

Abstract
Efficacy of candidate antibacterial treatments must be demonstrated in animal models of infection as part of the discovery and development process, preferably in models which mimic the intended clinical indication. A method for inducing robust lung infections in immunocompetent rats and mice is described which allows for the assessment of treatments in a model of serious pneumonia caused by S. pneumoniae, H. influenzae, P. aeruginosa, K. pneumoniae or A. baumannii. Animals are anesthetized, and an agar-based inoculum is deposited deep into the lung via nonsurgical intratracheal intubation. The resulting infection is consistent, reproducible, and stable for at least 48 h and up to 96 h for most isolates. Studies with marketed antibacterials have demonstrated good correlation between in vivo efficacy and in vitro susceptibility, and concordance between pharmacokinetic/pharmacodynamic targets determined in this model and clinically accepted targets has been observed. Although there is an initial time investment when learning the technique, it can be performed quickly and efficiently once proficiency is achieved. Benefits of the model include elimination of the neutropenic requirement, increased robustness and reproducibility, ability to study more pathogens and isolates, improved flexibility in study design and establishment of a challenging infection in an immunocompetent host.
Keywords: Infection, Issue 119, in vivo, pneumonia, lung, infection, model, mouse, rat, bacteria, microbiology, efficacy, drug, discovery
Introduction
Evaluating the efficacy of potential drug candidates in animal models of infection is a critical component of the antibacterial drug discovery process. In vivo efficacy studies provide important data for decision-making across the span of discovery efforts, from elucidating structure-activity relationships (SAR) and optimizing lead chemical series through determining pharmacokinetic-pharmacodynamic (PK/PD) characteristics of compounds and supporting dose selection for clinical development and regulatory approval. Numerous animal infection models are described in the literature for these purposes. One of the most common and widely used models is the neutropenic mouse thigh infection, which was pioneered by Eagle1, 2 and later expanded by Vogelman and Craig3, 4. This model has been invaluable for supporting determination of bacterial susceptibility breakpoints and for providing PK/PD targets to guide human dosing. There are many examples where the predictive capability of this model has been proven4-7. However, one of the drawbacks of the thigh infection model is the lack of direct relevance for lung infections. There is now a greater emphasis on matching the site of infection in animal models to the site of infection in humans; and thus, to study compounds for the treatment of pneumonia, it is ideal to utilize a lung infection model in animals.
Several methods for inducing lung infections in laboratory animals have been described and used for antibacterial efficacy studies including intranasal inhalation, aspiration via the oropharyngeal route, aerosol exposure, and surgical intratracheal inoculation8. In many cases, the animals (particularly mice) must first be rendered neutropenic in order to achieve a robust lung infection. Despite this severely immunocompromised state, the number of bacterial pathogens and strains which produce viable infections in rodents is limited. For example, it can be difficult to successfully establish Haemophilus influenzae or Acinetobacter baumannii in pneumonia models9, 10, and even more virulent pathogens, such as Streptococcus pneumoniae, Pseudomonas aeruginosa and Klebsiella pneumoniae, can pose challenges 11-13.
In 1991, Smith described a lung infection model in immunocompetent weanling rats in which infection was established by instilling bacteria directly into the lungs via a simple nonsurgical intratracheal intubation method14. Berry et al. later modified the method for infecting mice15. Using an agar-based inoculum, this inoculation technique induces robust lung infections in both immunocompetent rats and mice with S. pneumoniae, H. influenzae, K. pneumoniae, P. aeruginosa and A. baumannii. It is amenable to a wide range of isolates, including those harboring various resistance determinants and those which do not produce a viable infection via other inoculation routes. It also allows efficacy to be determined in immunocompetent animals, a condition which is more relevant than the traditional neutropenic mouse model for patient populations that are not severely immunocompromised. The consistency and reliability of this model across multiple pathogens and isolates makes it well-suited for antibacterial efficacy studies.
Protocol
S. pneumonie
All procedures are in accordance with protocols approved by the GSK Institutional Animal Care and Use Committee (IACUC), and meet or exceed the standards of the American Association for the Accreditation of Laboratory Animal Care (AAALAC), the United States Department of Health and Human Services and all local and federal animal welfare laws.
NOTE: Unless otherwise specified, all procedures should be performed using aseptic technique.
1. Culture Bacterial Isolates
- Prepare 3 to 6 agar plates with S. pneumoniae or H. influenzae, as broth culture generally does not provide enough material for inoculation.
- Remove a frozen stock culture from storage at -80 °C, thaw completely, and streak up to 100 µL per plate onto tryptic soy agar supplemented with sheep blood (S. pneumoniae) or onto chocolate agar (H. influenzae) using standard microbiological techniques. Incubate overnight at 37 °C with or without 5% CO2.
- Prepare broth cultures with K. pneumoniae, P. aeruginosa or A. baumannii.
- Remove a frozen stock culture from storage at -80 °C, thaw completely, and aliquot 100 µL of the stock into 50 mL brain-heart infusion (BHI) broth. Incubate overnight at 37 °C with gentle shaking (approximately 120 rpm).
- [Optional] If a log phase culture is desired, aliquot 1 mL from the resultant overnight culture into 50 mL fresh BHI broth. Incubate for 3 h at 37 °C with gentle shaking (approximately 120 rpm).
2. Prepare Saline Bacterial Suspensions
For all suspension preparation steps (2.1 to 2.3), utilize sterile saline at a temperature between ambient air and 37 °C. Harvest overnight growth of S. pneumoniae or H. influenzae from the agar plates by scraping colonies from the surface with a sterile loop. Transfer the harvested material into 5 mL of sterile saline until a cloudy, opaque suspension is obtained and gently vortex until homogeneous.
Centrifuge 50 mL of the K. pneumoniae, P. aeruginosa or A. baumannii final broth cultures intended for inoculation (e.g. overnight or log phase cultures) for 5 min at 4,500 x g. Remove supernatant with a pipette and discard. Resuspend the pellet in at least 5 mL sterile saline, and gently vortex to achieve a homogeneous suspension.
If necessary, dilute the suspension further using sterile saline to obtain a density in the range of 8 to 9 log10 colony-forming units (CFU)/mL. Remove an aliquot for determination of CFU/mL. Serially dilute and plate the aliquot as described in Sections 7.5 and 7.6.
3. Prepare the Agar-based Inoculum
Prepare a small bottle (approximately 50 mL) of nutrient agar according to the manufacturer's instructions or melt a bottle of solid nutrient agar that was previously prepared and stored. Allow it to cool to approximately 50 °C.
Transport the liquid agar and all necessary tools and equipment for the infection process to the procedure area where the animals will be infected. Maintain a small water bath set at 42 °C in the area.
Allow the agar to reach a temperature of approximately 50 °C, and then aliquot 9 mL into a sterile tube appropriately sized to fit into the water bath. Leave this tube in the water bath until the agar equilibrates to approximately 42 °C. Ensure the water is at a depth which will cover the surface of the agar in its container but not touch the cap or lid.
Add 1 mL of the saline bacterial suspension from Step 2.3 into the tube containing 9 mL agar in the water bath. Cap the tube, invert several times to mix, and return it to the water bath.
4. Anesthetize, Intubate and Inoculate Animals
NOTE: Specific pathogen free (SPF) immunocompetent male Sprague-Dawley rats weighing 100 - 120 g or male CD-1 mice weighing 20 - 25 g are recommended. Provide all animals with food and water ad libitum and house them socially on wood chips or other absorbent bedding with standard 12 h light-dark cycles.
- Prior to conducting any experiments, obtain and sterilize a metal cannula and polyethylene tubing of the appropriate size for the animal species. Obtain sterile 1 mL disposable syringes and sterile disposable 25-gauge needles. Caution: Always dispose of these syringes and needles in a sharps container.
- For rats, purchase or manufacture a metal cannula that is approximately 12 cm in length, has an ID of 0.9 - 1.0 mm and OD of 1.0 - 1.2 mm, and is bent to an approximate 45° angle at one end. Cut an 11- to 12-inch length of polyethylene tubing with an ID of 0.4 mm and OD 0.8 mm. (Refer to Figure 1A.)
- For mice, purchase a #20 animal feeding tube. Remove the ball from the end of the tube by grasping it with pliers and pulling sharply, and then gently bend the end to an approximate 45° angle. Cut an 11- to 12-inch length of polyethylene tubing with an ID of 0.28 mm and OD 0.61 mm. Also purchase and sterilize a glass 100 µL micro-injection syringe and a 30-gauge metal micro-injection needle. (Refer to Figure 1B.)
- Place the metal cannula in a glass, screw-cap tube and autoclave by standard methods to sterilize. Soak and store cut lengths of the polyethylene tubing in a covered beaker containing alcohol.
- Prepare the work surface and intubation device.
- Place a clean disposable mat onto the work surface, close to the water bath. Transfer the metal cannula, in its glass storage tube, to this surface. Remove the cap from the storage tube and slide the "handle" end of the cannula to the open end of the tube.
- Using sterile forceps, remove one length of polyethylene tubing from the beaker and insert it through the inside of the sterile metal cannula, making sure it moves freely. Fit a sterile disposable 25-gauge needle (for rats) or the sterilized 30-gauge metal needle of a glass micro-injection syringe (for mice) onto the free end of the polyethylene tubing by sliding the needle several mm into the tubing. (Refer to Figure 1A for rat and Figure 1B for mice).
- Place guide marks on both the metal cannula and polyethylene tubing to indicate depth of insertion in the animal by following the intubation procedure described below on a single euthanized animal. Open the chest cavity for observation, then place the metal cannula properly and advance the polyethylene tubing appropriately (as described in Section 4.7). Using an indelible pen, mark the metal cannula where it meets the mouth of the animal and the polyethylene tubing where it meets the top of the metal cannula to aid proper placement in remaining animals.
Within a fume hood (or using an appropriate scavenging system), anesthetize up to 6 animals at a time in a closed chamber supplied with ≤5% isoflurane in 1.5 L/min oxygen until the gag reflex is inhibited (approximately 1 min). NOTE: Prior to conducting any experiments, establish the safe dose and exposure time for isoflurane under the specific conditions being used (e.g. size/type of chamber and size/species/strain of animal) to prevent inadvertent lethal exposure.
- Fill a sterile disposable 1 mL syringe (for rats) with sterile saline by placing the sterile tip into the saline and pulling back on the plunger. Fill the sterile glass micro-injection syringe (for mice) as described below. Attach the syringe to the needle fitted onto the polyethylene tubing, and flush the entire volume of saline through the tubing until the syringe is emptied.
- To fill the micro-injection syringe (for mice), completely remove the metal plunger and needle (fitted onto the polyethylene tubing) from the syringe and set both aside.
- Fill a sterile disposable 1 mL syringe with sterile saline (as described above) and attach a sterile disposable 25-gauge needle. Insert the needle into the top of the micro-injection syringe (where the metal plunger will be inserted), and depress the plunger of the disposable syringe until it fills the micro-injection syringe with saline.
- Discard the disposable syringe and needle that were used to fill the micro-injection syringe into a sharps container. Re-attach the metal micro-injection needle (fitted onto the polyethylene tubing) to the tip of the micro-injection syringe and re-insert the metal plunger, depressing until the entire volume of saline has been flushed through the syringe and tubing.
- Fill the syringe with the agar-based inoculum from the container in the water bath, and flush agar through the tubing just until the tubing is primed (e.g. completely filled with the agar).
- For rats, remove the 25-gauge needle (fitted onto the polyethylene tubing) from the disposable syringe. Discard the used syringe in a sharps container. Fill a new, sterile disposable 1 mL syringe with the agar-based inoculum from the container in the water bath by placing the sterile tip into the inoculum and pulling back on the plunger. Re-attach needle (fitted onto the polyethylene tubing) and flush agar through the tubing by depressing the plunger just until tubing is completely filled with agar.
- For mice, remove the metal micro-injection needle (fitted onto the polyethylene tubing) and metal plunger from the micro-injection syringe and set aside. Using a sterile disposable 1 mL syringe and sterile disposable 25-gauge needle, fill the micro-injection syringe with the agar-based inoculum from the container in the water bath using the filling procedure described in Section 4.4. Re-attach the metal micro-injection needle (fitted onto the polyethylene tubing), insert the metal plunger, and flush agar through the tubing just until tubing is completely filled with agar.
Remove one animal from the anesthetic chamber. Place the animal in a supine position on the disposable mat with the head to the right and tail to the left as shown in Figure 1C.
- Using the intubation device (i.e. the metal cannula fitted with polyethylene tubing), intubate the animal and insert the cannula into the large lobe of the left lung (see Figure 1D).
- Insert the free end of the metal cannula into the animal's mouth. Turn the cannula so that the free end is angled upward and gently advance it into the trachea, carefully bypassing the laryngeal structures with a slight twisting motion. Confirm insertion into the trachea as opposed to the esophagus by gently sliding the cannula slightly forward and back several times while palpating the tracheal rings with the left forefinger as shown in Figure 1C.
- When the cannula reaches the bifurcation where the trachea splits into the left and right bronchi, make a slight twisting motion toward the animal's left side to ensure that the cannula is inserted into the left bronchus. Advance the metal cannula until the end is halfway to three-quarters down the left lung, using the previously placed guide mark to confirm that the appropriate depth has been reached.
- With the metal cannula in place, advance the polyethylene tubing several mm. Use the previously placed guide mark on the tubing to ensure it is advanced only far enough to exit the end of the metal cannula and not puncture the lung.
With both cannulae in place, use the attached syringe to instill 100 µL (for rats) or 20 µL (for mice) of the agar suspension deep into the large lobe of the left lung (see Figure 1D). Withdraw several mm of the polyethylene tubing, and then gently remove the intact intubation device. Set it back into the glass storage tube, and move the animal into a fresh cage to recover.
- Continue anesthetizing, intubating and inoculating the remaining animals.
- Between batches of anesthetized animals, remove the needle (fitted onto the tubing) from the syringe. For rats, discard the used disposable syringe in a sharps container and fill a new, sterile disposable syringe from the inoculum in the water bath. For mice, refill the same glass micro-injection syringe from the inoculum in the water bath using the fill technique described in Section 4.4.
- If the agar begins to thicken in the intubation device, flush all remaining agar through the tubing. Remove the needle from the syringe (leaving the needle fitted onto the polyethylene tubing). Follow the procedures described in Section 4.4 to flush warm, sterile saline through the intubation device, repeating as necessary to completely clear any blockage. Empty all saline from the syringe and prepare the agar inoculum again as described in Section 4.5.
- Calculate the final bacterial inoculum per animal using the CFU determined from the saline suspension (see Section 2.3), the final dilution factor of the saline suspension into agar (e.g. ten-fold), and the volume instilled into the animals (e.g. 100 µL for rats or 20 µL for mice).
5. Assess Animals for Potential Mis-inoculation or Other Adverse Events
Carefully observe all animals during intubation, instillation of the agar and recovery from anesthesia to assess for potential mis-inoculation. Immediately euthanize (according to local IACUC guidelines) any animal which bleeds, exhibits abnormal respiration (such as labored breathing or gasping), does not move normally about the cage, or does not appear bright-eyed and healthy upon recovery from anesthesia.
If spontaneous cessation of breathing occurs (usually when the inoculum is not placed deep enough and blocks the airway), confirm death by observation and perform a secondary method of euthanasia (such as cervical dislocation or thoracotomy).
Minimize the length of time animals are exposed to isoflurane, as anesthesia is only required for several minutes to perform the procedure. If injectable anesthetics are used, ensure the animals are under observation until fully awake. Place animals, especially mice, in a warmed environment for recovery when using injectable anesthetics.
Observe animals at least twice daily during the study period for the following signs of respiratory disease: pronounced breathing, piloerection, reduced activity and reduced body temperature. Immediately euthanize (according to local IACUC guidelines) any animal that is moribund, experiences labored breathing or respiratory distress, has a noticeably reduced body temperature upon handling, is unable or unwilling to move, or cannot reach food and water.
6. Administer Test Treatments
Prepare and administer test treatments according to planned study design. Begin administration of treatments at 1 or 2 h post infection (or delay treatment further if a higher baseline bacterial burden is desired). Specific details for preparation and administration of test treatments cannot be given as it is dependent on the objective of the study as well as the properties of each individual treatment. (Refer to Figure 3 as an example.)
Set aside one group of infected, untreated animals to serve as baseline controls. Enumerate the bacterial burden in the lungs of these animals at the time treatment is initiated for the other groups, following the procedures outlined in Section 7.
For PK/PD analyses, assess the pharmacokinetic profile of the administered treatment(s) in the blood and/or tissues of the infected animals. Specific procedures for pharmacokinetic studies are outside the scope of this article.
7. Enumerate Viable Bacteria from the Lungs
Euthanize one group of untreated animals at the time treatment is initiated for the other groups (baseline) and all remaining animals at the end of the study period (usually 24, 48 or 96 h post infection) by inhalation exposure to gradually rising levels of carbon dioxide within a closed chamber or an alternative method as recommended by local IACUC guidelines. Verify death by observation, and perform a secondary method of euthanasia (such as cervical dislocation or thoracotomy).
Place the animals in a supine position on a clean disposable mat, and thoroughly wet the fur over the chest with alcohol.
Using sterile scissors, cut through the skin, underlying muscle layer and ribcage parallel to the sternum to open the chest cavity. Gently grasp the lungs with sterile forceps and pull to remove, using the scissors to separate them from the trachea if necessary. Place lungs onto sterile gauze to absorb excess blood. If the heart has also been removed, dissect it away and discard.
Make small snips in the lungs with sterile scissors to expose interior sections and use sterile forceps to place lungs (plus any pieces that were inadvertently snipped off) into the bottom of a lab blender bag. Briefly roll a thick round object (such as a pen or marker) over the outside of the bag to mash the tissue. Pipette 1 mL of sterile saline into the bag, place the bag into a lab blender, and run the lab blender at high speed for 2 min.
Transfer the homogenated samples from the blended bags into tubes or plates using a pipette. Dilute the homogenate ten-fold by aliquotting 1-part homogenate into 9-parts saline (such as 100 µL homogenate into 900 µL saline). Dilute the resultant suspension by ten-fold again in a similar manner. Continue diluting each new resultant suspension by another ten-fold factor until at least 6 serial dilutions have been prepared from the original homogenate.
Pipette triplicate aliquots of 20 µL each from all dilutions onto trypticase soy agar plates supplemented with sheep blood (S. pneumoniae, K. pneumoniae, P. aeruginosa or A. baumannii) or onto chocolate agar plates (H. influenzae). Incubate overnight at 37 °C with or without CO2. (See Figure 1E for an example of a resultant plate.)
Count the colonies in each of the three replicates at the first dilution that contains a "countable" number (i.e. not too many colonies to reasonably count). Calculate the average value of the three replicates. Determine the CFU per lungs by multiplying the average value by 50 (to account for plating 20 µL of the total 1 mL homogenate) and factoring in the final dilution factor from the original homogenate at which the colonies were counted.
Representative Results
The tools required, orientation of the animal, and depth of intubation are shown in Figure 1. Also shown is a representative example of colonies growing on an agar plate after serial dilution and triplicate plating of a lung homogenate sample. An increase in bacterial burden of several log10 CFU above baseline controls is typically observed in infected lungs for most bacterial isolates, even at 96 h post infection, and variability between animals is low (Figure 2). For some H. influenzae, there may be little growth; however, the infection should not begin to self-resolve within a 48 or 96 h time period (Figure 2B). This lung infection model can be used during optimization of lead chemical series to support SAR, as shown in Figure 3 for representative compounds of two different series16, 17. It provides a consistent, reproducible method for assessing PK/PD in immunocompetent animals and was used to determine that free drug area under the concentration-time curve over the minimum inhibitory concentration (fAUC/MIC) correlated with efficacy of a novel polypeptide deformylase (PDF) inhibitor against S. pneumoniae and H. influenzae (Figure 4). Additionally, the fAUC/MIC targets for stasis and a 1-log10 reduction in CFU from baseline controls were determined across 11 S. pneumoniae and 5 H. influenzae isolates (Table 1)18. Coupling this infection model with a drug delivery system to recreate human pharmacokinetic profiles in rats creates a powerful tool for evaluating human dosing regimens prior to clinical studies. An example of this is summarized in Table 2, which demonstrates that an extended-release formulation of amoxicillin-clavulanate was more effective than existing dose regimens for organisms with elevated MIC values19.
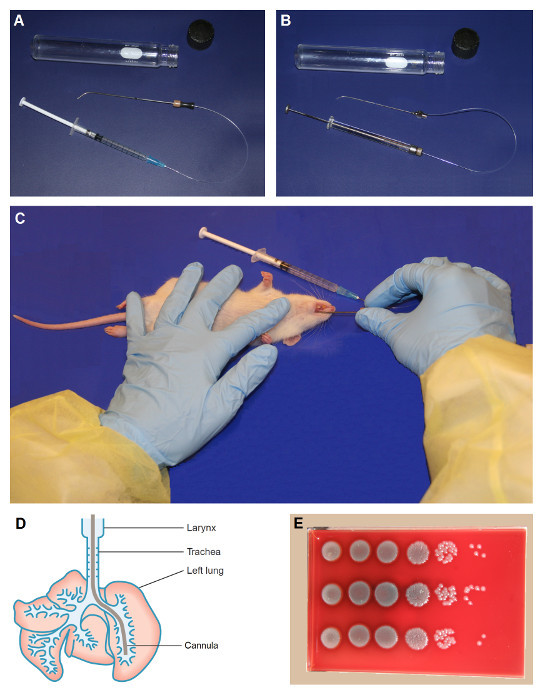 Figure 1:Nonsurgical Intratracheal Intubation. Tools are shown for intubating the rat (A) and the mouse (B). Once anesthetized, the animal should be positioned as shown in C, with the head to the right and tail to the left if the individual performing the technique is right-handed. Place the left forefinger gently on the throat to help palpate the tracheal rings for confirmation of correct cannula placement. The inoculum should be placed deep into the left lung, as shown in the ventral view of a rat lung (D). Colonies growing on agar after serial dilution and plating of a sample should appear similar to that shown in E. Figure 1D, Copyright © Gill Smith [Laboratory Animals, Volume 25 (1991), G. Smith, A simple non-surgical method of intrabronchial instillation for the establishment of respiratory infections in the rat, pages 46 - 49]14. Reprinted with permission. Please click here to view a larger version of this figure.
Figure 1:Nonsurgical Intratracheal Intubation. Tools are shown for intubating the rat (A) and the mouse (B). Once anesthetized, the animal should be positioned as shown in C, with the head to the right and tail to the left if the individual performing the technique is right-handed. Place the left forefinger gently on the throat to help palpate the tracheal rings for confirmation of correct cannula placement. The inoculum should be placed deep into the left lung, as shown in the ventral view of a rat lung (D). Colonies growing on agar after serial dilution and plating of a sample should appear similar to that shown in E. Figure 1D, Copyright © Gill Smith [Laboratory Animals, Volume 25 (1991), G. Smith, A simple non-surgical method of intrabronchial instillation for the establishment of respiratory infections in the rat, pages 46 - 49]14. Reprinted with permission. Please click here to view a larger version of this figure.
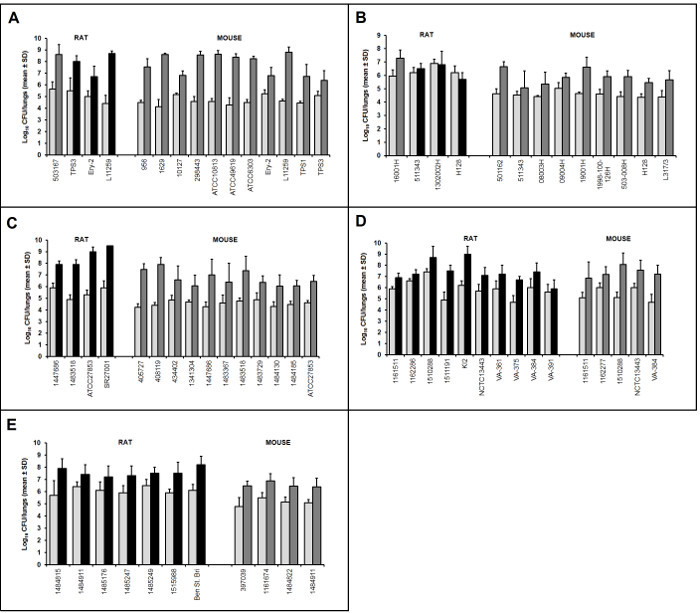 Figure 2:Representative Examples of Bacterial Growth in the Lungs of Infected Animals. Mean ± standard deviation CFU data from N=5 animals/group is shown for various isolates of S. pneumoniae
(A), H. influenzae
(B), P. aeruginosa
(C), K. pneumoniae
(D) and A. baumannii
(E). The first bar of each pair (light gray) is the baseline bacterial burden at 1 or 2 h post infection, while the second bar is the end-of-study growth control at 48 h post infection (dark gray) or 96 h post infection (black). No data censoring methods were applied; thus, these results include data from all 5 animals per group. Please click here to view a larger version of this figure.
Figure 2:Representative Examples of Bacterial Growth in the Lungs of Infected Animals. Mean ± standard deviation CFU data from N=5 animals/group is shown for various isolates of S. pneumoniae
(A), H. influenzae
(B), P. aeruginosa
(C), K. pneumoniae
(D) and A. baumannii
(E). The first bar of each pair (light gray) is the baseline bacterial burden at 1 or 2 h post infection, while the second bar is the end-of-study growth control at 48 h post infection (dark gray) or 96 h post infection (black). No data censoring methods were applied; thus, these results include data from all 5 animals per group. Please click here to view a larger version of this figure.
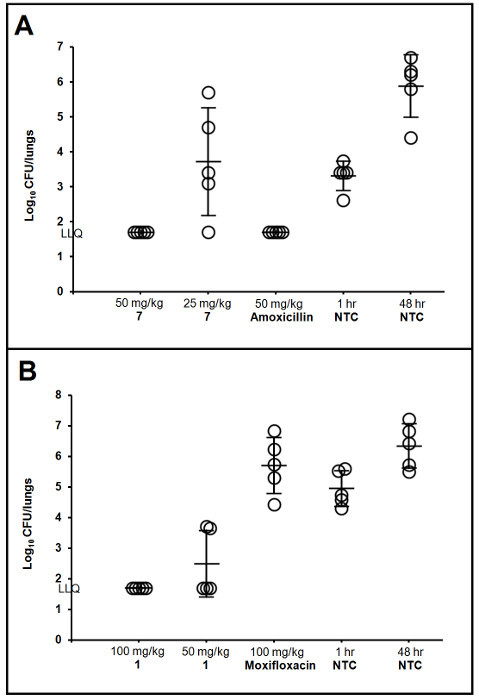 Figure 3:Evaluating the Efficacy of Various Chemical Series during Lead Optimization. Lung infection models using the described techniques were utilized to explore structure-activity relationships as part of abacterial type IIA topoisomerase program. Promising compounds 7 and 1 were evaluated against quinolone-susceptible (A)16 or quinolone-resistant (B)17 isolates of S. pneumoniae, respectively. Each symbol represents CFU determined from the lungs of one rat with lines representing the group mean and standard deviation. Lower limit of quantification (LLQ) was 1.7 log10 CFU/lungs. Compounds were weighed (56 or 112 mg of pure free parent molecule), dissolved in 9 mL of sterile water, diluted 3 mL into 3 mL water (1:1) and administered by oral gavage of 1 mL/125 g rat twice daily (6 - 7 h apart) for 2 days, starting at 1 h post infection. Nontreated controls (NTC) were sampled at 1 h post infection for baseline or treated with saline and sampled at the end of the study (48 h) for growth controls. Figure 3A reprinted from Bioorganic & Medicinal Chemistry Letters, Volume 21, T.J. Miles et al., Novel cyclohexyl-amides as potent antibacterials targeting bacterial type IIA topoisomerases, Pages 7483 - 7488, Copyright (2011)16, with permission from Elsevier. Figure 3B reprinted from Bioorganic & Medicinal Chemistry Letters, Volume 26, T.J. Miles et al., Novel tricyclics (e.g. GSK945237) as potent inhibitors of bacterial type IIA topoisomerases, Pages 2464 - 2469, Copyright (2016)17, with permission from Elsevier. Please click here to view a larger version of this figure.
Figure 3:Evaluating the Efficacy of Various Chemical Series during Lead Optimization. Lung infection models using the described techniques were utilized to explore structure-activity relationships as part of abacterial type IIA topoisomerase program. Promising compounds 7 and 1 were evaluated against quinolone-susceptible (A)16 or quinolone-resistant (B)17 isolates of S. pneumoniae, respectively. Each symbol represents CFU determined from the lungs of one rat with lines representing the group mean and standard deviation. Lower limit of quantification (LLQ) was 1.7 log10 CFU/lungs. Compounds were weighed (56 or 112 mg of pure free parent molecule), dissolved in 9 mL of sterile water, diluted 3 mL into 3 mL water (1:1) and administered by oral gavage of 1 mL/125 g rat twice daily (6 - 7 h apart) for 2 days, starting at 1 h post infection. Nontreated controls (NTC) were sampled at 1 h post infection for baseline or treated with saline and sampled at the end of the study (48 h) for growth controls. Figure 3A reprinted from Bioorganic & Medicinal Chemistry Letters, Volume 21, T.J. Miles et al., Novel cyclohexyl-amides as potent antibacterials targeting bacterial type IIA topoisomerases, Pages 7483 - 7488, Copyright (2011)16, with permission from Elsevier. Figure 3B reprinted from Bioorganic & Medicinal Chemistry Letters, Volume 26, T.J. Miles et al., Novel tricyclics (e.g. GSK945237) as potent inhibitors of bacterial type IIA topoisomerases, Pages 2464 - 2469, Copyright (2016)17, with permission from Elsevier. Please click here to view a larger version of this figure.
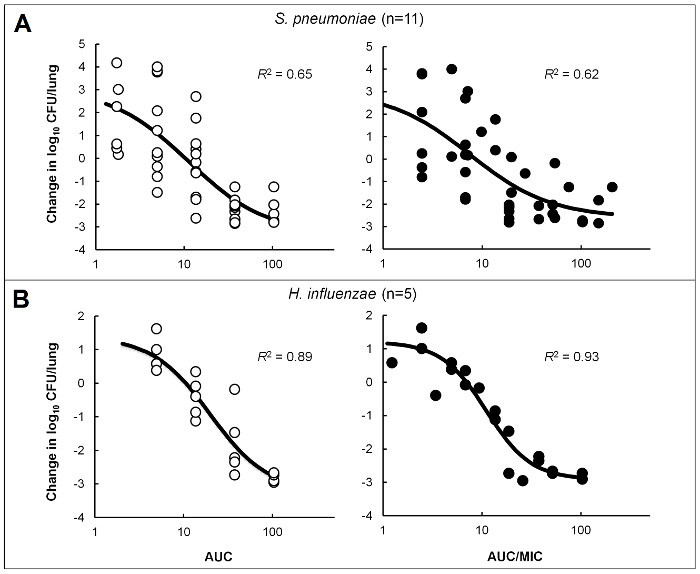 Figure 4:PK/PD Characterization of a Novel PDF Inhibitor. Emax models were created using dose ranging efficacy data in immunocompetent mice infected in the lung with isolates of S. pneumoniae (A) or H. influenzae (B) to determine PK/PD targets for a novel PDF inhibitor18. Circles represent the mean bacterial burden from 4-5 mice/group treated with varying doses of compound. Analyses were performed to correlate efficacy with free-drug 24 h AUC (open circles) or AUC/MIC (filled circles). Reprinted with permission, Copyright © American Society for Microbiology, [Antimicrobial Agents and Chemotherapy, volume 60, 2016, pages 180 - 189]18. Please click here to view a larger version of this figure.
Figure 4:PK/PD Characterization of a Novel PDF Inhibitor. Emax models were created using dose ranging efficacy data in immunocompetent mice infected in the lung with isolates of S. pneumoniae (A) or H. influenzae (B) to determine PK/PD targets for a novel PDF inhibitor18. Circles represent the mean bacterial burden from 4-5 mice/group treated with varying doses of compound. Analyses were performed to correlate efficacy with free-drug 24 h AUC (open circles) or AUC/MIC (filled circles). Reprinted with permission, Copyright © American Society for Microbiology, [Antimicrobial Agents and Chemotherapy, volume 60, 2016, pages 180 - 189]18. Please click here to view a larger version of this figure.
| Stasis | 1-log10 reduction | ||||||
| Organism | MIC (µg/mL) | R2 for line fit (%) | fAUC | fAUC/MIC | fAUC | fAUC/MIC | Max killing (log10 reduction) |
| S. pneumoniae | |||||||
| 10127 | 0.25 | 99.9 | 2.0 | 8.1 | 3.6 | 14.4 | 2.8 |
| 1316009S | 0.25 | 96.1 | 6.5 | 26.0 | 15.1 | 60.3 | 1.8 |
| ATCC10813 | 0.5 | 100 | 8.4 | 16.9 | 22.2 | 44.5 | 1.2 |
| 1307007S | 1 | 95.8 | 16.1 | 16.1 | 20.1 | 20.1 | 2.7 |
| ATCC6303 | 1 | 99.8 | 19.8 | 19.8 | 24.8 | 24.8 | 2.8 |
| 1629 | 2 | 100 | 15.8 | 7.9 | 21.2 | 10.6 | 2.4 |
| 298443 | 2 | 99.9 | 26.5 | 13.2 | 31.0 | 15.5 | 2.3 |
| 336808 | 2 | 86 | 8.2 | 4.2 | 19.0 | 9.5 | 2.6 |
| 338860 | 2 | 99.5 | 2.9 | 1.5 | 6.7 | 3.4 | 2.8 |
| 340449 | 2 | 94.6 | 8.0 | 4.0 | 14.9 | 7.4 | 2.1 |
| L11259 | 2 | 100 | 7.1 | 3.5 | 9.0 | 4.5 | 2.0 |
| Mean ± SD | NAa | NA | 11.0 ± 7.5 | 11.0 ± 7.9 | 17.0 ± 8.2 | 19.5 ± 17.8 | 2.3 ± 0.5 |
| Median | NA | NA | 8.2 | 8.1 | 19.0 | 14.4 | 2.4 |
| H. influenzae | |||||||
| 1998-100-126H | 1 | 99.7 | 7.3 | 7.3 | 13.4 | 13.4 | 2.9 |
| 503-008H | 1 | 99.8 | 7.1 | 7.1 | 13.9 | 13.9 | 2.7 |
| 08003H | 2 | 99.9 | 14.5 | 7.2 | 16.7 | 8.3 | 2.7 |
| H128 | 2 | 96.6 | 15.3 | 7.6 | 26.0 | 13.0 | 2.7 |
| 19001H | 4 | 91.9 | 14.8 | 3.7 | 37.3 | 9.3 | 3.0 |
| Mean ± SD | NA | NA | 11.8 ± 4.2 | 6.6 ± 1.6 | 21.5 ± 10.2 | 11.6 ± 2.6 | 2.8 ± 0.1 |
| Median | NA | NA | 14.5 | 7.2 | 16.7 | 13.0 | 2.7 |
| a NA, not applicable. |
Table 1: PK/PD Targets for a Novel PDF Inhibitor against S. pneumoniae and H. influenzae Isolates. Free-drug AUC and AUC/MIC targets for stasis and a 1-log10 reduction in CFU from baseline were determined for 11 S. pneumoniae and 5 H. influenzae in immunocompetent mice infected in the lung and treated with compound18. The data was used to help select human doses for clinical development. This table has been adapted and reprinted with permission, Copyright © American Society for Microbiology, [Antimicrobial Agents and Chemotherapy, volume 60, 2016, pages 180 - 189]18.
| Treatment Groupa | Mean log10S. pneumoniae (CFU/lung)b ± SD for strain (AMX/CA MIC): | |||||||
| 05010S (2 µg/mL) | 16001Sc (4 µg/mL) | 16001S (4 µg/mL) | 30005S (4 µg/mL) | 20009S (4 µg/mL) | 05003S (8 µg/mL) | 404053 (8 µg/mL) | 47003S (8 µg/mL) | |
| Control | 7.3 ± 0.4 | 7.0 ± 0.8 | 5.7 ± 1.2 | 7.1 ± 0.7 | 7.1 ± 0.4 | 6.4 ± 0.6 | 6.8 ± 0.4 | 6.0 ± 0.3 |
| AMX/CA | ||||||||
| 2,000/125 mg b.i.d. (ratio 16:1) | ≤ 1.7** | 2.8 ± 1.2** | 2.2 ± 0.8** | 2.3 ± 0.9** | 2.5 ± 0.9** | 2.0 ± 0.9** | 3.8 ± 1.4** | 1.8 ± 0.2** |
| 1,000/125 mg t.i.d. (ratio 8:1) | NT | NT | 2.8 ± 0.5** | 2.7 ± 1.3** | 3.3 ± 0.9** | 4.9 ± 0.5** | 6.6 ± 1.3 | 4.7 ± 0.7** |
| 875/125 mg t.i.d. (ratio 7:1) | NT | NT | 3.0 ± 1.3* | 2.1 ± 0.7** | 3.2 ± 0.4** | 5.2 ± 0.9* | 6.4 ± 0.6 | 4.9 ± 0.4** |
| 875/125 mg b.i.d. (ratio 7:1) | NT | NT | 4.5 ± 1.2* | 5.6 ± 1.3* | 5.2 ± 0.7** | 6.3 ± 0.7 | 6.4 ± 0.7 | 5.5 ± 0.4 |
| 500/125 mg t.i.d. (ratio 4:1) | 3.1 ± 1.3** | 4.5 ± 0.9** | NT | NT | NT | NT | NT | NT |
| Azithromycin, 1,000/500 mg o.d. | NT | NT | NT | 5.8 ± 2.0 | 7.1 ± 0.9 | 3.1 ± 1.0** | 6.6 ± 0.4 | 6.0 ± 0.6 |
| Levofloxacin, 500 mg o.d. | NT | NT | 4.2 ± 0.8* | 5.7 ± 0.8* | 4.9 ± 0.8** | 3.0 ± 1.3** | 3.5 ± 0.2** | 3.9 ± 0.8** |
| Treatment Groupd | Mean log10H. influenzae (CFU/lung) ± SD for strain: | |||||||
| H128 (β-lactamase positive) | Chesterfield (β-lactamase negative, ampicillin resistant) | |||||||
| Control | 6.4 ± 0.6 | 6.7 ± 0.6 | ||||||
| AMX/CA | ||||||||
| 2,000/125 mg b.i.d. (ratio 16:1) | 2.0 ± 0.7** | 3.1 ± 0.9** | ||||||
| 1,000/125 mg t.i.d. (ratio 8:1) | 2.1 ± 1.0** | 3.6 ± 1.1** | ||||||
| 875/125 mg t.i.d. (ratio 7:1) | 2.8 ± 1.3** | 3.8 ± 1.3** | ||||||
| 875/125 mg b.i.d. (ratio 7:1) | 2.0 ± 0.8** | 5.1 ± 1.1** | ||||||
| Azithromycin, 1,000/500 mg o.d. | 3.2 ± 1.7* | 5.8 ± 1.1 | ||||||
| Levofloxacin, 500 mg o.d. | ≤ 1.7** | ≤ 1.7** | ||||||
| a AMX/CA, amoxicillin-clavulanate; b.i.d., twice a day; t.i.d., three times a day; o.d., once a day. | ||||||||
| b The limit of detection was <1.7; *, significantly different from the control (P ≤ 0.05); **, significantly different from the control (P ≤ 0.01); NT, not tested. | ||||||||
| c Strain 16001S was tested in two separate experiments. | ||||||||
| d Amoxicillin-clavulanate 500/125 mg (4:1) three times a day was not tested against the H. influenzae strains. |
Table 2: Efficacy of Recreated Human Plasma Exposure Profiles for Different Amoxicillin/Clavulanate Dosing Regimens. Data generated in immunocompetent rats infected in the lung with isolates of S. pneumoniae or H. influenzae using this model demonstrated that an enhanced formulation of amoxicillin-clavulanate (2,000/125 mg twice daily) was more effective than standard dosing regimens against isolates with elevated in vitro susceptibility19. This table has been adapted and reprinted with permission, Copyright © American Society for Microbiology, [Antimicrobial Agents and Chemotherapy, volume 49, 2005, pages 908 - 915]19.
Discussion
For optimal success in reproducing this infection model, the following additional suggestions should be considered. Virulence of new isolates can be enhanced by passaging 2 - 3 times in vivo prior to utilization in a study. Frozen bacterial stocks should always be prepared from a primary in vivo-derived source with as few passages as possible from the original, and refreezing or reuse of thawed stock is discouraged. Using recent clinical isolates recovered from patients with pneumonia, and/or preparing cultures in log phase growth can also help to improve establishment of the infection. A five-fold dilution into the agar (e.g. 2 mL saline suspension added to 8 mL agar) can be used instead of ten-fold to increase the bacterial inoculum, and the inoculum volume can be adjusted based on the size of the animal (e.g. 200 µL/animal is usually inoculated into 250 g rats). Prior to adding the saline bacterial suspension into the agar (i.e. at Step 2.3), the bacterial density can be predicted or estimated by visual inspection, MacFarland standards or optical density measurements. It is helpful to become familiar with the in vitro growth characteristics for each isolate prior to conducting in vivo experiments. Consistency in growth and handling enables the most accurate estimation of the bacterial density for any given isolate. Standard microbiological methods that differ from those described, such as alternative media and tissue homogenizers, may be used as appropriate for preparing inocula and enumerating bacteria from infected tissues.
It is highly recommended to prepare or melt the agar the day before the experiment and store it overnight in a separate water bath set to 50 °C. This will simplify the process and help guard against the agar being too hot at the time of infection. A temperature of 41 - 42 °C achieves a good balance between maintaining the agar in liquid state without being too hot for short-term bacterial survival. As a possible alternative to nutrient agar, noble agar has been successfully used in some experiments. One tube of agar inoculum is usually sufficient for an entire experiment (and is recommended), but multiple tubes can be prepared for infecting a large number of animals (e.g. more than 60) or when the infecting process takes longer than 30 - 45 min If multiple inoculum tubes are required, add the saline bacterial suspension to each tube of agar as it is needed. This reduces the risk that bacterial survival and/or fitness will be affected by extended exposure to an elevated temperature prior to inoculation. It should be noted that using multiple inoculum tubes may also require additional animal randomization procedures. Whenever possible, infect all animals from the same inoculum preparation. It is recommended to tailor the size of experiments to the current level of proficiency with the technique.
A skilled scientist can intubate and inoculate an animal in less than 30 s and inoculate up to 5 - 6 animals per batch (i.e. with one syringe full of agar inoculum). For those learning the technique, speed is important as the agar will begin to solidify; however, accurate placement of the inoculum is more important. Begin with 1 or 2 animals per batch, and increase as the technique becomes more familiar. The animals continue to breathe during intubation; thus, the time window for inoculation depends on how quickly the animal recovers from isoflurane, which should be approximately 2 - 3 min. Animals that awaken during the process can be re-anesthetized and attempted a second time, but it is not recommended to do so repeatedly. Successful inoculation on the first attempt is expected in almost 100% of animals once proficiency is achieved. Note that it is easier to learn the technique using rats first; mice are more delicate and the smaller tools are more prone to blockage with solidified agar if not manipulated quickly. Pre-warming syringes, tubing and saline as well as keeping the intubation device on a warm (not hot) surface, such as a heating pad on low setting, can help prevent solidification of the agar. When learning the technique, it is also helpful to practice proper placement of the inoculum using a dark-colored dye (such as concentrated methylene blue) instead of the bacterial suspension in agar. Perform the procedure as described, but euthanize the animal immediately following inoculation of the dye (not allowing the animal to recover between anesthesia and euthanasia). Dissect to determine where the dye has been placed, and adjust technique accordingly.
The primary endpoint in this model is CFU from infected lungs. Survival is not a good indicator of bacterial burden and is not a recommended endpoint. For efficacy studies, the suggested N is 5 - 6 animals per group as this is predicted to detect ≥1 log10 CFU differences between groups with at least 90% power. Animals can be infected in groups such that cage mates remain together, or a true randomization process can be followed. Note that if groups are assigned by cage, the groups should be infected in a random sequence with the end-of-study growth controls infected last (to confirm that bacterial survival/fitness was not affected by the length of time exposed to an elevated temperature in the water bath). No data censoring methods should be necessary, and none are recommended with the exception of immediate removal of any animal that has been obviously mis-inoculated at the beginning of the study (prior to initiation of any treatments). Animals that are euthanized prior to the end of the study should be sampled and results included in the data set unless a valid reason for exclusion was identified prospectively (i.e. an event unrelated to the infection or treatment). Drug carryover may affect some samples and is an important consideration across all in vivo infection models. If a compound is given frequently, close to the time of euthanasia or has a long half-life, it can be present in the tissue homogenate at a high enough concentration to continue killing bacteria ex vivo during the bacterial enumeration process (dilution and plating of the samples or on the agar plates during overnight incubation). To prevent this, activated charcoal and/or an additive which degrades the active molecule without harming the bacterial cells can be added to the sample prior to homogenization. Other methods include centrifuging the samples to remove the majority of the active compound (which should remain in the supernatant) and employing different dilution and plating schemes to sufficiently dilute the active compound to a non-inhibitory concentration.
As evidenced by the examples shown in Figure 2, this model successfully induces lung infections in rodents with a wide range of organisms including those that do not grow well in other models (e.g. H. influenzae). These infections are consistent and reproducible, reducing the likelihood that experiments will need to be repeated due to model failure and/or poor performance with a given isolate. Although the animals are immunocompetent, they are unable to rapidly resolve the infection, if at all. This allows for increased flexibility in study length, as many isolates maintain a viable infection through at least 96 h without the need for repeated injections to maintain neutropenia. The potential benefit of studying antibacterial efficacy in immunocompetent animals has been noted previously20, 21, and there is evidence that, for some compounds (e.g. oxazolidinones), data from non-neutropenic rodents may more accurately predict human exposure targets compared with those rendered neutropenic5. The multi-purpose utility of the model is demonstrated in Figures 3 and 4 and Tables 1 and 2. These studies are part of a large collection of published and unpublished data that has been generated with this model to support lead optimization efforts16, 17, 22-24, for comparison and confirmation of proposed human dosing regimens19, 25-29 and for PK/PD characterization18, 30-32.While this model is initially more complex to conduct compared with the intranasal inhalation method, there are many benefits as described above. With practice and routine use, the techniques should become straightforward to perform.
It may be noted in some studies that the bacterial burden at baseline was lower than that typically targeted in other lung infection models. This is due in large part to the required dilution into agar and the small challenge volume, particularly in mice. However, it should also be noted that bacterial growth was seen in all cases even when the initial burden was relatively low. The target baseline bacterial load is 6 to 6.5 log10 CFU/lungs (6.8 to 7.3 log10 CFU/g of tissue in mice based on average lung weights) which corresponds to densities estimated in human patients with severe pneumonia33. A higher baseline burden can be achieved by further concentrating the bacterial inoculum or by delaying the start of compound administration to allow additional bacterial growth; however, increasing the challenge load too much can lead to abnormally severe and acute disease (i.e. death in less than 24 h) that is refractory to all antibacterial treatments regardless of susceptibility. Although the inoculum is initially placed preferentially into the left lung of the animal, the infection generally spreads throughout both lungs. Progressive lung disease, dissemination of the bacteria to other organs, and eventual morbidity is often observed with S. pneumoniae, K. pneumoniae and P. aeruginosa. Of interest, infections with H. influenzae and A. baumannii are usually more contained and rarely lead to death at the prescribed bacterial inocula.
Efficacy results obtained from this model have correlated well with in vitro susceptibility profiles as well as defined PK/PD targets. Reductions in bacterial burden are routinely observed for isolates considered susceptible to the agent being tested, while those considered resistant exhibit no change or bacterial growth above baseline16, 17, 19, 24-29. Studies in rats evaluating two quinolones and a macrolide using this model32 have shown that the PK/PD target required for a 1-log10 reduction in S. pneumoniae compared with baseline correlated with the target determined in neutropenic mice and, more importantly, with clinical targets for community-acquired bacterial pneumonia6, 7, 34, 35. Gepotidacin, a novel mechanism antibiotic, was tested against multiple S. pneumoniae and required a similar PK/PD target for a 1-log10 reduction in this lung infection model31 as that required for a 1-log10 reduction in the neutropenic thigh model36 when lung penetration was taken into account (data on file). Similarly, the PK/PD targets determined in mice for GSK2251052 against P. aeruginosa were consistent for stasis, 1- and 2-log10 reductions in CFU between this lung model30 and the neutropenic thigh infection model when lung penetration was considered37, 38. Correlation with a neutropenic lung infection model using intranasal inoculation was poor; however, GSK2251052 did not produce more than a static response in that study38. This may be attributable to the higher bacterial inoculum, which was 8 log10 CFU/mouse as compared with 6 log10 CFU/mouse in the neutropenic thigh37 and intratracheal intubation30 models. Collection of data such as this is critical for benchmarking, as it allows direct comparison with existing models and correlation with clinical data to assess the translational predictive capability. More widespread use of the intratracheal intubation lung infection model will provide additional data for these types of analyses.
There are several limitations of this immunocompetent pneumonia model. First, it is not well-suited to evaluating emergence of spontaneous resistance because the high bacterial inoculum generally required for such studies results in too severe an infection. Following attempts to achieve bacterial burdens of 7 or 8 log10 CFU at baseline, rapid morbidity has been observed (i.e. animals becoming moribund in less than 24 h) despite thorough washing of the inocula to remove pre-existing toxins (data on file). It may be possible to overcome this issue by using larger rodents. Rats, especially heavier rats (> 250 g), appear to better tolerate higher inocula as compared with mice and may be suitable for these types of studies. A second limitation is that the use of agar as an infection-enhancer may preclude using the model for evaluation of certain host:pathogen interactions. It is believed that the agar provides a protected focal point until infection is fully established within the lung. It is possible to deliver the inoculum in saline rather than agar to help overcome this issue; however, it should be noted that this will only produce infection with some isolates. Thirdly, there is a steep learning curve required to become proficient in the technique. The procedure can be delicate, especially in mice. It is easy to mistakenly place the metal cannula into the esophagus, and excessive force can lead to puncture of either the esophagus or the trachea. Careful placement of the inoculum is also required or animals may either not recover or not be adequately infected. However, with patience and perseverance, one can become highly proficient and perform the technique quickly, smoothly and accurately.
By removing the requirement for neutropenia, increasing robustness and reproducibility, allowing investigators to study more pathogens and isolates, improving the flexibility of experimental design and providing a challenging infection to characterize pharmacodynamics for pneumonia, this immunocompetent lung infection model adds substantial value to the antibiotic discovery community. Increased use of this model by additional investigators will help provide the necessary benchmarking for it to gain more widespread acceptance and continue providing supportive information for appropriate interpretation.
Disclosures
The authors are all paid employees of GlaxoSmithKline Pharmaceuticals and own shares of stock within the company.
Acknowledgments
JH wishes to acknowledge and thank mentor, friend and former manager Valerie Berry for first teaching us this model; and Gary Woodnutt, who taught us the foundations of and inspired our commitment to the field of antibacterial PK/PD. All the authors wish to thank David Payne for his support and appreciation for our science. We would like to acknowledge our former in vivo microbiology colleagues who contributed to the body of data generated using these methods, especially Pete DeMarsh, Rob Straub, Roni Page and Nerissa Simon. Finally, our thanks go to our in vitro microbiology colleagues for their assistance, especially for providing all the MICs we request (Lynn McCloskey, Josh West and Sharon Min); and for our clinical microbiology team who provides expert advice and support (Linda Miller, Nicole Scangarella-Oman and Deborah Butler).
References
- Eagle H, Fleischman R, Levy M. Continuous' vs. 'discontinuous' therapy with penicillin: the effect of the interval between injections on therapeutic efficacy. N Engl J Med. 1953;248(12):481–488. doi: 10.1056/NEJM195303192481201. [DOI] [PubMed] [Google Scholar]
- Eagle H, Fleischman R, Musselman AD. The bactericidal action of penicillin in vivo: the participation of the host, and the slow recovery of the surviving organisms. Ann Intern Med. 1950;33(3):544–571. doi: 10.7326/0003-4819-33-3-544. [DOI] [PubMed] [Google Scholar]
- Vogelman B, et al. Correlation of antimicrobial pharmacokinetic parameters with therapeutic efficacy in an animal model. J Infect Dis. 1988;158(4):831–847. doi: 10.1093/infdis/158.4.831. [DOI] [PubMed] [Google Scholar]
- Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998;26(1):1–12. doi: 10.1086/516284. [DOI] [PubMed] [Google Scholar]
- Ambrose PG, et al. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin Infect Dis. 2007;44(1):79–86. doi: 10.1086/510079. [DOI] [PubMed] [Google Scholar]
- Ambrose PG, Bhavnani SM, Owens RC. Clinical pharmacodynamics of quinolones. Infect Dis Clin North Am. 2003;17(3):529–543. doi: 10.1016/s0891-5520(03)00061-8. [DOI] [PubMed] [Google Scholar]
- Andes D. Pharmacokinetic and pharmacodynamic properties of antimicrobials in the therapy of respiratory tract infections. Curr Opin Inf Dis. 2001;14(2):165–172. doi: 10.1097/00001432-200104000-00010. [DOI] [PubMed] [Google Scholar]
- Mizgerd JP, Skerrett SJ. Animal models of human pneumonia. Am J Physiol Lung Cell Mol Physiol. 2008;294(3):L387–L398. doi: 10.1152/ajplung.00330.2007. [DOI] [PubMed] [Google Scholar]
- Lorian V, editor. Antibiotics in laboratory medicine, 5th edition. Chapter 15: Evaluation of antimicrobials in experimental animal infections. Philadelphia: Lippincott Williams & Wilkins; 2005. [Google Scholar]
- McConnell MJ, Actis L, Pachón J. Acinetobacter baumannii: human infections, factors contributing to pathogenesis and animal models. FEMS Microbiol Rev. 2013;37(2):130–155. doi: 10.1111/j.1574-6976.2012.00344.x. [DOI] [PubMed] [Google Scholar]
- Nuermberger E. Murine models of pneumococcal pneumonia and their applicability to the study of tissue-directed antimicrobials. Pharmacotherapy. 2005;25(12 Pt 2):134S–139S. doi: 10.1592/phco.2005.25.12part2.134S. [DOI] [PubMed] [Google Scholar]
- Mehrad B, Standiford TJ. Use of animal models in the study of inflammatory mediators of pneumonia. ILAR J. 1999;40(4):167–174. doi: 10.1093/ilar.40.4.167. [DOI] [PubMed] [Google Scholar]
- De Simone M, et al. Host genetic background influences the response to the opportunistic Pseudomonas aeruginosa infection altering cell-mediated immunity and bacterial replication. PLoS ONE. 2014;9(9) doi: 10.1371/journal.pone.0106873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G. A simple non-surgical method of intrabronchial instillation for the establishment of respiratory infections in the rat. Lab Anim. 1991;25(1):46–49. doi: 10.1258/002367791780808130. [DOI] [PubMed] [Google Scholar]
- Berry V, Ferreira-Cornwell MC, Singley C, Woodnutt G. Development of experimental murine infections caused by H. influenzae and H. parainfluenzae. Clin Microbiol Infect. 2001;7(S1):322. [Google Scholar]
- Miles TJ, et al. Novel cyclohexyl-amides as potent antibacterials targeting bacterial type IIA topoisomerases. Bioorg Med Chem Lett. 2011;21(24):7483–7488. doi: 10.1016/j.bmcl.2011.09.114. [DOI] [PubMed] [Google Scholar]
- Miles TJ, et al. Novel tricyclics (e.g. GSK945237) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg Med Chem Lett. 2016;26(10):2464–2469. doi: 10.1016/j.bmcl.2016.03.106. [DOI] [PubMed] [Google Scholar]
- Hoover J, et al. Pharmacokinetics/pharmacodynamics of peptide deformylase inhibitor GSK1322322 against Streptococcus pneumoniae, Haemophilus influenzae, and Staphylococcus aureus in rodent models of infection. Antimicrob Agents Chemother. 2016;60(1):180–189. doi: 10.1128/AAC.01842-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry V, Hoover J, Singley C, Woodnutt G. Comparative bacteriological efficacy of pharmacokinetically enhanced amoxicillin-clavulanate against Streptococcus pneumoniae with elevated amoxicillin MICs and Haemophilus influenzae. Antimicrob Agents Chemother. 2005;49(3):908–915. doi: 10.1128/AAC.49.3.908-915.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czock D, Markert C, Hartman B, Keller F. Pharmacokinetics and pharmacodynamics of antimicrobial drugs. Expert Opin Drug Metab Toxicol. 2009;5(5):475–487. doi: 10.1517/17425250902913808. [DOI] [PubMed] [Google Scholar]
- Drusano GL, Fregeau C, Liu W, Brown DL, Louie A. Impact of burden on granulocyte clearance of bacteria in a mouse thigh infection model. Antimicrob Agents Chemother. 2010;54(10):4368–4372. doi: 10.1128/AAC.00133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles TJ, et al. Novel hydroxyl tricyclics (e.g. GSK966587) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg Med Chem Lett. 2013;23(19):5437–5441. doi: 10.1016/j.bmcl.2013.07.013. [DOI] [PubMed] [Google Scholar]
- Hunt E. Pleuromutilin antibiotics. Drugs Future. 2000;25(11):1163–1168. [Google Scholar]
- Jin Q, et al. Novel LpxC inhibitors with broad-spectrum gram-negative activity; Proceedings of the 44th National Organic Chemistry Symposium.; College Park, MD. 2015. Abstract T-31. [Google Scholar]
- Singley C, Page R, Hoover J, Elefante P, DeMarsh P. Efficacy of a LRS inhibitor GSK2251052 against Enterobacteriaceae isolates using a computer-controlled infusion system to recreate human PK profiles in rats. Clin Microbiol Infect. 2011;17(S4):S429–S430. [Google Scholar]
- Berry V, et al. Comparative in vivo activity of gemifloxacin in a rat model of respiratory tract infection. J Antimicrob Chemother. 2000;45(S1):79–85. doi: 10.1093/jac/45.suppl_3.79. [DOI] [PubMed] [Google Scholar]
- Tsuji M, et al. S-649266, a Novel Siderophore Cephalosporin: Efficacy against Klebsiella pneumoniae producing NDM-1 or KPC in rat lung infection model with recreated humanized exposure profile of 2 gram dose with 1 hour and 3 hours infusion. Open Forum Infect Dis. 2014;1(S1):S106–S107. [Google Scholar]
- Tsuji M, et al. Use of Iron Depleted Mueller Hinton Broth (IDMHB) for microdilution testing of S-649266, a novel siderophore cephalosporin. 26th European Congress of Clinical Microbiology and Infectious Diseases; Amsterdam, Netherlands. 2016. Abstract P0808. [Google Scholar]
- Singley C, Hoover J, DeMarsh PJ, Elefante P, Zalacain M. Efficacy of PDF Inhibitor GSK1322322 Against Abscess Infections Caused by MRSA Using a Computer-Controlled Infusion System to Recreate Human PK Profiles in Rats. Proceedings of the 60th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; Boston, MA. 2010. Abstract F1-2114. [Google Scholar]
- Hoover J, Mininger C, Rittenhouse S. GSK2251052, a novel LeuRS inhibitor, is effective against multi-drug resistant Pseudomonas aeruginosa in a mouse pneumonia model; Proceedings of the 52nd Interscience Conference of Antimicrobial Agents and Chemotherapy; San Francisco, CA. 2012. Abstract B-1308. [Google Scholar]
- Hoover J, Mininger C, Novick S, Rittenhouse S. Efficacy of GSK2140944 against Streptococcus pneumoniae in a non-neutropenic mouse lung infection model; Proceedings of the 53rd Interscience Conference of Antimicrobial Agents and Chemotherapy; Denver, CO. 2013. Abstract A-014. [Google Scholar]
- Hoover JL, et al. Selection of a lung infection model to predict efficacy in community-acquired pneumonia (CAP) caused by S. pneumoniae (SP); Proceedings of the 47th Interscience Conference of Antimicrobial Agents and Chemotherapy; Chicago, Illinois. 2007. Abstract A-17. [Google Scholar]
- Drusano GL, et al. Saturability of granulocyte kill of Pseudomonas aeruginosa in a murine model of pneumonia. Antimicrob Agents Chemother. 2011;55(6):2693–2695. doi: 10.1128/AAC.01687-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric M, Browne FA, Jacobs MR, Applebaum PC. Activity of nine oral agents against gram-positive and gram-negative bacteria encountered in community-acquired infections: use of pharmacokinetic/pharmacodynamic breakpoints in the comparative assessment of beta-lactam and macrolide antimicrobial agents. Clin Ther. 2003;25(1):169–177. doi: 10.1016/s0149-2918(03)90021-x. [DOI] [PubMed] [Google Scholar]
- Calbo E, Garau J. Application of pharmacokinetics and pharmacodynamics to antimicrobial therapy of community-acquired respiratory tract infections. Respiration. 2005;72(6):561–571. doi: 10.1159/000089567. [DOI] [PubMed] [Google Scholar]
- Bulik CC, et al. Evaluation of the pharmacokinetics-pharmacodynamics of GSK2140944 against Staphylococcus aureus and Streptococcus pneumoniae in a murine-thigh infection model; Proceedings of the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy; Washington, DC. 2014. Abstract A-680. [Google Scholar]
- Bulik CC, et al. Evaluation of the pharmacokinetics-pharmacodynamics of GSK2251052 against gram-negative bacilli in a murine-thigh infection model; Proceedings of the 52nd Interscience Conference of Antimicrobial Agents and Chemotherapy; San Francisco, CA. 2012. Abstract A-1270. [Google Scholar]
- Andes DR, et al. Evaluation of the pharmacokinetics-pharmacodynamics of GSK2251052 against gram-negative bacilli using data from a neutropenic murine-pneumonia infection model; Proceedings of the 52nd Interscience Conference of Antimicrobial Agents and Chemotherapy; San Francisco, CA. 2012. Abstract A-1271. [Google Scholar]