was also identified as an HMGA1 gene target in vascular smooth muscle cells [discussed below; ref. 204]. As noted previously, cMYC encodes a potent oncogenic transcription factor that also regulates HMGA1 expression [88, 205]. Studies in hESCs and induced pluripotent stem cells also indicate that HMGA1 regulates cMYC expression [see below; ref. 98]. This regulatory circuit could orchestrate proliferative and oncogenic pathways in tumors overexpressing HMGA1, cMYC, JUNB, and cFOS.
10.4.5. NANOG, OCT4, SOX2, cMYC, and LIN28
Studies in hESCs and cellular reprogramming discovered a novel role for HMGA1 in maintaining a de-differentiated state in pluripotent stem cells [98]. In hESCs induced to differentiate, HMGA1 expression falls and parallels that of known pluripotency genes. Silencing HMGA1 in hESCs results in repression of OCT4, SOX2, cMYC, and LIN28, while forced expression of HMGA1 in hESCs cultured under differentiation conditions maintains expression OCT4, SOX2, cMYC and NANOG. HMGA1 binds directly to the promoters of SOX-2, cMYC, and LIN28 in vivo in hESCs, as demonstrated by chromatin immunoprecipitation. In experiments of induced pluripotency with the “Yamanka” reprogramming factors (Oct4, Sox2, Klf4, cMyc or OSKM), HMGA1 enhances cellular reprogramming, yielding an increased number of fully reprogrammed, induced pluripotent stem cell colonies that were larger in size. HMGA1 also up-regulates expression of SOX2, cMYC, and LIN28 during cellular reprogramming. Moreover, interfering with HMGA1 expression (via short hairpin RNA) or function (via a dominant-negative construct) completely abrogates reprogramming of somatic cells to induced pluripotent stem cells by the Yamanaka factors. These findings demonstrate a novel function for HMGA1 as a key regulator of the stem cell state by directly inducing networks involved in pluripotency [98].
10.4.6. Signal Transducer and Activator of Transcription 3 (STAT3)
Global gene expression profile analysis in fibroblasts identified STAT3 as a critical downstream target of HMGA1 [91]. STAT3 is up-regulated in cultured cells and transgenic mice engineered to overexpress Hmga1a. Similar to other HMGA1-regulated promoters, HMGA1 binds to an AT-rich region near an NF-κB binding site and activates STAT3 expression. HMGA1 occupies this promoter in cultured cells from diverse hematopoietic malignancies, including myeloid and lymphoid leukemia cells. Moreover, a STAT3 small molecule inhibitor induces apoptosis in leukemic cells from Hmga1 transgenics, but not in normal lymphoid cells, indicating that this pathway could be a rational therapeutic target in some malignancies [91]. STAT3 small molecule inhibitors also result in decreased tumor burdens in the Hmga1a transgenic mice crossed onto an Ink4a/Arf null background [206]. Recent studies also showed that a small molecule inhibitor to STAT3 disrupted lymphoid tumor cell proliferation in vitro and xenograft tumors in vivo [206]. Because STAT3 is a key mediator of inflammatory signals and molecular pathways that contribute to cancer initiation and progression, including proliferation, angiogenesis, metastatic progression, survival, and immune evasion [207–209], this pathway could be important in diverse tumor types.
10.4.7. Fox-P1
Fox-P1 is repressed in lymphoid cells from Hmga1 transgenic mice, both early and late in tumorigenesis [108]. This gene is also repressed by HMGA1 in human Jurkat T-cells. Fox-P1 is a member of the “forkhead box or FOX” transcription factors [108]. The P subfamily of Fox transcription factors regulate tissue and cell-type specific genes during development and differentiation [210]. The FOX-P1 protein has both DNA binding and protein-protein binding domains. FOX-P1 maps to chromosomal region 3p14.1, which is lost in several tumor types, and FOX-P1 is therefore thought to function as a tumor suppressor [210].
10.4.8. Eomesodermin (EOMES)
In the same gene expression analysis, the Eomes gene (also known as T-box brain protein 1 or TBR1 gene) was up-regulated both early and late in lymphoid transformation [108]. Knock-down of HMGA1 in human Jurkat T-cell acute lymphoblastic leukemic cells represses EOMES expression, indicating that the HMGA1-EOMES network is also activated in human T-leukemia cells. EOMES encodes a transcription factor that functions in endodermal and mesodermal specification during development [211–213]. Disruption of the Eomes gene in mice demonstrate that it is necessary for trophoblastic development and gastrulation. Studies in human T cells indicate the EOMES is critical for IFN-γ production in CD8+ cells, and possibly in CD4+ cells [213, 214]. Although further studies are needed to determine if EOMES is directly regulated by HMGA1, this pathway could promote inflammatory signaling, EMT and other stem-like properties in cells overexpressing HMGA1.
10.4.9. TWIST
Increasing evidence suggests that cancer cells metastasize by co-opting stem cell transcription networks that mediate changes resembling an epithelial-mesenchymal transition (EMT) during tumor progression [58, 68, 214]. In poorly differentiated colon cancer cells, HMGA1 induces the EMT genes, TWIST and VIMENTIN, while repressing the epithelial gene, E-CADHERIN [68; VIMENTIN and E-CADHERIN are discussed in detail later]. TWIST encodes a bHLH transcription factor protein involved in EMT during embryogenesis and in tumor progression. Concurrent with repressing TWIST and VIMENTIN, and inducing E-CADHERIN, silencing HMGA1 in the high-grade colon cancer cells also blocks three dimensional colonosphere formation, a cancer stem cell property [68]. Moreover, there is depletion in tumor-initiator cells in limiting dilution tumorigenesis experiments in the HMGA1 knock-down cells. In addition, interfering with HMGA1 expression blocks anchorage-independent cell growth, migration, and invasion in vitro as well as metastatic progression in vivo [68]. These results indicate that HMGA1 promotes tumor progression through transcriptional networks that facilitate EMT, metastatic progression, and a de-differentiated, stem-like state.
10.4.10. HAND1 (Heart and Neural Crest Derivatives Expressed 1)
In addition to activating cellular programs that mediate tumor growth and progression, HMGA1 represses factors required for differentiation. To illustrate, HMGA1 was reported to repress HAND1 (Heart and Neural Crest Derivatives expressed 1), a member of the Twist subfamily of basic helix-loop-helix (bHLH) transcription factors [215]. Studies in Hand1 null mice indicate that it plays a critical role in placenta formation and cardiac morphogenesis in the ventricular chambers [215] Hand1 expression was reported to be enhanced in Hmga1 null mice and HMGA1 binds to the Hand1 promoter, both in vitro and in vivo, as demonstrated by in gel shift analysis and chromatin immunoprecipitation. Transfection experiments showed that HMGA1 represses HAND1 promoter expression. In addition, there was an inverse correlation between expression of HMGA1 and HAND1 in primary thyroid tumors. Moreover, HMGA1 overexpression was reported to be associated with DNA methylation of the HAND1 promoter in anaplastic thyroid carcinomas in advanced tumors. HAND1 is also silenced in gastric, colorectal, and pancreatic cancers [215–218].
10.4.11. Transcription Factor EB (Tfeb)
The Tfeb gene is also reported to be induced by Hmga1 in wild-type mESCs compared to those deficient in Hmga1 in microarray gene expression profile analysis and RT-PCR experiments [198]. This gene encodes a protein that regulates genes involved in lysosomal biogenesis and autophagy [219]. The protein is phosphorylated by ERK2 which responds to levels of extracellular nutrients, suggesting a link between HMGA1, TFEB, nutrient sensing, and autophagy [220]. Increasing evidence also suggests an important role for nutrient metabolism and autophagy in tumor cells [155–158]. TFEB activates the expression of CD40L in T-cells; thus, it is linked to cell-dependent antibody responses in activated CD4 T-cells and cancers associated with abnormal T-cell growth [175].
10.5. Genes Involved in Repair of DNA Damage and Cell Cycle Progression
10.5.1. Xeroderma Pigmentosa Complementation Group A (XPA)
HMGA1 participates in genomic instability in cancer cells by modulating expression of DNA repair genes [61, 110–112]. The gene encoding the nuclear excision repair factor, XPA, is repressed in MCF-7 cells engineered to overexpress HMGA1a [112]. Nuclear excision repair (NER) is responsible for the detection and repair of large, DNA helix distorting lesions, such as cyclobutane pyrimidine, photoproducts, and cisplatin adducts that arise following UV radiation and chemical carcinogens. Chromatin immunoprecipitation experiments show that HMGA1 binds upstream of the XPA gene. In transfection experiments, HMGA1a no longer represses the XPA promoter when the consensus HMGA1 binding site is mutated. MCF-7-HMGA1 cells also exhibit an increase in cyclobutane pyrimidine dimers, consistent with the hypothesis that HMGA1 is associated with DNA helix distorting lesions. Another study discovered an increase in unbalanced translocations using spectral karyotype analysis of prostate cancer cells engineered to overexpress HMGA1 [61]. Together, these studies suggest that cancer cells with high expression of HMGA1 are predisposed to chromosomal instability, at least in part, by repressing XPA.
10.5.2. Breast Cancer 1 (BRCA1)
The BRCA1 tumor suppressor gene was also reported to be repressed by HMGA1 [110]. BRCA1 was originally isolated as the gene mutated in familial breast and ovarian cancer [175]. It is important in regulating cell cycle progression, processing DNA damage, and maintaining chromosomal stability, including repair of double-stranded breaks. It associates with other tumor suppressors, DNA damage sensors, and signal transducers to form a large protein complex known as the BRCA1-associated genome surveillance complex (BASC). Mutations in BRCA1 are associated >40% of inherited breast cancer and >80% of cases of with inherited breast and ovarian cancer. Prior studies report that Brca1 is up-regulated mESCs null for Hmga1, while it was down-regulated in cells and tissues overexpressing HMGA1 [110]. This group found that HMGA1 binds directly to the BRCA1 promoter/enhancer region (gel shift assays; chromatin immunoprecipitation) and represses its expression in transfection experiments. They also reported that HMGA1 blocks induction of the BRCA1 gene by estrogen. In a pilot study of 14 human breast cell lines and primary tumors, HMGA1 and BRCA1 mRNA and expression were inversely correlated [111]. These findings suggest that repression of the BRCA1 tumor suppressor by HMGA1 could contribute to tumorigenesis in cases where BRCA1 is not mutated.
10.6. Genes Involved in Pro-Survival/Anti-Apoptotic Pathways
10.6.1. B-Cell Lymphoma 2 Gene (BCL-2)
A prior study reported that HMGA1 promotes a survival/anti-apoptotic response by directly inducing BCL-2 expression [221]. In breast cancer cells (MCF-7) engineered to overexpress HMGA1b, HMGA1b was reported to bind to an AT-rich region, as shown in vitro and in vitro in gel shift analysis and chromatin immunoprecipitation. Transient transfection experiments showed that HMGA1 interferes with the repression of the BCL-2 promoter by p53 and functional studies revealed that HMGA1 blocks p53-induced apoptosis. HMGA1 was also reported to bind to the Brn-3a transcription factor by co-immunoprecipitation. Brn-3a is known to bind to both p53 and HIPK2 to repress BCL-2 expression, thereby promoting apoptosis. Co- expression of HMGA1 with p53 in transfection experiments blocks repression of BCL-2 and functional studies show that apoptosis is also blocked by HMGA1. In addition, the expression of HMGA1 and BCL-2 correlates in breast cancer cell lines. Together, these studies suggest that HMGA1 interferes with p53-mediated transcriptional repression of the anti-apoptotic genes (BCL-2), thereby up-regulating its expression.
10.7. Genes Involved in RAS/ERK Signaling
10.7.1. c-KIT Ligand/Stem Cell Factor, SCF
The RAS/ERK signaling pathway is disrupted in many cancers and thought to be a key mechanism that drives uncontrolled cell growth in tumors. This pathway mediates extracellular signaling by mitogens, which result in receptor tyrosine kinase activity and phosphorylation events that promote the removal of GDP from RAS to enable GTP binding and activation, subsequently activating protein kinases and ultimately activating oncogenic transcription factors. Thus, activation of RAS signaling could promote oncogenic transformation by HMGA1. Genes involved in RAS/ERK (MAPK) signaling were identified using global gene expression analysis in breast (MCF-7) cells transduced to overexpress HMGA1a [222, 223]. The gene encoding c-KIT Ligand (also known as Stem Cell Factor or SCF) was identified in this screen [222]. c-Kit ligand binds to its receptor to activate RAS/ERK signaling. HMGA1 binds to an AT-rich region upstream of the c-KIT Ligand gene in vitro and in vivo. c-Kit ligand is essential for hematopoiesis during embryogenesis and important for maintenance of hematopoietic stem cells in the niche during adult hematopoiesis. Thus, overexpression of HMGA1 could enforce hematopoietic stem cell-like programs during leukemic transformation through c-KIT pathways and also help to maintain leukemic cells in a protective niche.
10.7.2. Caveolin-1 and Caveolin-2 (CAV1 and CAV2)
The CAV1 and CAV2 genes are also repressed by HMGA1 in the MCF-7 model [222]. The caveolin protein family (caveolin-1, -2, -3) are membrane proteins involved in receptor-independent endocytosis. The caveolins form “cave-like” structures that associate with cholesterol and sphingolipids in the cell membrane. Caveolin-1 functions in cellular signaling and accumulating evidence suggests that it is important in survival of androgen-insensitive prostate cancer, by promoting metastases, and the development of multidrug resistance [223]. Caveolin-1 links integrin subunits to the tyrosine kinase FYN to initiate the Ras-ERK pathway in cell cycle progression [224]. CAV1 and CAV2 expression are also associated with a basal-like, triple negative phenotype in breast cancer [225]. Caveolins also inhibit some components of RAS/ERK signaling and repression by HMGA1 could enhance RAS/ERK function [222]. Further work is needed to determine if HMGA1 directly down-regulates CAV1 and CAV2.
10.7.3. p96/Differentially Expressed in Ovarian Cancer 2 (DOC2)
The p96 gene was reported to be repressed in murine embryonic stem cells deficient in Hmga1 [198]. p96 is a putative mitogen-responsive gene that is repressed in human ovarian and mouse mammary tumors [226–228]. Expressing the p96 human homolog, DOC2, in human ovarian carcinoma cells inhibits both cell growth and xenograft tumorigenesis, consistent with a tumor suppressor function [228]. In mice, deficiency of Doc2 (disabled-2 or Dab2) results in embryonic lethality with endodermal disorganization. In murine embryonic stem cells, Doc2 appears to repress cFos expression and could act as a negative regulator of MAPK/Ras/ERK mitogenic signaling [228].
10.7.4. Gly96 or Immediate Early Response Gene 1 (Iex-1, Ier3)
Gly96 (Iex-1 or Ier3) was also reported to be repressed by Hmga1 in gene expression studies (microarray and quantitative RT-PCR) comparing wildtype MEFs to those deficient in Hmga1 [198]. Gly96 is an immediate-early gene that is rapidly induced following various cellular stressors, including growth factors, irradiation, viral infection, inflammatory cytokines, chemical carcinogens and hormones [229]. Gly96 encodes a short-lived glycoprotein and is also transcriptionally regulated by NF-κB, cMyc, Ap1, Sp1, and p53. Transgenic mice expressing Gly96 in T cells develop T-lymphomas [229]. Gly96 can either positively or negatively regulate proliferation, possibly by acting as a substrate of ERK or an inhibitor to ERK phosphorylation and function. Gly96 also plays an indirect role in protecting the cell from Fas- or tumor necrosis factor type alpha-induced apoptosis pathways [175]. It is not yet known if Hmga1 directly regulates gly96/IEX-1.
10.8 Genes Involved in Vascular Smooth Muscle Proliferation and Function
10.8.1. Inducible Nitric-Oxide Synthase (iNOS or NOS2)
i-NOS or NOS2 is an HMGA1 gene target with a promoter similar to that of the IFN-β. In vascular endothelial cells, HMGA1 binds to an AT-rich region in the enhancer, thus facilitating the binding of NF-κB to its cognate DNA binding site to form a complex, which activates transcription of NOS2 [230–232]. HMGA1 also binds further upstream (at positions −3,506 to −3,375) [230]. Activation of NOS2 is blunted by a dominant-negative HMGA1 construct, the small molecule inhibitor, distamycin [172], or RNA interference to HMGA1 [230–232]. The gene product, inducible nitric oxide synthase, is the enzyme that catalyzes the conversion of L-arginine to nitric oxide (NO) following induction by cytokines or other inflammatory signals. NO functions as a gaseous signaling molecule released from the endothelium to cause vasodilation of blood vessels [232]. It is also generated by granulocytes, monocytes, and macrophages. In cancer, it is thought to contribute to angiogenesis. In addition, NOS2 enhances Cyclooxygenase-2 (COX-2) activity by S-nitrosylation [233–236] which also participates in angiogenesis and many oncogenic pathways. Thus, the HMGA1-NOS2-COX-2 pathway could serve as another inflammatory signal that promotes angio-genesis and multiple, diverse oncogenic pathways.
10.8.2. SMOOTH MUSCLE SPECIFIC GENE (SM22) TRANSGELIN (TRGLN), and cFOS
Studies in vascular smooth muscle cells showed that HMGA1 proteins are dramatically induced during their proliferation. In addition, HMGA1 enhances expression of both the SM22, (TRGLN), and the cFOS proto-oncogene [237]. In gel shift experiments, HMGA1 binds to an AT-rich region within the DNA binding site of the Serum Response Factor (SRF), a protein known to function during proliferation to activate vascular smooth muscle genes. HMGA1 interacts with SRF and enhances its binding to the SM22 and cFOS promoters, thereby augmenting SRF-dependent trans-activation of these genes. The activation of vascular smooth muscle genes could induce angiogenesis during tumor development. cFOS functions as a proto-oncogene and regulator of cell proliferation, differentiation, and transformation. It could cooperate with HMGA1 during transformation. Of note, SRF is also elevated in Alzheimer’s disease and a recent study suggests [238] that this results in increased accumulation of amyloid β. Thus, these SRF-dependent smooth muscle genes could work together with HMGA1 in cancer, Alzheimer’s disease, and embryonic development.
10.8.3. Cd44
The Cd44 gene is a downstream transcriptional target of HMGA1 that is up-regulated in vascular smooth muscle cells to promote proliferation following arterial wall injury [239]. CD44 encodes a cell adhesion molecule that is induced during inflammation, wound healing, tumorigenesis, and metastatic progression. Previous studies identified CD44 as a marker for progenitor/stem cells in breast tissue, breast cancer, and other solid tumors [240–242]. More recently, CD44 was found to be a key factor in maintaining acute myeloid leukemic stem cells in the bone marrow niche; HMGA1 could indirectly facilitate this interaction by inducing CD44 expression [242]. The murine Cd44 promoter includes an AP-1 site adjacent to an AT-rich HMGA1 DNA binding site and the AP-1 transcription factors, cJun and cFos, along with Hmga1, bind to these sites in gel shift assays [239]. In transfection experiments, HMGA1 potentiates AP-1 transactivation of the murine Cd44 promoter, suggesting that AP-1 proteins and HMGA1 cooperate in inducing Cd44. Interestingly, Cd44/CD44 is subject to proteolytic cleavage by MMPs, which are also regulated by HMGA1 (see below), and this could provide a negative feedback loop whereby HMGA1 induces CD44 expression and also promotes degradation of the CD44 protein once translated. Given the link between CD44 and stem cell qualities [240–242], the HMGA1-CD44 pathway could maintain a stem cell phenotype in cancer and normal stem cells.
10.8.4. Vascular Smooth Muscle Actin, Vsm-Actin
Vascular smooth muscle actin, Vsm-actin was also reported to be induced by Hmga1 in MEFs, both in microarray gene expression studies and qRT-PCR [198]. The protein encoded by Vsm-actin is involved the cytoskeleton, which is responsible for the structure and integrity of the cell [175]. Alterations in cellular cytoskeleton could contribute to cellular mobility and metastatic progression.
10.9. Genes Encoding Helix-Loop-Helix (HLH) Factors Regulated by HMGA1
10.9.1. Id3
The inhibitors of differentiation (Id or inhibitors of DNA binding proteins) represent another class of helix-loop-helix (HLH) proteins that are reported to be regulated by HMGA1 [243]. Id proteins bind cellular proteins to regulate cell growth, angiogenesis, and differentiation of adult and embryonic cells [243, 244]. In contrast to bHLH proteins like HAND1, Id proteins lack the distinctive region of basic amino acids found in other HLH proteins (bHLH) that are critical for DNA binding. As a result, heterodimers between Id proteins and other bHLH transcription factors are not capable of interacting with DNA and the Id protein thereby exerts a dominant-negative effect that results in sequestration of factors including, E2A and RB, to inhibit their transcriptional activity. Similar to HMGA1, both Id1 and Id3 are overexpressed in proliferating and neoplastic tissues, but decreased in differentiated cells. Id3 was reported to be a potential HMGA1 target gene in an investigation of gene expression profiles by microarray analysis from mESCs deficient in Hmga1 [198]. Id3 gene expression by qRT-PCR was reported to correlate with HMGA1 expression in MEFs and spleen, but not in liver or heart from the mice with wildtype or deficient (homozygous or heterozygous) Hmga1. Id3 protein levels by Western analysis also correlated positively with Hmga1 in the spleen, kidney, and lung, but not in MEFs, liver, brain, or testes from the wildtype or Hmga1 deficient mice [198]. The basis for the variation in mRNA and protein levels is unclear, but could relate to differentiation status and cellular context. Gel shift experiments showed that HMGA1 binds to an AT rich sequence in the Id3 promoter in vitro and chromatin immunoprecipitation showed that HMGA1 occupies this region in vivo in mouse splenocytes. Because Id proteins inhibit differentiation, the HMGA1-Id3 pathway could help to maintain cells in an undifferentiated state during development and cancer as well as in specific tissues which require undifferentiated progenitors.
10.9.2. T-Cell Leukemia Homeobox 3 (TLX3, HOX11L2) and Nirenberg-KIM 2 Homeobox 5 (NKX2-5, CSX) Genes
HMGA1, together with PU.1, also induce expression of fusion genes that encode HLH proteins involved in T-cell acute lymphoblastic leukemia [245]. These fusion genes encode either the T-cell leukemia homeobox 3 protein (TLX3) or the highly related Nirenberg Kim homeobox protein (NKX2-5, also known as the cardiac-specific homeobox or CSX protein). Dysregulation of the TLX3 gene results from a translocation event at 5q35 via the recurrent t(5;14) (q35;q32) rearrangement, while the neighboring homeobox gene, NKX2-5, is activated by a variant t(5;14)(q35;q32) or by t(5;14)(q35;q11.2) involving the the T-cell receptor D locus at 14q11.2. These translocations juxtapose the 3′ noncoding region of the BCL11B gene to the NKX2-5 or TLX3 genes. BCL11B normally encodes a Kruppel family zinc finger gene located at 14q32. To determine how the translocation events lead to dysregulation of the TLX or NKX2-5 genes, the 3′-BCL11B noncoding sequence involved in the translocations was investigated and binding sites for both HMGA1 and PU.1 were identified by the TRANSFAC database. Interfering with expression of either HMGA1 or PU.1 decreases expression of TLX3/NKX2-5 fusions, suggesting that HMGA1 and PU.1 bind to the 3′-BCL11B noncoding sequence to enhance expression of the fusion proteins. The TLX and NKX2-5 homeobox domain proteins function as key regulators in cell fate decisions and proliferation and their expression is often cell-type specific. NKX2-5 is normally expressed during embryogenesis in cardiac progenitors and involved in cardiac development. TLX3 is expressed in the dorsal and ventral medulla oblongata during development, a region in the brainstem important in controlling respiration. Mice deficient in TLX3 die within 24 hours after birth due to central respiratory failure. While HMGA1 regulates other embryonic genes involved in malignancy, TLX and NKX2-5 are embryonic genes that become translocated to other genes, and the resultant fusion genes are regulated by HMGA1.
10.10. HMGA1 Target Genes Involved in Adipo-genesis
10.10.1. Obese, Leptin
The obese/leptin gene was identified as transcriptional targets induced by Hmga1 and involved in adipocyte differentiation [103, 244]. The first clue linking Hmga1 to adipogenesis was the observation that Hmga1 expression increases when cultured, preadipocytes (3T3-L1 cells) are induced to differentiate [244]. Experiments using an antisense approach to inhibit Hmga1 expression block differentiation of pre-adipocytes while enhancing their proliferation rates. In addition, HMGA1 interacts with the CCAAT/enhancer-binding protein β (C/EBPβ) and potentiates C/EBPβ-mediated transactivation of the obese gene [244]. Obese is an adipocyte-specific gene that encodes the adipocyte-derived hormone, leptin. Leptin plays a key role in regulating energy intake and expenditure, including appetite and metabolism, suggesting that Hmga1 is an important regulator in these processes. Prior studies also suggest that leptin is involved in blastocyst development during murine embryogenesis [246].
10.11. Putative Targets Involved in Invasion and Metastatic Progression
10.11.1. Matrix Metalloproteinases
10.11.1.1. MMP-2, MMP-9, MMP-13 and Integrin-β1
Several independent groups studying HMGA1 identified at least three different matrix metalloproteinases (MMP) as HMGA1 transcriptional targets [29, 50, 60, 70]. This family of zinc-dependent proteinases were originally characterized based on their ability to degrade the extracellular matrix and basement membrane proteins. By degrading the basement membrane, some members of the MMP family are thought to enhance cell mobility in a stationary tumor cell and promote metastases. In fact, MMP activity correlates with cellular invasiveness and metastatic potential in a subset of solid tumors [247]. More recently, MMPs were shown to exert other important biologic effects relevant to cancer by cleaving critical proteins involved in angiogenesis, apoptosis, chemotaxis, cell migration, and cell proliferation [247]. Even tumor suppressor functions have been demonstrated for MMP family members [248]. Thus, it has become clear that MMPs not only function in cancer progression and metastasis, but also in early steps of cancer development. In the first study linking HMGA1 to MMPs, the MMP-13 gene was among the list of differentially expressed genes in MCF-7 breast cells overexpressing HMGA1a or HMGA1b identified from a microarray that included almost 1,200 cDNAs [29]. Differential expression was confirmed using qRT-PCR in these cells. Integrin-β1 was also identified in this screen and confirmed by qRT-PCR. Using a similar strategy, MMP-2 was identified as a differentially expressed gene in human prostate cancer cells engineered to express HMGA1a as compared to cells engineered with an empty, control vector [60]. Differential expression of the pre-form of the MMP-2 protein was confirmed by Western analysis. HMGA1 also up-regulates MMP-2 expression in lung cancer, but only in large cell, undifferentiated lung carcinoma cells [50]. This study showed that HMGA1 occupies the MMP-2 promoter in vivo in undifferentiated, large cell carcinoma cell lines by chromatin immunoprecipitation, but not in cultured cells from other lung cancer subtypes. In addition, both HMGA1a and MMP-2 expression were significantly correlated in cultured, large cell carcinoma cells. Functional studies showed that knock-down of HMGA1 or MMP-2 blocked anchorage-independent cell growth. Inhibiting MMP-2 also disrupted migration, and invasion, suggesting that this pathway could mediate these tumor progression phenotypes in undifferentiated, large cell lung carcinomas. Another study found that HMGA1 increases invasiveness and up-regulates MMP-9 expression in cultured, pancreatic adenocarcinoma cells [70]. Together, these studies suggest that HMGA1 drives the expression of matrix metalloproteinases in diverse tumor cells. Because many MMP promoters have AT-rich regions and putative HMGA1 binding sites, HMGA1 could orchestrate MMP function in diverse settings, such as cancer and development, and the specific MMP pathway likely depends upon the cellular milieu.
The ability of HMGA1 to activate MMPs could also be relevant in the pathophysiology of Alzheimer’s disease. Interestingly, the release of both MMP-2 and MMP-9 proteins is increased in the brain microvessels of patients with Alzheimer’s disease and could contribute to angiogenesis [249]. In fact, recent work suggests that aberrant angiogenesis leads to disruption in the blood brain barrier and disease progression [249]. MMP-9 protein was also found to be elevated in postmortem brain tissue of Alzheimer’s patients [250]. As discussed earlier, HMGA1 is increased in the hippocampus of patients with Alzheimer’s and HMGA1 is linked to the production of aberrantly spliced, presenilin-2V (PS2V) [39–42], which appears to play a role in Alzheimer’s disease [159–162]. Together, these findings implicate HMGA1 in the up-regulation of MMP-2 and MMP-9 in Alzheimer’s, which could lead aberrant angiogenesis along with increases in PSV2.
10.12. Other Protease Genes Regulated by HMGA1
10.12.1. Cathepsin H (CtsH) and Legumain
CtsH and Legumain were reported to be repressed by Hmga1 in studies comparing wildtype mESCs to those deficient in Hmga1 [198]. Differential expression of these genes was confirmed by RT-PCR experiments. The CtsH protein functions as both an amino- an endo-petidase that degrades proteins in lysosomes [251]. CtsH also represses apoptosis and induces angiogenesis. Legumain encodes another cysteine protease that is localized to the lysosomal/endosomal system and functions in antigen presentation. Legumain is also overexpressed in diverse tumors and linked to invasion and migration in vitro [252, 253].
10.12.2. Carboxypeptidase E (CpE)
The carboxypeptidase E (CpE) gene was reported to be induced by Hmga1 in the same study of mESCs [198]. CpE encodes the enzyme carboxypeptidase that cleaves C-terminal amino acid residues to yield biologically active neuropeptides and peptide hormones, such as insulin [254]. Mutations in CpE are implicated in type II diabetes; mice lacking Cpe have diabetes, infertility, obesity, osteopenia, and deficits in memory and learning. An alternatively spliced form induces tumor growth and is a biomarker for metastases in a subset of human tumors [254].
10.13. Putative Target Genes Involved in Multiple Transformation Pathways
10.13.1. Inflammation, Proliferation, Angiogenesis
10.13.1.1. Cyclooxygenase-2 (COX-2)
COX-2 was originally identified as a putative HMGA1 target gene in hypoxic vascular endothelial cells [255]. HMGA1’s role in regulating this promoter was investigated because the promoter includes an NF-kB site adjacent to an AT-rich region and NF-κB protein or mRNA levels were not induced under hypoxic conditions. In contrast, HMGA1 mRNA and protein increase in low oxygen tension. Further, HMGA1 binds to an AT-rich region near an NF-κB binding site in the COX-2 promoter in vitro and up-regulates COX-2 expression in transfection experiments. A subsequent study found that HMGA1 activates COX-2 expression during uterine tumorigenesis [75, 256]. Uterine sarcomas from Hmga1a transgenics have increased Cox-2. Treatment of Hmga1 transgenics with sulindac (a COX-1/COX-2 inhibitor) impairs uterine tumor growth [256]. Both HMGA1 and COX-2 are also overexpressed in leiomyosarcomas, a rare and frequently aggressive human uterine sarcoma. Sulindac or celecoxib (a more selective COX-2 inhibitor) disrupt anchorage-independent cell growth in vitro and xenograft tumorigenesis in vivo in human uterine sarcoma cells, but only in cells with high HMGA1 levels [256]. This suggests that COX-2 inhibitors could be effective in tumors that dysregulate the HMGA1-COX-2 pathway. A subsequent study also found that this pathway is important in pancreatic adenocarcinoma [257]. COX-2 is induced by inflammatory signals, and, like HMGA1, could cooperate to elicit a number of cellular pathways involved in neoplastic transformation, including proliferation, angiogenesis, metastasis, and inhibition of apoptosis. Further studies are therefore warranted to elucidate the role of this pathway in other cancers.
10.13.1.2. Pim-2 and Pim 1
The Pim-2 proto-oncogene was also reported to be induced by Hmga1 in mESCs, based on gene expression microarray and qRT-PCR studies [198]. Pim-2 is a member of the Pim family of genes, which encode Pim-1 and Pim-2 proto-oncoproteins. Pim-1 and Pim-2 are serine/threonine kinases that function in cell survival and proliferation [175]. Pim-2 phosphorylates cMyc, which enhances its stability and ability to regulate transcription. Pim-2 also phosphorylates BAD, a pro-apoptotic protein, resulting in the release of the anti-apoptotic protein, Bcl-XL. Pim-2 promotes proliferation by phosphorylating cyclin-dependent kinases, such as Cdkn1a and Cdkn1b [175]. PIM-1 was also proposed to activate cMYC by phosphorylation and thereby induce HMGA1 in human prostate cancers associated with trichomonas infection [258]. PIM-1/Pim-1 also promote cell survival and interferes with apoptosis by inhibiting the JNK/p38MAPK signaling pathway, thereby reducing caspase-3 activation [175]. The HMGA1-PIM pathways could therefore contribute to oncogenesis through cell survival and proliferation pathways. This pathway also further links HMGA1 to cMyc function.
10.14. Putative HMGA1 Targets Involved in Ocular Function
10.14.1. Rhodopsin (RHO)
Although HMGA1 is expressed predominantly in highly proliferative cancer cells or during embryonic development, high levels of Hmga1 expression were also discovered in terminally differentiated murine photoreceptor cells (rods and cones) of the retina at both the mRNA and protein level [259]. High Hmga1 protein levels could contribute to the ability of the photoreceptor cells to maintain high rates of metabolism and translation. Retinoblastoma cells were also reported to express high levels of HMGA1 and inhibition of HMGA1 expression by antisense technology results in repression of the Rhodopsin (RHO) gene [259]. Rho protein belongs to a family of G-protein coupled receptors, which are extremely sensitive to light and required for the perception of light. By footprint analysis, HMGA1 binds to an AT-rich region in the rho promoter that coincides with the BAT-1 cis element, which is also bound by the paired-like homeodomain protein, Crx [259]. Crx is a photoreceptor-specific transcription factor that is thought to be important for photoreceptor cell functioning and differentiation. Mutations in Crx are associated with human diseases such as night blindness characterized by photoreceptor dysfunction, including photoreceptor degeneration, rod-cone dystrophy, retinitis pigmentosa, and Leber congenital amaurosis [175, 259, 260]. In retinoblastoma cells HMGA1 binds to the BAT-1 cis element in gel shift assays and transactivates the rho promoter in transfection assays. HMGA1 also physically interacts with the Crx protein, shown by GST pulldown assays [259]. Taken together, these findings suggest that HMGA1 is important in regulating the retinal photoreceptor gene rho and functions by enhancing transcription through interactions with Crx.
10.14.2. CRYAB
HMGA1 induces expression of the CRYAB gene, which encodes the αβ-Crystallin (CRYAB) protein, a heat-shock protein that is highly expressed in the ocular lens of vertebrates [261]. Deficiencies in CRYAB and other crystalline proteins are associated with cataracts. CRYAB also plays a role in cell survival by encoding proteins that function as chaperones for misfolded proteins, thus preventing these abnormal proteins from inducing apoptosis [175]. In vitro gel shift assays and in vivo chromatin immunoprecipitation demonstrate that HMGA1 binds to a Brahma-Related Gene 1 (BRG1) response element within the CRYAB promoter and transfection experiments show that HMGA1 induces CRYAB expression. Nucleosome mapping experiments show that this response element is located adjacent to a nucleosome and immediately upstream of the transcription start site. HMGA1 along with the HMGA1 binding sequences are required for maximal CRYAB induction, suggesting that HMGA1 helps to orchestrate the optimal chromatin state and assembly of BRG1 to this promoter. Notably, BRG1 is a member of the SWI/SNF chromatin remodeling family. Like other SWI/SNF family members, BRG1 has both ATPase and helicase activity, which facilitate nucleosome sliding for transcription factor assembly at specific regulatory regions of DNA. How CRYAB relates to HMGA1 function will require further study, although emerging evidence indicates that heat shock proteins are important in both cancer and development.
10.15. Genes Involved in Diabetes, and Insulin Resistance
10.15.1. Insulin Receptor
As noted earlier in this review, increasing evidence links HMGA1 to glucose metabolism and many recent studies underscore the importance of glucose metabolism in cancer and embryogenesis. HMGA1 regulates expression of several genes that encode proteins involved in glucose homeostasis. In addition, patients with Type 2 diabetes [35–38] were found to have genetic alterations that affect HMGA1 expression, further implicating HMGA1 in glucose homeostasis. Type 2 diabetes is characterized by hyperglycemia and reduced sensitivity to insulin. After insulin binds the insulin receptor (IR), a tyrosine kinase receptor, glucose is translocated intracellularly by the glucose transporter type 4 (GLUT4) which mediates the uptake of glucose into adipose tissues and striated muscle cells. HMGA1 was reported to bind to the IR promoter at an AT-rich in vitro, as shown by gel shift analysis [262]. Transfection experiments showed that the IR promoter is repressed when HMGA1 expression is silenced (in HepG2 hepatoma cells or IM-9 human lymphocytes). Silencing HMGA1 also decreases radio-labeled insulin binding to HepG2 and IM-9 cells, while overexpression of HMGA1 increases insulin binding [262]. Subsequent studies found that HMGA1 up-regulates IR expression as a complex with SP1 and C/EBPβ [263]. Co-immunoprecipitation and gel shift experiments show that HMGA1 interacts directly with SP1 and enhances SP1 binding to the IR promoters. Similarly, HMGA1 interacts with C/EBPβ and enhances its binding to the IR promoter. In addition, co-transfection of plasmids expressing HMGA1 and SP1 activates the IR promoter, while transfection of HMGA1 or SP1 alone results in only modest activation of the promoter. Consistent with these findings, Hmga1-deficient mice were reported to have a marked reduction in Ir expression and phosphorylation in the major downstream targets of the insulin pathway resulting in impaired insulin signaling and decreased insulin secretion. This results in a type 2-like diabetes phenotype with defects in pancreatic beta-cell insulin secretion. As above, four individuals were reported with genetic variants resulting in decreased HMGA1 expression have a type 2 diabetes phenotype [35, 264]. In addition, alterations in expression of HMGA1 mediated by expression of the HMGA1 pseudogene can also lead to post-transcriptional silencing of HMGA1 and has been implicated in type 2 diabetes in humans [37, 264]. In contrast to type-2 diabetes in humans, however, the Hmga1-deficient mice have an increased sensitivity to insulin, requiring higher baseline serum glucose infusions to maintain euglycemia [36].
10.15.2. Retinol Binding Protein 4 (RBP4)
Further studies of HMGA1 and glucose metabolism reveal that HMGA1 is required for both the basal and cAMP-induced expression of the gene encoding the retinol-binding protein 4 (RBP4), a protein secreted by adipocytes (thus designated an adipokine) that is involved in glucose metabolism and insulin resistance [36]. In transfection experiments in HepG2 cells, HMGA1 was reported to transactivate the RPB4 promoter, while inhibiting HMGA1 expression represses the RPB4 promoter [36, 37]. HMGA1 binds to this promoter in vivo, as shown by chromatin immunoprecipitation experiments. In addition, the Hmga1-deficient mice were reported to have reduced expression of Rpb4 mRNA and lower serum Rpb4 protein levels [36]. In cultured cells, both HMGA1 and RBP4 expression are induced by increases in intracellular cAMP, which is known to induce RBP4 expression. This study also showed that the intraperitoneal injection of glucagon, a hormone that increases intracellular cAMP, results in increased expression of Hmga1 and Rbp4 in the liver and fat of wildtype mice. In contrast, the induction of Rbp4 was attenuated in the Hmga1-deficient mice, providing further evidence that HMGA1 induces RBP4 expression. The Hmga1-deficient mice were also reported to have lower levels of serum free fatty acids, which could contribute to increased sensitivity to insulin. To further assess the role of HMGA1 and RBP4 in glucose homeostasis, the Hmga1-deficient mice were injected with recombinant human RBP4, which attenuated the fall in glucose following insulin administration [36]. Taken together, these findings indicate a potential role for HMGA1 in the regulation of RBP4 and other genes involved in glucose metabolism. By directly regulating RBP4 expression, HMGA1 could enhance peripheral insulin sensitivity and ensure adequate glucose uptake in skeletal muscle.
10.15.3. Glut4
As noted previously, the Hmga1-deficient mice were reported to have an increased sensitivity to insulin, despite decreased expression of Ir. To investigate the mechanisms underlying the increased insulin sensitivity, expression of the glucose transporter, Glut4, was assessed in the skeletal muscle and fat of the Hmga1-deficient mice [35]. Glut4 mRNA and protein were reported to be increased in quantitative RT-PCR and Western assays, respectively. The Akt2 protein kinase is a downstream target of PI3-kinase that is required for insulin-induced translocation of Glut4 to the plasma membrane. Phospospho-Akt protein was reported to be increased in the skeletal muscle and adipose tissues of Hmga1-deficient mice compared to wildtype mice and paralleled that of Glut4 protein [263].
10.15.4. GLUT3
HMGA1 also induces expression of the gene encoding the GLUT3 glucose transporter and promotes aerobic glycolysis in colorectal cancer cells [264]. Chromatin immunoprecipitation in human colon cancer (HCT116 and LoVo) showed that HMGA1 binds directly to the GLUT3 promoter and transient transfection experiments demonstrate that HMGA1 induces expression of this promoter [264]. In primary human colorectal tumors, there was a positive correlation between HMGA1 and GLUT3 expression. This study also found that Caveolin-1 induces HMGA1 expression in colon cancer cells. Interestingly, CAV-1 and CAV-2 appear to be repressed by HMGA1 in an ovarian cancer cell line [as discussed earlier; see ref. 222]. It is possible that there is a negative feedback loop whereby HMGA1 inhibits CAV expression is some cancer cells, while Caveolin could stimulate HMGA1 expression in colon cancer and other tumors. Additional work is needed to dissect the role of these pathways in cancer. Nonetheless, these findings further underscore an important role for HMGA1 in regulating glucose metabolism that could be relevant to abnormal tumor metabolism.
10.15.5. Insulin-like Growth Factor Binding Protein-1 and -3 (IGFBP-1 and IGFBP-3)
IGF binding proteins (IGFBPs) bind to insulin-like growth factors (IGFs) and regulate their bioavailability and distribution. IGF/IGFBPs function, not only in maintaining glucose homeostasis, but recent studies point to a key role for IGFs in cancer [265]. In experimental models, IGFs promote cell proliferation and other oncogenic pathways [265]. In humans, epidemiologic studies suggest that obesity and type 2 diabetes are associated with an increased risk for diverse cancers. HMGA1 also affects IGF levels by regulating genes encoding IGF binding protein [266]. For example, HMGA1 was reported to bind to the insulin response element (IRE) to induce expression of both the IGFBP-1 and IFGBP-3 genes [266, 267]. The IRE is a critical element in hormonal regulation of many genes and HMGA1 binds to this element together with one or more hepatic nuclear factor proteins. Gel shift and DNase protection assays with recombinant proteins showed that HMGA binds to the IRE in the IGFBP-1 promoter and suggest that hepatic nuclear factor 3 proteins (HNF3α and HNF3β) form a complex with HMGA1 [266]. Because the IGFBP-1 and IGFBP-3 genes are contiguously arranged on chromosome 7, regulation of both of these genes by HMGA1 was also investigated. Studies using gel shift and chromatin immunoprecipitation analyses in HepG2 cells found that HMGA1 interacts with C/EBP, SP1, and HNF-1α when binding to the IGFBP-1 and IGFBP-3 promoters, suggesting that it orchestrates the assembly of these complexes at the IREs [266]. These findings point to yet another pathway regulated by HMGA1 that is involved in glucose metabolism, in this case by inducing expression of proteins that bind to and decrease functional IGF [266, 267].
10.15.6. Mac25, Insulin-Like Growth Factor-Binding Protein-Related Protein-7 or IGFBP-7
The Mac25 or IGFBP-7 gene was also reported to be induced by Hmga1 in mESCs compared to mESCs deficient in Hmga1 [198]. Increased expression of IGFBP-7 in the Hmga1-expressing cells was detected in both microarray gene expression and qRT-PCR experiments [198]. In colorectal cancer cell lines, both IGFBP-3 and IGFBP-7 are linked to proliferation and metastatic progression [268]. Silencing of IGFBP-7 results in decreased proliferation, colony formation, and metastatic progression to the liver. These results further suggest that HMGA1 and IGF pathways play an important role in cancer. Because prior studies found that HMGA1 regulates the IR [35, 262, 263], OBESE [244], RBP4 [36], IGFBP-1 [35, 267], IGFBP-3 [268], IGFBP-7 [198] and GLUT4 [263, 269] genes, it is likely that HMGA1 regulates additional genes important in energy utilization, including glucose and fat metabolism. Additional studies are needed to better elucidate its role in metabolism, particularly given recent evidence that alterations in metabolic pathways are critical adaptations in cancer and stem cells that could be targeted in cancer therapy [148–158].
10.16. Other Genes Involved in Metabolism
10.16.1. Cubulin
The Cubulin gene encodes the intrinsic factor cobalamin receptor which acts as a receptor for the intrinsic factor-vitamin B12 complex. Cubulin was reported to be repressed by Hmga1 in wildtype mESCs compared to cells deficient in Hmga1, as demonstrated by gene expression profile analysis and RT-PCR experiments [198]. Intrinsic factor-vitamin B12 is involved in purine metabolism and DNA synthesis.
10.16.2. Parathyroid Hormone Receptor (Pthr)
The Pthr gene encodes a receptor for parathyroid hormone and for parathyroid hormone-related peptide [175]. This receptor is a member of calcium-sensing receptor (CaSR) family of G protein coupled receptors that is activated by elevated levels of calcium and other divalent cations. The CaSR family functions as a multimodal nutrient sensor to maintain calcium homeostasis by sensing extracellular levels of calcium. The receptors can also be activated by other compounds, including polyamines such as spermine, which are increased in some cancers. A prior study in thyroid cancer cells found that PTHR promotes cellular adhesion and migration by coupling to members of the integrin family of extracellular matrix proteins [270]. This gene was reported to be induced by Hmga1 in wildtype mESCs compared to mESCs deficient in Hmga1 [198]. Although it is not yet known whether Hmga1 directly regulates Pthr expression, this pathway could promote migration and metastatic progression in cancer.
10.17. Development/Cell Fate, Stem Cell & Epithelial-Mesenchymal Transition Genes
10.17.1. Jagged-1 (JAG1)
Notch signaling is an evolutionarily conserved, signaling pathway essential for proper development of diverse organisms, from insects and nematodes to mammals [271–273]. The JAG1 gene encodes Jagged-1, a ligand for the NOTCH1 receptor. Jagged-1 binding to Notch1 leads to a cascade of proteolytic cleavage events that result in the translocation of the intracellular part of the Notch receptor to the nucleus where it binds to DNA to regulate expression of transcription factors involved in cell fate and morphogenesis [271–273]. In humans, mutations in JAG1 cause Alagille syndrome, a disorder characterized by development abnormalities affecting which heart, liver, skeleton, and central nervous system [271]. JAG1 is downstream of canonical WNT signaling and important in normal intestinal stem cell function. JAG1 is also frequently up-regulated in diverse cancers and thought to contribute to tumor progression and angiogenesis [273]. JAG1 was discovered in gene expression profile analysis from MCF-7 cells engineered to overexpress HMGA1a or HMGA1b [29]. Although further studies are needed to determine if HMGA1 directly regulates JAG1 expression, this pathway could represent a network involved in both development and cancer.
10.17.2. VIMENTIN, E-CADHERIN
When tumor cells metastasize, emerging evidence suggests that they usurp stem cell transcription networks that mediate an epithelial-mesenchymal transition (EMT) and other changes necessary for tumor progression [58, 68, 117]. In poorly differentiated colon cancer cells (HCT116 and SW480), HMGA1 induces expression of EMT genes, including VIMENTIN and TWIST (described above under Transcription Factors), while it represses the epithelial gene, E-CADHERIN. TWIST, along with SNAIL and SLUG, were previously shown to be up-regulated by the HMGA2 family member in mouse mammary epithelial cells induced to undergo EMT [274]. Since HMGA1 and HMGA2 induce similar phenotypes when ectopically expressed in cultured cells [28], it is plausible that they share a subset of transcriptional targets. VIMENTIN is also up-regulated with the progression from an epithelial to mesenchymal transition [29]. As noted above, silencing HMGA1 in the high-grade colon cancer cells blocks stem cell properties (three dimensional colonosphere formation and limiting dilution tumorigenesis) as well as oncogenic/tumor progression phenotypes (anchorage-independent cell growth, migration, invasion, and metastatic progression) [68]. These results suggest that HMGA1 promotes tumor progression through transcriptional networks that facilitate EMT, metastatic progression, and a stem-like state.
10.18. Spindle-Assembly Checkpoint (SAC) Genes
10.18.1. BUB1, Bub1b, Ttk, Mad2/1
The Bub1b gene was first discovered to be induced by HMGA1 in an Hmga1a transgenic model of leukemia [108]. BUB1 proteins function in activation of mitotic spindle check points. In addition, BUB1 was identified as a “gene signature” or set of genes associated with poor outcomes and early death in diverse cancers. This 11 gene signature was therefore dubbed the “death from cancer” signature, and was derived by identifying common genes expressed in normal stem cells and metastatic lesions in a transgenic mouse model of prostate cancer [275, 276]. Interestingly, the expression of this signature correlates with highly malignant tumors, metastatic progression and poor responses to therapy. The HMGA1-BUB1b pathway further links HMGA1 to transcriptional networks in tumors with poor outcomes, stem cells, and possibly to chromosomal integrity given the role of BUB1 in mitosis. More recent studies reported that multiple genes involved in the spindle assembly checkpoint, including Bub1, Bub1b, Ttk, Mad2/1, are up-regulated by Hmga1b [277]. This group reported that ectopic expression of HMGA1 increased expression of all of these genes in colon cancer cell lines and protein levels in mouse fibroblasts and a human colon cancer cell line. They also reported that HMGA1 binding is enriched at the promoters for these genes by chromatin immunoprecipitation in colon cancer cell lines and reporter gene activation downstream of the promoter regions was enhanced by HMGA1 in transfection experiments. In addition, aberrant mitosis in MEFs deficient in Hmga1 was reported. In data from The Cancer Genome Atlas, they found that HMGA1 gene expression correlates with that of spindle-assembly checkpoint genes in colorectal cancer. Together, these data indicate that HMGA1 may be an important regulator of mitosis in cancer cells through these genes.
10.19. Extracellular Matrix and Cell Structure Genes
10.19.1. Collagen Pro-Alpha1 Type 1 or Col1a1
Col1a1 was reported to be induced by Hmga1 in mESCs, and increased levels in Hmga1-expressing cells were found in microarray gene expression analyses and by qRT-PCR studies [198]. Col1a1 encodes the major component of type 1 collagen, which is found in most connective tissues, including cartilage. A study of colorectal cancer identified type 1 collagen at the invasive front of colorectal cancer in histopathologic studies [278]. This group also found that culturing colorectal cancer cells in the presence of type 1 collagen induced an epithelial-to-mesenchymal (EMT) transition and stem cell markers. These findings suggest that the HMGA1-COLIA1 pathway could promote tumor progression, EMT, and a stem like phenotype.
10.19.2. Sparc, Secreted Protein, Acidic, Cysteine-Rich (or Osteonectin)
Sparc, secreted protein, acidic, cysteine-rich (or osteonectin) was reported to be repressed by Hmga1 in wildtype mESCs compared to mESCs deficient in Hmga1 [198] Repression of Sparc was found by microarray gene expression studies and qRT-PCR analysis [198]. The protein encoded by Sparc functions in regulating cell growth by mediating interactions with the extracellular matrix and cytokines. SPARC has been associated with both tumor suppression and metastasis, the latter of which is thought to relate to its role in changing cell shape which could promote tumor cell invasion [175].
10.19.3. Cyr61, Cysteine-Rich Angiogenic Inducer 61
The same group reported that Cyr61 (cysteine-rich angiogenic inducer 61) is induced by Hmga1. This gene encodes a growth-factor inducible protein that promotes endothelial cell adhesion and angiogenesis. It was identified in gene expression profile analysis of mESCs wildtype for Hmga1 compared to mESCs deficient in Hmga1, and confirmed by qRT-PCR [198]. Cyr61 is a secreted protein that promotes proliferation, chemotaxis, angiogenesis, and endothelial cell adhesion by up-regulating genes involved in inflammation, angiogenesis, matrix remodeling and EMT [279].
11. AREAS FOR FUTURE INVESTIGATION
Although prior studies have uncovered downstream genes regulated by HMGA1, it is likely that these results provide only a glimpse of HMGA1 transcriptional networks in tumorigenesis and normal embryogenesis. Given the central role for HMGA1 in cancer and development, a global investigation of HMGA1 function using state-of-the-art technology with global chromatin immunoprecipitation coupled with sequencing technology in different cellular contexts is needed to further elucidate the molecular pathways induced by HMGA1 in these settings. Of note, many HMGA1 targets reported to date include NF-κB regulatory elements in their promoter regions near the HMGA1 DNA binding sites, suggesting that HMGA1 and NF-κB function together to regulate gene expression [23]. In addition, many NF-κB-HMGA1 gene targets encode important proteins involved in inflammatory pathways associated with cancer. It will be important to determine if NF-κB is required for HMGA1 function in cancer, particularly because NF-κB inhibitors are available and could be exploited in cancer therapy. Both HMGA1 overexpression and NF-κB activation are prominent in diverse high-grade cancers and further efforts to target these pathways are likely to advance therapy for multiple tumor types. Because HMGA1 is a chromatin remodeling protein that modulates histone and transcription factor binding to chromatin, it is likely that it plays an important role in multiple epigenetic modifications that occur in cancer and development, and this area has not been well-studied.
Studies are also needed to identify, not only transcriptional gene targets, but also noncoding RNAs, regulated by HMGA1. MicroRNAs are non-protein coding small RNA molecules that function as critical regulators of mammalian gene expression [27, 280]. Recent studies indicate that aberrant expression of miRNAs plays a causal role in tumorigenesis. In fact, most high-grade tumors are characterized by global repression of miRNAs and down-regulation of tumor suppressor miRNAs is emerging as a key feature in refractory tumors. Thus, it will be important to identify miRNAs that are regulated by HMGA1 proteins as well as miRNAs that modulate HMGA1 expression. Such studies should also reveal potential therapeutic targets relevant to diverse tumors. Emerging evidence indicates that pseudogenes play a role in regulating gene expression and HMGA1 pseudogenes alter HMGA1 expression and function [37]. Further studies to identify long non-coding RNAs that affect HMGA1 expression or function should provide important insight on HMGA1 in cancer and development. Similarly, it is possible that noncoding regions of HMGA1 could regulate expression of microRNAs, thereby imparting functions independent of the protein coding domains.
Recent studies have uncovered important metabolic adaptations in tumor cells that could be exploited in therapy [148–158]. Indeed, most, if not all, oncogenic proteins and tumor suppressor appear to alter or “reprogram” tumor cell metabolism. As noted, the cMYC oncogenic transcription factor metabolically reprograms cells, forcing them to become “addicted to glucose” via aerobic glycolysis (also known as the Warburg effect) and glutaminolysis [154–158]. Interestingly, the dependence upon aerobic glycolysis is also characteristic of early embryogenesis up to the 16 cell stage, after which both aerobic glycolysis and oxidative phosphorylation occur concurrently. The rapid growth of early embryos could recapitulate growth patterns observed in some cancers [155]. Precisely how oncogenic pathways contribute to metabolic reprogramming is an area of active investigation that is likely to uncover new approaches to target cancer cells. Recent studies also suggest that tumor cells can reprogram the surrounding stroma, and conversely, the tumor stroma can release factors and compounds that help to reprogram the tumor cells themselves. This complexity and how HMGA1 affects tumor and stromal cell metabolism requires further study.
As outlined here, prior studies found that HMGA1 induces expression of many nuclear genes involved in metabolism, including the insulin receptor [263, 264], RBP4 [36, 37], GLUT3 [269], IGFBP-1, IGFBP-3 [266, 267], and IGFBP-7 [198], and leptin/obese [103, 245] genes, indicating that it is involved in both glucose and fat metabolism. Reports of mice deficient in Hmga1 describe some features present in type 2 diabetes and a small number of patients with type 2 diabetes have genetic lesions that interfere with HMGA1 expression, further implicating HMGA1 in glucose metabolism [35]. In addition, HMGA1 induces expression of mitochondrial genes [32–34]; how this affects normal and tumor metabolism is still incompletely understood. Thus, studies are needed to dissect the role of HMGA1 in metabolism in tumors, stroma, and other cells within the tumor microenvironment.
HMGA1 is also enriched in human embryonic stem cells [58, 97, 98], adult stem cells, such as hematopoietic stem cells [96, 97], intestinal stem cells [120], and high-grade or poorly differentiated tumors [27, 281, 282], suggesting that it plays a role in driving the undifferentiated, stem-like state. In addition, its high expression during embryogenesis with absent or markedly decreased levels in differentiated, adult tissues further implicates HMGA1 in enforcing a stem-like state [98]. Although numerous studies in tumors demonstrate a positive correlation between HMGA1 and poor differentiation status, there are few studies to elucidate its function in embryonic stem cells. Recent studies highlight a role for HMGA1 as a master regulator in pathways involved in normal stem cells and epithelial-mesenchymal transition, a process important in development and tumor progression [29, 68, 117]. These studies found that HMGA1 orchestrates transcriptional networks and epigenetic pathways involved in maintaining an undifferentiated, pluripotent state in embryonic stem cells and induced pluripotent stem cells [98]. Further work is needed to better understand these networks, not only in normal development, but also in cancer. Such studies are likely to uncover novel roles that could be targeted in cancer and possibly regenerative medicine. Because emerging evidence indicates that stem cells and stem-like cancer cells have epigenetic alterations and changes in chromatin structure, a more detailed understanding of the role of HMGA1 in epigenetic modifications and nuclear chromatin structure should also enhance our understanding of HMGA1 in these settings.
We now know that HMGA1 also has a role in regulating genes involved in DNA repair, such as the XPA gene [112]. HMGA1 proteins can bind to 4-way DNA structures in vitro and these structures resemble the Holiday junctions formed as intermediate DNA structures during homologous recombination events [61]. HMGA1 proteins also enhance integration of the HIV genome into host DNA [116]. These activities suggest that HMGA1 could participate in translocation and recombination events. HMGA1 expression is associated with an increased number of unbalanced translocations in prostate cancer cells, suggesting that HMGA1 promotes such chromosomal aberrations [61]. Given the high levels of HMGA proteins in cancer cells and embryonic stem cells, coupled with its location at the nuclear matrix where recombination events occur, additional studies are needed to better understand the role of HMGA1 in chromosomal integrity and chromosomal structure.
Another largely unanswered question in the field is how HMGA1 expression is regulated, both during development, postnatally, and in cancer. Although previous studies found that HMGA1 is up-regulated by hypoxia [256], growth factors stimulation [44] oncogenic factors, such as cMyc [88, 204, 283] AP1 [142, 283, 284] or Wnt/Tcf-4 [285], HMGA1 expression is up-regulated in cancer cells lacking up-regulation of these pathways, suggesting that there are additional, unknown mechanisms that induce its expression. Preliminary data from our group indicates that HMGA1 expression is regulated by additional epigenetic mechanisms in stem cells and possibly cancer [97–98] and studies to elucidate these alterations might uncover additional targets for therapeutic interventions in tumors overexpressing HMGA1. For example, emerging data also implicates HMGA1 in microRNA networks and further dissection of miRNA regulation of HMGA1 should reveal additional pathways relevant to HMGA1 function. Expression of pseudogenes or noncoding regions of HMGA1 could also act as a sponge for microRNAs that normally down-regulate HMGA1 and thereby contribute to overexpression of HMGA1 [37]. This area requires further research because it could reveal additional pathways that could be exploited in cancer therapy.
A more detailed investigation of the function of each protein domain should provide insight into HMGA1 and novel therapeutic targets. The high degree of conservation of the domains that are required for DNA and protein-protein interactions suggest that these regions are key to its overall function. Indeed, a construct with mutations in the AT-hook DNA binding domains results in dominant-negative function in oncogenic assays [29, 98], which further underscores the importance of DNA binding for HMGA1 activity. While previous research has identified interacting proteins that are important in HMGA1 function [163], the discovery of additional protein-protein interactions should also advance our understanding of HMGA in cancer, development, and other cellular processes.
Substantial evidence also indicates that posttranslational modifications serve as important modulators of HMGA1 function and the specific regulators and sites of modification is an area of active investigation. Since their discovery in 1983 in HeLa cervical cancer cells, HMGA1a and HMGA1b were known to be highly phosphorylated [3, 286–291]. Indeed, HMGA1 proteins, along with histone H1, are among the most highly phosphorylated proteins in the nucleus of cancer cells [286–291]. Subsequent studies found that the AT-hook DNA binding domain sequence matches the consensus sequence for mammalian cyclin dependent kinase 1 or cdk1 (also known as cell division control protein 2 or cdc2 kinase) and phosphorylation of HMGA1 by cdk1 significantly decreases the binding of HMGA1 to DNA [289–291]. This kinase phosphorylates HMGA1b at serine-43 and serine-58, which impairs DNA binding. More detailed analysis showed that phosphorylation at additional sites (threonine-41 and threonine-67) also disrupts the interaction of the amino-terminal DNA binding domain of HMGA1b to the IFN-β promoter at PRDIII-I element [28]. The homeodomain-interacting protein kinase-2 (HIPK2) phosphorylates HMGA1a (at serine-35, threonine-52, and threonine-77) and HMGA1b (at threonine-41 and threonine-66) [291]. Although phosphorylation by HIPK is similar to that observed for cdk1, the kinases exhibit different site preferences for phosphorylation. Another study found that phosphorylation by HIPK reduces the binding affinity of HMGA1 for the promoter of target genes [290]. The effects of cdk1 were more pronounced than those observed for HIPK. Because cdk1 has a greater effect on DNA binding of the second AT-hook, these results suggest that the second AT-hook is a key determinant of DNA binding for HMGA1. Cdk1 also phosphorylates HMGA proteins at the serines near the carboxy-termini, resulting in a more compact protein that may have less oncogenic capabilities [290].
In addition to phosphorylation, acetylation and methylation are important posttranslational modifications that alter HMGA1 function [130, 292–295]. In fact, emerging evidence indicates that HMGA proteins undergo posttranslational modifications (methylation, acetylation and phosphorylation) similar to histones, suggesting that there is an HMGA1 posttranslational modification code (or “PTM code”) similar to the modifications that occur on histones. As noted above, HMGA1 proteins in plants and lower organisms are highly homologous to histone H1, suggesting that they evolved from the same protein. Thus, it is not surprising that similar PTMs are found in histones and HMGA1 proteins. As discussed above, acetylation of lysine-65 by PCAF/GCN5 stabilizes the HMGA1 enhanceosome at the IFN-β promoter, while acetylation of lysine-71 by CBP disrupts the enhanceosome and halts transcription of IFN-β [130]. More recently, both mono- and di-methylation of arginine-25 (in the first AT-hook) has been reported [293, 294]. Indeed, up to 50% of the HMGA1 was found to be monomethylated at arginine-25 in tumor cells. This posttranslational modification was also observed in cells undergoing apoptosis. Protein arginine methyltransferase 6 (PRMT6) was identified as the enzyme responsible for methylation of HMGA1 (at arginine-57 and arginine-59) in cells undergoing apoptosis [293, 294]. These two residues are embedded within the second AT-hook, a key DNA binding and protein-protein interacting domain. A more recent study utilizing innovative technology to monitor posttranslational modifications in intact proteins (called “top-down” proteomics because intact proteins rather than digested proteins are investigated) discovered a striking up-regulation of methylated forms of di- and tri-phosphorylated HMGA1a, but not of its splice variant, HMGA1b, in cancer cells (B16F10 melanoma cells and H1299 lung cancer cells) during stress-induced senescence [295]. Further studies are needed to elucidate the functional consequences of these posttranslational modifications.
CONCLUSION
In summary, numerous studies have established a clear link and functional role for HMGA1 over-expression and high-grade, aggressive cancers, from its initial discovery in the extraordinarily proliferative HeLa human cervical cancer cells [3] to the more recent identification of HMGA1 as a key transcription factor enriched in poorly differentiated cancers with adverse outcomes [55, 58, 74, 79]. Recent advances in technology to globally assess chromatin binding proteins and epigenetic modifications provides unprecedented opportunities to investigate HMGA1 in cancer and normal development. In addition, new approaches to elucidate metabolic adaptations to HMGA1 should advance our knowledge of tumor metabolism. The discovery of noncoding RNAs as key regulators opens up additional avenues of investigation relevant to HMGA1. Moreover, improvements in protein analytical approaches will lead to a better understanding of posttranslational modifications that regulate HMGA1 function as well as proteins that interact with HMGA1. Identification of additional cellular pathways and chromatin modifications involving HMGA1 promises to have a major impact in our ability to understand and treat cancer. These insights may also have relevance to diabetes, cardiac hypertrophy, Alzheimers disease, and other, as yet undiscovered, diseases linked to HMGA1.
The ultimate goal of further work in this field is to identify therapeutic strategies to target HMGA1 in human cancer [reviewed in reference 282]. While many studies cited here underscore the fundamental role of HMGA1 in tumor progression, there has been relatively little work focused on targeting HMGA1 in cancer [176]. To date, disrupting the function of oncogenic transcription factors has been challenging given the hurdles of delivery and specificity. Nonetheless, efforts to target critical downstream pathways have shown some promise in preclinical models, such as inhibitors to COX-2 [75, 256, 257] or STAT3 [91, 206, 296]. For this reason, a better understanding of the HMGA1 transcriptome and downstream pathways may reveal novel therapeutic targets. Alternatively, disrupting pathways that induce HMGA1 in cancer should be an effective strategy and studies to dissect these pathways are needed. In addition, further insight on protein-protein and protein-DNA interactions should reveal new interactions that could be targeted to disrupt HMGA1 chromatin binding and function. The identification of critical posttranslational modifications could also lead to the development of specific inhibitors that impair HMGA1 function. Advances in the stabilization and delivery of short hairpin RNAs or tumor suppressor microarrays that repress HMGA1 expression may also be effective in cancer therapy. Finally, a better understanding of HMGA1’s role in epigenetic alterations and global nuclear structure could also reveal approaches to interfere with its function in malignant transformation. Conversely, studies on HMGA1 in pluripotent stem cells suggest that enhancing its function will improve stem cell function and may be beneficial in regenerative medicine. Thus, studies to better elucidate the role of HMGA1 in adult stem cells and approaches to manipulate its levels in this setting are also warranted.
Acknowledgments
This review is dedicated to Dr. Barbara Migeon, a scientist, colleague, mentor and friend without parallel. TFS and LR conceived of this review; TFS and LR wrote the first draft; TFS, LX, TH, MK, CI, DR, LR helped to complete the Table and reviewed MatInspector results, TFS, LX, TH, Y-T C, LC, TN reviewed Gene Cards and helped to write the manuscript, LR edited and finalized the manuscript and figures.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.Goodwin G, Walker JH, Johns EW. The cell nucleus. New York: Academic Press; 1978. The high mobility group (HMG) nonhistone chromosomal proteins; pp. 181–219. [Google Scholar]
- 2.Goodwin GH, Sanders C, Johns EW. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur J Biochem. 1973;38:14–9. doi: 10.1111/j.1432-1033.1973.tb03026.x. [DOI] [PubMed] [Google Scholar]
- 3.Lund T, Holtlund J, Fredriksen M, Laland SG. On the presence of two new high mobility group-like proteins in Hela S3 cells. FEBS Lett. 1983;152:163–7. doi: 10.1016/0014-5793(83)80370-6. [DOI] [PubMed] [Google Scholar]
- 4.Giancotti V, Berlingieri MT, DiFiore PP, Fusco A, Vecchio G, Crane-Robinson C. Changes in nuclear proteins on transformation of rat epithelial thyroid cells by a murine sarcoma retrovirus. Cancer Res. 1985;45:6051–7. [PubMed] [Google Scholar]
- 5.Giancotti V, Pani B, D’Andrea P, et al. Elevated levels of a specific class of nuclear phosphoproteins in cells transformed with v-ras and v-mos oncogenes and by cotransfection with c-myc and polyoma middle t genes. EMBO J. 1987;6:1981–7. doi: 10.1002/j.1460-2075.1987.tb02461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson KR, Lehn DA, Elton TS, Barr PJ, Reeves R. Complete murine cdna sequence, genomic structure, and tissue expression of the high mobility group protein HMG-I(Y) J Biol Chem. 1988;263:18338–42. [PubMed] [Google Scholar]
- 7.Friedmann M, Holth LT, Zoghbi HY, Reeves R. Organization, inducible-expression and chromosome localization of the human HMG-I(Y) nonhistone protein gene. Nucleic Acids Res. 1993;21:4259–67. doi: 10.1093/nar/21.18.4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giancotti V, Buratti E, Perissin L, et al. Analysis of the HMGI nuclear proteins in mouse neoplastic cells induced by different procedures. Exp Cell Res. 1989;184:538–45. doi: 10.1016/0014-4827(89)90352-2. [DOI] [PubMed] [Google Scholar]
- 9.Bustin M, Lehn DA, Landsman D. Structural features of the hmg chromosomal proteins and their genes. Biochim Biophys Acta. 1990;1049:231–43. doi: 10.1016/0167-4781(90)90092-g. [DOI] [PubMed] [Google Scholar]
- 10.Giancotti V, Bandiera A, Buratti E, et al. Comparison of multiple forms of the high mobility group I proteins in rodent and human cells. Identification of the human High Mobility Group I-C protein. Eur J Biochem. 1991;198:211–6. doi: 10.1111/j.1432-1033.1991.tb16003.x. [DOI] [PubMed] [Google Scholar]
- 11.Manfioletti G, Rustighi A, Mantovani F, Goodwin GH, Giancotti V. Isolation and characterization of the gene coding for murine high-mobility-group protein HMGI-C. Gene. 1995;167:249–53. doi: 10.1016/0378-1119(95)00666-4. [DOI] [PubMed] [Google Scholar]
- 12.Manfioletti G, Giancotti V, Bandiera A, et al. cDNA cloning of the HMGI-C phosphoprotein, a nuclear protein associated with neoplastic and undifferentiated phenotypes. Nucleic Acids Res. 1991;19:6793–7. doi: 10.1093/nar/19.24.6793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solomon MJ, Strauss F, Varshavsky A. A mammalian high mobility group protein recognizes any stretch of six A. T base pairs in duplex DNA. Proc Natl Acad Sci U S A. 1986;83:1276–80. doi: 10.1073/pnas.83.5.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reeves R, Nissen MS. The AT-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J Biol Chem. 1990;265:8573–82. [PubMed] [Google Scholar]
- 15.Geierstanger BH, Volkman BF, Kremer W, Wemmer DE. Short peptide fragments derived from HMG-I/Y proteins bind specifically to the minor groove of DNA. Biochemistry. 1994;33:5347–55. doi: 10.1021/bi00183a043. [DOI] [PubMed] [Google Scholar]
- 16.Maher JF, Nathans D. Multivalent DNA-binding properties of the HMG-1 proteins. Proc Natl Acad Sci U S A. 1996;93:6716–20. doi: 10.1073/pnas.93.13.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bustin M, Reeves R. High-mobility-group chromosomal proteins: Architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 18.Banks GC, Mohr B, Reeves R. The HMG-I(Y) AT-hook peptide motif confers DNA-binding specificity to a structured chimeric protein. J Biol Chem. 1999;274:16536–44. doi: 10.1074/jbc.274.23.16536. [DOI] [PubMed] [Google Scholar]
- 19.Bustin M. Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol Cell Biol. 1999;19:5237–46. doi: 10.1128/mcb.19.8.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reeves R, Beckerbauer L. HMGI/Y proteins: Flexible regulators of transcription and chromatin structure. Biochim Biophys Acta. 2001;1519:13–29. doi: 10.1016/s0167-4781(01)00215-9. [DOI] [PubMed] [Google Scholar]
- 21.Hock R, Furusawa T, Ueda T, Bustin M. HMG chromosomal proteins in development and disease. Trends Cell Biol. 2007;17:72–9. doi: 10.1016/j.tcb.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 23.Resar LM. The high mobility group A1 gene: Transforming inflammatory signals into cancer? Cancer Res. 2010;70:436–9. doi: 10.1158/0008-5472.CAN-09-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah SN, Resar LM. High mobility group A1 and cancer: Potential biomarker and therapeutic target. Histol Histopathol. 2012;27:567–79. doi: 10.14670/HH-27.567. [DOI] [PubMed] [Google Scholar]
- 25.Di Cello F, Hillion J, Hristov A, et al. HMGA2 participates in transformation in human lung cancer. Mol Cancer Res. 2008;6:743–50. doi: 10.1158/1541-7786.MCR-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hristov AC, Cope L, Reyes MD, et al. HMGA2 protein expression correlates with lymph node metastasis and increased tumor grade in pancreatic ductal adenocarcinoma. Mod Pathol. 2009;22:43–9. doi: 10.1038/modpathol.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Resar LM, Brodsky RA. “Let”-ing go with clonal expansion? Blood. 2011;117:5788–90. doi: 10.1182/blood-2011-04-346668. [DOI] [PubMed] [Google Scholar]
- 28.Wood LJ, Maher JF, Bunton TE, Resar LM. The oncogenic properties of the HMG-I gene family. Cancer Res. 2000;60:4256–61. [PubMed] [Google Scholar]
- 29.Reeves R, Edberg DD, Li Y. Architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Mol Cell Biol. 2001;21:575–94. doi: 10.1128/MCB.21.2.575-594.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reeves R. Molecular biology of HMGA proteins: Hubs of nuclear function. Gene. 2001;277:63–81. doi: 10.1016/s0378-1119(01)00689-8. [DOI] [PubMed] [Google Scholar]
- 31.Moliterno AR, Resar LM. AKNA: Another AT-hook transcription factor “hooking-up” with inflammation. Cell Res. 2011;21:1528–30. doi: 10.1038/cr.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dement GA, Treff NR, Magnuson NS, Franceschi V, Reeves R. Dynamic mitochondrial localization of nuclear transcription factor HMGA1. Exp Cell Res. 2005;307:388–401. doi: 10.1016/j.yexcr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Dement GA, Maloney SC, Reeves R. Nuclear HMGA1 nonhistone chromatin proteins directly influence mitochondrial transcription, maintenance, and function. Exp Cell Res. 2007;313:77–87. doi: 10.1016/j.yexcr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao L, Wertzler KJ, Maloney SC, Wang Z, Magnuson NS, Reeves R. HMGA1 levels influence mitochondrial function and mitochondrial DNA repair efficiency. Mol Cell Biol. 2009;29:5426–40. doi: 10.1128/MCB.00105-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foti D, Chiefari E, Fedele M, et al. Lack of the architectural factor HMGA1 causes insulin resistance and diabetes in humans and mice. Nat Med. 2005;11:765–73. doi: 10.1038/nm1254. [DOI] [PubMed] [Google Scholar]
- 36.Chiefari E, Paonessa F, Iiritano S, et al. The cAMP-HMGA1-RBP4 system: A novel biochemical pathway for modulating glucose homeostasis. BMC Biol. 2009;7:24. doi: 10.1186/1741-7007-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiefari E, Iiritano S, Paonessa F, et al. Pseudogene-mediated posttranscriptional silencing HMGA1 can result in insulin resistance and type 2 diabetes. Nat Commun. 2010;1:1–7. doi: 10.1038/ncomms1040. [DOI] [PubMed] [Google Scholar]
- 38.Semple RK. From bending DNA to diabetes: The curious case of HMGA1. J Biol. 2009;8:64. doi: 10.1186/jbiol164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manabe T, Katayama T, Sato N, et al. Induced HMGA1a expression causes aberrant splicing of presenilin-2 pre-mRNA in sporadic alzheimer’s disease. Cell Death Differ. 2003;10:698–708. doi: 10.1038/sj.cdd.4401221. [DOI] [PubMed] [Google Scholar]
- 40.Katayama T, Imaizumi K, Manabe T, Hitomi J, Kudo T, Tohyama M. Induction of neuronal death by ER stress in alzheimer’s disease. J Chem Neuroanat. 2004;28:67–78. doi: 10.1016/j.jchemneu.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Manabe T, Ohe K, Katayama T, et al. HMGA1a: Sequence-specific RNA-binding factor causing sporadic alzheimer’s disease-linked exon skipping of presenilin-2 pre-mRNA. Genes Cells. 2007;12:1179–91. doi: 10.1111/j.1365-2443.2007.01123.x. [DOI] [PubMed] [Google Scholar]
- 42.Ohe K, Mayeda A. HMGA1a trapping of u1 snrnp at an authentic 5′ splice site induces aberrant exon skipping in sporadic Alzheimer’s disease. Mol Cell Biol. 2010;30:2220–8. doi: 10.1128/MCB.00114-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giancotti V, Bandiera A, Ciani L, et al. High-mobility-group (HMG) proteins and histone h1 subtypes expression in normal and tumor tissues of mouse. Eur J Biochem. 1993;213:825–32. doi: 10.1111/j.1432-1033.1993.tb17825.x. [DOI] [PubMed] [Google Scholar]
- 44.Lanahan A, Williams JB, Sanders LK, Nathans D. Growth factor-induced delayed early response genes. Mol Cell Biol. 1992;12:3919–29. doi: 10.1128/mcb.12.9.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiappetta G, Tallini G, De Biasio MC, et al. Detection of high mobility group I HMGI(Y) protein in the diagnosis of thyroid tumors: HMGI(Y) expression represents a potential diagnostic indicator of carcinoma. Cancer Res. 1998;58:4193–8. [PubMed] [Google Scholar]
- 46.Chiappetta G, Bandiera A, Berlingieri MT, et al. The expression of the high mobility group HMGI (Y) proteins correlates with the malignant phenotype of human thyroid neoplasias. Oncogene. 1995;10:1307–14. [PubMed] [Google Scholar]
- 47.Dal Cin P, Fusco A, Belge G, et al. Involvement of the HMGI(Y) gene in a microfollicular adenoma of the thyroid. Genes Chromosomes Cancer. 1999;24:286–9. doi: 10.1002/(sici)1098-2264(199903)24:3<286::aid-gcc16>3.3.co;2-5. [DOI] [PubMed] [Google Scholar]
- 48.Czyz W, Balcerczak E, Jakubiak M, Pasieka Z, Kuzdak K, Mirowski M. Hmgi(y) gene expression as a potential marker of thyroid follicular carcinoma. Langenbecks Arch Surg. 2004;389:193–7. doi: 10.1007/s00423-004-0479-6. [DOI] [PubMed] [Google Scholar]
- 49.Sarhadi VK, Wikman H, Salmenkivi K, et al. Increased expression of high mobility group A proteins in lung cancer. J Pathol. 2006;209:206–12. doi: 10.1002/path.1960. [DOI] [PubMed] [Google Scholar]
- 50.Hillion J, Wood LJ, Mukherjee M, et al. Upregulation of MMP-2 by HMGA1 promotes transformation in undifferentiated, large-cell lung cancer. Mol Cancer Res. 2009;7:1803–12. doi: 10.1158/1541-7786.MCR-08-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kettunen E, Anttila S, Seppanen JK, et al. Differentially expressed genes in nonsmall cell lung cancer: Expression profiling of cancer-related genes in squamous cell lung cancer. Cancer Genet Cytogenet. 2004;149:98–106. doi: 10.1016/S0165-4608(03)00300-5. [DOI] [PubMed] [Google Scholar]
- 52.Holth LT, Thorlacius AE, Reeves R. Effects of epidermal growth factor and estrogen on the regulation of the HMG-I/Y gene in human mammary epithelial cell lines. DNA Cell Biol. 1997;16:1299–309. doi: 10.1089/dna.1997.16.1299. [DOI] [PubMed] [Google Scholar]
- 53.Liu WM, Guerra-Vladusic FK, Kurakata S, Lupu R, Kohwi-Shigematsu T. HMG-I(Y) recognizes base-unpairing regions of matrix attachment sequences and its increased expression is directly linked to metastatic breast cancer phenotype. Cancer Res. 1999;59:5695–703. [PubMed] [Google Scholar]
- 54.Dolde CE, Mukherjee M, Cho C, Resar LM. HMG-I/Y in human breast cancer cell lines. Breast Cancer Res Treat. 2002;71:181–91. doi: 10.1023/a:1014444114804. [DOI] [PubMed] [Google Scholar]
- 55.Flohr AM, Rogalla P, Bonk U, et al. High mobility group protein HMGA1 expression in breast cancer reveals a positive correlation with tumour grade. Histol Histopathol. 2003;18:999–1004. doi: 10.14670/HH-18.999. [DOI] [PubMed] [Google Scholar]
- 56.Chiappetta G, Botti G, Monaco M, et al. HMGA1 protein overexpression in human breast carcinomas: Correlation with erbb2 expression. Clin Cancer Res. 2004;10:7637–44. doi: 10.1158/1078-0432.CCR-04-0291. [DOI] [PubMed] [Google Scholar]
- 57.Zou Y, Wang Y. Mass spectrometric analysis of high-mobility group proteins and their post-translational modifications in normal and cancerous human breast tissues. J Proteome Res. 2007;6:2304–14. doi: 10.1021/pr070072q. [DOI] [PubMed] [Google Scholar]
- 58.Ben-Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chiappetta G, Ottaiano A, Vuttariello E, et al. HMGA1 protein expression in familial breast carcinoma patients. Eur J Cancer. 2010;46:332–9. doi: 10.1016/j.ejca.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 60.Takaha N, Resar LM, Vindivich D, Coffey DS. High mobility group protein HMGI(Y) enhances tumor cell growth, invasion, and matrix metalloproteinase-2 expression in prostate cancer cells. Prostate. 2004;60:160–7. doi: 10.1002/pros.20049. [DOI] [PubMed] [Google Scholar]
- 61.Takaha N, Hawkins AL, Griffin CA, Isaacs WB, Coffey DS. High mobility group protein I(Y): A candidate architectural protein for chromosomal rearrangements in prostate cancer cells. Cancer Res. 2002;62:647–51. [PubMed] [Google Scholar]
- 62.Takeuchi I, Takaha N, Nakamura T, et al. High mobility group protein at-hook 1 (HMGA1) is associated with the development of androgen independence in prostate cancer cells. Prostate. 2012;72:1124–32. doi: 10.1002/pros.22460. [DOI] [PubMed] [Google Scholar]
- 63.Fedele M, Bandiera A, Chiappetta G, et al. Human colorectal carcinomas express high levels of high mobility group HMGI (Y) proteins. Cancer Res. 1996;56:1896–901. [PubMed] [Google Scholar]
- 64.Kim DH, Park YS, Park CJ, et al. Expression of the HMGI(Y) gene in human colorectal cancer. Int J Cancer. 1999;84:376–80. doi: 10.1002/(sici)1097-0215(19990820)84:4<376::aid-ijc8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 65.Chiappetta G, Manfioletti G, Pentimalli F, et al. High mobility group HMGI(Y) protein expression in human colorectal hyperplastic and neoplastic diseases. Int J Cancer. 2001;91:147–51. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1033>3.3.co;2-m. [DOI] [PubMed] [Google Scholar]
- 66.Cleynen I, Huysmans C, Sasazuki T, Shirasawa S, Van de Ven W, Peeters K. Transcriptional control of the human high mobility group a1 gene: Basal and oncogenic ras-regulated expression. Cancer Res. 2007;67:4620–9. doi: 10.1158/0008-5472.CAN-06-4325. [DOI] [PubMed] [Google Scholar]
- 67.Grade M, Hormann P, Becker S, et al. Gene expression profiling reveals a massive, aneuploidy-dependent transcriptional deregulation and distinct differences between lymph node-negative and lymph node-positive colon carcinomas. Cancer Res. 2007;67:41–56. doi: 10.1158/0008-5472.CAN-06-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belton A, Gabrovsky A, Bae YK, et al. HMGA1 induces intestinal polyposis in transgenic mice and drives tumor progression and stem cell properties in colon cancer cells. PLoS One. 2012;7:e30034. doi: 10.1371/journal.pone.0030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abe N, Watanabe T, Masaki T, et al. Pancreatic duct cell carcinomas express high levels of high mobility group I(Y) proteins. Cancer Res. 2000;60:3117–22. [PubMed] [Google Scholar]
- 70.Liau SS, Jazag A, Whang EE. HMGA1 is a determinant of cellular invasiveness and in vivo metastatic potential in pancreatic adenocarcinoma. Cancer Res. 2006;66:11613–22. doi: 10.1158/0008-5472.CAN-06-1460. [DOI] [PubMed] [Google Scholar]
- 71.Liau SS, Jazag A, Ito K, Whang EE. Overexpression of HMGA1 promotes anoikis resistance and constitutive akt activation in pancreatic adenocarcinoma cells. Br J Cancer. 2007;96:993–1000. doi: 10.1038/sj.bjc.6603654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liau SS, Rocha F, Matros E, Redston M, Whang E. High mobility group at-hook 1 (HMGA1) is an independent prognostic factor and novel therapeutic target in pancreatic adenocarcinoma. Cancer. 2008;113:302–14. doi: 10.1002/cncr.23560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liau SS, Whang E. Hmga1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin Cancer Res. 2008;14:1470–7. doi: 10.1158/1078-0432.CCR-07-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hristov AC, Cope L, Di Cello F, et al. HMGA1 correlates with advanced tumor grade and decreased survival in pancreatic ductal adenocarcinoma. Mod Pathol. 2010;23:98–104. doi: 10.1038/modpathol.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tesfaye A, Di Cello F, Hillion J, et al. The high-mobility group A1 gene up-regulates cyclooxygenase 2 expression in uterine tumorigenesis. Cancer Res. 2007;67:3998–4004. doi: 10.1158/0008-5472.CAN-05-1684. [DOI] [PubMed] [Google Scholar]
- 76.Bandiera A, Bonifacio D, Manfioletti G, et al. Expression of HMGI(Y) proteins in squamous intraepithelial and invasive lesions of the uterine cervix. Cancer Res. 1998;58:426–31. [PubMed] [Google Scholar]
- 77.Takaha N, Sowa Y, Takeuchi I, Hongo F, Kawauchi A, Miki T. Expression and role of HMGA1 in renal cell carcinoma. J Urol. 2012;187:2215–22. doi: 10.1016/j.juro.2012.01.069. [DOI] [PubMed] [Google Scholar]
- 78.Rho YS, Lim YC, Park IS, et al. High mobility group HMGI(Y) protein expression in head and neck squamous cell carcinoma. Acta Otolaryngol. 2007;127:76–81. doi: 10.1080/00016480600740571. [DOI] [PubMed] [Google Scholar]
- 79.Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–42. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 80.Giannini G, Cerignoli F, Mellone M, et al. High mobility group A1 is a molecular target for MYCN in human neuroblastoma. Cancer Res. 2005;65:8308–16. doi: 10.1158/0008-5472.CAN-05-0607. [DOI] [PubMed] [Google Scholar]
- 81.Giannini G, Cerignoli F, Mellone M, et al. Molecular mechanism of HMGA1 deregulation in human neuroblastoma. Cancer Lett. 2005;228:97–104. doi: 10.1016/j.canlet.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 82.Cerignoli F, Ambrosi C, Mellone M, et al. HMGA molecules in neuroblastic tumors. Ann N Y Acad Sci. 2004;1028:122–32. doi: 10.1196/annals.1322.013. [DOI] [PubMed] [Google Scholar]
- 83.Giannini G, Kim CJ, Di Marcotullio L, et al. Expression of the HMGI(Y) gene products in human neuroblastic tumours correlates with differentiation status. Br J Cancer. 2000;83:1503–9. doi: 10.1054/bjoc.2000.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giannini G, Di Marcotullio L, Ristori E, et al. HMGI(Y) and HMGI-C genes are expressed in neuroblastoma cell lines and tumors and affect retinoic acid responsiveness. Cancer Res. 1999;59:2484–92. [PubMed] [Google Scholar]
- 85.Rahman MM, Qian ZR, Wang EL, et al. Frequent overexpression of hmga1 and 2 in gastroenteropancreatic neuroendocrine tumours and its relationship to let-7 downregulation. Br J Cancer. 2009;100:501–10. doi: 10.1038/sj.bjc.6604883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Akaboshi S, Watanabe S, Hino Y, et al. HMGA1 is induced by wnt/beta-catenin pathway and maintains cell proliferation in gastric cancer. Am J Pathol. 2009;175:1675–85. doi: 10.2353/ajpath.2009.090069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chuma M, Saeki N, Yamamoto Y, et al. Expression profiling in hepatocellular carcinoma with intrahepatic metastasis: Identification of high-mobility group I(Y) protein as a molecular marker of hepatocellular carcinoma metastasis. Keio J Med. 2004;53:90–7. doi: 10.2302/kjm.53.90. [DOI] [PubMed] [Google Scholar]
- 88.Wood LJ, Mukherjee M, Dolde CE, et al. HMG-I/Y, a new cmyc target gene and potential oncogene. Mol Cell Biol. 2000;20:5490–502. doi: 10.1128/mcb.20.15.5490-5502.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pierantoni GM, Agosti V, Fedele M, et al. High-mobility group A1 proteins are overexpressed in human leukaemias. Biochem J. 2003;372:145–50. doi: 10.1042/BJ20021493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu Y, Sumter TF, Bhattacharya R, et al. The HMG-I oncogene causes highly penetrant, aggressive lymphoid malignancy in transgenic mice and is overexpressed in human leukemia. Cancer Res. 2004;64:3371–5. doi: 10.1158/0008-5472.CAN-04-0044. [DOI] [PubMed] [Google Scholar]
- 91.Hillion J, Dhara S, Sumter TF, et al. The high-mobility group A1a/signal transducer and activator of transcription-3 axis: An achilles heel for hematopoietic malignancies? Cancer Res. 2008;68:10121–7. doi: 10.1158/0008-5472.CAN-08-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karp JE, Smith BD, Resar LS, et al. Phase 1 and pharmacokinetic study of bolus-infusion flavopiridol followed by cytosine arabinoside and mitoxantrone for acute leukemias. Blood. 2011;117:3302–10. doi: 10.1182/blood-2010-09-310862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nelson DM, Joseph B, Hillion J, Segal J, Karp JE, Resar LM. Flavopiridol induces bcl-2 expression and represses oncogenic transcription factors in leukemic blasts from adults with refractory acute myeloid leukemia. Leuk Lymphoma. 2011;52:1999–2006. doi: 10.3109/10428194.2011.591012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tarbe N, Evtimova V, Burtscher H, Jarsch M, Alves F, Weidle UH. Transcriptional profiling of cell lines derived from an orthotopic pancreatic tumor model reveals metastasis-associated genes. Anticancer Res. 2001;21:3221–8. [PubMed] [Google Scholar]
- 95.Chiappetta G, Avantaggiato V, Visconti R, et al. High level expression of the HMGI (Y) gene during embryonic development. Oncogene. 1996;13:2439–46. [PubMed] [Google Scholar]
- 96.Zhou G, Chen J, Lee S, Clark T, Rowley JD, Wang SM. The pattern of gene expression in human CD34(+) stem/progenitor cells. Proc Natl Acad Sci U S A. 2001;98:13966–71. doi: 10.1073/pnas.241526198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chou BK, Mali P, Huang X, et al. Efficient human ips cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011;21:518–29. doi: 10.1038/cr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shah SN, Kerr C, Cope L, et al. HMGA1 reprograms somatic cells into pluripotent stem cells by inducing stem cell transcriptional networks. PLoS One. 2012;7:e48533. doi: 10.1371/journal.pone.0048533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Somervaille TC, Matheny CJ, Spencer GJ, et al. Hierarchical maintenance of mll myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell. 2009;4:129–40. doi: 10.1016/j.stem.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fedele M, Pentimalli F, Baldassarre G, et al. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene. 2005;24:3427–35. doi: 10.1038/sj.onc.1208501. [DOI] [PubMed] [Google Scholar]
- 101.Fedele M, Fusco A. Role of the high mobility group A proteins in the regulation of pituitary cell cycle. J Mol Endocrinol. 2010;44:309–18. doi: 10.1677/JME-09-0178. [DOI] [PubMed] [Google Scholar]
- 102.Rothermund K, Rogulski K, Fernandes E, et al. C-myc-independent restoration of multiple phenotypes by two c-myc target genes with overlapping functions. Cancer Res. 2005;65:2097–107. doi: 10.1158/0008-5472.CAN-04-2928. [DOI] [PubMed] [Google Scholar]
- 103.Pierantoni GM, Battista S, Pentimalli F, et al. A truncated HMGA1 gene induces proliferation of the 3T3-L1 pre-adipocytic cells: A model of human lipomas. Carcinogenesis. 2003;24:1861–9. doi: 10.1093/carcin/bgg149. [DOI] [PubMed] [Google Scholar]
- 104.Pierantoni GM, Fedele M, Pentimalli F, et al. High mobility group I (Y) proteins bind HIPK2, a serine-threonine kinase protein which inhibits cell growth. Oncogene. 2001;20:6132–41. doi: 10.1038/sj.onc.1204635. [DOI] [PubMed] [Google Scholar]
- 105.Fedele M, Pierantoni GM, Berlingieri MT, et al. Overexpression of proteins HMGA1 induces cell cycle deregulation and apoptosis in normal rat thyroid cells. Cancer Res. 2001;61:4583–90. [PubMed] [Google Scholar]
- 106.Reeves R, Nissen MS. Cell cycle regulation and functions of HMG-I(Y) Prog Cell Cycle Res. 1995;1:339–49. doi: 10.1007/978-1-4615-1809-9_28. [DOI] [PubMed] [Google Scholar]
- 107.Kolb S, Fritsch R, Saur D, Reichert M, Schmid RM, Schneider G. HMGA1 controls transcription of insulin receptor to regulate cyclin D1 translation in pancreatic cancer cells. Cancer Res. 2007;67:4679–86. doi: 10.1158/0008-5472.CAN-06-3308. [DOI] [PubMed] [Google Scholar]
- 108.Schuldenfrei A, Belton A, Kowalski J, et al. HMGA1 drives stem cell, inflammatory pathway, and cell cycle progression genes during lymphoid tumorigenesis. BMC Genomics. 2011;12:549. doi: 10.1186/1471-2164-12-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Baldassarre G, Belletti B, Battista S, et al. HMGA1 protein expression sensitizes cells to cisplatin-induced cell death. Oncogene. 2005;24:6809–19. doi: 10.1038/sj.onc.1208831. [DOI] [PubMed] [Google Scholar]
- 110.Baldassarre G, Battista S, Belletti B, et al. Negative regulation of brca1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol Cell Biol. 2003;23:2225–38. doi: 10.1128/MCB.23.7.2225-2238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maloney SC, Adair JE, Smerdon MJ, Reeves R. Gene-specific nucleotide excision repair is impaired in human cells expressing elevated levels of high mobility group A1 nonhistone proteins. DNA Repair (Amst) 2007;6:1371–9. doi: 10.1016/j.dnarep.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Adair JE, Maloney SC, Dement GA, Wertzler KJ, Smerdon MJ, Reeves R. High-mobility group A1 proteins inhibit expression of nucleotide excision repair factor xeroderma pigmentosum group A. Cancer Res. 2007;67:6044–52. doi: 10.1158/0008-5472.CAN-06-1689. [DOI] [PubMed] [Google Scholar]
- 113.Satou W, Suzuki T, Noguchi T, Ogino H, Fujii M, Ayusawa D. AT-hook proteins stimulate induction of senescence markers triggered by 5-bromodeoxyuridine in mammalian cells. Exp Gerontol. 2004;39:173–9. doi: 10.1016/j.exger.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 114.Narita M, Narita M, Krizhanovsky V, et al. A novel role for high-mobility group A proteins in cellular senescence and heterochromatin formation. Cell. 2006;126:503–14. doi: 10.1016/j.cell.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 115.Markowski DN, Bartnitzke S, Belge G, Drieschner N, Helmke BM, Bullerdiek J. Cell culture and senescence in uterine fibroids. Cancer Genet Cytogenet. 2010;202:53–7. doi: 10.1016/j.cancergencyto.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 116.Farnet CM, Bushman FD. HIV-1 cDNA integration: Requirement of HMG I(Y) protein for function of preintegration complexes in vitro. Cell. 1997;88:483–92. doi: 10.1016/s0092-8674(00)81888-7. [DOI] [PubMed] [Google Scholar]
- 117.Shah SN, Cope L, Poh W, et al. Hmga1: A master regulator of tumor progression in triple-negative breast cancer cells. PLoS One. 2013;8:e63419. doi: 10.1371/journal.pone.0063419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Di Cello F, Dhara S, Hristov AC, et al. Inactivation of the Cdkn2a locus cooperates with HMGA1 to drive T-cell leukemogenesis. Leuk Lymphoma. 2013;54:1762–8. doi: 10.3109/10428194.2013.764422. [DOI] [PubMed] [Google Scholar]
- 119.Roy S, Di Cello F, Kowalski J, et al. Hmga1 overexpression correlates with relapse in childhood B-lineage acute lymphoblastic leukemia. Leuk Lymphoma. 2013;54:2565–7. doi: 10.3109/10428194.2013.782610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Munoz J, Stange DE, Schepers AG, et al. The Lgr5 intestinal stem cell signature: Robust expression of proposed quiescent ‘+4′ cell markers. EMBO J. 2012;31:3079–91. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saitoh Y, Laemmli UK. Metaphase chromosome structure: Bands arise from a differential folding path of the highly AT-rich scaffold. Cell. 1994;76:609–22. doi: 10.1016/0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- 122.Girard F, Bello B, Laemmli UK, Gehring WJ. In vivo analysis of scaffold-associated regions in drosophila: A synthetic high-affinity SAR binding protein suppresses position effect variegation. EMBO J. 1998;17:2079–85. doi: 10.1093/emboj/17.7.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhao K, Kas E, Gonzalez E, Laemmli UK. SAR-dependent mobilization of histone H1 by HMG-I/Y in vitro: Hmg-I/Y is enriched in H1-depleted chromatin. EMBO J. 1993;12:3237–47. doi: 10.1002/j.1460-2075.1993.tb05993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Strick R, Laemmli UK. SARS are cis DNA elements of chromosome dynamics: Synthesis of a SAR repressor protein. Cell. 1995;83:1137–48. doi: 10.1016/0092-8674(95)90140-x. [DOI] [PubMed] [Google Scholar]
- 125.Mirkovitch J, Mirault ME, Laemmli UK. Organization of the higher-order chromatin loop: Specific DNA attachment sites on nuclear scaffold. Cell. 1984;39:223–32. doi: 10.1016/0092-8674(84)90208-3. [DOI] [PubMed] [Google Scholar]
- 126.Falvo JV, Thanos D, Maniatis T. Reversal of intrinsic DNA bends in the IFN beta gene enhancer by transcription factors and the architectural protein HMG I(Y) Cell. 1995;83:1101–11. doi: 10.1016/0092-8674(95)90137-x. [DOI] [PubMed] [Google Scholar]
- 127.Thanos D, Maniatis T. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell. 1995;83:1091–100. doi: 10.1016/0092-8674(95)90136-1. [DOI] [PubMed] [Google Scholar]
- 128.Du W, Maniatis T. The high mobility group protein HMG I(Y) can stimulate or inhibit DNA binding of distinct transcription factor ATF-2 isoforms. Proc Natl Acad Sci U S A. 1994;91:11318–22. doi: 10.1073/pnas.91.24.11318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Thanos D, Maniatis T. The high mobility group protein HMG I(Y) is required for NF-kappa b-dependent virus induction of the human IFN-beta gene. Cell. 1992;71:777–89. doi: 10.1016/0092-8674(92)90554-p. [DOI] [PubMed] [Google Scholar]
- 130.Munshi N, Agalioti T, Lomvardas S, Merika M, Chen G, Thanos D. Coordination of a transcriptional switch by HMGI(Y) acetylation. Science. 2001;293:1133–6. doi: 10.1126/science.293.5532.1133. [DOI] [PubMed] [Google Scholar]
- 131.Parekh BS, Maniatis T. Virus infection leads to localized hyperacetylation of histones H3 and H4 at the IFN-beta promoter. Mol Cell. 1999;3:125–9. doi: 10.1016/s1097-2765(00)80181-1. [DOI] [PubMed] [Google Scholar]
- 132.Panne D, Maniatis T, Harrison SC. Crystal structure of ATF-2/c-jun and IRF-3 bound to the interferon-beta enhancer. Embo J. 2004;23:4384–93. doi: 10.1038/sj.emboj.7600453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kim TK, Maniatis T. The mechanism of transcriptional synergy of an in vitro assembled interferon-beta enhanceosome. Mol Cell. 1997;1:119–29. doi: 10.1016/s1097-2765(00)80013-1. [DOI] [PubMed] [Google Scholar]
- 134.Munshi N, Merika M, Yie J, Senger K, Chen G, Thanos D. Acetylation of HMG I(Y) by CBP turns off IFN beta expression by disrupting the enhanceosome. Mol Cell. 1998;2:457–67. doi: 10.1016/s1097-2765(00)80145-8. [DOI] [PubMed] [Google Scholar]
- 135.Merika M, Williams AJ, Chen G, Collins T, Thanos D. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol Cell. 1998;1:277–87. doi: 10.1016/s1097-2765(00)80028-3. [DOI] [PubMed] [Google Scholar]
- 136.Yie J, Merika M, Munshi N, Chen G, Thanos D. The role of HMG I(Y) in the assembly and function of the IFN-beta enhanceosome. Embo J. 1999;18:3074–89. doi: 10.1093/emboj/18.11.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dragan AI, Carrillo R, Gerasimova TI, Privalov PL. Assembling the human IFN-beta enhanceosome in solution. J Mol Biol. 2008;384:335–48. doi: 10.1016/j.jmb.2008.09.015. [DOI] [PubMed] [Google Scholar]
- 138.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–23. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Apostolou E, Thanos D. Virus infection induces NF-kappab-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell. 2008;134:85–96. doi: 10.1016/j.cell.2008.05.052. [DOI] [PubMed] [Google Scholar]
- 140.Ford E, Thanos D. The transcriptional code of human IFN-beta gene expression. Biochim Biophys Acta. 2010;1799:328–36. doi: 10.1016/j.bbagrm.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 141.Krech AB, Wulff D, Grasser KD, Feix G. Plant chromosomal HMGI/Y proteins and histone H1 exhibit a protein domain of common origin. Gene. 1999;230:1–5. doi: 10.1016/s0378-1119(99)00067-0. [DOI] [PubMed] [Google Scholar]
- 142.Dhar A, Hu J, Reeves R, Resar LM, Colburn NH. Dominant-negative c-jun (tam67) target genes: HMGA1 is required for tumor promoter-induced transformation. Oncogene. 2004;23:4466–76. doi: 10.1038/sj.onc.1207581. [DOI] [PubMed] [Google Scholar]
- 143.Scala S, Portella G, Fedele M, Chiappetta G, Fusco A. Adenovirus-mediated suppression of HMGI(Y) protein synthesis as potential therapy of human malignant neoplasias. Proc Natl Acad Sci U S A. 2000;97:4256–61. doi: 10.1073/pnas.070029997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Battista S, Pentimalli F, Baldassarre G, et al. Loss of HMGA1 gene function affects embryonic stem cell lympho-hematopoietic differentiation. FASEB J. 2003;17:1496–8. doi: 10.1096/fj.02-0977fje. [DOI] [PubMed] [Google Scholar]
- 145.Fedele M, Fidanza V, Battista S, et al. Haploinsufficiency of the HMGA1 gene causes cardiac hypertrophy and myelo-lymphoproliferative disorders in mice. Cancer Res. 2006;66:2536–43. doi: 10.1158/0008-5472.CAN-05-1889. [DOI] [PubMed] [Google Scholar]
- 146.Visone R, Iuliano R, Palmieri D, et al. HMGA1 null mice are less susceptible to chemically induced skin carcinogenesis. Eur J Cancer. 2008;44:318–25. doi: 10.1016/j.ejca.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 147.Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heteroxygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A. 1995;92:4481–6. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 149.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70. [PubMed] [Google Scholar]
- 150.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 151.Williams MD, Reeves R, Resar LMS, Hill HH., Jr Metabolomics of colorectal cancer (CRC): past and current analytical platforms. Analytical Bioanalytical Chem. 2013;405:5013–30. doi: 10.1007/s00216-013-6777-5. [DOI] [PubMed] [Google Scholar]
- 152.Williams MD, Zhang X, Belton AS, et al. HMGA1 drives metabolomics reprogramming of intenstinal epithelium during hyperproliferation, polyposis and colorectal carcinogenesis. J Proteome Res. 2015;14:1420–31. doi: 10.1021/pr501084s. [DOI] [PubMed] [Google Scholar]
- 153.Williams MD, Zhang X, Park J-J, Siems WF, et al. Characterizing metabolic changes in human colorectal cancer. Anal Bioanal Chem. 2015;407:4581–95. doi: 10.1007/s00216-015-8662-x. [DOI] [PubMed] [Google Scholar]
- 154.Dang CV, Le A, Gao P. Myc-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009;23:537–48. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dang CVHM, Sun P, Le A, Gao P. Therapeutic targeting of cancer cell metabolism. J Mol Med (Berl) 2001;89:205–12. doi: 10.1007/s00109-011-0730-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–90. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107:2037–42. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Alzheimer’s Association. 2010 Alzheimer’s disease facts and figures. Alzheimers Dement. 2010;6:158–94. doi: 10.1016/j.jalz.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 160.White AR, Du T, Laughton KM, et al. Degradation of the alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity. J Biol Chem. 2006;281:17670–80. doi: 10.1074/jbc.M602487200. [DOI] [PubMed] [Google Scholar]
- 161.Chiang PK, Lam MA, Luo Y. The many faces of amyloid beta in alzheimer’s disease. Curr Mol Med. 2008;8:580–4. doi: 10.2174/156652408785747951. [DOI] [PubMed] [Google Scholar]
- 162.Selkoe DJ, Wolfe MS. Presenilin: Running with scissors in the membrane. Cell. 2007;131:215–21. doi: 10.1016/j.cell.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 163.Sgarra R, Tessari MA, Di Bernardo J, et al. Discovering high mobility group a molecular partners in tumour cells. Proteomics. 2005;5:1494–506. doi: 10.1002/pmic.200401028. [DOI] [PubMed] [Google Scholar]
- 164.Young NL, Plazas-Mayorca MD, DiMaggio PA, et al. Collective mass spectrometry approaches reveal broad and combinatorial modification of high mobility group protein a1a. J Am Soc Mass Spectrom. 2010;21:960–70. doi: 10.1016/j.jasms.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Coelho LF, Magno de Freitas Almeida G, Mennechet FJ, Blangy A, Uze G. Interferon-alpha and beta differentially regulate osteoclastogenesis: Role of differential induction of chemokine CXCL11 expression. Proc Natl Acad Sci U S A. 2005;102:11917–22. doi: 10.1073/pnas.0502188102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chau KY, Keane-Myers AM, Fedele M, et al. IFN-gamma gene expression is controlled by the architectural transcription factor HMGA1. Int Immunol. 2005;17:297–306. doi: 10.1093/intimm/dxh209. [DOI] [PubMed] [Google Scholar]
- 167.Himes SR, Reeves R, Attema J, Nissen M, Li Y, Shannon MF. The role of high-mobility group I(Y) proteins in expression of IL-2 and T cell proliferation. J Immunol. 2000;164:3157–68. doi: 10.4049/jimmunol.164.6.3157. [DOI] [PubMed] [Google Scholar]
- 168.Shannon MF, Himes SR, Attema J. A role for the architectural transcription factors HMGI(Y) in cytokine gene transcription in T cells. Immunol Cell Biol. 1998;76:461–6. doi: 10.1046/j.1440-1711.1998.00770.x. [DOI] [PubMed] [Google Scholar]
- 169.Chuvpilo S, Schomberg C, Gerwig R, et al. Multiple closely-linked NFAT/octamer and HMG I(Y) binding sites are part of the interleukin-4 promoter. Nucleic Acids Res. 1993;21:5694–704. doi: 10.1093/nar/21.24.5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Trentin L, Zambello R, Agostini C, et al. Expression and regulation of tumor necrosis factor, interleukin-2, and hematopoietic growth factor receptors in B-cell chronic lymphocytic leukemia. Blood. 1994;84:4249–56. [PubMed] [Google Scholar]
- 171.Kay NE, Pittner BT. Il-4 biology: Impact on normal and leukemic CLL B cells. Leuk Lymphoma. 2003;44:897–903. doi: 10.1080/1042819031000068007. [DOI] [PubMed] [Google Scholar]
- 172.Ma A, Koka R, Burkett P. Diverse functions of Il-2, IL-15, and Il-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–79. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- 173.Whitley MZ, Thanos D, Read MA, Maniatis T, Collins T. A striking similarity in the organization of the E-selectin and beta interferon gene promoters. Mol Cell Biol. 1994;14:6464–75. doi: 10.1128/mcb.14.10.6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Olofsson AM, Arfors KE, Ramezani L, Wolitzky BA, Butcher EC, von Andrian UH. E-selectin mediates leukocyte rolling in interleukin-1-treated rabbit mesentery venules. Blood. 1994;84:2749–58. [PubMed] [Google Scholar]
- 175.Safran M, Dalah I, Alexander J, et al. GeneCards Version 3: the human gene integrator. Database (Oxford) 2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed]
- 176.Baron RM, Lopez-Guzman S, Riascos DF, et al. Distamycin A inhibits HMGA1-binding to the P-selectin promoter and attenuates lung and liver inflammation during murine endotoxemia. PLoS One. 2010;5:e10656. doi: 10.1371/journal.pone.0010656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.John S, Reeves RB, Lin JX, et al. Regulation of cell-type-specific interleukin-2 receptor alpha-chain gene expression: Potential role of physical interactions between ELF-1, HMG-I(Y), and NF-kappa B family proteins. Mol Cell Biol. 1995;15:1786–96. doi: 10.1128/mcb.15.3.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.John S, Robbins CM, Leonard WJ. An IL-2 response element in the human IL-2 receptor alpha chain promoter is a composite element that binds STAT5, ELF-1, HMG-I(Y) and a gata family protein. EMBO J. 1996;15:5627–35. [PMC free article] [PubMed] [Google Scholar]
- 179.Reeves R, Leonard WJ, Nissen MS. Binding of HMG-I(Y) imparts architectural specificity to a positioned nucleosome on the promoter of the human interleukin-2 receptor alpha gene. Mol Cell Biol. 2000;20:4666–79. doi: 10.1128/mcb.20.13.4666-4679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Milani P, Marilley M, Sanchez-Sevilla A, et al. Mechanics of the IL2ra gene activation revealed by modeling and atomic force microscopy. PLoS One. 2011;6:e18811. doi: 10.1371/journal.pone.0018811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fashena SJ, Reeves R, Ruddle NH. A poly(da-dt) upstream activating sequence binds high-mobility group I protein and contributes to lymphotoxin (tumor necrosis factor-beta) gene regulation. Mol Cell Biol. 1992;12:894–903. doi: 10.1128/mcb.12.2.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Welniak LA, Kuprash DV, Tumanov AV, et al. Peyer patches are not required for acute graft-versus-host disease after myeloablative conditioning and murine allogeneic bone marrow transplantation. Blood. 2006;107:410–2. doi: 10.1182/blood-2004-11-4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Drutskaya MS, Efimov GA, Kruglov AA, Kuprash DV, Nedospasov SA. Tumor necrosis factor, lymphotoxin and cancer. IUBMB Life. 2010;62:283–9. doi: 10.1002/iub.309. [DOI] [PubMed] [Google Scholar]
- 184.Wood LD, Farmer AA, Richmond A. HMGI(Y) and Sp1 in addition to NF-kappa B regulate transcription of the MGSA/GRO alpha gene. Nucleic Acids Res. 1995;23:4210–9. doi: 10.1093/nar/23.20.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen J, Petrus M, Bamford R, et al. Increased serum soluble IL-15R alpha levels in T-cell large granular lymphocyte leukemia. Blood. 2012;119:137–43. doi: 10.1182/blood-2011-04-346759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci. 2012;33:35–41. doi: 10.1016/j.tips.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lin SC. Identification of an NF-Y/HMG-I(Y)-binding site in the human IL-10 promoter. Mol Immunol. 2006;43:1325–31. doi: 10.1016/j.molimm.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 188.Kang YJ, Yang SJ, Park G, et al. A novel function of interleukin-10 promoting self-renewal of hematopoietic stem cells. Stem Cells. 2007;25:1814–22. doi: 10.1634/stemcells.2007-0002. [DOI] [PubMed] [Google Scholar]
- 189.Himes SR, Coles LS, Reeves R, Shannon MF. High mobility group protein I(Y) is required for function and for c-rel binding to CD28 response elements within the GC-CSF and IL-2 promoters. Immunity. 1996;5:479–89. doi: 10.1016/s1074-7613(00)80503-8. [DOI] [PubMed] [Google Scholar]
- 190.Schweizerhof M, Stosser S, Kurejova M, et al. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat Med. 2009;15:802–7. doi: 10.1038/nm.1976. [DOI] [PubMed] [Google Scholar]
- 191.Bagga R, Emerson BM. An hmg i/y-containing repressor complex and supercoiled DNA topology are critical for long-range enhancer-dependent transcription in vitro. Genes Dev. 1997;11:629–39. doi: 10.1101/gad.11.5.629. [DOI] [PubMed] [Google Scholar]
- 192.Kim J, Reeves R, Rothman P, Boothby M. The non-histone chromosomal protein HMG-I(Y) contributes to repression of the immunoglobulin heavy chain germ-line epsilon RNA promoter. Eur J Immunol. 1995;25:798–808. doi: 10.1002/eji.1830250326. [DOI] [PubMed] [Google Scholar]
- 193.Lewis RT, Andreucci A, Nikolajczyk BS. PU. 1-mediated transcription is enhanced by HMG-I(Y)-dependent structural mechanisms. J Biol Chem. 2001;276:9550–7. doi: 10.1074/jbc.M008726200. [DOI] [PubMed] [Google Scholar]
- 194.Andreucci A, Reeves R, McCarthy KM, Nikolajczyk BS. Dominant-negative hmga1 blocks mu enhancer activation through a novel mechanism. Biochem Biophys Res Commun. 2002;292:427–33. doi: 10.1006/bbrc.2002.6672. [DOI] [PubMed] [Google Scholar]
- 195.McCarthy KM, McDevit D, Andreucci A, Reeves R, Nikolajczyk BS. HMGA1 co-activates transcription in B cells through indirect association with DNA. J Biol Chem. 2003;278:42106–14. doi: 10.1074/jbc.M308586200. [DOI] [PubMed] [Google Scholar]
- 196.Skalnik DG, Neufeld EJ. Sequence-specific binding of HMG-I(Y) to the proximal promoter of the gp91-phox gene. Biochem Biophys Res Commun. 1992;187:563–9. doi: 10.1016/0006-291x(92)91231-e. [DOI] [PubMed] [Google Scholar]
- 197.Efremov DG, Laurenti L. The syk kinase as a therapeutic target in leukemia and lymphoma. Expert Opin Investig Drugs. 2011;20:623–36. doi: 10.1517/13543784.2011.570329. [DOI] [PubMed] [Google Scholar]
- 198.Martinez Hoyos J, Fedele M, Battista S, et al. Identification of the genes up- and down-regulated by the high mobility group A1 (HMGA1) proteins: Tissue specificity of the HMGA1-dependent gene regulation. Cancer Res. 2004;64:5728–35. doi: 10.1158/0008-5472.CAN-04-1410. [DOI] [PubMed] [Google Scholar]
- 199.Molkentin JD. The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem. 2000;275:38949–52. doi: 10.1074/jbc.R000029200. [DOI] [PubMed] [Google Scholar]
- 200.Kodo K, Nishizawa T, Furutani M, et al. GATA6 mutations cause human cardiac outflow tract defects by disrupting semaphorin-plexin signaling. Proc Natl Acad Sci U S A. 2009;106:13933–8. doi: 10.1073/pnas.0904744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wani MA, Means RT, Jr, Lingrel JB. Loss of LKLF function results in embryonic lethality in mice. Transgenic Res. 1998;7:229–38. doi: 10.1023/a:1008809809843. [DOI] [PubMed] [Google Scholar]
- 202.Saavedra MT, Patterson AD, West J, et al. Abrogation of anti-inflammatory transcription factor LKLF in neutrophil-dominated airways. Am J Respir Cell Mol Biol. 2008;38:679–88. doi: 10.1165/rcmb.2007-0282OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gitenay D, Baron VT. Is EGR1 a potential target for prostate cancer therapy? Future Oncol. 2009;5:993–1003. doi: 10.2217/fon.09.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Chin MT, Pellacani A, Wang H, et al. Enhancement of serum-response factor-dependent transcription and DNA binding by the architectural transcription factor HMG-I(Y) J Biol Chem. 1998;273:9755–60. doi: 10.1074/jbc.273.16.9755. [DOI] [PubMed] [Google Scholar]
- 205.Dang CV, Resar LM, Emison E, et al. Function of the c-myc oncogenic transcription factor. Exp Cell Res. 1999;253:63–77. doi: 10.1006/excr.1999.4686. [DOI] [PubMed] [Google Scholar]
- 206.Hillion J, Belton AM, Shah SN, et al. Nanoparticle delivery of inhibitory signal transducer and activator of transcription 3 (STAT3) G-quartet oligonucleotides blocks tumor growth in HMGA1 transgenic model of T-cell leukemia. Leuk Lymphoma. 2014;55:1194–7. doi: 10.3109/10428194.2013.821202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Turkson J, Zhang S, Palmer J, et al. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol Cancer Ther. 2004;3:1533–42. [PubMed] [Google Scholar]
- 208.Yu H, Pardoll D, Jove R. Stats in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yue P, Turkson J. Targeting STAT3 in cancer: How successful are we? Expert Opin Investig Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Katoh M, Katoh M. Human FOX gene family (review) Int J Oncol. 2004;25:1495–500. [PubMed] [Google Scholar]
- 211.Kimura N, Nakashima K, Ueno M, Kiyama H, Taga T. A novel mammalian T-box-containing gene, Tbr2, expressed in mouse developing brain. Brain Res Dev Brain Res. 1999;115:183–93. doi: 10.1016/s0165-3806(99)00064-4. [DOI] [PubMed] [Google Scholar]
- 212.Yang Y, Xu J, Niu Y, Bromberg JS, Ding Y. T-bet and eomesodermin play critical roles in directing T cell differentiation to Th1 versus Th17. J Immunol. 2008;181:8700–10. doi: 10.4049/jimmunol.181.12.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Narayanan S, Silva R, Peruzzi G, et al. Human Th1 cells that express CD300a are polyfunctional and after stimulation up-regulate the T-box transcription factor eomesodermin. PLoS One. 2010;5:e10636. doi: 10.1371/journal.pone.0010636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Martinez-Hoyos J, Ferraro A, Sacchetti S, et al. HAND1 gene expression is negatively regulated by the high mobility group A1 proteins and is drastically reduced in human thyroid carcinomas. Oncogene. 2009;28:876–85. doi: 10.1038/onc.2008.438. [DOI] [PubMed] [Google Scholar]
- 216.Chang MS, Uozaki H, Chong JM, et al. Cpg island methylation status in gastric carcinoma with and without infection of Epstein-Barr virus. Clin Cancer Res. 2006;12:2995–3002. doi: 10.1158/1078-0432.CCR-05-1601. [DOI] [PubMed] [Google Scholar]
- 217.Oue N, Mitani Y, Motoshita J, et al. Accumulation of DNA methylation is associated with tumor stage in gastric cancer. Cancer. 2006;106:1250–9. doi: 10.1002/cncr.21754. [DOI] [PubMed] [Google Scholar]
- 218.Maekita T, Nakazawa K, Mihara M, et al. High levels of aberrant DNA methylation in helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–95. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 219.Imura M, Yamashita S, Cai LY, et al. Methylation and expression analysis of 15 genes and three normally-methylated genes in 13 ovarian cancer cell lines. Cancer Lett. 2006;241:213–20. doi: 10.1016/j.canlet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 220.Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Esposito F, Tornincasa M, Chieffi P, De Martino I, Pierantoni GM, Fusco A. High-mobility group A1 proteins regulate p53-mediated transcription of Bcl-2 gene. Cancer Res. 2010;70:5379–88. doi: 10.1158/0008-5472.CAN-09-4199. [DOI] [PubMed] [Google Scholar]
- 222.Treff NR, Pouchnik D, Dement GA, Britt RL, Reeves R. High-mobility group A1a protein regulates Ras/ERK signaling in MCF-7 human breast cancer cells. Oncogene. 2004;23:777–85. doi: 10.1038/sj.onc.1207167. [DOI] [PubMed] [Google Scholar]
- 223.Treff NR, Dement GA, Adair JE, et al. Human KIT ligand promoter is positively regulated by HMGA1 in breast and ovarian cancer cells. Oncogene. 2004;23:8557–62. doi: 10.1038/sj.onc.1207926. [DOI] [PubMed] [Google Scholar]
- 224.Quest AF, Gutierrez-Pajares JL, Torres VA. Caveolin-1: An ambiguous partner in cell signalling and cancer. J Cell Mol Med. 2008;12:1130–50. doi: 10.1111/j.1582-4934.2008.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Elsheikh SE, Green AR, Rakha EA, et al. Caveolin 1 and caveolin 2 are associated with breast cancer basal-like and triple-negative immunophenotype. Br J Cancer. 2008;99:327–34. doi: 10.1038/sj.bjc.6604463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Schwahn DJ, Medina D. p96, a MAPK-related protein, is consistently downregulated during mouse mammary carcinogenesis. Oncogene. 1998;17:1173–8. doi: 10.1038/sj.onc.1202038. [DOI] [PubMed] [Google Scholar]
- 227.Albertsen HM, Smith SA, Melis R, et al. Sequence, genomic structure, and chromosomal assignment of human DOC-2. Genomics. 1996;33:207–13. doi: 10.1006/geno.1996.0185. [DOI] [PubMed] [Google Scholar]
- 228.Mok SC, Chan WY, Wong KK, et al. DOC-2, a candidate tumor suppressor gene in human epithelial ovarian cancer. Oncogene. 1998;16:2381–7. doi: 10.1038/sj.onc.1201769. [DOI] [PubMed] [Google Scholar]
- 229.Zhang Y, Finegold MJ, Porteu F, Kanteti P, Wu MX. Development of T-cell lymphomas in Emu-IEX-1 mice. Oncogene. 2003;22:6845–51. doi: 10.1038/sj.onc.1206707. [DOI] [PubMed] [Google Scholar]
- 230.Perrella MA, Pellacani A, Wiesel P, et al. High mobility group-I(Y) protein facilitates nuclear factor-kappab binding and transactivation of the inducible nitric-oxide synthase promoter/enhancer. J Biol Chem. 1999;274:9045–52. doi: 10.1074/jbc.274.13.9045. [DOI] [PubMed] [Google Scholar]
- 231.Takamiya R, Baron RM, Yet SF, Layne MD, Perrella MA. High mobility group A1 protein mediates human nitric oxide synthase 2 gene expression. FEBS Lett. 2008;582:810–4. doi: 10.1016/j.febslet.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Pellacani A, Wiesel P, Razavi S, et al. Down-regulation of high mobility group-I(Y) protein contributes to the inhibition of nitric-oxide synthase 2 by transforming growth factor-beta1. J Biol Chem. 2001;276:1653–9. doi: 10.1074/jbc.M008170200. [DOI] [PubMed] [Google Scholar]
- 233.Darville MI, Terryn S, Eizirik DL. An octamer motif is required for activation of the inducible nitric oxide synthase promoter in pancreatic beta-cells. Endocrinology. 2004;145:1130–6. doi: 10.1210/en.2003-1200. [DOI] [PubMed] [Google Scholar]
- 234.Rao CV. Nitric oxide signaling in colon cancer chemoprevention. Mutat Res. 2004;555:107–19. doi: 10.1016/j.mrfmmm.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 235.Ye Y, Martinez JD, Perez-Polo RJ, Lin Y, Uretsky BF, Birnbaum Y. The role of eNOS, iNOS, and NF-kappaB in upregulation and activation of cyclooxygenase-2 and infarct size reduction by atorvastatin. Am J Physiol Heart Circ Physiol. 2008;295:H343–51. doi: 10.1152/ajpheart.01350.2007. [DOI] [PubMed] [Google Scholar]
- 236.Kim SF, Huri DA, Snyder SH. Inducible nitric oxide synthase binds, s-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–70. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- 237.Chin MT, Pellacani A, Wang H, et al. of serum-response factor-dependent transcription and DNA binding by the architectural transcription factor hmg-I(Y) J Biol Chem. 1998;273:9755–60. doi: 10.1074/jbc.273.16.9755. [DOI] [PubMed] [Google Scholar]
- 238.Bell RD, Deane R, Chow N, et al. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–53. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Foster LC, Wiesel P, Huggins GS, et al. Role of activating protein-1 and high mobility group-I(Y) protein in the induction of CD44 gene expression by interleukin-1beta in vascular smooth muscle cells. FASEB J. 2000;14:368–78. doi: 10.1096/fasebj.14.2.368. [DOI] [PubMed] [Google Scholar]
- 240.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 242.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 243.Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410–8. doi: 10.1016/s0962-8924(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 244.Melillo RM, Pierantoni GM, Scala S, et al. Critical role of the HMGI(Y) proteins in adipocytic cell growth and differentiation. Mol Cell Biol. 2001;21:2485–95. doi: 10.1128/MCB.21.7.2485-2495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 245.Nagel S, Scherr M, Kel A, et al. Activation of TLX3 and NKX2–5 in t(5;14)(q35;q32) t-cell acute lymphoblastic leukemia by remote 3′-BCL11B enhancers and coregulation by PU. Cancer Res. 2007;67:1461–71. doi: 10.1158/0008-5472.CAN-06-2615. [DOI] [PubMed] [Google Scholar]
- 246.Yang YJ, Cao YJ, Bo SM, Peng S, Liu WM, Duan EK. Leptin-directed embryo implantation: Leptin regulates adhesion and outgrowth of mouse blastocysts and receptivity of endometrial epithelial cells. Anim Reprod Sci. 2006;92:155–67. doi: 10.1016/j.anireprosci.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 247.Zucker S, Cao J. Selective matrix metalloproteinase (MMP) inhibitors in cancer therapy: Ready for prime time? Cancer Biol Ther. 2009;8:2371–3. doi: 10.4161/cbt.8.24.10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Palavalli LH, Prickett TD, Wunderlich JR, et al. Analysis of the matrix metalloproteinase family reveals that MMP8 is often mutated in melanoma. Nat Genet. 2009;41:518–20. doi: 10.1038/ng.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Thirumangalakudi L, Samany PG, Owoso A, Wiskar B, Grammas P. Angiogenic proteins are expressed by brain blood vessels in alzheimer’s disease. J Alzheimers Dis. 2006;10:111–8. doi: 10.3233/jad-2006-10114. [DOI] [PubMed] [Google Scholar]
- 250.Lorenzl S, Albers DS, Relkin N, et al. Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer’s disease. Neurochem Int. 2003;43:191–6. doi: 10.1016/s0197-0186(03)00004-4. [DOI] [PubMed] [Google Scholar]
- 251.Reiser J, Adair B, Reinheckel T. Specialized roles for cysteine cathepsins in health and disease. J Clin Invest. 2010;120:3421–31. doi: 10.1172/JCI42918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Wang L, Chen S, Zhang M, et al. Legumain: A biomarker for diagnosis and prognosis of human ovarian cancer. J Cell Biochem. 2012;113:2679–86. doi: 10.1002/jcb.24143. [DOI] [PubMed] [Google Scholar]
- 253.Wolk K, Grutz G, Witte K, Volk HD, Sabat R. The expression of legumain, an asparaginyl endopeptidase that controls antigen processing, is reduced in endotoxin-tolerant monocytes. Genes Immun. 2005;6:452–6. doi: 10.1038/sj.gene.6364224. [DOI] [PubMed] [Google Scholar]
- 254.Lee TK, Murthy SR, Cawley NX, et al. An N-terminal truncated carboxypeptidase E splice isoform induces tumor growth and is a biomarker for predicting future metastasis in human cancers. J Clin Invest. 2011;121:880–92. doi: 10.1172/JCI40433. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 255.Ji YS, Xu Q, Schmedtje JF., Jr Hypoxia induces high-mobility-group protein I(Y) and transcription of the cyclooxygenase-2 gene in human vascular endothelium. Circ Res. 1998;83:295–304. doi: 10.1161/01.res.83.3.295. [DOI] [PubMed] [Google Scholar]
- 256.Di Cello F, Hillion J, Kowalski J, et al. Cyclooxygenase inhibitors block uterine tumorigenesis in HMGA1a transgenic mice and human xenografts. Mol Cancer Ther. 2008;7:2090–5. doi: 10.1158/1535-7163.MCT-07-2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Hillion J, Smail SS, Di Cello F, et al. The HMGA1-COX-2 axis: A molecular pathway leading to tumor progression in human pancreatic adenocarcinoma. Pancreatology. 2012;12:372–9. doi: 10.1016/j.pan.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Sutcliffe S, Neace C, Magnuson NS, Reeves R, Alderete JF. Trichomonosis, a common curable STI, and prostate carcinogenesis--a proposed molecular mechanism. PLoS Pathog. 2012;8:e1002801. doi: 10.1371/journal.ppat.1002801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Chau KY, Munshi N, Keane-Myers A, et al. The architectural transcription factor high mobility group I(Y) participates in photoreceptor-specific gene expression. J Neurosci. 2000;20:7317–24. doi: 10.1523/JNEUROSCI.20-19-07317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Chen S, Wang QL, Nie Z, et al. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron. 1997;19:1017–30. doi: 10.1016/s0896-6273(00)80394-3. [DOI] [PubMed] [Google Scholar]
- 261.Duncan B, Zhao K. HMGA1 mediates the activation of the CRYAB promoter by BRG1. DNA Cell Biol. 2007;26:745–52. doi: 10.1089/dna.2007.0629. [DOI] [PubMed] [Google Scholar]
- 262.Brunetti A, Manfioletti G, Chiefari E, Goldfine ID, Foti D. Transcriptional regulation of human insulin receptor gene by the high-mobility group protein HMGI(Y) FASEB J. 2001;15:492–500. doi: 10.1096/fj.00-0190com. [DOI] [PubMed] [Google Scholar]
- 263.Foti D, Iuliano R, Chiefari E, Brunetti A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol Cell Biol. 2003;23:2720–32. doi: 10.1128/MCB.23.8.2720-2732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Chiefari E, Tanyolac S, Paonessa F, et al. Functional variants of the HMGA1 gene and type 2 diabetes mellitus. JAMA. 2011;305:903–12. doi: 10.1001/jama.2011.207. [DOI] [PubMed] [Google Scholar]
- 265.Arnaldez FI, Helman LJ. Targeting the insulin growth factor receptor 1. Hematol Oncol Clin North Am. 2012;26:527–42. vii–viii. doi: 10.1016/j.hoc.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Allander SV, Durham SK, Scheimann AO, Wasserman RM, Suwanichkul A, Powell DR. Hepatic nuclear factor 3 and high mobility group I/Y proteins bind the insulin response element of the insulin-like growth factor-binding protein-1 promoter. Endocrinology. 1997;138:4291–300. doi: 10.1210/endo.138.10.5268. [DOI] [PubMed] [Google Scholar]
- 267.Iiritano S, Chiefari E, Ventura V, et al. The HMGA1-IGF-I/IGFBP system: A novel pathway for modulating glucose uptake. Mol Endocrinol. 2012;26:1578–89. doi: 10.1210/me.2011-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Georges RB, Adwan H, Hamdi H, Hielscher T, Linnemann U, Berger MR. The insulin-like growth factor binding proteins 3 and 7 are associated with colorectal cancer and liver metastasis. Cancer Biol Ther. 2011;12:69–79. doi: 10.4161/cbt.12.1.15719. [DOI] [PubMed] [Google Scholar]
- 269.Ha TK, Her NG, Lee MG, et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. 2012;72:4097–109. doi: 10.1158/0008-5472.CAN-12-0448. [DOI] [PubMed] [Google Scholar]
- 270.Tharmalingam S, Daulat AM, Antflick JE, et al. Calcium-sensing receptor modulates cell adhesion and migration via integrins. J Biol Chem. 2011;286:40922–33. doi: 10.1074/jbc.M111.265454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Gridley T. Notch signaling and inherited disease syndromes. Hum Mol Genet. 2003;12(Spec No 1):R9–13. doi: 10.1093/hmg/ddg052. [DOI] [PubMed] [Google Scholar]
- 272.Chillakuri CR, Sheppard D, Lea SM, Handford PA. Notch receptor-ligand binding and activation: Insights from molecular studies. Semin Cell Dev Biol. 2012;23:421–8. doi: 10.1016/j.semcdb.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ferrando AA. The role of NOTCH1 signaling in T-ALL. Hematology Am Soc Hematol Educ Program. 2009:353–61. doi: 10.1182/asheducation-2009.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–83. doi: 10.1083/jcb.200512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Glinsky GV. Death-from-cancer signatures and stem cell contribution to metastatic cancer. Cell Cycle. 2005;4:1171–5. doi: 10.4161/cc.4.9.2001. [DOI] [PubMed] [Google Scholar]
- 276.Glinsky GV. Genomic models of metastatic cancer: Functional analysis of death-from-cancer signature genes reveals aneuploid, anoikis-resistant, metastasis-enabling phenotype with altered cell cycle control and activated polycomb group (PcG) protein chromatin silencing pathway. Cell Cycle. 2006;5:1208–16. doi: 10.4161/cc.5.11.2796. [DOI] [PubMed] [Google Scholar]
- 277.Pierantoni GM, Conte A, Rinaldo C, et al. Deregulation of HMGA1 expression induces chromosome instability through regulation of spindle assembly checkpoint genes. Oncotarget. 2015;6:17342–53. doi: 10.18632/oncotarget.3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kirkland SC. Type 1 collagen inhibits differentiation and promotes a stem cell-like phenotype in human colorectal carcinoma cells. Br J Cancer. 2009;101:320–6. doi: 10.1038/sj.bjc.6605143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Haque I, Mehta S, Majumder M, et al. Cyr61/CCN1 signaling is critical for epithelial-mesenchymal transition and stemness and promotes pancreatic carcinogenesis. Mol Cancer. 2011;10:8. doi: 10.1186/1476-4598-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 281.Yanagisawa BL, Resar LM. Hitting the bull’s eye: Targeting HMGA1 in cancer stem cells. Expert Rev Anticancer Ther. 2014;14:23–30. doi: 10.1586/14737140.2013.859988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Huso TH, Resar LM. The high mobility group A1 molecular switch: turning on cancer - can we turn it off? Expert Opin Ther Targets. 2014;18:541–53. doi: 10.1517/14728222.2014.900045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Pedulla ML, Treff NR, Resar LM, Reeves R. Sequence and analysis of the murine HMGIY (HMGA1) gene locus. Gene. 2001;271:51–8. doi: 10.1016/s0378-1119(01)00500-5. [DOI] [PubMed] [Google Scholar]
- 284.Hommura F, Katabami M, Leaner VD, et al. HMG-I/Y is a c-jun/activator protein-1 target gene and is necessary for c-jun-induced anchorage-independent growth in rat1a cells. Mol Cancer Res. 2004;2:305–14. [PubMed] [Google Scholar]
- 285.Bush BM, Brock AT, Deng JA, Nelson RA, Sumter TF. The Wnt/beta-catenin/T-cell factor 4 pathway up-regulates high-mobility group A1 expression in colon cancer. Cell Biochem Funct. 2013;31:228–36. doi: 10.1002/cbf.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Lund T, Holtlund J, Laland SG. On the phosphorylation of low molecular mass HMG (high mobility group) proteins in Ehrlich ascites cells. FEBS Lett. 1985;180:275–9. doi: 10.1016/0014-5793(85)81085-1. [DOI] [PubMed] [Google Scholar]
- 287.Elton TS, Reeves R. Purification and postsynthetic modifications of friend erythroleukemic cell high mobility group protein HMG-I. Anal Biochem. 1986;157:53–62. doi: 10.1016/0003-2697(86)90195-8. [DOI] [PubMed] [Google Scholar]
- 288.Reeves R, Langan TA, Nissen MS. Phosphorylation of the DNA-binding domain of nonhistone high-mobility group I protein by cdc2 kinase: Reduction of binding affinity. Proc Natl Acad Sci U S A. 1991;88:1671–5. doi: 10.1073/pnas.88.5.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Nissen MS, Langan TA, Reeves R. Phosphorylation by cdc2 kinase modulates DNA binding activity of high mobility group I nonhistone chromatin protein. J Biol Chem. 1991;266:19945–52. [PubMed] [Google Scholar]
- 290.Piekielko A, Drung A, Rogalla P, et al. Distinct organization of DNA complexes of various HMGI/Y family proteins and their modulation upon mitotic phosphorylation. J Biol Chem. 2001;276:1984–92. doi: 10.1074/jbc.M004065200. [DOI] [PubMed] [Google Scholar]
- 291.Zhang Q, Wang Y. Homeodomain-interacting protein kinase- 2 (HIPK2) phosphorylates HMGA1a at ser-35, thr-52, and thr-77 and modulates its DNA binding affinity. J Proteome Res. 2007;6:4711–9. doi: 10.1021/pr700571d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Sgarra R, Diana F, Bellarosa C, et al. During apoptosis of tumor cells HMGA1a protein undergoes methylation: Identification of the modification site by mass spectrometry. Biochemistry. 2003;42:3575–85. doi: 10.1021/bi027338l. [DOI] [PubMed] [Google Scholar]
- 293.Maurizio E, Cravello L, Brady L, et al. Conformational role for the C-terminal tail of the intrinsically disordered high mobility group A (HMGA) chromatin factors. J Proteome Res. 2011;10:3283–91. doi: 10.1021/pr200116w. [DOI] [PubMed] [Google Scholar]
- 294.Sgarra R, Lee J, Tessari MA, et al. The AT-hook of the chromatin architectural transcription factor high mobility group A1a is arginine-methylated by protein arginine methyltransferase 6. J Biol Chem. 2006;281:3764–72. doi: 10.1074/jbc.M510231200. [DOI] [PubMed] [Google Scholar]
- 295.Tran JC, Zamdborg L, Ahlf DR, et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature. 2011;480:254–8. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Belton A, Xian L, Huso T, et al. STAT3 inhibitor has potent anti-tumor activity in B-lineage acute lymphoblastic leukemia cells overexpressing the High Mobility Group A1 (HMGA1)-STAT3 pathway. Leuk Lymphoma. 2016:1–4. doi: 10.3109/10428194.2016.1153089. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Cartharius K, Frech K, Grote K, et al. MatInspector and beyong: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–42. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]