

Abstract
Global warming and eutrophication make some aquatic ecosystems behave as true bioreactors that trigger rapid and massive cyanobacterial growth; this has relevant health and economic consequences. Many cyanobacterial strains are toxin producers, and only a few cells are necessary to induce irreparable damage to the environment. Therefore, water-body authorities and administrations require rapid and efficient early-warning systems providing reliable data to support their preventive or curative decisions. This manuscript reports an experimental protocol for the in-field detection of toxin-producing cyanobacterial strains by using an antibody microarray chip with 17 antibodies (Abs) with taxonomic resolution (CYANOCHIP). Here, a multiplex fluorescent sandwich microarray immunoassay (FSMI) for the simultaneous monitoring of 17 cyanobacterial strains frequently found blooming in freshwater ecosystems, some of them toxin producers, is described. A microarray with multiple identical replicates (up to 24) of the CYANOCHIP was printed onto a single microscope slide to simultaneously test a similar number of samples. Liquid samples can be tested either by direct incubation with the antibodies (Abs) or after cell concentration by filtration through a 1- to 3-μm filter. Solid samples, such as sediments and ground rocks, are first homogenized and dispersed by a hand-held ultrasonicator in an incubation buffer. They are then filtered (5 - 20 μm) to remove the coarse material, and the filtrate is incubated with Abs. Immunoreactions are revealed by a final incubation with a mixture of the 17 fluorescence-labeled Abs and are read by a portable fluorescence detector. The whole process takes around 3 h, most of it corresponding to two 1-h periods of incubation. The output is an image, where bright spots correspond to the positive detection of cyanobacterial markers.
Keywords: Environmental Sciences, Issue 120, cyanobacteria, CYANOCHIP, environmental monitoring, antibody microarray, portable fluorescent immunoassay, freshwater reservoir monitoring
Introduction
The detection and monitoring of microorganisms in complex natural microbial communities are crucial in many fields, including biomedicine, environmental ecology, and astrobiology. Cyanobacteria are prokaryotic microorganisms well-known for their ability to form blooms (excessive proliferation) of cells in fresh water. They are ubiquitous, and many species are able to produce toxins, leading not only to a potential risk for human health, but also to an ecological impact. In this regard, it is essential to develop rapid and sensitive methods for the early detection of cyanobacteria and/or their toxins in soil and water. For this purpose, a multiplex fluorescent sandwich microarray immunoassay (FSMI) has been developed as a tool for water managers to help them in making decisions and, consequently, in implementing proper water management programs.
A diverse range of methods has been developed to detect and identify cyanobacterial cells and cyanotoxins in soil and water, including optical microscopy, molecular biology, and immunological techniques. These methods can vary greatly in the information they provide. Microscopic techniques are based on cell morphology and the detection of in vivo fluorescence from cyanobacterial pigments, such as phycocyanin or chlorophyll a1. Although they are quick and cheap methods for real-time and frequent monitoring that inform about the type and number of cyanobacteria present in a sample, they do not give information about the potential toxicity. In addition, they require a certain level of expertise, considering that it is often very difficult to distinguish between closely related species2. To overcome these limitations, light microscopy must be accompanied by both biological and biochemical screening assays and physicochemical methods for the identification and quantification of cyanotoxins.
Enzyme-linked immunosorbent assays (ELISA), protein phosphate inhibition assays (PPIA), and neurochemical tests in mice are examples of biochemical screening assays for the detection of cyanotoxins. While the first two are rapid and sensitive methodologies, false positives have been described when using ELISA and PPIA tests are restricted to three types of toxins. The mouse bioassay is a qualitative technique with low sensitivity and precision, and special licensing and training is required. In addition, it does not give information about the type of toxins present in a sample. Cyanotoxins can be identified and quantified by other analytical methods, such as high-performance liquid chromatography (HPLC), liquid chromatography-mass spectrometry (LC-MS), gas chromatography (GC), gas chromatography-mass spectrometry (GC-MS), or matrix-assisted laser desorption/ionization time of flight (MALDI-TOF). However, this is only possible if reference standards, which are needed to determine individual toxin concentrations in complex samples, are available3,4. Moreover, these methods are time-consuming; require costly equipment, supplies, and sample preparation; and must be performed by experienced and specialized staff.
Molecular-based methods have been applied for decades to detect, identify, and quantify cyanobacteria and their corresponding cyanotoxins thanks to the sequence information published in the genome databases (e.g., National Center for Biotechnology Information, NCBI). Among these methods are those based on the polymerase chain reaction (PCR), which requires the design of sets of primers for DNA amplification and depend on previous knowledge of DNA sequences of different cyanobacterial species. While gene detection, like the phycocyanin operon, leads to accurate identification at the genus level, some species or strains are undetected with this method. However, toxin-encoding genes, such as those belonging to the microcystin operon, facilitate the identification of toxins in samples where the producers are scarce5. Nonetheless, the detection of toxin markers by PCR does not necessarily imply toxicity in the environment. Furthermore, the set of primers developed to analyze the whole range of species of cyanobacteria and toxin producers present in a sample is still incomplete, and further studies must be done to identify unknown species. Other molecular techniques are non-PCR-based, such as fluorescence in situ hybridization (FISH) and DNA microarrays.
In the last two decades, microarray technology has gained importance in many fields of application, particularly in environmental monitoring. DNA microarrays allow for discrimination between species and analytes4,6,7,8,9,10, but they are considered very laborious and time-consuming tasks that involve multiple steps (e.g., microarray performance, DNA extraction, PCR amplification, and hybridization). For that reason, less time-consuming assays based on antibodies, such as sandwich and competitive immunological microarrays, have become an essential and reliable high-throughput method for the detection of multiple environmental analytes11,12,13. The capability of antibodies to specifically recognize their target compounds and to detect small amounts of analytes and proteins, along with the possibility of producing antibodies against almost any substance, make antibody microarrays a powerful technique for environmental purposes. In addition, the capability of achieving multiple analyses in a single assay, with limits of detection ranging from ppb to ppm, is one of the main advantages of this method14.
Antibody-based biosensors have proved to be sensitive and rapid tools for the detection of a wide range of pathogens and toxins in environmental monitoring15,16,17,18,19,20,21. While DNA methods involve several steps, the antibody-based microarrays only require a small sample preparation that is mainly based on a short lysis step in an appropriate solution buffer. Delehanty and Ligler15 reported the simultaneous detection of proteins and bacterial analytes in complex mixtures based on an antibody sandwich immunoassay capable of detecting a protein concentration of 4 ppb and 104 cfu/mL of cells. Szkola et al.21 have developed a cheap and reliable multiplex microarray for the simultaneous detection of proteotoxins and small toxins, compounds that might be used in biological warfare. They detected concentrations of ricin toxin, with a limit of detection of 3 ppb, in less than 20 min. Recently, the CYANOCHIP, an antibody microarray-based biosensor for the in situ detection of toxic and nontoxic cyanobacteria, has been described22. This microarray allows for the identification of potential cyanobacterial blooms, mostly in aquatic environments, which are difficult to identify microscopically. The limit of detection of the microarray is 102 - 103 cells for most species, turning this biosensor into a cost-effective tool for the multiplex detection and identification of cyanobacteria, even at the species level. All these properties make the antibody microarray technique, and particularly the method presented in this work, a quicker and simpler method compared to the aforementioned techniques.
This work presents two examples of experiments that use an antibody microarray-based biosensor to detect the presence of cyanobacteria in soil and water samples. It is a simple and reliable method based on a sandwich immunoassay format that requires very small sample volumes and very basic sample preparation. The method requires a short time and can be easily performed in the field.
Protocol
1. Preparation of the Immunogens
Grow each cyanobacterial strain in the corresponding culture medium under conditions described in Table 1. NOTE: Growth medium and culture conditions for each cyanobacteria strain are listed in Table 1. All the cyanobacterial strains, with the exception of K17, belong to Antonio Quesada's group from Autonoma University (Madrid, Spain). The antibody against Planktothrix rubescens was generated from a natural sample of the monospecific bloom of this cyanobacterium from Vilasouto reservoir (northern Spain).
Quantify the number of cells using a cell counting chamber by optical microscopy to obtain approximately 108 cells/mL from a late exponential or stationary growth phase culture.
Harvest cells from 5 mL of culture by centrifugation at 2,000 x g for 5 min.
Discard the supernatant and resuspend the cells in a 10-mL tube with 5 mL of 1x phosphate-buffered saline (1x PBS) to obtain around 108 cells/mL.
Homogenize and lyse the cells of the suspension by sonication for 5 cycles, 30 s each, with a 30- to 60-s pause on ice, using a portable, hand-held ultrasonic processor or by dipping the tube into the water bath of a cell disruptor at maximum amplitude (30 kHz).
Repeat steps 1.3 - 1.5 for each strain of cyanobacterium. NOTE: Sonication produces cell disruption and cellular content release. While proteins and polysaccharides are good immunogens, specific molecules, such as lipids and nucleic acids, do not induce a humoral immunogenic response by themselves. Therefore, they must bind to carriers, such as polysaccharides or proteins, to increase the molecular complexity. In addition, sonication releases intracellular material that can trigger antibody production.
2. Production of Polyclonal Antibodies
Prepare the first immunogen dose by mixing 0.5 mL of the ultrasonicated cell lysate obtained in step 1.5 with 0.5 mL of complete Freund's adjuvant. Deliver it to the operator of an animal facility for polyclonal rabbit antibody production.
Prepare three more doses as before for further use as memory boosts in the antibody production process by mixing 0.5 mL of the same homogenate/lysate obtained in step 1.5 with 0.5 mL of incomplete Freund's adjuvant. Give them to the animal facility to fulfill the antibody production process.
Repeat steps 2.1 and 2.2 for each new antibody production process. NOTE: Normally, antibody production is entrusted to specialized animal facilities or companies, because appropriate licensing and training are required to work with animals. The companies supply a relative enzyme-linked immunosorbent assay (ELISA) measurement of the quantity of antigen-specific antibodies present in the serum sample.
3. Antibody Purification
Purify the immunoglobulin G (IgG) fraction from both the immune and the pre-immune serum collected in step 2 by protein-A affinity chromatography. Some commercial purification kits, based on cartridge systems, work well; follow the provider instructions.
When the purification kit does not provide a desalting system, change the buffer after the elution of the purified antibodies to 0.1x PBS, either by dialysis or by using centrifugal filter devices with a 100-kDa (or lower) membrane pore size.
Determine the antibody concentration by measuring the absorbance at 280 nm or by using colorimetric methods, such as Bradford23, Lowry24, or Bicinchoninic acid (BCA)25. NOTE: It is important to avoid including amine groups in the elution buffer (e.g., Tris buffers) because they compete with the antibody for binding to solid surfaces activated with epoxy groups. After purification, it is important to test the antibody activity with ELISA.
4. Fluorescence Antibody Labeling
Label the purified antibodies obtained in section 3 with a fluorochrome (e.g., a far-red fluorescent dye) by dissolving a commercial vial containing dye for labeling 1 mg of protein into 100 µL of dimethyl sulfoxide (DMSO). Add 2 µL of the dissolved dye to each antibody preparation at a concentration of 2 mg/mL in a final volume of 50 µL in 50 mM phosphate-buffered saline (pH 8.5).
Maintain the labeling reactions under continuous agitation for 1 h at ambient temperature and 1,200 rpm on a vibrating platform.
Purify the labeled antibodies by size exclusion chromatography (e.g., using a gel with a fractionation range between 1.5 and 30 kDa trapped into a column), following supplier recommendations.
Measure the absorbance at 280 nm and at 650 nm in eluates and calculate the labeling efficiencies following supplier recommendations. NOTE: Up to 50 x 50-µL antibody-labeling reactions can be done with a single vial of the fluorescent dye for labeling 1 mg of protein. It is recommended to cover the tubes with aluminum foil or, alternatively, to use opaque 0.5-mL tubes to avoid quenching processes after step 4.3. For IgG antibodies, optimal labeling is achieved with 3-7 mol of the dye per mol of antibody.
5. CYANOCHIP Production
- Antibodies and controls in printing solution
- Prepare 30 µL of each purified antibody in printing solution by mixing each antibody at 1 mg/mL in a commercial 1x protein printing buffer with 0.01% (v/v) Tween 20 (a non-ionic detergent), all as final concentrations. NOTE: Alternatively, an antibody solution may contain 20% glycerol, 1% (w/v) sucrose (or trehalose, as a preservative), and 0.01% (v/v) Tween 20 in carbonate buffer (pH 8.5).
- Prepare 30 µL of the printing solution as controls: (a) 1x protein printing buffer with 0.01% (v/v) Tween 20, (b) protein-A purified pre-immune serum at 1 mg/mL in 1x protein printing buffer with 0.01% (v/v) Tween 20, and (c) bovine serum albumin (BSA) at 1 mg/mL in 1x protein printing buffer with 0.01% (v/v) Tween 20.
- Prepare 30 µL of a fluorescently labeled, purified pre-immune serum at different concentrations (e.g., from 50 µg/mL to 1 µg/mL) in 1x protein printing buffer with Tween 20, as in step 5.1.1. These samples will be used as fluorescent frame markers and for relative fluorescence quantification after spotting.
- Add 30 µL per well of the printing solutions prepared in steps 5.1.1, 5.1.2, and 5.1.3 to the highest-quality 384-well microplates for microarray manufacturing, such as polypropylene. NOTE: Protein printing buffer increases the quality and stability of the antibodies, and Tween 20 homogenizes spot morphology and builds up protein coupling up. It is recommended to maintain the 384-well microplate at 4 °C before use. Store it at -20 °C for long periods of time. Polypropylene has low DNA, protein, cell extract, and small-molecule intrinsic binding.
- Antibody printing onto a microscope slide NOTE: Print the antibodies onto activated microscope slides by using different array platforms, like contact, split-needle, or non-contact devices, such as piezoelectric printers or "ink jet" technologies. In this work, the CYANOCHIP has been routinely printed by contact using a robotic system (arrayer) capable of spotting nL quantities of the antibodies at the µm scale.
- Set the environmental conditions of the printing room to 20 °C and 40 - 50% relative humidity.
- Set up the slide substrates (e.g., 75- x 25-mm epoxy-activated microscope glass slides) to perform several identical antibody arrays on each slide.
- Spot each purified antibody, including controls and the reference frame, in a triplicate spot pattern; under these conditions, the spots are 180 - 200 µm in diameter.
- After printing, leave the slides for at least 30 min at ambient temperature to let them dry, and then store them at 4 °C; for working in the field, the slides can be transported and stored at ambient temperature for several months. NOTE: The CYANOCHIP is printed in a triplicate spot microarray format with 3 x 8 identical microarrays per slide or 9 identical arrays in a 1 x 9 microarray format. Each array size must not be higher than the reaction chamber dimensions for a 24-well gasket (usually 7.5 x 6.5 mm in a 3 x 8 hybridization chamber).
- Before using the microarray to analyze environmental samples, use FSMI to determine the working dilution for each antibody in a titration curve. For each antibody, use a standard concentration of the corresponding immunogen (103 - 104 cells/mL) and serial dilutions of the fluorescent antibody (between 1:500 and 1:32,000). The optimal antibody concentration corresponds to 50% of the maximum signal intensity obtained in the titration curve. Also, the sensitivity and specificity for each antibody must be determined, as described in Blanco et al.22.
6. Preparation of Environmental Multianalyte Extracts for the Fluorescent Sandwich Microarray Immunoassay (FSMI)
- Multianalyte extract from a liquid sample
- Take 1 - 100 mL of the liquid sample with a sterile syringe (e.g., water from the shore of a water reservoir); the amount of sample is greatly dependent upon the potential concentration of the targets.
- Concentrate the cells by passing the water sample through a 3-µm pore size, 47-mm diameter polycarbonate filter; a cellular concentration between 103 - 108 cells/mL is desirable for positive detection, but the actual concentration is unknown.
- Recover the biomass collected in the filter with 1 mL of a modified Tris-buffered saline, Tween 20-reinforced buffer (TBSTRR; 0.4 M Tris-HCl (pH 8), 0.3 M NaCl, and 0.1% Tween 20) by scraping it with a spatula into a 15-mL tube.
- Homogenize and disaggregate by using a hand-held ultrasonic processor, as described in step 1.5, or just by pipetting up and down multiple times; this prepares the sample for analysis by the microarray.
- Multianalyte extract from a solid sample
- Weigh up to 0.5 g of the solid sample (e.g., rock, soil, or sediments) into a 10-mL tube and add up to 2 mL of TBSTRR.
- Sonicate by immersing the sonicator probe in the tube, by dipping the tube into the water bath of a powerful sonicator horn, or by using a hand-held sonicator. Perform at least 5 x 30-s cycles at 30 kHz, stopping for 30 s while on ice .
- Filter to remove sand, clay, and other coarse material with a 10-mL syringe coupled to a 10- to 12-mm diameter, 5- to 20-µm pore size nylon filter holder. Push the sample through the filter into a 1.5-mL tube. If the filter saturates, agitate the suspension in the syringe and take it to a new one; this prepares the filtrate material for the immunoassay (step 7). NOTE: The buffering capacity of TBSTRR depends on the type of sample. It is important to carry out the immunoassay immediately after the preparation of the environmental extract to avoid the effect of enzymatic degradation on the analytes. Alternatively, add protease inhibitors into the environmental extract and freeze at -80 °C until the next step.
7. Fluorescent Sandwich Microarray Immunoassay (FSMI)
- Blocking the CYANOCHIP NOTE: Immediately before use, treat the printed slides to block all free epoxy groups on the slide and to remove the excess of non-covalently bound antibodies.
- Immerse the microarray into 0.5 M Tris-HCl (pH 9) with 5% (w/v) BSA solution on a clean surface (e.g., Petri dish or a 50-mL tube) with mild agitation from a rocker platform for 5 min. Alternatively, lay the slide down onto a 100- to 200-µL drop of the above solution with the microarray spots facing it. Leave for 3 - 5 min and then proceed.
- Carefully pick up the slide using plastic-tipped forceps; try to avoid touching microarray zones. Eliminate the excess of liquid by softly knocking the slide onto a paper towel. Immerse it in 0.5 M Tris-HCl (pH 8) with 2% (w/v) BSA solution for 30 min with mild agitation from a rocker platform.
- Dry the slide by performing a short centrifugation (200 - 300 x g for 1 min) using a commercial microcentrifuge adapted for microscope slides. Alternatively, dry the slide by softly knocking onto a paper towel.
- Incubation of the multianalyte sample extract with the microarray
- Set up the slide in a commercial microarray hybridization cassette with 24 wells for multiple microarrays; follow the provider instructions.
- After slide and cassette assembly, pipette up to 50 µL of the sample extract or a dilution of it in TBSTRR into each well of the cassette.
- Repeat step 7.2.2 for each sample to be analyzed.
- As a blank control, pipette 50 µL of TBSTRR buffer into at least two separate wells of the cassette.
- Incubate at ambient temperature for 1 h with mixing by pipetting every 15 min or by leaving it under mild shaking. Alternatively, incubate for 12 h at 4 °C. NOTE: Use other incubation gaskets as a function of the microarray pattern. The time and temperature of the incubation in step 7.2.5 are empirical parameters that normally depend on the affinity and binding kinetics of each paired antigen-antibody.
- Washing
- Remove the samples by putting the cassette down and carefully knocking it onto a clean, absorbent paper.
- Wash the wells by adding 150 µL of TBSTRR to each one, and eliminate the buffer, as above.
- Repeat step 7.3.2 three more times.
- Incubation with fluorescent detector antibodies
- Add 50 µL of an antibody mixture containing the 17 anti-cyanobacterial-strain antibodies, each labeled with the fluorochrome in TBSTRR with 1% (w/v) BSA. Determine the concentration of each fluorescent antibody in the mixture (from 0.7 to 2 µg/mL)by performing titration experiments of each antigen/antibody pair22.
- Incubate for 1 h at ambient temperature, as described in step 7.2.5, or for 12 h at 4 °C.
- Washing out the fluorescent antibodies
- Remove the fluorescent unbound antibodies, as in step 7.3.
- Disassemble the cassette and immerse the slide into 0.1x PBS (e.g., in a 50-mL tube) for a quick rinse.
- Dry the slide as in step 7.1.3.
8. Scanning for Fluorescence
Scan the slide for fluorescence at the maximum emission fluorescence peak for far-red fluorescent dye in a scanner for fluorescence. Take several images of the microarrays at different scanning parameters, generally by lowering the laser gain value. NOTE: Avoid saturated spots (> 65,000 fluorescence counts)—they are out of scale and may introduce quantification errors.
9. Image Processing and Data Analysis
Use a commercial software for image analysis and quantification; the software provides measurements of the fluorescence intensity (FI; median or mean of all pixels from a single spot) for each spot of the whole microarray. Subtract the local background around the spots: FI = FIspot - FIlocal background.
Save the FI data and open them using a spreadsheet program.
Use the values obtained for the blank arrays as the negative control to identify and discard the false positives. Apply the following equation to calculate the FI for each antibody spot: FI = (FIsample - FIblank), where FIsample = FIspot - FIlocal background in the microarrays run with sample extracts and FIblank = FIspot - FIlocal background in the blank microarrays.
Apply an additional cut-off threshold of 2- to 3-fold the average of the FI of the whole microarray to minimize the probability of false positives; this is especially relevant for poor-quality microarray images and low signal-to-noise ratios.
Create plots and/or perform further analysis as necessary.
Representative Results
This work describes a multiplex immunoassay test for the simultaneous identification of the most relevant freshwater cyanobacterial species (Table 1) using the CYANOCHIP antibody microarray. The microarray can be a 3 x 8 microarray format printed onto microscope slides. Each microarray is made up of a set of 17 antibodies printed in a triplicate spot pattern, their corresponding pre-immune antibodies and BSA as negative controls. The microarrays also include a fluorescent frame, using a fluorescently labeled pre-immune antibody to easily localize the microarray pattern22 (Figure 1).
The microarray was tested in the field for the in situ analysis of water samples collected at the shore of the freshwater Lozoya Reservoir, which supplies the city of Madrid. Samples were processed as described above, and the main positive immunoreactions corresponded to antibodies to planktonic cyanobacteria, such as Microcystis spp. (K4 and K5), and to benthic Oscillatoriales, such as Leptolyngbya spp. (K10 and K15). Lower fluorescence signals were obtained from Nostocales (K6 and K12, two planktonic Aphanizomenon spp.), benthic Rivularia sp. (K8), Anabaena sp. (K1), and planktonic Microcystis flos-aquae sp. (K3). Optical microscope observations (not shown) showed a diverse cyanobacterial community within abundant terrigenous debris from the shore. Several species of Anabaena dominated the community, but Microcystis spp. and Pseudanabaena spp. were also present.
Additionally, the microarray was also validated by assaying a dry, nearly 1,000-year-old microbial mat collected in the McMurdo Ice Shelf in Antarctica to test for the presence of cyanobacterial markers. Figure 1 shows highly fluorescent, positive reactions in antibodies produced to benthic cyanobacteria isolated from other Antarctic mats, including Anabaena sp. and Leptolyngbya spp. (K14 and K15, respectively). Additional positive immunoreactions were detected with antibodies to planktonic cyanobacteria, such as Microcystis spp. (K4 and K5), Aphanizomenon spp. (K6 and K12), and Planktothrix rubescens sp. (K17). Low signals were obtained from antibodies to other benthic species isolated in an Antarctic mat, including Tolypothrix sp. (K16) and Anabaena sp. (K1). Fluorescent optical microscopy revealed the presence of cells structurally similar to cyanobacteria and green algae (not shown). No filamentous cyanobacteria were identified. Nevertheless, multiple amorphous fluorescent structures of about 1 µm in size and copious diffuse fluorescence were detected, which could be attributed to the presence of cell remains (broken and dead cells) and extracellular polymeric substances (EPS), respectively. Accordingly, the biochemical analysis showed that the mat consisted of profuse amounts of biopolymers and cell remains (complex biological matter) that could be the targets for the antibodies in the microarray.
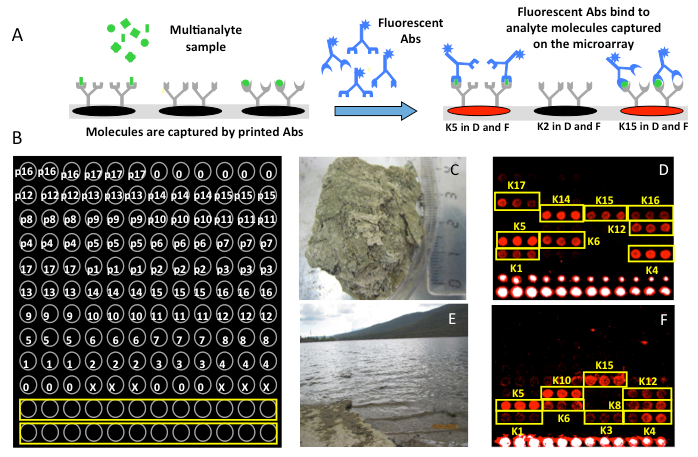 Figure 1:Quick and Reliable Multiplex Microarray Immunoassay for DetectingCyanobacteria. A) Scheme showing the main steps of the FSMI for the analysis of multi-target samples with a CYANOCHIP. B) Schematic of a printing pattern layout (by triplicate) of the anti-cyanobacteria antibody collection: (0) BSA, bovine serum albumin; (X) only printing buffer; (1-17) each of the antibodies as in Table 1 in Blanco et al. 2015 (K1 to K17). From p1 to p17, the corresponding pre-immune antibodies act as controls. The yellow rectangles correspond to a fluorescent spot gradient as a frame reference. C and D) Picture showing the top part of a 1,000-year-old dry microbial mat from the McMurdo Ice Shelf (Antarctica) and the CYANOCHIP image detecting cyanobacteria in this mat, respectively. E) Panoramic view of the Lozoya Reservoir showing transparent water with no visible green particles at the time of sampling. F) microarray image after the in situ analysis of the water sample (modified from reference 22).
Figure 1:Quick and Reliable Multiplex Microarray Immunoassay for DetectingCyanobacteria. A) Scheme showing the main steps of the FSMI for the analysis of multi-target samples with a CYANOCHIP. B) Schematic of a printing pattern layout (by triplicate) of the anti-cyanobacteria antibody collection: (0) BSA, bovine serum albumin; (X) only printing buffer; (1-17) each of the antibodies as in Table 1 in Blanco et al. 2015 (K1 to K17). From p1 to p17, the corresponding pre-immune antibodies act as controls. The yellow rectangles correspond to a fluorescent spot gradient as a frame reference. C and D) Picture showing the top part of a 1,000-year-old dry microbial mat from the McMurdo Ice Shelf (Antarctica) and the CYANOCHIP image detecting cyanobacteria in this mat, respectively. E) Panoramic view of the Lozoya Reservoir showing transparent water with no visible green particles at the time of sampling. F) microarray image after the in situ analysis of the water sample (modified from reference 22).
| Ab code | Immunogen (strain) | Order | Habitat | Medium | Culture conditions |
| K1 | Anabaena sp. | Nostocales | unknown | BG11 and nitrate | 30 ºC, continuous light |
| K2 | Anabaena sp. | Nostocales | unknown | BG11o | 30 ºC, continuous light |
| K3 | Microcystis flos-aquae | Chroococcales | planktonic | BG11 | 28 ºC, continuous light |
| K4 | Microcystis novacekii | Chroococcales | planktonic | BG11 | 28 ºC, continuous light |
| K5 | Microcystis aeruginosa | Chroococcales | planktonic | BG11 | 28 ºC, continuous light |
| K6 | Aphanizomenon ovalisporum | Nostocales | planktonic | BG11o | 28 ºC, continuous light |
| K7 | Phormidium sp. | Oscillatoriales | benthic | BG11 | 18 ºC, 16-8 photoperiod |
| K8 | Rivularia sp. | Nostocales | benthic | CHU-D | 18 ºC, 16-8 photoperiod |
| K9 | Chamaesiphon sp. | Chroococcales | benthic | BG11 | 18 ºC, 16-8 photoperiod |
| K10 | Leptolyngbya boryana | Oscillatoriales | benthic | BG11 | 18 ºC, 16-8 photoperiod |
| K11 | Tolypothrix distorta | Nostocales | benthic | BG11o | 18 ºC, 16-8 photoperiod |
| K12 | Aphanizomenon aphanizomenoides | Nostocales | planktonic | BG11o | 28 ºC, continuous light |
| K13 | Nostoc sp. (Antarctica) | Nostocales | benthic | BG11o | 13 ºC, 16-8 photoperiod |
| K14 | Anabaena sp. | Nostocales | benthic | BG11o | 13 ºC, 16-8 photoperiod |
| K15 | Leptolyngbya sp. | Oscillatoriales | benthic | BG11 | 13 ºC, 16-8 photoperiod |
| K16 | Tolypothrix sp. | Nostocales | benthic | BG11 | 13 ºC, 16-8 photoperiod |
| K17 | Planktothrix rubescens | Oscillatoriales | planktonic | none | none |
Table 1. List of the Antibodies (Abs) and the Cyanobacterial Strains Used to Produce the CYANOCHIP22.
Discussion
Here, a multiplex fluorescent sandwich immunoassay using the CYANOCHIP, a 17-antibody microarray for the detection and identification of a wide range of cyanobacterial genera, is described22. These cyanobacteria represent the most frequent benthic and planktonic genera in freshwater habitats, some of them being toxin producers. Recently, the fluorescent sandwich immunoassay format has been used to identify microorganisms and/or bioanalytes in environmental applications26,27,28. The protocol is primarily based on two steps: (i) immobilizing or capturing antibodies to specifically bind analytes from a test sample and (ii) detecting analyte-antibody pairs by using fluorescently labeled antibodies (tracer or detector antibodies). Because the sandwich assay requires at least two accessible antibody binding sites (epitopes) in the analyte for the reaction to take place, any positive fluorescent signal indicates the presence of relatively large and complex oligo- or multimeric analytes that are identical or highly similar to the ones used to produce the capturing antibodies.
Even though this method requires small volumes and basic sample preparation, without the need for special expertise or knowledge, several drawbacks could ruin the assay. Low fluorescent signals or a complete lack thereof may be due to poor antibody purification or poor labeling efficiency. For the isolation of rabbit IgG, protein A is the best choice, because it specifically binds with high efficiency to the Fc region of immunoglobulins. As indicated in section 4, fluorescent antibodies with a labeling range between 3-7 mol of dye per mole of antibody must be used. Furthermore, a lack of fluorescent spots can be explained by the concentration of analytes lying under the limits of detection. In this case, the sample can be concentrated before incubating it with the microarray, or greater amounts can be used for a new extraction. High fluorescent backgrounds are the result of inefficient chip blocking or/and washing steps and of extracts from samples composed of minerals and complex organic matter that stick onto the chip. When complex samples are used as analytes, it is desirable to increase the salt and/or the detergent concentration to favor the specific interaction between analyte-antibody pairs.
In recent years, antibody microarray technology has been developed for environmental applications. Nonetheless, the use of this technique does involve some limitations. Polyclonal antibodies are faster and cheaper to produce and, more importantly, the possibility to bind any target epitope in a complex environmental sample are theoretically higher than with monoclonal antibodies. However, the fact that they can recognize different epitopes increases the number of cross-reactions in FSMI. To gain high specificity, the use of methods to disentangle these cross-reactivity events from the true cognate antigen-antibody reactions by using deconvolution methods26,27, for example, is highly desirable. Basically, the microarray is considered a qualitative biosensor for the multiple detection and classification of cyanobacteria. In this regard, it is essential to determine the limit of detection for each antibody one-by-one to use their optimal working concentrations in the assay. Although this microarray, together with FSMI, is not conceived as a quantitative method, the biosensor implies high sensitivity, because the lower detection limit of most of the antibodies contained in the CYANOCHIP22 is from 102 to 103 cells/mL.
Despite these limitations, this methodology has several advantages against other techniques used in environmental monitoring. Antibody-biosensors allow the possibility of recognizing different molecules simultaneously in a single analysis, the possibility of detecting low concentration of analytes, and the possibility of producing antibodies against almost any substance. Furthermore, the microarray can achieve up to 24 analyses in a single assay, with limits of detection from 102 cells/mL. In comparison with other methods, antibodies can detect living or dead cells, extracellular material, and cellular debris. Although it takes at least 4 h to complete the whole assay, it is faster than other analytical methods applied to environmental monitoring. The CYANOCHIP was originally conceived for the identification of cyanobacterial strains. This biosensor was not formally designed for that purpose, so it can identify potential toxin producers and could be improved in the future by adding antibodies against a wide range of new strains. Biochemical assays, such as ELISA, PPIA, and neurochemical tests in mice, allow the identification of cyanotoxins, but they are restricted to a few known toxins and can give false positives. In addition, using the CYANOCHIP does not require special training, while optical microscopy and mouse bioassays require trained personnel or labor-intensive work with live animals, and they do not give information about the type of toxins present in a sample. Cyanotoxins can also be identified and quantified by other analytical methods, such as HPLC, GC-MS, or MALDI-TOF. In these methods, the sample must be purified, and the lack of reference standards limits the identification of cyanotoxins. Furthermore, analytical methods require costly equipment and supplies and specialized training. Molecular methods are based on DNA extraction from the samples, while FSMI only requires an environmental extract prepared in a couple of very simple steps, without cyanobacteria purification.
The high performance of the CYANOCHIP for the in situ detection of cyanobacteria in freshwater reservoirs makes it a new tool for the early warning of water administrators. In addition, the microarray is also interesting for the field of astrobiology, particularly for searching for microbial markers as evidence of life. The study of extremophiles can help us to understand the origin of life on Earth and how life could survive in the extreme environments present in our solar system and beyond. As several environments on Earth are very similar to places on other planets, such as Mars, it might be possible to find remains of photosynthetic prokaryotes as evidences of extinct life. The microarray was able to identify cyanobacterial markers from living or nonliving cells; from population remains and/or extracellular material in old microbial mats (Figure 1); and from water, soil, and rocks collected in extreme environments, such as the Antarctica, Atacama, the Andean lakes, the High Arctic, or the Rio Tinto area in Spain (not shown). Considering that cyanobacteria are the primeval microorganisms on Earth, there is reason to believe that they might once have lived on other planets.
In conclusion, the fact that CYANOCHIP-FSMI can identify in situ cyanobacterial markers and can even associate them to different phylotypes or groups, and that the microarray covers a broad range of habitats, including those of plankton, benthos, and endoliths, demonstrates that this technique could a tool for environmental monitoring. Future improvements to the microarray could be to increase the number of antibodies to new strains so that relevant phylogenetic groups still pending are included. Additionally, the chip can be implemented with antibodies to specific cyanobacterial compounds, such as toxins or cyanophicin polymer. This is especially relevant for monitoring fresh water reservoirs, pipes, and installations in human facilities. The current microarray and future versions will be very useful in the field of astrobiology, either for life detection or for monitoring human space facilities (e.g., monitoring water reservoirs or life support systems). In fact, we routinely use the microarray in field campaigns to extreme environments as a method for the "on-site" detection of life remains. The microarray is part of the so-called Life Detector Chip (LDChip), a microarray with more than 300 antibodies for the search for life in planetary exploration missions29,30. The microarray alone or as part of the LDChip will be implemented in the Signs of Life Detector (SOLID) instrument29 to validate the SOLID-LDChip concept for planetary exploration in multiple field campaigns to terrestrial analogues. The microarray will provide useful information by identifying cyanobacteria and/or their toxins. By determining the strains, it will give information about the environments and the habitats in which they developed.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Dr. Antonio Quesada from the Universidad Autónoma de Madrid for providing cyanobacterial strains. This work was funded by the Subdirección General de Proyectos de Investigación of the Spanish Ministerio de Economía y Competitividad (MINECO), grants no. AYA2011-24803 and ESP2014-58494-R.
References
- Simis SGH, Peters SWM, Gons HJ. Remote sensing of the cyanobacterial pigment phycocianin in turbid inland water. Limnol. Oceanogr. 2005;50(1):237–245. [Google Scholar]
- Zamyadi A, McQuaid N, Prevost M, Dorner S. Monitoring of potentially toxic cyanobacteria using an online multi-probe in drinking water sources. J. Environ. Monit. 2012;14:579–588. doi: 10.1039/c1em10819k. [DOI] [PubMed] [Google Scholar]
- Msagati TAM, Siame BA, Shushu DD. Evaluation of methods for the isolation, detection and quantification of cyanobacterial hepatotoxins. Aquat. Toxicol. 2006;78(4):382–397. doi: 10.1016/j.aquatox.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Weller MG. Immunoassays and biosensors for the detection of cyanobacterial toxins in water. Sensors. 2013;13(11):15085–15112. doi: 10.3390/s131115085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JA, Neilan BA, Entsch B, McKay DB. Identification of cyanobacteria and their toxigenicity in environmental samples by rapid molecular analysis. Environ. Toxicol. 2001;16(6):472–482. [PubMed] [Google Scholar]
- Li H, Singh AK, McIntyre LM, Sherman LA. Differential gene expression in response to hydrogen peroxide and the putative PerR regulon of Synechocystis sp. Strain PCC 6803. J. Bacteriol. 2004;186(11):3331–3345. doi: 10.1128/JB.186.11.3331-3345.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Cembella AD, Hallegraeff GM. Progress in understanding harmful algal blooms: paradigm shifts and new technologies for research, monitoring, and management. Ann. Rev. Mar. Sci. 2012;4:143–176. doi: 10.1146/annurev-marine-120308-081121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittami SM, Pazos Y, Laspra M, Medlin LK. Microarray testing for the presence of toxic algae monitoring programme in Galicia (NW Spain) Environ. Sci. Pollut. Res. Int. 2013;20:6778–6793. doi: 10.1007/s11356-012-1295-0. [DOI] [PubMed] [Google Scholar]
- Kegel JU, Del Amo , Medlin Y, K L. Introduction to Project MIDTAL: its methods and samples from Arcachon Bay, France. Environ. Sci. Pollut. Res. Int. 2013;20(10):6690–6704. doi: 10.1007/s11356-012-1299-9. [DOI] [PubMed] [Google Scholar]
- Marcheggiani S, et al. Detection of emerging and re-emerging pathogens in surface waters close to an urban area. Int. J. Environ. Res. Public Health. 2015;12(5):5505–5527. doi: 10.3390/ijerph120505505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Calvo P, Näke C, Rivas LA, García-Villadangos M, Gómez-Elvira J, Parro V. A multi-array competitive immunoassay for the detection of broad-range molecular size organic compounds relevant for astrobiology. Planet. Space Sci. 2006;54(815):1612–1621. [Google Scholar]
- Parro V. Antibody microarrays for environmental monitoring. Handbook of Hydrocarbon and Lipid Microbiology. Berlin Heidelberg, Germany: Springer-Verlag; 2010. pp. 2699–2710. [Google Scholar]
- Fan Z, Keum YS, Li QX, Shelver WL, Guo LH. Sensitive immunoassay detection of multiple environmental chemicals on protein microarrays using DNA/dye conjugate as a fluorescent label. J. Environ. Monit. 2012;14(5):1345–1352. doi: 10.1039/c2em10956e. [DOI] [PubMed] [Google Scholar]
- Rivas LA, García-Villadangos M, Moreno-Paz M, Patricia Cruz-Gil P, Gómez-Elvira J, Parro V. A 200-Antibody microarray biochip for environmental monitoring: Searching for universal microbial biomarkers through immunoprofiling. Anal. Chem. 2008;80(21):7970–7979. doi: 10.1021/ac8008093. [DOI] [PubMed] [Google Scholar]
- Delehanty JB, Ligler FS. A microarray immunoassay for simultaneous detection of proteins and bacteria. Anal. Chem. 2002;74(21):5681–5687. doi: 10.1021/ac025631l. [DOI] [PubMed] [Google Scholar]
- Ligler FS, Taitt CR, Shriver-Lake LC, Sapsford KE, Shubin Y, Golden JP. Array biosensor for detection of toxins. Anal. Bioanal. Chem. 2003;377(3):469–477. doi: 10.1007/s00216-003-1992-0. [DOI] [PubMed] [Google Scholar]
- Rucker VC, Havenstrite KL, Herr AE. Antibody microarrays for native toxin detection. Anal. Biochem. 2005;339(2):262–270. doi: 10.1016/j.ab.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Kang J, Kim S, Kwon Y. Antibody-based biosensors for environmental monitoring. Toxicol. Environ. Health. Sci. 2009;1(3):145–150. [Google Scholar]
- Herranz S, Marazuela MD, Moreno-Bondi MC. Automated portable array biosensor for multisample microcystin analysis in freshwater samples. Biosens. Bioelectron. 2012;33(1):50–55. doi: 10.1016/j.bios.2011.12.016. [DOI] [PubMed] [Google Scholar]
- Shlyapnikov Y, et al. Rapid simultaneous ultrasensitive immunodetection of five bacterial toxins. Anal. Chem. 2012;84(13):5596–5603. doi: 10.1021/ac300567f. [DOI] [PubMed] [Google Scholar]
- Szkola A, et al. Rapid and simultaneous detection of ricin, staphylococcal enterotoxin B and saxitoxin by chemiluminescence-based microarray immunoassay. Analyst. 2014;139:5885–5892. doi: 10.1039/c4an00345d. [DOI] [PubMed] [Google Scholar]
- Blanco Y, Quesada A, Gallardo-Carreño I, Jacobo Aguirre , Parro J, V CYANOCHIP: An antibody microarray for high-taxonomical-resolution cyanobacterial monitoring. Environ. Sci. Technol. 2015;49(3):1611–1620. doi: 10.1021/es5051106. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Chem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- Smith PK, et al. Measurement of protein using Bicinchoninic acid. Anal. Biochem. 1985;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Rivas LA, Aguirre J, Blanco Y, González-Toril E, Parro V. Graph-based deconvolution analysis of multiplex sandwich microarray immunoassays: applications for environmental monitoring. Environ. Microbiol. 2011;13(6):1421–1432. doi: 10.1111/j.1462-2920.2011.02442.x. [DOI] [PubMed] [Google Scholar]
- Blanco Y, et al. Deciphering the prokaryotic community and metabolisms in south african deep-mine biofilms through antibody microarrays and graph theory. PloS One. 2014;9(2):e114180. doi: 10.1371/journal.pone.0114180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gas F, Baus B, Queré J, Chapelle A, Dreanno C. Rapid detection and quantification of the marine toxic algae, Alexandrium minutum, using a super-paramagnetic immunochromatographic strip test. Talanta. 2016;147:581–589. doi: 10.1016/j.talanta.2015.10.036. [DOI] [PubMed] [Google Scholar]
- Parro V, et al. SOLID3: a multiplex antibody microarray-based optical sensor instrument for in situ life detection in planetary exploration. Astrobiology. 2011;11:15–28. doi: 10.1089/ast.2010.0501. [DOI] [PubMed] [Google Scholar]
- McKay CP, et al. The Icebreaker Life Mission to Mars: a search for biomolecular evidence for life. Astrobiology. 2013;13:334–353. doi: 10.1089/ast.2012.0878. [DOI] [PubMed] [Google Scholar]