

Abstract
Induced Pluripotent Stem Cells (iPSCs) hold great promise for disease modeling and regenerative therapies. We previously reported the use of Episomal Vectors (EV) to generate integration-free iPSCs from peripheral blood mononuclear cells (PB MNCs). The episomal vectors used are DNA plasmids incorporated with oriP and EBNA1 elements from the Epstein-Barr (EB) virus, which allow for replication and long-term retainment of plasmids in mammalian cells, respectively. With further optimization, thousands of iPSC colonies can be obtained from 1 mL of peripheral blood. Two critical factors for achieving high reprogramming efficiencies are: 1) the use of a 2A "self-cleavage" peptide to link OCT4 and SOX2, thus achieving equimolar expression of the two factors; 2) the use of two vectors to express MYC and KLF4 individually. Here we describe a step-by-step protocol for generating integration-free iPSCs from adult peripheral blood samples. The generated iPSCs are integration-free as residual episomal plasmids are undetectable after five passages. Although the reprogramming efficiency is comparable to that of Sendai Virus (SV) vectors, EV plasmids are considerably more economical than the commercially available SV vectors. This affordable EV reprogramming system holds potential for clinical applications in regenerative medicine and provides an approach for the direct reprogramming of PB MNCs to integration-free mesenchymal stem cells, neural stem cells, etc.
Keywords: Developmental Biology, Issue 119, peripheral blood mononuclear cells (PB MNC), hematopoietic progenitor cells (HPCs), reprogramming, episomal vectors, integration-free, induced pluripotent stem cells (iPSCs)
Introduction
After forced expression of several transcription factors(i.e. OCT4, SOX2, MYC and KLF4), somatic cells can be reprogrammed to induced Pluripotent Stem Cells (iPSCs), which hold great promise for applications in regenerative medicine and cell replacement therapy1-3. To date, diverse methods have been developed to increase the success rate of reprogramming4-7. Viral vectors-induced reprogramming is widely used for efficient generation of iPSCs, because viral integration leads to a high-level, stable expression of the reprogramming factors. However, permanent integration of the vector DNA into the cell genome may induce insertional mutagenesis5. In addition, insufficient inactivation of reprogramming factors may disturb iPSCs differentiation8. As such, the use of iPSCs without integration of reprogramming factors is imperative, especially for use in cell therapy applications.
Episomal Vectors (EVs) are widely used in the generation of integration-free iPSCs. The most commonly used EV is a plasmid containing two elements, origin of viral replication (oriP) and EB Nuclear Antigen 1 (EBNA1), from the Epstein-Barr (EB) virus9. The oriP element promotes plasmid replication in mammalian cells, while the EBNA1 element tethers the oriP-containing plasmid DNA to the chromosomal DNA that allows for the partitioning of the episome during division of the host cell. In comparison to other integration-free approaches, including Sendai Virus (SV) and RNA transfection, EVs possess multiple advantages5,6,10. As plasmid DNA, EVs can be readily produced and modified in house, making them extremely affordable. In addition, reprogramming with EV is a less labor-intensive process since a single transfection with EVs is sufficient for iPSC generation, whereas several RNA transfections are necessary for successful reprogramming.
Dermal fibroblasts have been used in many reprogramming studies. However, skin biopsy is not only an invasive and painful process, but also time-consuming for expanding cells to sufficient quantities for reprogramming. Of greater concern, skin cells of adult donors have often been exposed to long-term UV light radiation, which may lead to mutations associated with tumors, thus limiting the applications for iPSCs derived from skin fibroblasts11,12. Recently, it has been reported that normal human skin cells accumulate somatic mutations and multiple cancer genes, including most of the key drivers of cutaneous squamous cell carcinomas, are under strong positive selection13.
In contrast to skin fibroblasts, peripheral blood (PB) cells are a preferable source of cells for reprogramming because 1) blood cells can be easily obtained through a minimally invasive process, 2) peripheral blood cells are the progeny of hematopoietic stem cells residing in bone marrow, thus protected from harmful radiation. Peripheral blood mononuclear cells (PB MNCs) can be collected in an hour from the buffy coat layer following a simple gradient centrifugation using Ficoll-Hypaque (1.077 g/mL). The obtained PB MNCs are composed of lymphocytes, monocytes and a few Hematopoietic Progenitor Cells (HPCs) 14. Although human T lymphocytes are one of the major cell types in PB, mature T cells contain rearrangements of the T cell receptor (TCR) genes and lack an intact genome thus limiting their potential for applications15,16. However, rejuvenation of T cells via iPSC generation may have potential applications in Chimeric Antigen Receptor (CAR) T-cell therapy 17-19. In comparison, HPCs have an intact genome and are readily reprogrammable. Although only 0.01 - 0.1% cells in peripheral circulation are HPCs, these cells can be expanded ex vivo in erythroid medium that favors proliferation of erythroid progenitor cells14.
In our previous study, we used the factor BCL-XL in addition to the Yamanaka factors (OCT4, SOX2, MYC and KLF4), which resulted in a 10x increase in PB reprogramming efficency20. BCL-XL, also known as BCL2L1, is a potent inhibitor of cell death, by inhibiting activation of caspases21,22. But, BCL-XL may also play an important role in maintaining pluripotency21,22. Recently, we have further optimized our EV reprogramming system by separately expressing MYC and KLF4 with two vectors, which leads to an approximately 100x increase in reprogramming efficiency23. Using this method, the reprogramming efficiency, defined by colony number divided by starting cell number at transfection, is 0.2 - 0.5% from healthy donors. As follows, we describe the detailed experimental procedure for generating integration-free iPSCs from PB.
Protocol
All of the human PB samples were obtained from anonymous adult donors with no identification information available from Tianjin Blood Center with approval of the local research ethics committee.
1. Endo-free Plasmid Preparation
Use a commercial Plasmid Purification Maxi Kit to extract episomal vectors from E. coli according to manufacturer's protocol. For the final step, substitute TE buffer with endotoxin-free sterile water to dissolve the DNA pellet.
Measure DNA concentration using a commercial UV/Vis spectrophotometer. The concentration is usually greater than 1 µg/µL, with A260/A280 and A260/A230 ratios greater than 1.8 and 2.0, respectively.
2. Culture Media
Prepare erythroid medium: Hematopoietic Stem Cell Expansion Medium supplemented with 100 ng/mL human Stem Cell Factor (SCF), 10 ng/mL Interleukin-3 (IL3), 2 U/mL Erythropoietin (EPO), 20 ng/mL Insulin Growth Factor-1 (IGF1), 1 µM dexamethasone and 0.2 mM 1-thioglycerol. Filter sterilize with a 0.22 µm syringe filter. Erythroid medium can be stored at 4 °C for up to one month.
Prepare iPSC medium: DMEM/F12 medium (Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12) supplemented with 1x L-glutamine, 1x penicillin/streptomycin, 1x non-essential amino acids solution, 50 ng/mL Fibroblast Growth Factor 2 (FGF2), 1x Insulin-Transferrin-Selenite supplement (ITS), and 50 mg/mL ascorbic acid. Filter sterilize with a 0.22 µm syringe filter. iPSC medium can be stored at 4 °C for up to one month.
Prepare MEF medium: Dulbecco's Modified Eagle Medium (DMEM; high glucose) supplemented with 1x penicillin/streptomycin and 10% Fetal Bovine Serum (FBS). Filter sterilize with a 0.22 µm syringe filter. MEF medium can be stored at 4 °C for up to one month or freshly add 1x L-glutamine before use.
Prepare iPSC cryopreservation medium (2x): Dissolve 5 g of D-trehalose in 30 mL of sterile water in a 37 °C water bath. Bring the temperature to 4 °C and then add 10 mL of FBS and 10 mL of dimethylsulfoxide (DMSO). Filter sterilize with a 0.22 µm syringe filter. Store at 4 °C for up to 3 months24.
3. Isolation of Peripheral Blood Mononuclear Cells (PB MNCs)
Combine 10 mL fresh PB sample and 10 mL PBS buffer into a 50 mL tube and mix well. Bring PBS to RT or 37 °C prior to use. NOTE: Approximately 1 - 3 x 106 MNCs (fresh or cryopreserved) can be obtained from 1 mL of PB. After 6 d of culture, expect to have 0.5 - 1 x 106 total cells due to death of mature cells during culture. Approximately 1 x 107 cells can be obtained from 10 mL of fresh PB.
Add 10 mL Ficoll to the bottom of the tube using a 10 mL syringe attached to a long needle. This process should be done slowly to ensure that the Ficoll layer does not mix with the PB.
Centrifuge at 400 x g for 30 min at a low acceleration and deceleration rate. After centrifugation, the PB MNCs are in the white layer (buffy coat) located between the PB plasma and the Ficoll.
Slowly aspirate ~10 mL PB plasma without disturbing the buffy coat layer. Carefully harvest the white layer using a 1 mL pipette tip to a new 50 mL tube. The total volume of collected cells from the white layer will be between 3 - 6 mL.
Add PBS to bring the total volume to 30 mL and mix well with a 10 mL pipette. Centrifuge at 400 x g for 10 min.
Remove the supernatant and resuspend cell pellet in 20 mL culture medium (i.e. Iscove's Modified Dulbecco's Medium, IMDM) and mix well. Centrifuge at 400 x g for 10 min to remove the majority of the platelets.
Remove the supernatant and resuspend the cell pellet in 1 - 2 mL IMDM. Cells may be used for immediate culture, or frozen-down for later use. For cryopreservation, add an equal amount of cryopreservation medium. Aliquot cells in cryovials (0.5 - 1 mL per vial) and transfer cryovials to a -80 °C freezer immediately. PB MNCs may be stored in -80 °C freezer for several weeks or transferred to a liquid nitrogen tank for long-term storage. NOTE: When thawing the frozen cells, though our cryopreservation medium may sustain cell viability, the cell number may decrease due to cell death during the thawing process. It is recommended that 1 - 10 x 107 cells will be frozen in each vial.
4. Expansion of PB MNCs in Erythroid Medium
Prepare 5 mL IMDM medium in a 15 mL tube. Quickly thaw the frozen PB MNCs in a 37 °C water bath and then transfer them to the tube with the IMDM medium. NOTE: The length of time in water bath depends on the volume of the cryopreserved cells and the cryovial used. Usually, the frozen cell will thaw within 1 min.
Centrifuge at 400 x g for 5 - 10 min. Remove the supernatant and resuspend the cell pellet in erythroid medium. Add 10 µL Trypan Blue solution to 10 µL cell suspension and mix well. Trypan Blue stains dead cells within 1 - 3 min. Count the cells under the microscope using a hemocytometer.
Culture PB MNCs in a non-tissue culture (non-TC) treated 6-well plate with 2 mL medium per well, at a cell density of ~5 x 106 cells/mL, at 37 °C in a 5% CO2 humidified incubator.
Add 1 mL fresh erythroid medium directly to each well without changing the medium at D 3 and 5.
At D 6, harvest the PB MNCs for nucleofection.
5. PB MNC Nucleofection and Reprogramming
One day before nucleofection, pre-coat TC-treated 6-well plates with 0.1% gelatin at 37 °C for 20 min. Thaw inactivated Murine Embryonic Fibroblast (MEF) feeder cells in a 37 °C water bath and immediately transfer them to a 15 mL tube containing 5 mL of MEF medium.
Centrifuge at 400 x g for 5 min and remove the supernatant. Remove the gelatin from the 6-well plate, resuspend the cell pellet in MEF medium, and transfer them to the gelatin pre-treated 6-well plate. In each well, seed 2 - 4 x 105 inactivated MEF cells suspended in 2 mL MEF medium.
Culture the MEF feeder cells at 37 °C in a 5% CO2 humidified incubator.
On the day of nucleofection, aspirate the MEF medium and replace it with 1 mL erythroid medium. Pre-equilibrate the culture plate for 10 - 30 min at 37 °C in a 5% CO2 humidified incubator.
Add 2 µg pEV-OCT4-2A-SOX2, 1 µg pEV-MYC, 1 µg pEV-KLF4, and 0.5 µg pEV-BCL-XL in a 1.5 mL sterile Eppendorf tube. Heat the tube at 50 °C for 5 min to prevent contamination, and cool down to RT. Add 57 µL nucleofection buffer and 13 µL supplement.
Harvest 2 x 106 cultured PB MNCs to a 5 mL tube by centrifuging at 200 x g for 7 min. Remove the supernatant and add the plasmid and nucleofection buffer mix to the cell pellet. Mix well by flicking the tube with your finger. NOTE: As low as 2 x 105 cells may be used for nucleofection, but a lower reprogramming efficiency should be expected as well.
Transfer the DNA and cell suspension to the cuvette provided in the kit and cap the cuvette. Select the Program U-008 on the nucleofection device. Insert the cuvette into the holder and press OK to apply the U-008 program.
Take the cuvette out of the holder, add 0.5 - 1 mL pre-warmed erythroid medium in each cuvette, and transfer cells to the pre-equilibrated MEF plate immediately. Due to the high reprogramming efficiency and donor variation, seeding different numbers of cells ranging from 1-10 x 105 cells per well is highly encouraged.
Transfer the plate to a hypoxia chamber, and flush the chamber with a mixed gas composed of 92% N2, 5% CO2 and 3% O2, for 1 - 2 min at a rate of 20 L/min. After sealing the chamber, culture the cells at 37 °C.
At D 2 post-nucleofection, add 2 mL iPSC medium directly to each well.
At D 4 post-nucleofection, remove 3 mL of medium and add 2 mL fresh iPSC medium. At D 4 post-nucleofection, most of the live cells will have attached to the feeder layer.
After D 6 post-nucleofection, change the medium every 2 d by leaving 500 µL spent medium and adding 2 mL fresh E8 medium supplemented with 0.25 mM Sodium Butyrate until D 14 - 18. NOTE: Additional feeder cells may be added in the culture wells whenever observing that the feeder cells have been detached. After thawing MEFs (see 5.1 and 5.2), resuspend the cell pellet in a small volume of E8 medium (i.e. ~0.2 mL for one well of a 6-well plate) and then add MEFs into the culture wells. Approximately 2% FBS may be added to increase cell attachment. Alternatively, MEF-conditioned medium can also be used, but it is more laborious.
6. Expansion of iPSCs
NOTE: In most cases, large numbers of iPSC colonies appear at D 8 - 10 post nucleofection. After D 14, big iPSC colonies are usually ready for picking.
- Before picking colonies, add 500 µL E8 medium supplemented with ROCK inhibitor, Y27632 (10 µM), in one well of a feeder or Matrigel coated 24-well plate. Use a 10 or 20 µL pipette to scratch and pick colonies under the microscope. Transfer the pieces of each colony to the wells containing the culture medium. NOTE: The concentration of Matrigel differs from one batch to another. Usually, we use 1:100 dilution of Matrigel.
- In order to avoid cross-contamination, pick colonies that are large and well separated from others. Culture iPSCs at 37 °C in a 5% CO2 humidified incubator. NOTE: It should be note that the iPSC population may be polyclonal due to cell migration when there are dozens or hundreds of colonies in each well of a 6-well plate.
Do not change medium for the first 2 d. ROCK inhibitor can promote survival of small colonies25.
From D 2 onwards, change medium every day by removing spent medium and adding 500 µL fresh E8 medium in each well.
Approximately 1 week later, when the colonies are large enough, remove the medium and treat the cells with 300 µL cell detachment solution (0.5 mM EDTA in PBS) at 37 °C for 1 - 3 min. Remove the cell detachment solution and add E8 medium containing ROCK inhibitor (10 µM) to suspend iPSCs. Do not break down colonies into single cells, which may lead to excessive cell death. The cell clumps with 5 - 50 cells are suitable for iPSC passage.
Transfer 50 - 100% of the cell suspension to each well of a 12- or 6-well plate that has been precoated with feeders or Matrigel.
For long-term culture in Matrigel-coated plates (see note in 6.1), change the medium every day. When the cell confluency reaches 40 - 60%, treat the cells with 1 mL cell detachment solution (0.5 mM EDTA in PBS) at 37 °C for ~3 min. When the colonies start to curl up, remove the cell detachment solution and add E8 medium containing ROCK inhibitor (10 µM) to suspend the iPSCs. Passage cells with a splitting factor of 4 - 8. the ROCK inhibitor should be added when passaging cells to increase survival, and removed 1 d later to prevent differentiation.
To freeze-down iPSCs, treat cells with cell detachment solution at 37 °C for 3 - 5 min according to manufacturer's protocol. When iPSC colonies start to curl up, aspirate the buffer and add 0.5 - 1 mL E8 medium.
Add an equal volume of cryopreservation medium to the cell suspension and mix well. Frozen iPSCs can be stored in a -80 °C freezer for several weeks or in liquid nitrogen for long-term storage.
7. Selection of iPSCs without Residual Episomal Plasmids
After passage 5, harvest iPSCs and extract genomic DNA. NOTE: Usually after 5 passages of culture, residual episomal plasmids are undetectable. Thus, almost all of the iPSC colonies at passages above 5 (i.e. passage 10) are lacking residual episomal plasmids.
Use genomic DNA from untransfected PB MNCs as a negative control. Add 1.6 pg pEV-OCT4-2A-SOX2 plasmid into 1 µg negative control DNA to mimic cells with one copy of pEV plasmid per cell.
Add 9 µL PCR-grade water including 100 ng genomic DNA and 1 µL specific primers (10 µM each of forward and reverse primers) into 10 µL High-Fidelity PCR Master Mix. Normalize the amount of DNA by GAPDH. Use the following primers for PCR: EBNA1-F TTTAATACGATTGAGGGCGTCT, EBNA1-R GGTTTTGAAGGATGCGATTAAG, WPRE-F GGTTTAAACGCGTCGACAAT, WPRE-R GTTGCGTCAGCAAACACAGT, GAPDH-F GAGTCCACTGGCGTCTTC, GAPDH-R GACTGTGGTCATGAGTCCTTC.
Incubate the reaction mixture at 98 °C for 60 s; followed by 98 °C for 10 s, 60 °C for 30 s, and 72 °C for 30 s. After 30 cycles; extend the reaction at 72 °C for 5 min.
Load 5 µL PCR products in each well of a 1% agarose gel. Run the gel for 20 - 30 min at 100 V. Choose the iPSC clones with undetectable bands for EBNA1 and WPRE for further culture and downstream analysis.
8. Flow Cytometry
Harvest iPSCs by treating them with Accutase at 37 °C for 3 - 5 min to obtain a single cell suspension. Add 1 µL PE-conjugated anti-TRA-1-60 or eFluor 570-conjugated anti-SSEA4 or Isotypic antibody to 100 µL of cell suspension (1 - 5 x 105 cells) in 5 mL tubes. Incubate the tubes in a dark location at RT for 20 min.
After staining for 20 min at RT, add 2 mL PBS and centrifuge at 400 x g for 5 min. Resuspend cells in 300 µL PBS for Fluorescence-activated Cell Sorting (FACS) analysis using a flow cytometry cell analyzer.23,26
9. Confocal Imaging
Seed iPSCs in Matrigel coated chamber slides.
After 3 - 4 d of culture, remove the growth medium and fix with 4% paraformaldehyde (PFA) at RT for 30 min. Wash cells twice with PBS. Note: PFA is toxic and harmful.
Treat the cells with 0.1% Triton X-100 in PBS (PBS-T) at RT for 30 min. Wash the cells twice with PBS.
Block the cells with blocking buffer (5% goat serum in PBS, V:V) at RT for 1 h.
During the blocking step, dilute the primary antibody (100x) in blocking buffer. NOTE: The antibodies information is listed in the Table of Materials/Equipment.
Incubate cells with diluted antibody at 4 °C O/N.
Wash twice with PBS-T for 15 min, followed by twice with PBS for 15 min.
Incubate cells with a diluted fluorophore-conjugated secondary antibody at RT for 2 h.
Wash the cells twice with PBS-T for 15 min, followed by twice with PBS for 15 min.
Stain the nucleus with DAPI (1 µg/mL) in PBS at RT for 10 min.
Wash cells twice with PBS for 15 min.
Capture images with a confocal microscope.23,27
10. Teratoma Assay
Harvest 1 x 106 iPSCs with cell detachment solution (0.5 mM EDTA in PBS) and resuspend cells in 200 µL DMEM/F12 diluted (1:1) Matrigel.
Subcutaneously inject cells into the rear haunch of NOD/SCID immunodeficient mice.
At 8 - 12 week after iPSC injection, dissect and fix teratomas in 10% formalin.28
After microsectioning and staining with hematoxylin and eosin (H&E), analyze samples.29
Representative Results
Using this protocol, we can obtain hundreds of colonies from 1 x 105 nucleofected PB MNCs (Figures 1A and 1B). The reprogramming efficiency is approximately 0.2 - 0.5% and the colonies express pluripotency markers (Figures 1C and 1D). iPSCs generated using the described protocol are integration-free and have the ability to form teratoma composing the 3 germ layers (Figures 1E and 1F).
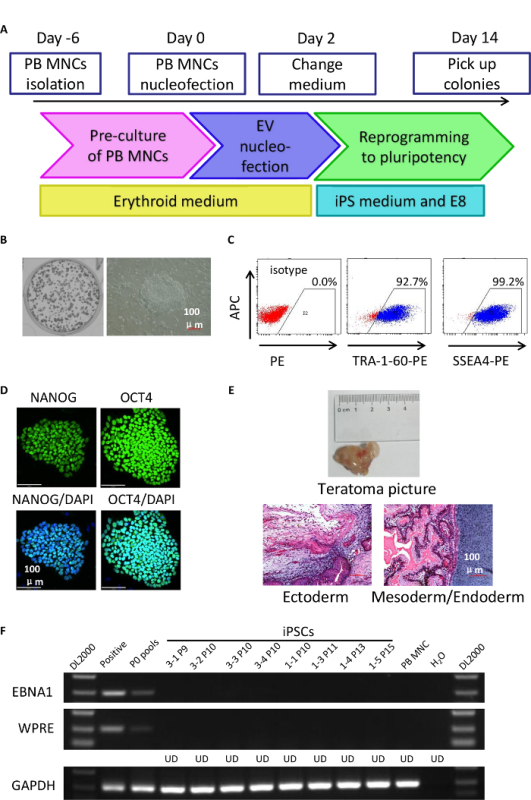 Figure 1:Generation of Integration-free iPSCs from Peripheral Blood Mononuclear Cells. (A): A schematic of the protocol for reprogramming peripheral blood cells. (B): AP staining in bulk (left) and a typical ESC-like iPSC colony (right) 14 d after PB MNCs nucleofection. Scale bar: 100 µm. (C): Representative FACS diagrams of iPSCs at passage 5 expressing TRA-1-60 or SSEA4. (D): Representative confocal images of iPSC colonies expressing NANOG and OCT4. Scale bar: 100 µm. (E): Representative image of the total picture and H&E staining of teratoma comprising all three germ layers. Scale bar: 100 µm. (F): Copy numbers of residual EV plasmids in iPSCs after five passages. Specific primers for EBNA1 and WPRE were used to amplify episomal vectors. GAPDH was used as a DNA loading control. UD, undetectable. The positive lane indicates a one-copy control. Please click here to view a larger version of this figure.
Figure 1:Generation of Integration-free iPSCs from Peripheral Blood Mononuclear Cells. (A): A schematic of the protocol for reprogramming peripheral blood cells. (B): AP staining in bulk (left) and a typical ESC-like iPSC colony (right) 14 d after PB MNCs nucleofection. Scale bar: 100 µm. (C): Representative FACS diagrams of iPSCs at passage 5 expressing TRA-1-60 or SSEA4. (D): Representative confocal images of iPSC colonies expressing NANOG and OCT4. Scale bar: 100 µm. (E): Representative image of the total picture and H&E staining of teratoma comprising all three germ layers. Scale bar: 100 µm. (F): Copy numbers of residual EV plasmids in iPSCs after five passages. Specific primers for EBNA1 and WPRE were used to amplify episomal vectors. GAPDH was used as a DNA loading control. UD, undetectable. The positive lane indicates a one-copy control. Please click here to view a larger version of this figure.
Discussion
Acquiring blood samples from healthy donors or patients is convenient and noninvasive, making it an attractive cell source for basic research and clinical cell therapy. Here we have described a protocol for highly efficient generation of integration-free iPSCs from peripheral blood samples. This reproducible and affordable approach should benefit the iPSC field.
We have reported that there are two critical factors responsible for the highly efficient PB reprogramming30. One is equimolar expression of OCT4 and SOX2 by using a 2A linker31. To achieve this, pEV-OCT4-2A-SOX2 is used in this protocol. The other is the use of two plasmids pEV-MYC and pEV-KLF4 to express MYC and KLF4 instead of pEV-MYC-2A-KLF4 that we previously have used30. The seemingly simple change leads to an approximately 100X increase in PB reprogramming efficiencies, which is largely due to a gradual and greater increase in the MYC:KLF4 ratio during the course of the process.
Several points should be considered for achieving high-efficiency iPSC generation from PB. PB MNCs are cultured for ~1 week in erythroid medium, which promotes proliferation and expansion of erythroid progenitor cells and thus increases reprogramming efficiency20. However, for some samples, especially for blood cells from leukemia patients, cells survive poorly when cultured in erythroid medium. In this case, it would be helpful to decrease culturing time. It is also encouraged to use 1 - 2 µg pmaxGFP vector to verify the nucleofection efficiency, which should be greater than 50%. In addition, the plasmid quality is important. It is recommended to use plasmids without endotoxin contamination (i.e. using Endo-free Plasmid Maxi Kit to extract plasmids). Poor plasmid quality may lead to significant cell death and thus reduce reprogramming efficiency. We and others have shown that hypoxia promotes iPSC generation14,32. We flush the hypoxia chamber with a mixed gas composed of 92% N2, 5% CO2 and 3% O2. If normoxia is used instead, an up to 80% reduction in reprogramming efficiency may be observed. Once the reprogramming is complete, iPSCs may be cultured in a regular humidified 5% CO2 incubator. When changing medium, it is handy to leave a small amount of spent medium (i.e. 500 µL per well in a 6-well plate), and then add fresh medium. Change medium only every other day during reprogramming, as too frequent medium change may reduce iPSCs generation33.
The new protocol we have developed has achieved a high-level PB reprogramming that is comparable to the SV reprogramming system. The EV system has several obvious advantages. For one, EV is more affordable than SV. The EV system can also be further modified by including additional factors or different combinations of multiple factors, whereas the SVs vectors are a commercial product that cannot be modified by investigators. Our current protocol includes the use of MEF feeder cells and animal-derived products, which may limit the clinical applications, but can be easily removed by further protocol development34,35.
iPSCs generated with this system can be used not only for disease modeling and drug screening, but also for clinical therapy, because GMP-level EV plasmids can be easily generated and no iPSCs with considerable EV integration has been identified. In clinical applications, patient cells usually harbor a disease-inducing gene, which needs to be corrected in the iPSCs before differentiation into functional cells for therapy. A recent report showed that the EV reprogramming system can be easily integrated with the CRISPR/Cas9 system to achieve simultaneous reprogramming and genome editing after a simple nucleofection36. This is another advantage of EV over the SV system. Our unpublished data have shown that up to a 20% genome editing efficiency can be achieved when piggybacking genome editing upon EV reprogramming. It is our hope that integration of PB reprogramming with CRISP/Cas9R genome editing may have potential applications in treating multiple diseases that are otherwise incurable in the coming decades.
In summary, our improved peripheral blood cell reprogramming protocol should be useful for a wide-spectrum of investigators in basic research and clinical applications. This efficient EV reprogramming system can also be employed, after modifications, to generate integration-free Mesenchymal Stem Cells (MSCs)37, or Neural Stem Cells (NSCs)38, directly from adult peripheral blood cells without passing through an iPSC stage.
Disclosures
The authors have no competing or conflicting interests to disclose.
Acknowledgments
This work was supported by the Ministry of Science and Technology of China (2015CB964902, 2013CB966902 and 2012CB966601), the National Natural Science Foundation of China (81500148, 81570164 and 81421002), the Loma Linda University School of Medicine GCAT grant (2015), and Telemedicine and Advanced Technology Research Center (W81XWH-08-1-0697).
References
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Trounson A, McDonald C. Stem Cell Therapies in Clinical Trials: Progress and Challenges. Cell Stem Cell. 2015;17:11–22. doi: 10.1016/j.stem.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Hayes M, Zavazava N. Strategies to generate induced pluripotent stem cells. Methods Mol Biol. 2013;1029:77–92. doi: 10.1007/978-1-62703-478-4_6. [DOI] [PubMed] [Google Scholar]
- Zhang XB. Cellular reprogramming of human peripheral blood cells. Genomics Proteomics Bioinformatics. 2013;11:264–274. doi: 10.1016/j.gpb.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaeger TM, et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015;33:58–63. doi: 10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, et al. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013;31:458–466. doi: 10.1002/stem.1293. [DOI] [PubMed] [Google Scholar]
- Carey BW, et al. Reprogramming factor stoichiometry influences the epigenetic state and biological properties of induced pluripotent stem cells. Cell Stem Cell. 2011;9:588–598. doi: 10.1016/j.stem.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Dorigo O, et al. Development of a novel helper-dependent adenovirus-Epstein-Barr virus hybrid system for the stable transformation of mammalian cells. J Virol. 2004;78:6556–6566. doi: 10.1128/JVI.78.12.6556-6566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowey SN, Huang X, Chou BK, Ye Z, Cheng L. Generation of integration-free human induced pluripotent stem cells from postnatal blood mononuclear cells by plasmid vector expression. Nat Protoc. 2012;7:2013–2021. doi: 10.1038/nprot.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms PW, et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod Pathol. 2016;29:240–248. doi: 10.1038/modpathol.2015.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abyzov A, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su RJ, Neises A, Zhang XB. Generation of iPS Cells from Human Peripheral Blood Mononuclear Cells Using Episomal Vectors. Methods Mol Biol. 2016;1357:57–69. doi: 10.1007/7651_2014_139. [DOI] [PubMed] [Google Scholar]
- Seki T, et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–14. doi: 10.1016/j.stem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Loh YH, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010;7:15–19. doi: 10.1016/j.stem.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino Y, Seki T, Yuasa S, Fujita J, Fukuda K. Generation of Induced Pluripotent Stem Cells from Human Peripheral T Cells Using Sendai Virus in Feeder-free Conditions. J Vis Exp. 2015. [DOI] [PMC free article] [PubMed]
- Seki T, Yuasa S, Fukuda K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat Protoc. 2012;7:718–728. doi: 10.1038/nprot.2012.015. [DOI] [PubMed] [Google Scholar]
- Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunological reviews. 2015;263:68–89. doi: 10.1111/imr.12243. [DOI] [PubMed] [Google Scholar]
- Su RJ, et al. Efficient generation of integration-free ips cells from human adult peripheral blood using BCL-XL together with Yamanaka factors. PLoS One. 2013;8:e64496. doi: 10.1371/journal.pone.0064496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. The p53-PUMA axis suppresses iPSC generation. Nat Commun. 2013;4:2174. doi: 10.1038/ncomms3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Youle RJ. SnapShot: BCL-2 proteins. Cell. 2009;138:404. doi: 10.1016/j.cell.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W, et al. Enhanced Generation of Integration-free iPSCs from Human Adult Peripheral Blood Mononuclear Cells with an Optimal Combination of Episomal Vectors. Stem Cell Reports. 2016;6:873–884. doi: 10.1016/j.stemcr.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XB, et al. Trehalose ameliorates the cryopreservation of cord blood in a preclinical system and increases the recovery of CFUs, long-term culture-initiating cells, and nonobese diabetic-SCID repopulating cells. Transfusion. 2003;43:265–272. doi: 10.1046/j.1537-2995.2003.00301.x. [DOI] [PubMed] [Google Scholar]
- Watanabe K, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 2007;25:681–686. doi: 10.1038/nbt1310. [DOI] [PubMed] [Google Scholar]
- Basu S, Campbell HM, Dittel BN, Ray A. Purification of specific cell population by fluorescence activated cell sorting (FACS) J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Castiel A, et al. Cell death associated with abnormal mitosis observed by confocal imaging in live cancer cells. J Vis Exp. 2013. p. e50568. [DOI] [PMC free article] [PubMed]
- Peterson SE, et al. Teratoma generation in the testis capsule. J Vis Exp. 2011. p. e3177. [DOI] [PMC free article] [PubMed]
- Ritner C, Bernstein HS. Fate mapping of human embryonic stem cells by teratoma formation. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Wen W, et al. Enhanced Generation of Integration-free iPSCs from Human Adult Peripheral Blood Mononuclear Cells with an Optimal Combination of Episomal Vectors. Stem Cell Reports. 2016. [DOI] [PMC free article] [PubMed]
- Lo CA, et al. Quantification of Protein Levels in Single Living Cells. Cell Rep. 2015;13:2634–2644. doi: 10.1016/j.celrep.2015.11.048. [DOI] [PubMed] [Google Scholar]
- Liu SP, et al. An improved method for generating integration-free human induced pluripotent stem cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2014;22:580–587. doi: 10.7534/j.issn.1009-2137.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Luni C, et al. High-efficiency cellular reprogramming with microfluidics. Nat Methods. 2016;13:446–452. doi: 10.1038/nmeth.3832. [DOI] [PubMed] [Google Scholar]
- Chou BK, et al. A facile method to establish human induced pluripotent stem cells from adult blood cells under feeder-free and xeno-free culture conditions: a clinically compliant approach. Stem Cells Transl Med. 2015;4:320–332. doi: 10.5966/sctm.2014-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci Rep. 2014;4:3594. doi: 10.1038/srep03594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden SE, et al. Simultaneous Reprogramming and Gene Correction of Patient Fibroblasts. Stem Cell Reports. 2015;5:1109–1118. doi: 10.1016/j.stemcr.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, et al. Rapid and efficient reprogramming of human fetal and adult blood CD34+ cells into mesenchymal stem cells with a single factor. Cell research. 2013;23:658–672. doi: 10.1038/cr.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, et al. Direct Conversion of Cord Blood CD34+ Cells Into Neural Stem Cells by OCT4. Stem cells translational medicine. 2015;4:755–763. doi: 10.5966/sctm.2014-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]