

Abstract
When novel proteins are identified through affinity-based isolation and bioinformatics analysis, they are often largely uncharacterized. Antibodies against specific peptides within the predicted sequence allow some localization experiments. However, other possible interactions with the antibodies often cannot be excluded. This situation provided an opportunity to develop a set of assays dependent on the protein sequence. Specifically, a construct containing the gene sequence coupled to the GFP coding sequence at the C-terminal end of the protein was obtained and employed for these purposes. Experiments to characterize localization, ligand affinity, and gain of function were originally designed and carried out to confirm the identification of TMEM184A as a heparin receptor1. In addition, the construct can be employed for studies addressing membrane topology questions and detailed protein-ligand interactions. The present report presents a range of experimental protocols based on the GFP-TMEM184A construct expressed in vascular cells that could easily be adapted for other novel proteins.
Keywords: Genetics, Issue 120, GFP, TMEM184A, Heparin, Vascular Cells, Exogenous Expression, Co-localization
Introduction
Identification of candidate proteins for novel functions often depends upon affinity-based isolation protocols followed by partial sequence determination. Recent examples of newly identified proteins include transmembrane protein 184A (TMEM184A), a heparin receptor identified after heparin affinity interactions1, and TgPH1, a pleckstrin homology domain protein that binds phosphoinositide PI(3,5)P22. Other novel protein identification involves direct sequence analysis of peptides such as that by Vit, et al. who used transmembrane peptides to identify protein products from previously uncharacterized genes3. Similarly, identification of novel protein sequences can be accomplished using bioinformatics searching of previously characterized protein families such as identification of new 4TM proteins4. Examination of aquaporin family gene sequences has also yielded the identification of new members with novel functions5. After identification, analysis of protein function is typically a next step which can sometimes be examined using a specific assay of protein function such as in the aquaporin case.
When possible, function of a newly identified protein can be examined with specific enzymatic or similar in vitro function assays. Because many functions of novel proteins depend on complex interactions that occur only in intact cells or organisms, in vitro assays are not always effective. However, the in vivo assays must be designed in such a way that they depend on the gene sequence. In cell culture, and/or simple model organisms, knockdown can provide supporting evidence for the protein/function identification6. With novel proteins identified as noted above, it is often insufficient to simply knock down a protein to confirm function, and the design of in vivo functional assays that depend on gene sequence becomes important for the characterization of novel proteins.
The recent identification of TMEM184A as a heparin receptor (that modulates proliferation in vascular smooth muscle and inflammatory responses in endothelial cells) using affinity chromatography and MALDI MS1,7 provided an opportunity to develop a collection of assays after knockdown yielded results consistent with the identification. A recent review confirmed that heparin interacts specifically with many growth factors, their receptors, extracellular matrix components, cell adhesion receptors, and other proteins8. In the vascular system, heparin and heparan sulfate proteoglycans (containing heparan sulfate chains similar in structure to heparin) interact with several hundred proteins9. To functionally confirm that TMEM184A was involved with heparin uptake and binding, techniques that employed the gene construct for TMEM184A were developed. The present report includes a collection of assays based on a GFP-TMEM184A construct for use in confirming the identity of TMEM184A as a heparin receptor.
Protocol
1. Design of a GFP-protein Construct
Purchase, or design and build, a GFP-tagged construct based on the protein in question. NOTE: For a purchased construct, standard vectors are available from commercial laboratories that include some or all of the following suggestions: For a membrane protein, select a C-terminal location of the GFP because it is less likely to interfere with membrane protein trafficking. Consider an extension between the gene of interest and the GFP if there is reason to believe the C-terminus of the protein in question is compactly folded into a domain of the protein. Select the construct for general eukaryotic cell expression, but allow construct production in bacterial systems. Include a cleavage site (such as for the TEV protease) by which GFP can be removed if desired for some experiments where the GFP could interfere with the protein activity. Add other inserts such as an additional tag for localization or affinity interactions (e.g., His6). This was not necessary for the assays described herein, but might facilitate other assays, or be useful directly adjacent to the gene, before the GFP, if removal of GFP is desired.
2. GFP-TMEM184A Expression in Vascular Cells
Culture endothelial or vascular smooth muscle cells on 0.2% gelatin coated tissue culture dishes as reported previously1,6,7.
Add 5 mL cell culture trypsin solution (0.5% w/v) to a rinsed 100 mm, or larger, confluent dish of cells. Incubate at 37 °C until cells are just released from the plate, and transfer the cells to a sterile polypropylene centrifuge tube.
Add a trypsin inhibitor (e.g., an equal volume of regular culture medium), as used in routine culture, to the cell/trypsin solution to limit trypsin activity. Pellet the cells for 5 min at approximately 600 x g (or appropriate speed and time to just pellet the cells for standard tissue culture). Aspirate the supernatant.
Resuspend cells in 1 mL of HeBS (Hepes-buffered Saline) electroporation buffer, limiting the amount of time cells are in the pellet or suspension. Place the cell suspension on ice and carry out remaining steps on ice. NOTE: Pellet can be washed one time with HeBS prior to resuspension.
Add sufficient volume of GFP-tagged TMEM184A plasmid to the cells to achieve a final concentration of 20 µg/mL DNA. Use an identical protocol for a GFP construct.
Place approximately 0.4 mL of the cell solution in HeBS in an electroporation cuvette. Pre-chill the cuvettes prior to use.
- Electroporate the cells using the following conditions: Exponential Decay, 500 µF, ∞ ohm, and 170 V. NOTE: The voltage for a given cell type should be optimized. Preliminary studies with endothelial and smooth muscle cell types were accomplished with a GFP-vinculin construct to determine the optimal voltage value noted here, e.g.10.
- To optimize electroporation, start with the recommendations of the equipment manufacturer (which include conditions for some standard cell types). Use the desired construct or a control fluorescent protein construct similar in size and fluorescent characteristics to the desired construct. NOTE: Small nucleic acids appear to enter cells more readily than larger constructs, so a construct with only a fluorescent protein may be easier to express than a larger one.
- Use fluorescence microscopy to determine cell viability, the percentage of cells in which the fluorescent construct is expressed after 24 and 48 h, and the intensity of expression. NOTE: Decrease the voltage and/or time slightly if survival is low. Aim for the shortest time possible between release of cells from the culture surface and returning cells to culture to enhance survival. Slight increases in time or a second pulse can improve construct uptake if survival is high and expression is low. Optimum conditions and expression will vary between cell types. If the percentage of cells expressing the construct is high, but intensity is low, increasing the DNA concentration and monitoring the transfected cells over time for optimal intensity, which will vary based on protein half-life as well as expression, may improve intensity. Staining intensity can be magnified for some assays, if necessary, by immunofluorescence staining of GFP with anti-GFP antibodies
After electroporation, seed the cells into six 30 mm tissue culture wells with coverslips for imaging or into tissue culture dishes. Culture the cells using standard cell culture procedures.
Optional: Replace the culture medium after 24 h to remove any HeBS electroporation buffer.
3. Visualization of GFP-TMEM184A Localization
Rinse the cells with PBS and gentle shaking after culturing for at least 24 h. Limit exposure to bright light to avoid GFP fading.
Fix cells with 4% paraformaldehyde (PFA) in PBS for 15 min at RT with gentle shaking. Avoid using any methanol-containing reagents which may permeabilize the cells. Fixation with methanol can be used if it is not necessary to assess ligand interactions. Caution: Paraformaldehyde is toxic. Wear appropriate personal protective equipment. Use in a fume hood, with care, and discard properly.
Rinse with PBS as above. For antibody based staining, see 3.6 below.
Mount coverslips to slides using Mowiol or other suitable mounting medium.
Image slides using a confocal or fluorescence microscope with the appropriate filter sets for GFP excitation and emission spectra. Confocal microscopy is preferred because of the ability to separate samples in the z-plane. Save images in grey scale for quantitation purposes in TIF format (in addition to full color images for illustrations).
For comparison with WT TMEM184A expressed by the cells, prepare other cells for immunofluorescence using primary antibodies prepared to a peptide(s) from the TMEM184A sequence, e.g.1. Identification of GFP-TEMEM184A can also be accomplished using antibodies against GFP. Evaluate both cell samples by identical microscopy techniques.
4. Rhodamine-Heparin Binding and Colocalization with GFP-TMEM184A-transfected Cells
Treat GFP-TMEM184A-expressing cells with 100 µg/mL rhodamine-heparin added to culture medium, and allow cells to incubate for a specific amount of time, typically less than 10 min for A7r5 cells. Rinse and fix as in 3.1 and 3.2. NOTE: The optimal time and concentration of ligand depend on cell response, fluorescence intensity of the ligand and image intensity obtained. This concentration of heparin was employed because it results in more than 50% response in other assays11. This concentration could be visualized using standard fluorescence microscopy techniques, so it was not necessary to increase it. Dilution of rhodamine-heparin with unlabeled heparin results in lower fluorescence in the cells, thus is more difficult to image and quantify.
To quantitate the rhodamine-heparin binding due to GFP-TMEM184A transfection, prepare identical cells without transfected GFP-TMEM184A.
Image the cells using a confocal microscope with the appropriate GFP (488 nm) and rhodamine (543 nm) excitation and emission (500 - 530 nm for GFP and greater than 560 nm for rhodamine) values to determine co-localization. Obtain images of at least 50 cells from at least three separate experiments to obtain statistics. Maintain identical settings within experiments, and employ a control sample(s) (e.g., transfected cells with no treatment) that can be used to standardize data for analysis of multiple experiments.
- Examine the images using a computer program to determine relative rhodamine binding/uptake in each cell for quantitation of binding. NOTE: TIF images should be employed as they are convenient for analysis and can be converted to grey scale in many computer programs if they were not initially saved in that format. Image J (freeware) was employed in the present analysis and allows area and pixel intensity to be measured for any user-defined area in an image.
- Use a freehand tool to circle a cell within a fluorescent image and use the measure tool to determine the intensity within that space. NOTE: These measurements provide the needed information to calculate total intensity for a user-defined area (whole cell, nucleus, etc.) providing a way to compare different cells with different geometries. Mean intensity over an area of background can be reported, and that facilitates collection of background intensity for analysis.
- Export to a spreadsheet to calculate the area multiplied by the intensity and subtract background for that same amount of area. Average the fluorescence/cell for enough cells/experiment to obtain statistical significance.
5. Fluorescence Resonance Energy Transfer from GFP-TMEM184A to Rhodamine-Heparin
Prepare transfected cells and incubate with 200 µg/mL rhodamine-tagged ligand as in 3.1. Note that the incubation time with fluorescent ligand is cell-type dependent. Fix with PFA only, and mount slides for imaging.
First, excite at 405 nm and image for rhodamine emission (566 - 685 nm; FRET). Second, excite at 488 for GFP (image at 493 - 530), and third, excite at 561 for rhodamine (image at 566 - 685). Controls without GFP-TMEM184A or without rhodamine-heparin are critical.
6. Live-cell Imaging of Rhodamine-Heparin Uptake
Seed the GFP-TMEM184A transfected cells into individual dishes designed for live-cell imaging and treat as in protocol 3. NOTE: The number of cells required depends on cell type and density desired. To minimize the amount of time cells are in suspension, do not count cells. Seed cells from a 100 mm confluent dish into six 35 mm live imaging dishes to obtain cells near confluence in 48 h.
After 48 h, replace the medium with culture medium without phenol red.
Transfer a dish of cells to a confocal microscope with a warming stage to maintain temperature, and focus the microscope.
Pipet 100 µg/mL rhodamine-heparin into the dish and gently mix.
Immediately begin recording live images. If no cell uptake of heparin occurs within the region in view, move the dish slightly to identify cells with at least one green vesicle containing the rhodamine label. NOTE: Imaging excitation and emission specifics are the same as for the heparin uptake protocol (section 5). The time period between images was approximately 16 s. The conditions of the experiment and the microscope will typically determine the speed at which images can be obtained.
7. Isolation of GFP-TMEM184A and GFP from Cultured Cells
After removing culture medium and rinsing with PBS, add 2 mL 0.2% (w/v) CHAPS 1x PBS solution with protease inhibitors to a 150 mm plate of cells expressing GFP-TMEM184A or GFP (for control binding assays). Scrape the cells from the dish, place the cells/CHAPS solution in a 15 mL polypropylene tube on ice, and mix well by tapping. Complete all steps in bright light to ensure the GFP tag is bleached for the binding assay below.
To specifically bind GFP-TMEM184A (or an appropriate GFP control), add 2 µg/mL biotinylated anti-GFP antibody to the solution and incubate overnight at 4 °C with rocking.
Add 500 µL of streptavidin-agarose beads to at least 5 mL 0.2% CHAPS/PBS and centrifuge at approximately 600 x g for 3 to 5 min. Remove the supernatant. Repeat two more times with fresh CHAPS/PBS each time. Add beads to the antibody and cell solution and incubate overnight at 4 °C with rocking to allow the beads to bind to the biotinylated anti-GFP antibody bound to the GFP or GFP-TMEM184A.
Pellet the beads by centrifuging (as in 7.3) and remove the unbound material. Add at least 5 mL 0.2% CHAPS/PBS/protease inhibitors to wash. Repeat washing and centrifugation at least 3x to effectively remove any unbound protein.
After removing the final wash, incubate the beads with 1 mL of 0.2 M glycine/0.2% CHAPS in PBS (pH to 2.0 using HCl) on ice for 3 min to dissociate the antibody-GFP binding (resulting in free GFP-TMEM184A or free GFP). Mix gently by tapping every 30 s.
Centrifuge at approximately 600 x g and transfer the supernatant containing the purified protein to a new 15 mL tube followed by immediate neutralization with 5 mL of 1 M sodium bicarbonate (bring the pH to 7).
Concentrate the sample using a 10,000 Dalton molecular weight cut-off centrifugal concentrator (employ centrifugation speed recommended by the supplier). When the volume reaches approximately 0.5 mL, add 0.2% CHAPS/PBS. Continue concentrating and adding more 0.2% CHAPS/PBS until at least ten volumes (times the starting sample volume) of the 0.2% CHAPS/PBS have been added.
To determine an estimate of the amount of isolated GFP or GFP-tagged protein, obtain an absorbance reading at 280 nm and compare it to a standard curve prepared from bovine serum albumin or other control protein in the same buffer. NOTE: Due to extinction coefficient differences, this concentration is an estimate, but can provide an approximate protein concentration to facilitate further analysis without wasting sample. Accurate protein concentration can be determined using a micro-Lowry protein assay making certain to prepare the standard protein in the same buffer as the sample. Further analysis of the isolated protein can be accomplished by western blotting of the isolated protein (using antibody detection for GFP) and comparison to the predicted molecular weight. Alternatively, use of the TMEM184A antibodies should result in a band of the predicted construct size, but might also result in a WT TMEM184A band if dimers (or higher-order oligomers) are present in the sample. An estimate of purity can be obtained by staining a blot for total protein from the isolated sample vs total protein from an aliquot of the starting material.
8. In Vitro Heparin Binding Assay
Prepare a black avidin-coated 96-well plate by washing with 200 µL 0.2% CHAPS/PBS three times for 5 min with shaking. Prepare an identical black noncoated 96-well plate employing the same washing procedure. Prepare a layout for the experimental samples (in triplicate) to follow during the assay. Include wells for standard heparin concentrations, buffer controls, and wells with GFP samples without heparin to confirm bleaching of GFP. NOTE: If optimized isolation or ligand interaction buffers are different for the protein in question, do all of the plate preparations and washing with the optimal buffer system.
- Add 100 µL of 60 pmol/well biotinylated anti-GFP to all wells in the avidin-coated plate where GFP binding is desired. The amount of antibody is sufficient to just saturate the high-affinity avidin in the wells.
- Add buffer only to all wells used for control purposes (no GFP or GFP-TMEM184A binding desired). Seal the wells with a plate cover to prevent evaporation. Incubate for 2 h at RT, with shaking. NOTE: Volumes added to wells and incubation times are based on recommendations from the provider.
Wash all wells three times for 5 min with 200 µL 0.2% CHAPS/PBS.
Add 100 µL of 5 nmol/well GFP-TMEM184A or GFP to appropriate wells in the avidin-coated plate and incubate for 1 h at RT with shaking. Wash again as in 8.3. NOTE: This concentration of protein was chosen to ensure that all sites were saturated with excess GFP or GFP-TMEM184A. This is important to ensure that the same amount of protein is bound to each well. Lower amounts of protein might also saturate the sites, a possibility that could be examined by evaluating unbound protein remaining after incubating several concentrations of protein with the plate and comparing unbound protein and ligand binding assays.
Prepare various concentrations of fluorescein-labeled heparin, e.g., 10, 25, 50, 75, 100, 200 µg/mL, in 0.2% CHAPS/PBS. Do this and all remaining work in the dark. NOTE: Prior to preparing concentrations, specific concentrations for other ligands should be determined for the protein target in question. The lowest concentration should be detectable in the plate reader. Therefore it is important to first determine the range that can be detected accurately in the buffer system employed. Then determine the range needed to obtain measurable binding. Concentrations of fluorescein-heparin below 10 µg/mL did not consistently register in the plate reader under the buffer conditions employed. Similarly, while rhodamine-heparin such as used in the other assays, could be visualized in the plate reader, in the buffer conditions and at the concentrations required for binding, the fluorescence readings for standards were not reproducibly different with that fluorophore.
Add 100 µL of fluorescein-heparin to appropriate test wells in the avidin-coated plate and concentration standard wells in both the avidin-coated and the non-coated plate. Incubate for 10 min at RT with shaking. Keep plates in the dark (or foil covered).
Using a plate reader, record the initial fluorescence emission from the heparin in the control wells of both plates and the initial fluorescence (total) emission from wells with immobilized GFP-TMEM184A or GFP in the avidin-coated plate. If necessary, adjust the instrument gain to ensure detection of fluorescent heparin between the lowest and highest concentrations. NOTE: Make certain the plate reader reads wells from above and reads at the optimum location in the wells if that is adjustable. For the study in question, the optimum location was about 75% down the wells. Information available with the plate reader should provide suggestions that are unique to the plate reader in question for reading assays of bound sample in black plates.
Remove unbound fluorescent heparin from wells with immobilized GFP-TMEM184A or GFP and place into corresponding wells in the non-coated black plate. Immediately read the fluorescence emission.
Add 100 µL fresh 0.2% CHAPS/PBS back into wells from which the fluorescein-heparin was removed, and read fluorescence emission.
Repeat steps 8.8 - 8.9 3x. NOTE: These readings from the coated plate indicate fluorescein heparin remaining in the GFP or GFP-TMEM184A wells. If significant fluorescence is found in the third unbound sample, an additional wash should occur. If fluorescence continues to be removed in each wash, binding of ligand might be low affinity, and the conditions should be re-evaluated. The final reading constitutes the bound heparin reading.
Record fluorescence emission from the removed, unbound, samples in the noncoated plate, obtaining numbers for each wash. These are the "unbound" readings.
Use total heparin emission values obtained in 8.7 to confirm the correct levels of heparin added to each well. Subtract average background readings from the values. NOTE: Wells without any fluorescein-heparin serve as background due to antibody, GFP and buffer. Average the background repeats to obtain the background value. Subtract average background readings from the bound and unbound readings to get actual values.
Employ a plot of emission vs. total fluorescein-heparin added to determine the scale of emission vs. amount of heparin. NOTE: Antibody alone, or antibody and antigen resulted in some quenching of fluorescence. Therefore, total heparin was determined based on the total added heparin as noted in 8.7.
Plot the corrected bound heparin vs. the added heparin for the triplicates in both the GFP-TMEM184A case and the GFP case.
Determine free ligand by adding the unbound readings from all washes for a specific concentration.
Representative Results
While, in theory, transfection of any DNA construct into cells could be accomplished with lipophilic transfection reagents, previous reports indicate more effective transfection of GFP constructs into endothelial cells using electroporation12. The protocol provided here typically achieved GFP-construct expression in greater than 80% of the primary-derived endothelial cells and smooth muscle cells used. Design of the construct employed used a commercially available system that could deliver this construct rapidly. The major intended use was focused on location issues, therefore correct delivery to the membrane, along with optimal eukaryotic protein expression and continued construct production were primary considerations. Other considerations could include: different location of the GFP for differently localized proteins or proteins with a C-terminus that is part of a folded region, the possibility of wanting to remove the GFP (adding a protease cleavage site), and a secondary affinity site. The latter would provide alternate ways of purifying the GFP construct and stabilizing it on a surface for the binding assay. To confirm staining patterns for different commercial antibodies against TMEM184A, vascular cells expressing GFP-TMEM184A were compared to identical cells stained with the commercial antibodies. The length of time after transfection impacts distribution of GFP-TMEM184A, which appeared to accumulate over at least 72 h. The length of time after transfection resulting in optimal expression varies by cell type and should be optimized for each technique. Over time, more GFP label was in the peri-nuclear region. Depending on handling, GFP bleaching also occurred. The results indicated GFP-TMEM184A at the cell surface and in peri-nuclear and other vesicle structures similar to localization observed with TMEM184A antibody staining (Figure 1). The primary cells in Figure 1 were imaged 24 h after transfection, while cloned A7r5 cells were imaged 48 h post transfection. The decision to fix GFP transfected cells was made to ensure that all cells were treated identically to compare localization. Fixation likely reduced cell surface GFP fluorescence emission to some extent. The staining pattern of GFP appeared most similar to that of antibody raised against a C-terminal peptide, a logical outcome given that the GFP is at the C-terminus of the protein. Staining with an antibody targeted against a peptide in the N-terminal region was also similar, but appeared slightly more concentrated in the peri-nuclear region. Apparent differences in localization with different antibodies could be due to binding of ligand (possibly near the C-terminus in this case), which could be fixed there and limit antibody access. Alternatively, the N- and C- termini could be on opposite sides of the membrane. Possibly, newly synthesized protein would stain more effectively with an antibody relative to protein modified post-translationally. To compare total GFP-TMEM184A localization to endogenous TMEM184A, staining with antibodies to GFP provided a more accurate picture of total GFP-TMEM184A than simply imaging GFP. Images of anti-GFP antibody staining are shown in Figure 5 where the technique was also used to evaluate membrane topology.
The use of a fluorescent ligand (rhodamine-heparin) facilitated evaluation of co-localization of ligand and receptor over time, and the two labels made it possible to carry out live imaging (Figure 2 and Movie 1). Figure 2C was imaged by exciting the GFP at 405 nm, but capturing emission from the rhodamine (middle images). GFP excitation is limited at 405 nm yielding limited emission as well, but this prevented excitation of rhodamine and a false FRET signal. Similarly, the FRET-induced rhodamine emission was faint. In controls with GFP-TMEM184A expression, but no rhodamine-heparin, and others with only rhodamine-heparin there was no rhodamine emission with 405 nm excitation. Afterwards, total GFP (left images) and total rhodamine (right images) were measured using standard excitation/emission wavelengths for each fluorophore. The data in C further support the co-localization of the ligand and the GFP-TMEM184A. Rhodamine-heparin uptake in RAOEC required longer incubation as compared to A7r5 cells. No evidence for FRET was observed in cells without rhodamine-heparin or in untransfected cells. An ideal experiment would include another surface GFP-construct that should have no co-localization with the ligand.
Acquisition or recovery of function could also be accomplished with GFP-protein constructs and rhodamine-heparin. This provided the opportunity to use GFP as evidence for expression level and ligand co-localization and to quantify the interactions. Specifically, heparin uptake was increased in GFP-TMEM184A construct-transfected A7r5 cells (Figure 3). Stable knockdown cells which take up very limited rhodamine-heparin were produced1, and these provided a system where gain of function could be evaluated. Transfection of GFP-TMEM184A construct resulted in cells that could again internalize rhodamine-heparin at the level of wild-type cells or above (Figure 3). It should be noted that the "no heparin" image is of stable knockdown cells, which do express GFP as a reporter for the knockdown construct. Background fluorescence was higher in these cells, as observed in the image shown.
It is typically helpful to use isolated receptor for determination of ligand specificity to decrease the likelihood of other proteins interacting with the ligand. The use of a GFP-protein construct provides a significant advantage in this regard as the GFP tag can serve as a handle for isolation of the protein and avoids possible interactions of antibodies or other affinity reagents with additional proteins. GFP can also be expressed in identical populations of cells and isolated using the same GFP affinity procedure. This provides a control protein for ligand interaction studies. In the TMEM184A system, the GFP-TMEM184A isolated using an anti-GFP antibody affinity procedure resulted in specific, saturable heparin binding, while the control GFP did not bind any heparin (Figure 4).
Typically, GFP tags on constructs are placed at an end of a protein. The C-terminal location facilitates correct protein trafficking to a membrane. Therefore, the availability of GFP on a particular side of a membrane can provide evidence for specific topology of a membrane protein if folding and topology are not already known. Simple immunofluorescence staining with antibodies against GFP (such as in Figure 5) can provide evidence of GFP location based on antibody access in non-permeabilized cells. The anti-GFP antibodies recognize GFP even if bleached. Immunofluorescent staining shown in Figure 5 suggests significant anti-GFP staining without permeabilization of the GFP-TMEM184A transfected cells providing preliminary evidence that the GFP at the plasma membrane is extracellular. Clearly, intracellular protein in vesicles also becomes stained in permeabilized cells. FRET images in Figure 2C are consistent with this proposed topology.
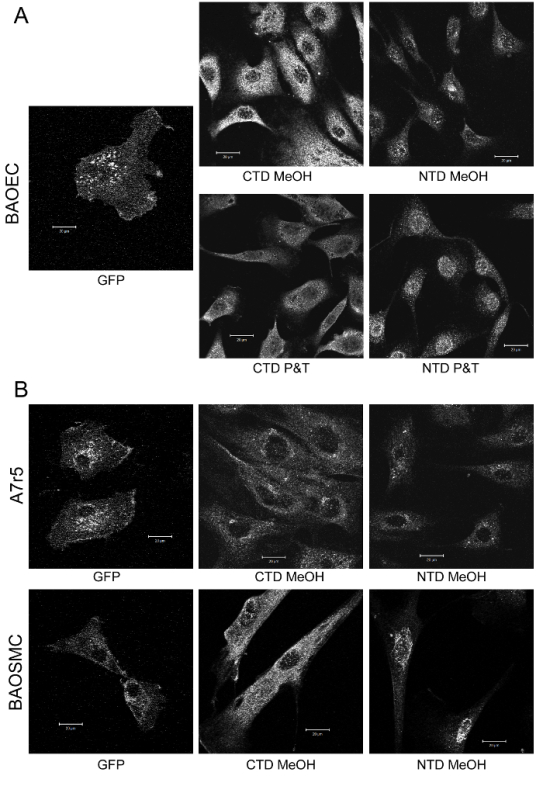 Figure 1: GFP-TEMEM184A Localization in Cells Confirms TMEM184A Localization Determined by Immunofluorescence. A) BAOECs transfected with GFP-TMEM184A were fixed with 4% PFA. Nontransfected cells were either processed with ice-cold methanol (MeOH) fixing and permeabilization or 4% PFA followed by Triton X-100 permeabilization (as noted). The nontransfected cells were stained using an antibody against a C-terminal peptide from TMEM184A (CTD) or an N-terminal peptide from TMEM184A (NTD) and a secondary anti-rabbit antibody tagged with TRITC. B) Examples of cloned (A7r5) and primary (BAOSMC) smooth muscle cells were processed similarly. Please click here to view a larger version of this figure.
Figure 1: GFP-TEMEM184A Localization in Cells Confirms TMEM184A Localization Determined by Immunofluorescence. A) BAOECs transfected with GFP-TMEM184A were fixed with 4% PFA. Nontransfected cells were either processed with ice-cold methanol (MeOH) fixing and permeabilization or 4% PFA followed by Triton X-100 permeabilization (as noted). The nontransfected cells were stained using an antibody against a C-terminal peptide from TMEM184A (CTD) or an N-terminal peptide from TMEM184A (NTD) and a secondary anti-rabbit antibody tagged with TRITC. B) Examples of cloned (A7r5) and primary (BAOSMC) smooth muscle cells were processed similarly. Please click here to view a larger version of this figure.
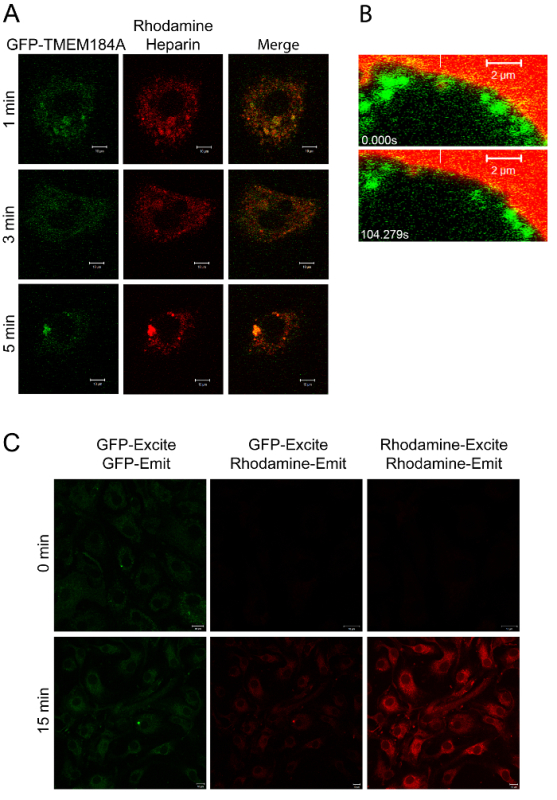 Figure 2: Imaging of GFP-TMEM184A with Rhodamine-Heparin. A) A7r5s electroporated with GFP-TMEM184A were treated with 100 µg/mL rhodamine-heparin for the times indicated and then fixed with 4% PFA. Images are representative of two separate experiments. B) A7r5 cells were transfected with GFP-TMEM184A and rhodamine-heparin was added with immediate live imaging. Selected frames of the movie are shown with a white vertical bar pointing to the initial localization of a GFP-coated vesicle containing rhodamine-heparin. The GFP label and the rhodamine move together. Scale bars = 2 µm. C) RAOECs treated as in A were imaged for fluorescence resonance energy transfer by exciting at 405 (GFP, for FRET) and compared to standard settings, see protocol 3.5. Panel A, and the two time points in B, were originally published in the Journal of Biological Chemistry. Pugh, R.P., et al.1. Copyright the American Society for Biochemistry and Molecular Biology. Please click here to view a larger version of this figure.
Figure 2: Imaging of GFP-TMEM184A with Rhodamine-Heparin. A) A7r5s electroporated with GFP-TMEM184A were treated with 100 µg/mL rhodamine-heparin for the times indicated and then fixed with 4% PFA. Images are representative of two separate experiments. B) A7r5 cells were transfected with GFP-TMEM184A and rhodamine-heparin was added with immediate live imaging. Selected frames of the movie are shown with a white vertical bar pointing to the initial localization of a GFP-coated vesicle containing rhodamine-heparin. The GFP label and the rhodamine move together. Scale bars = 2 µm. C) RAOECs treated as in A were imaged for fluorescence resonance energy transfer by exciting at 405 (GFP, for FRET) and compared to standard settings, see protocol 3.5. Panel A, and the two time points in B, were originally published in the Journal of Biological Chemistry. Pugh, R.P., et al.1. Copyright the American Society for Biochemistry and Molecular Biology. Please click here to view a larger version of this figure.
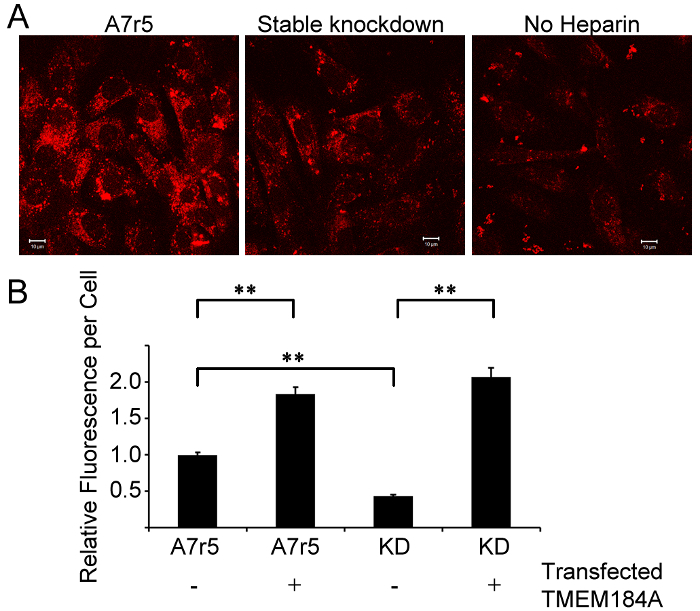 Figure 3: GFP-TMEM184A Transfection Provides Gain of Function. A) Significantly lower levels of rhodamine-heparin are seen in stable knockdown A7r5 cells compared to wild-type A7r5s, see example images. The "no heparin" image is of stable knockdown cells that express GFP as a marker of construct presence. Scale bars = 10 µm. B) Transfection of GFP-TMEM184A into both wild-type and stable knockdown cells significantly increases the rhodamine-heparin signal in these cells based on analysis of more than 50 cells/condition in three independent experiments. Error bars represent SEM. p <0.0001 based on a Tukey test. This figure was originally published in the Journal of Biological Chemistry. Pugh, R.P., et al.1. Copyright the American Society for Biochemistry and Molecular Biology. Please click here to view a larger version of this figure.
Figure 3: GFP-TMEM184A Transfection Provides Gain of Function. A) Significantly lower levels of rhodamine-heparin are seen in stable knockdown A7r5 cells compared to wild-type A7r5s, see example images. The "no heparin" image is of stable knockdown cells that express GFP as a marker of construct presence. Scale bars = 10 µm. B) Transfection of GFP-TMEM184A into both wild-type and stable knockdown cells significantly increases the rhodamine-heparin signal in these cells based on analysis of more than 50 cells/condition in three independent experiments. Error bars represent SEM. p <0.0001 based on a Tukey test. This figure was originally published in the Journal of Biological Chemistry. Pugh, R.P., et al.1. Copyright the American Society for Biochemistry and Molecular Biology. Please click here to view a larger version of this figure.
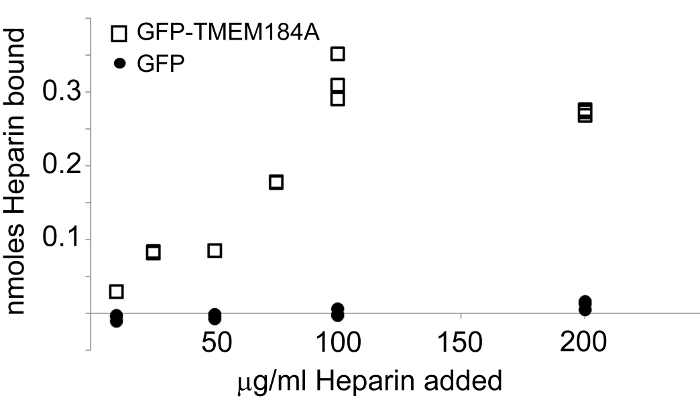 Figure 4: Isolated GFP-TMEM184A Binds Heparin. GFP-TMEM184A or GFP was isolated and bound to avidin-coated assay plates. Fluorescein-heparin was added at concentrations from 0.056 nmol through 1.111 nmol. Heparin bound was determined by measuring emission at 519 nm. GFP-TMEM184A (squares). GFP control (circles). This figure was originally published in the Journal of Biological Chemistry. Pugh, R.P., et al.1. Copyright the American Society for Biochemistry and Molecular Biology. Please click here to view a larger version of this figure.
Figure 4: Isolated GFP-TMEM184A Binds Heparin. GFP-TMEM184A or GFP was isolated and bound to avidin-coated assay plates. Fluorescein-heparin was added at concentrations from 0.056 nmol through 1.111 nmol. Heparin bound was determined by measuring emission at 519 nm. GFP-TMEM184A (squares). GFP control (circles). This figure was originally published in the Journal of Biological Chemistry. Pugh, R.P., et al.1. Copyright the American Society for Biochemistry and Molecular Biology. Please click here to view a larger version of this figure.
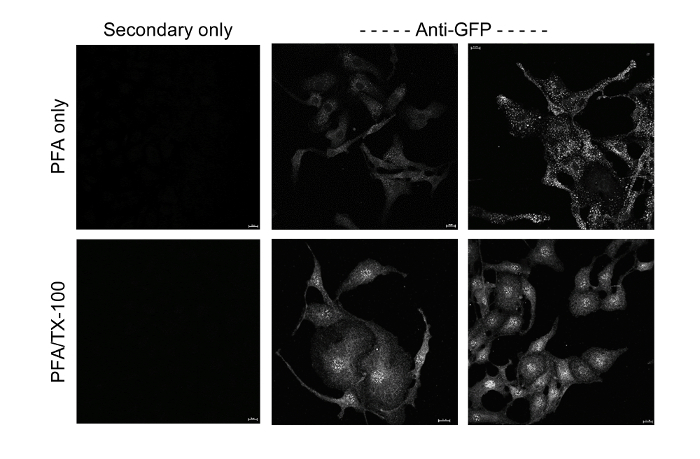 Figure 5: GFP-TMEM184A and Membrane Topology. GFP-TMEM184A transfected cells were either fixed with 4% PFA (top) or fixed with 4% PFA and permeabilized using Triton X-100 (bottom). Cells were stained identically with anti-GFP primary antibodies and Cy3-labeled secondary antibodies. Scale bars = 10 µm. Two different experiments are shown. Secondary only-stained cells showed essentially no staining. Please click here to view a larger version of this figure.
Figure 5: GFP-TMEM184A and Membrane Topology. GFP-TMEM184A transfected cells were either fixed with 4% PFA (top) or fixed with 4% PFA and permeabilized using Triton X-100 (bottom). Cells were stained identically with anti-GFP primary antibodies and Cy3-labeled secondary antibodies. Scale bars = 10 µm. Two different experiments are shown. Secondary only-stained cells showed essentially no staining. Please click here to view a larger version of this figure.
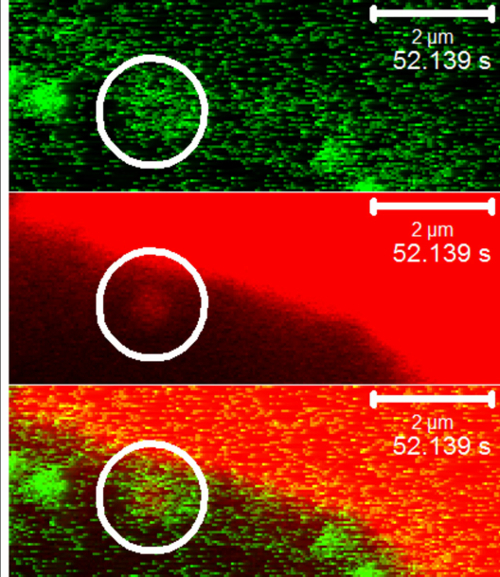 Movie 1: Live-imaging of GFP-TMEM184A with Rhodamine-Heparin (Right click to download). A7r5 cells were treated as in Figure 2B. The movie illustrates the GFP-TMEM184A and rhodamine-heparin vesicle at the top moving together. Separate color versions in addition to the merged version of the movie are shown. The vesicle with GFP and containing rhodamine is circled.
Movie 1: Live-imaging of GFP-TMEM184A with Rhodamine-Heparin (Right click to download). A7r5 cells were treated as in Figure 2B. The movie illustrates the GFP-TMEM184A and rhodamine-heparin vesicle at the top moving together. Separate color versions in addition to the merged version of the movie are shown. The vesicle with GFP and containing rhodamine is circled.
Discussion
The protocols reported here were designed to provide confirmatory evidence for the identification of TMEM184A as a heparin receptor in vascular cells1. Knockdown techniques are routinely used as one mechanism to confirm the identification of novel proteins. However, some functional loss after knockdown is typically not sufficient proof that a candidate protein is actually the correct receptor (or other functional protein). It is also important to have evidence that the candidate protein actually exhibits the function. Employing a construct for a novel gene tagged with GFP enables gain of function experiments and facilitates isolation of the tagged protein based on GFP affinity. Electroporation employed in this protocol facilitated very high transfection efficiency of the construct (greater than 80% of the cells expressed the construct). However, other transfection mechanisms could be used if electroporation is not available or not desired. There are several advantages to selecting a GFP-tagged construct for confirmation of a novel protein function. First, the GFP serves as a reporter for transfection and gene expression and it allows overexpression to be monitored through GFP if a cell line without expression of the protein is not available. Second, the GFP tag provides a tool for purification of the protein that does not depend on either ligand affinity or antibodies against peptides. Third, the GFP tag provides a tool to examine complex immunofluorescent localization visualized using antibodies generated against unique peptides in the target protein sequence to determine whether the actual gene product is similarly localized.
Gain of function assays with gene constructs are being used for a number of purposes including high-throughput screening for identification of novel protein function13. Ideally, gain of function assays would employ a well-studied cloned cell line that does not express the gene in question (the cells must still express cooperating gene products that allow function of the newly expressed gene). When using a specific GFP-tagged construct as in the present assays, it is critical to use cells in which the GFP-protein can function. Thus, the choice of cells depends on the function and, likely, on the cell type in which the function would normally occur to ensure cooperating proteins are present. GFP-tagging allows an additional way of monitoring function in the case where the fluorescence of the GFP-tagged protein can be followed as part of a functional assay (e.g., see Figures 2 and 3). One limitation to these studies would be if GFP somehow alters protein function. By linking the GFP to the C-terminus of the protein, large numbers of proteins have already been studied without an apparent functional alteration, but such a possibility does exist.
Radioisotopically labeled ligands have often been used to study ligand binding to intact cells11. However, those assays do not allow simple determination of ligand location after binding, nor do they facilitate comparisons of ligand and receptor together as in the present report. A GFP construct also provides further opportunities to examine a labeled membrane protein as here. Because of the C-terminal location of the GFP tag, it is possible to examine the location of the GFP tag in transfected cells throughout an assay. Immunofluorescence using antibodies to GFP allows comparison of GFP accessible to antibodies with and without permeabilization (Figure 5). Additional applications of GFP-TMEM184A take advantage of GFP imaging such as the colocalization with rhodamine-heparin (Figure 2). Employing FRET to allow emission from GFP to excite the rhodamine-heparin as shown in 2C provides an interesting way to evaluate binding and intracellular trafficking. Further, FRET data indicate that GFP is close enough (within 10 nm) to the rhodamine-ligand to transfer excitation. Evidence that such transfer from GFP to a ligand works has been published for parathyroid hormone tagged with tetramethyl-rhodamine14. Alternative FRET technology (photo-bleaching and/or computer-driven imaging patterns) can also be employed for more complex analysis than what is shown here as an example. Similarly, interactions of other proteins with the candidate might be examined using FRET with fluorescent tags optimized for FRET as in the EGFP/mCherry system reported by Albertazzi et al.15 and the CFP/YFP and GFP pairs used in potassium channel studies by Wang and colleagues16. Many new small fluorescent molecules are being designed, for example17. These molecules will increase the number of cases where FRET interactions between GFP-tagged proteins and fluorescent ligands can be examined. Future studies can also involve targeted changes to the gene, allowing the construct to help determine sequence specific aspects of gene function. One limitation for GFP-tagged membrane proteins is when the C-terminus of the protein is normally inside. In such cases, FRET assays with fluorescent water-soluble ligands may not be possible. However, alternative mechanisms to place GFP elsewhere in the construct might still be possible for some such proteins, and the other assays reported here could still be employed.
In addition to function, localization, and in vivo ligand interactions, a GFP-tagged protein can be isolated using an anti-GFP antibody and the isolated protein used to examine function employing in vitro assays where the GFP allows an unbiased isolation of the protein and a comparatively isolated control (free GFP). Together these assays can provide strong additional evidence needed to prove that the candidate protein functions as hypothesized.
Further, because GFP is protease resistant unless modified with sites that extend away from the compact structure18, limited protease treatment of cells should release extracellular GFP. The protease cleavage would occur in the protein to which GFP is attached or in a designed linkage region. The released product would be GFP with an extension N-terminal to the GFP sequence. Trypsin should not release GFP if the C-terminus of the protein to which GFP is attached is intracellular. A preliminary analysis of potential products could suggest a clear molecular weight pattern with a GFP-tagged protein released by trypsin. For many membrane proteins, such techniques would result in clear evidence for membrane topology by release of GFP, or lack thereof.
Together, the specific assays presented and/or proposed in this report provide a broad sampling of techniques that can be employed to examine novel proteins by starting with a fluorescently tagged construct. Such constructs can be easily obtained commercially. The opportunity to use them for a wide range of techniques makes them economically feasible for even small laboratories on limited budgets.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Research in the Lowe-Krentz lab is supported by research grant HL54269 from the National Institutes of Health to LLK.
References
- Pugh RJ, et al. Transmembrane Protein 184A Is a Receptor Required for Vascular Smooth Muscle Cell Responses to Heparin. J Biol Chem. 2016;291:5326–5341. doi: 10.1074/jbc.M115.681122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher W, et al. Identification of Toxoplasma TgPH1, a pleckstrin homology domain-containing protein that binds to the phosphoinositide PI(3,5)P. Mol Biochem Parasitol. 2016. [DOI] [PubMed]
- Vit O, et al. Large-scale identification of membrane proteins based on analysis of trypsin-protected transmembrane segments. J Proteomics. 2016. [DOI] [PubMed]
- Attwood MM, et al. Topology based identification and comprehensive classification of four-transmembrane helix containing proteins (4TMs) in the human genome. BMC genomics. 2016;17:268. doi: 10.1186/s12864-016-2592-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, et al. Genome-Wide Identification of Jatropha curcas Aquaporin Genes and the Comparative Analysis Provides Insights into the Gene Family Expansion and Evolution in Hevea brasiliensis. Front Plant Sci. 2016;7:395. doi: 10.3389/fpls.2016.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilotti AC, et al. Heparin responses in vascular smooth muscle cells involve cGMP-dependent protein kinase (PKG) J Cell Physiol. 2014;229:2142–2152. doi: 10.1002/jcp.24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell SL, et al. Heparin Decreases in Tumor Necrosis Factor alpha (TNFalpha)-induced Endothelial Stress Responses Require Transmembrane Protein 184A and Induction of Dual Specificity Phosphatase 1. J Biol Chem. 2016;291:5342–5354. doi: 10.1074/jbc.M115.681288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Esko JD. Demystifying heparan sulfate-protein interactions. Annu Rev Biochem. 2014;83:129–157. doi: 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiodelli P, Bugatti A, Urbinati C, Rusnati M. Heparin/Heparan sulfate proteoglycans glycomic interactome in angiogenesis: biological implications and therapeutical use. Molecules. 2015;20:6342–6388. doi: 10.3390/molecules20046342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee JB, Lowe-Krentz LJ. Actin realignment and cofilin regulation are essential for barrier integrity during shear stress. J Cell Biochem. 2013;114:782–795. doi: 10.1002/jcb.24416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton WA, et al. Identification of a heparin-binding protein using monoclonal antibodies that block heparin binding to porcine aortic endothelial cells. The Biochemical journal. 1995;311:461–469. doi: 10.1042/bj3110461. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett TM, Breslin JW. Study of the actin cytoskeleton in live endothelial cells expressing GFP-actin. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Skalamera D, et al. Generation of a genome scale lentiviral vector library for EF1alpha promoter-driven expression of human ORFs and identification of human genes affecting viral titer. PloS one. 2012;7:51733. doi: 10.1371/journal.pone.0051733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga JP. Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc Natl Acad Sci U S A. 2005;102:16084–16089. doi: 10.1073/pnas.0503942102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertazzi L, Arosio D, Marchetti L, Ricci F, Beltram F. Quantitative FRET analysis with the EGFP-mCherry fluorescent protein pair. Photochem Photobiol. 2009;85:287–297. doi: 10.1111/j.1751-1097.2008.00435.x. [DOI] [PubMed] [Google Scholar]
- Wang S, et al. Domain organization of the ATP-sensitive potassium channel complex examined by fluorescence resonance energy transfer. J Biol Chem. 2013;288:4378–4388. doi: 10.1074/jbc.M112.388629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen E, Hudson BD, Hansen AH, Milligan G, Ulven T. Development and Characterization of a Potent Free Fatty Acid Receptor 1 (FFA1) Fluorescent Tracer. J Med Chem. 2016;59:4849–4858. doi: 10.1021/acs.jmedchem.6b00202. [DOI] [PubMed] [Google Scholar]
- Chiang CF, Okou DT, Griffin TB, Verret CR, Green Williams MN. fluorescent protein rendered susceptible to proteolysis: positions for protease-sensitive insertions. Arch Biochem Biophys. 2001;394:229–235. doi: 10.1006/abbi.2001.2537. [DOI] [PubMed] [Google Scholar]