

Summary
The role of chromatin structure in lineage commitment of multipotent hematopoietic progenitors (HPCs) is presently unclear. We show here that CD34+ HPCs possess a post-replicative chromatin globally devoid of the repressive histone mark H3K27me3. This H3K27-unmodified chromatin is required for recruitment of lineage-determining transcription factors (TFs) C/EBPα, PU.1 and GATA-1 to DNA just after DNA replication upon cytokine-induced myeloid or erythroid commitment. Blocking DNA replication or increasing H3K27me3 levels prevents recruitment of these TFs to DNA and suppresses cytokine-induced erythroid or myeloid differentiation. However, H3K27me3 is rapidly associated with nascent DNA in more primitive human and murine HPCs. Treatment of these cells with instructive cytokines leads to a significant delay in accumulation of H3K27me3 in nascent chromatin due to activity of the H3K27me3 demethylase UTX. Thus, HPCs utilize special mechanisms of chromatin modification for recruitment of specific TFs to DNA during early stages of lineage specification.
Keywords: DNA replication, nascent DNA, nascent chromatin, H3K27me3, KDMs, HMTs, hematopoietic progenitors, myeloid and erythroid differentiation, transcription factors
Graphical Abstract
Introduction
The process of hematopoiesis is regulated by a network of transcription factors (TFs) acting cooperatively or antagonistically on multipotent or lineage-restricted hematopoietic progenitors/precursors (Friedman, 2007; Orkin and Zon, 2008). A critical step in differentiation of hematopoietic stem cells (HSCs) is the commitment of multipotent progenitors to lineage-restricted progenitors. The prevailing explanation for lineage commitment is that it depends on stochastic fluctuation in the levels of competing lineage-determining TFs that activate a lineage-specific gene expression program while suppressing those leading to alternative choices (Cantor and Orkin, 2001; Chang et al., 2008; Graf and Enver, 2009; Orkin, 2000; Till et al., 1964).
However, recent studies support an “instructive” model whereby lineage-specific cytokines (e.g., Granulocyte-colony stimulating factor (G-CSF), Macrophage-colony stimulating factor (M-CSF) or Erythropoietin (EPO)) act on multipotent progenitors to activate lineage-specific master TFs and promote the expansion of the myeloid or the erythroid lineage (Grover et al., 2014; Mossadegh-Keller et al., 2013; Rieger et al., 2009). On the other hand, single cell transcription profiling of primitive hematopoietic progenitors purified based on the expression of surface markers commonly used to define a “multipotent state” has revealed that truly multipotent progenitors may not exist and that these cells are highly heterogeneous and may be already committed to a specific lineage (Notta et al., 2016; Paul et al., 2015; Perie et al., 2015). Collectively, these findings raise the possibility that both “stochastic” and “instructive” mechanisms contribute to lineage specification and that “instructive” cytokines function in this process earlier than previously thought. Specification of myeloid cell fate requires the TFs PU.1 and C/EBPα, but macrophage versus granulocytic differentiation depends on the relative levels of PU.1 or C/EBPα, respectively (Dahl et al., 2003; Iwasaki et al., 2006; Laslo et al., 2006). EPO has an instructive role for erythroid commitment of stem cell-enriched cell subsets, a process that is, at least in part, GATA-1-dependent (Cui et al., 2009; Grover et al., 2014).
While C/EBPα, PU.1 or GATA-1 can promote unilineage differentiation by inducing new transcriptional programs, it remains unclear how these TFs cause changes in the existing epigenetic maintenance system. Upon EPO-induced erythroid differentiation of the multipotent CD133+CD34+ HPCs global changes in gene expression are in general correlated with the changes in association of the H3K4me3 or the H3K27me3 positive or negative histone marks respectively, although some “bivalent” genes lost or retained both marks (Cui et al., 2009). These changes in chromatin marks and gene expression profiles were detected in cells induced to erythroid differentiation for 9–11 days (Cui et al., 2009), suggesting that they may not reflect “commitment” to differentiation toward a specific lineage which is likely to occur much earlier. The dynamics of certain chromatin modifications was also globally examined in purified subsets representing distinct stages of murine hematopoietic differentiation (Lara-Astiaso et al., 2014). The results of this analysis are consistent with the existence of a transcription factor network that may control chromatin dynamics and lineage specification.
However, these studies are mostly correlative and do not provide a mechanistic explanation for how changes in transcriptional programs and chromatin structure occur. An essential issue is how newly induced TFs overcome the existing chromatin-based barrier when they are recruited to new target genes. It is also not clear whether chromatin marks play any role in allowing the access of lineage-determining TFs to the regulatory regions of the genes essential for differentiation, and whether chromatin changes occur before or after changes in the transcriptional programs. Thus, at present we only know the end results of lineage specification, but there is insufficient information on the molecular mechanisms that allow these changes during the early stages of lineage commitment. Knowledge of these mechanisms is essential in order to control lineage specification and may be important in understanding the mechanisms responsible for the differentiation arrest of leukemic cells.
In our study, we examined the assembly of chromatin in vivo at single-cell and gene-specific levels following DNA replication. We show that CD34+ HPCs possess a post-replicative chromatin globally devoid of the repressive histone mark H3K27me3. This type of chromatin is required for recruitment of C/EBPα, PU.1 and GATA-1 to DNA just after DNA replication upon treatment with instructive cytokines to induce erythroid or myeloid commitment. Blocking DNA replication or increasing H3K27me3 levels prevents recruitment of these TFs to DNA and suppresses cytokine-induced erythroid or myeloid differentiation. By contrast, H3K27me3 is rapidly associated with nascent DNA in primitive CD34+CD38− HPCs and treatment of these cells with instructive cytokines leads to a period of de-condensed, H3K27me3-unmodified nascent chromatin due to increase of the H3K27me3 demethylase UTX. These studies suggest that HPCs utilize special mechanisms of chromatin alteration for recruitment of specific TFs to DNA during initial stages of differentiation.
Results
Dynamics of H3K27me3 chromatin during lineage specification of multipotent hematopoietic progenitors
Our previous studies suggested that accumulation of major histone modifications, including H3K27me3, following DNA replication is delayed by at least 1 hr in cells of developing Drosophila embryos (Petruk et al., 2012). This suggests that modified histones do not directly carry epigenetic information, and implies that post-replicative chromatin may transiently consist of nucleosomes lacking H3K27me3 marks, which were not detected previously in bulk chromatin (Petruk et al., 2012). Since the absence of repressive H3K27me3 correlates with low density of nucleosomes (Bell et al., 2010; Shlyueva et al., 2014; Yuan et al., 2012), such unmodified histones may create a very de-condensed conformation of post-replicative chromatin. We hypothesize that this unusual H3K27me3-unmodified post-replicative chromatin may be essential during early stages of differentiation to create more favorable conditions for binding of newly induced lineage-determining TFs.
First, we examined whether chromatin lacking H3K27me3 is a common feature of human HPCs. We used the “Chromatin Assembly Assay (CAA)” (Petruk et al., 2012), to examine the rate of accumulation of H3K27me3 in human cord blood and G-CSF-mobilized CD34+ progenitor cells. DNA in proliferating CD34+ cells was pulse-labeled with EdU, which was then conjugated with biotin, and the proximity of H3K27me3 to nascent DNA was assessed by the Proximity Ligation Assay (PLA, Olink) using antibodies to biotin and H3K27me3. Following CAA, cells were immunostained with EdU and DAPI to assess the specificity of the assay. We found that in CD34+ cells H3K27me3 accumulates significantly at nascent DNA only at around 2 hr following DNA replication (Figure 1A).
Figure 1. Kinetics of H3K27me3 accumulation following DNA replication in cytokine-treated CD34+ HPCs.

(A) Left, accumulation of H3K27me3 on nascent DNA of G-CSF-mobilized CD34+ HPCs. DNA was labeled with EdU for 15 min and chased to 1, 2 and 4 hr. Following conjugation with biotin, CAA was performed between nascent DNA (biotin) and H3K27me3. PLA, red, EdU (biotin), green, DAPI, blue. Lower panels show PLA signals only. Right, quantification of the results of CAA experiments shown to the left by counting the number of PLA signals per EdU-labeled nuclei in 50 cells/each of the three independent experiments.
(B) Left, accumulation of post-replicative H3K27me3 during differentiation of CD34+HPCs. Cells were induced for myeloid differentiation with either G-CSF (upper panels) or M-CSF (lower panels) for 0, 6, 12 and 24 hr. DNA was labeled with EdU for 15 min and CAA performed between EdU (biotin) and H3K27me3. PLA, red, EdU (biotin), green, DAPI, blue. Lower panels show PLA signals only. Right, quantification of the results of CAA experiments shown to the left by counting the number of PLA signals per EdU-labeled nuclei in 50 cells/each of the three independent experiments. Error bars represent +/− standard deviation. p Values were determined by ANOVA. ns, non-significant; *, p<0.05.
Then, we examined whether the accumulation of post-replicative H3K27me3 undergoes changes during lineage specification of HPCs. Cells were induced toward granulocytic or macrophage differentiation by treatment with G-CSF or M-CSF, respectively, and accumulation of H3K27me3 on nascent DNA was analyzed after 15 min of EdU incorporation. No H3K27me3 was detected on 15 min EdU-labeled nascent DNA at 6 hr and 12 hr after G-CSF or M-CSF treatment. However, we detected significant accumulation of H3K27me3 on 15 min EdU-labeled nascent DNA at 24 hr after induction with each cytokine (Figure 1B). Thus, irrespective of lineage choice, the H3K27me3-unmodified post-replicative chromatin structure of HPCs persists for at least 12 hr following exposure to instructive cytokines. The post-replicative structure of chromatin may become more compact 1 day into HPC differentiation.
Lineage-determining transcription factors bind to post-replicative H3K27me3-unmodified chromatin upon cytokine-induced lineage commitment
Induction with ‘instructive’ cytokines leads to expression of key TFs that specifically change the transcriptional landscape of differentiating HPCs. It is not known whether there is a specific mechanism that allows these TFs to bind to DNA during cell differentiation. Previous EMSA analyses showed that C/EBPα is associated with promoters of target genes after 3 days of G-CSF-induced differentiation of myeloid precursor cells (Iida et al., 2008). To further investigate this, we used the CAA to examine the association of C/EBPα, PU.1 and GATA-1 with DNA following induction with G-CSF, M-CSF and EPO, respectively, of cord blood or G-CSF-mobilized peripheral blood CD34+ cells. As expected, cytokine treatment induced a decrease in CD34 positivity first detected at 72 hrs and the simultaneous appearance of lineage-specific differentiation markers (Figure 2A and Figure S1). C/EBPα and PU.1 are first detected on the 1 hr EdU-labeled nascent DNA at 6 hr following treatment with G-CSF and M-CSF, respectively, while GATA-1 is detected at 12 hr following induction with EPO (Figure 2B). Thus, irrespective of the induced differentiation program, specific TFs are recruited to DNA at 6–12 hr following treatment with lineage-determining cytokines, much earlier than previously thought. Importantly, the time of recruitment is within the period in which cytokine-treated HPCs carry the H3K27me3-unmodified post-replicative chromatin.
Figure 2. Kinetics of recruitment of lineage-determining TFs C/EBPα, PU.1 and GATA-1 to DNA.

(A) In vitro differentiation of G-CSF-mobilized CD34+ cells. Cells were treated with G-CSF, M-CSF or EPO for the indicated times. Differentiation was assessed by flow cytometry for lineage specific markers.
(B) G-CSF-mobilized CD34+ HPCs were induced toward myeloid or erythroid differentiation by treatment with G-CSF, M-CSF, or EPO respectively, for 6, 12 and 24 hr. DNA was pulse-labeled with EdU for 15 min and chased to 1 hr. CAA was performed between nascent DNA (biotin) and C/EBPα, PU.1, or GATA-1. PLA, red, EdU (biotin), green, DAPI, blue. Lower panel shows PLA signals only. Bottom, quantification of the results of CAA experiments shown above by counting the number of PLA signals per EdU-labeled nuclei in 50 cells/each of the three independent experiments. Error bars represent +/− standard deviation. p Values were determined by ANOVA. *, p<0.05.
These results are consistent with our hypothesis that recruitment of key lineage-determining TFs to DNA may be facilitated by the low-nucleosome density of post-replicative chromatin devoid of H3K27me3. This state of histone H3K27 modification may result from the activities of H3K27me3-specific homologous histone demethylases (KDMs) UTX and JMJD3. To examine this, we inhibited the activities of these KDMs by treatment with GSKJ4, a small molecule inhibitor of these enzymes (Kruidenier et al., 2012). Then, cord blood CD34+ cells were induced for 12 hr to myeloid or erythroid differentiation with G-CSF or M-CSF, or EPO, respectively, and accumulation of H3K27me3 was analyzed by CAA on nascent DNA labeled with EdU for 15 min. Similar to previous results with G-CSF and M-CSF (Figure 1), we did not detect H3K27me3 on 15 min EdU-labeled nascent DNA following 12 hr induction with EPO (Figure 3A, upper panels). Treatment of cells with GSKJ4 leads to significant accumulation of H3K27me3 on nascent DNA labeled with EdU for 15 min following 12 hr induction of these cells with all three lineage-determining cytokines (Figure 3A, lower panels). Then, we asked whether faster accumulation of post-replicative H3K27me3 affects recruitment of induced TFs in these cell lineages. The strong recruitment of C/EBPα, PU.1 and GATA-1, detected at 12 hr following induction with the corresponding cytokine (Figure 2 and 3A, upper panels), was completely blocked by treatment with GSKJ4 (Figure 3A, lower panels). This suggests that the H3K27me3-unmodified post-replicative chromatin is favorable for recruitment of C/EBPα, PU.1 and GATA-1 to their DNA binding sites.
Figure 3. H3K27me3-unmodified post-replicative chromatin is essential for DNA binding of lineage-determining TFs.

(A) Top, accumulation of post-replicative H3K27me3 leads to decreased recruitment of lineage-determining TFs. Cord blood CD34+ cells were induced toward myeloid differentiation with G-CSF (left) or M-CSF (middle), or erythroid differentiation with EPO (right) for 12 hr. During the induction period cells were left untreated (upper panels) or treated with the GSKJ4 inhibitor of KDMs UTX and JMJD3 (lower panels). DNA was pulse-labeled with EdU for 15 min and chased to 1 hr. CAA was performed between nascent DNA (biotin) and C/EBPα (left), PU.1 (middle) or GATA-1 (right). PLA, red, EdU (biotin), green, DAPI, blue. Lower panel shows PLA signals only. Bottom, quantification of the results of CAA experiments shown above by counting the number of PLA signals per EdU-labeled nuclei in 50 cells/each of the three independent experiments.
(B, C) G-CSF-mobilized CD34+ cells were cultured for 24 h in the presence of the CC100 cytokine cocktail (CTRL), or with EPO only (EPO) or with EPO and GSKJ4 (EPO+GSKJ4). B, Cells were immunostained with antibody to GATA-1 (red). DAPI, blue. Lower panel shows GATA-1 signals only. C, Western blot analysis with GATA1 and Actin antibodies
(D, E) Effect of UTX/JMJD3 inhibition on EPO-induced erythroid differentiation of CD34+ HPCs. Cells were cultured for 72 hr in the presence of the CC100 cytokine cocktail (CTRL), or in the presence of EPO only (EPO) or in the presence of EPO only and GSKJ4 (EPO + GSKJ4). D, real time PCR analysis (performed in triplicate) of GATA-1 target genes in untreated, EPO-, or EPO plus GSKJ4-treated CD34+CD38+ cells; E, erythroid differentiation monitored by flow cytometry. Error bars represent +/− standard deviation. p Values were determined by Student’s T test. ***, p<0.001.
By contrast, maintaining H3K27me3 accumulated on nascent chromatin may prevent association of key lineage-inducing TFs to DNA, leading to decreased induction of the gene targets of these TFs, and may ultimately result in a block of lineage specification.
In line with this, expression of GATA-1 was not affected by GSKJ4 treatment of EPO-stimulated CD34+ cells (Figure 3B); by contrast, inhibiting GATA-1 binding to DNA (Figure 3A, right panel) led to a marked decrease in the expression of GATA-1-regulated genes (Welch et al., 2004) and a complete block of EPO-induced differentiation revealed by loss of erythroid marker expression (Figures 3C, D). Likewise, treatment with GSKJ4 had no effect on G-CSF-induced C/EBPα expression but suppressed cytokine-induced myeloid differentiation of mobilized CD34+ cells, as indicated by its ability to block the increase in the expression of several C/EBPα-regulated genes (Figure S2A). Moreover, compared to treatment with G-CSF only, co-treatment with GSKJ4 resulted in a less differentiated phenotype revealed by the marked decrease in the proportion of cells co-expressing the CD33 and the CD15 differentiation antigens (27.5 vs 53.2%) (Figure S2B). Of interest, at the 72 hr time point chosen for the analysis of erythroid differentiation markers, CD34 positivity was essentially identical in cells treated with EPO only or with GSKJ4 and EPO (Figure S3). Thus, the H3K27me3 unmodified structure of nascent chromatin is essential for lineage specification of HPCs.
C/EBPα is recruited to post-replicative chromatin in the S phase of the cell cycle
It is not well established when lineage-specific TFs are recruited to DNA in undifferentiated cells; while some studies propose that this occurs during the G1 phase (Kapinas et al., 2013; Murray and Kirschner, 1989; Singh and Dalton, 2009), other studies dispute this idea (Filipczyk et al., 2007; Jonk et al., 1992; Li et al., 2012; Mummery et al., 1987). To examine this, CD34+ cells were labeled with EdU and then treated with G-CSF for 24 hrs. This allows detection of C/EBPα on labeled DNA (Figure 4A, left). However, blocking S phase entry in these cells by treatment with thymidine (Fig. 4B, left) prevented the accumulation of C/EBPα on EdU-labeled DNA (Fig. 4A, 0 hr). Release from thymidine block for 2 and 4 hr allows significant re-initiation of DNA replication (Figure 4B, middle and right), and leads to efficient accumulation of C/EBPα on nascent DNA (Figure 4A, 2 and 4 hr). These findings support our hypothesis that the initial association of TFs with DNA following cytokine treatment occurs primarily during early post-replicative periods in S phase.
Figure 4. C/EBPα is recruited to DNA only during post-replicative period.

(A) Cord blood CD34+ HPCs were labeled with EdU for 30 min and induced with G-CSF for 24 hr in the absence (Cont) or presence of 2.5 mM thymidine. After release from thymidine block, CAA was performed between nascent DNA (biotin) and C/EBPα at 0, 2 and 4 hr. PLA, red, EdU (biotin), green, DAPI, blue. Lower panel shows PLA signals only. A graph below shows quantification of the results of CAA experiments by counting the number of PLA signals per EdU-labeled nuclei in 50 cells/each of the three independent experiments.
(B) Following DNA labeling with EdU for 30 min and induction with G-CSF, CD34+ HPCs were grown for 24 hr in the presence of thymidine. Thymidine block was released for 0, 2 and 4 hr, and cells were labeled for 20 min with BrdU. Cells were double-immunostained for EdU (green) and BrdU (red). Error bars represent +/− standard deviation. p Values were determined by ANOVA. *, p<0.05.
Accumulation of H3K27me3-modified chromatin in primitive HPCs
To assess whether the H3K27me3-unmodified chromatin also exists in more primitive HPCs, we examined the loading of H3K27me3 on nascent DNA in the CD90+ subpopulation of CD34+ cells. Surprisingly, CD90+ cells exhibited high accumulation of H3K27me3 on DNA labeled with EdU for only 15 min (Figure 5A). Since most CD34+CD90+ cells are contained within the CD34+CD38− subset, we confirmed this fast mode of accumulation of H3K27me3 in uninduced CD34+CD38− cells (Figure 5B). However, 12 h induction of CD34+CD38− cells with G-CSF or M-CSF led to slow accumulation of post-replicative H3K27me3 (Figure 5B). Further treatment for 24 h restored fast accumulation of post-replicative H3K27me3. This suggests that compared to CD34+cells most of which carry H3K27me3-unmodified post-replicative chromatin, H3K27me3-containing chromatin forms very fast after DNA replication in the more primitive CD34+CD90+ and CD34+CD38− subsets. Induction with cytokines slows post-replicative accumulation of H3K27me3, making it similar to that of bulk CD34+ cells. Similar to CD34+ cells, further differentiation accelerates accumulation of post-replicative H3K27me3 in both types of cells (Figures 1B and 5A). The differences in the initial rate of accumulation of post-replicative H3K27me3 between CD34+, CD34+CD38−, and CD34+CD90+ cells may be explained by the fact that a significant proportion of bulk CD34+ cells likely consists of progenitors with the molecular profile of cells committed to a specific lineage but unable to further differentiate in the absence of instructive cytokines. Instead, the H3K27me3-modified chromatin of more primitive CD34+CD38− and CD34+CD90+ progenitors may reflect the undifferentiated status of hematopoietic stem cells (HSCs) and, upon cytokine treatment, exhibit significant changes in the rate of accumulation of H3K27me3 after DNA replication. Similar to that detected in bulk CD34+ cells (Figure 2B), G-CSF treatment of CD34+CD38− cells promotes the accumulation of the lineage-determining TF C/EBPα on nascent DNA (Figure S4).
Figure 5. H3K27me3-conatining nascent chromatin undergoes dynamic changes in cytokine-treated primitive HPCs.

(A) CD34+ HPCs were labeled with EdU for 15 min. CAA was performed for H3K27me3 antibody. Cells were then immunostained for CD90 (green) and EdU (blue).
(B) Induction of CD34+CD38− HPCs with G-CSF (top) or M-CSF (bottom) leads to de-methylation of H3K27me3. Purified CD34+CD38− HPCs were induced with G-CSF (upper panels) or M-CSF (lower panels) for 0, 12 and 24 hr. Cells were labeled with EdU for 15 min. CAA was performed between nascent DNA (biotin) and H3K27me3 antibodies. PLA, red, EdU (biotin), green, DAPI, blue. Lower panel shows PLA signals only. Quantification of the results of CAA experiments is shown under images and was estimated by counting the number of PLA signals per EdU-labeled nuclei in 50 cells/each of the three independent experiments. Error bars represent +/− standard deviation. p Values were determined by ANOVA. *, p<0.05.
We also assessed the loading of H3K27me3 on nascent DNA of primitive Lin-Sca-1+Kit+ mouse HPCs. Like human CD34+CD38− cells, these cells exhibited high accumulation of H3K27me3 on DNA labeled with EdU for only 15 min (Figure S5); moreover, treatment of Lin-Sca-1+Kit+ cells with recombinant murine G-CSF for 4 hr induced a state of globally de-condensed, H3K27me3-unmodified nascent chromatin. However, an 8 hr treatment of these cells with G-CSF was sufficient to induce the rapid accumulation of H3K27me3 on nascent DNA. Together, these data indicate that accumulation of H3K27me3 on nascent DNA is similar in primitive human and mouse HPCs, although in the latter cells the kinetics appears to be somewhat faster.
Accumulation of H3K27me3 is delayed on nascent chromatin of repressed genes
Our results suggest that following induction with cytokines accumulation of H3K27me3 is globally delayed following DNA replication. This implies that accumulation of H3K27me3 is delayed in regulatory regions of repressed genes that contain H3K27me3 during transcriptional interphases. To examine this, we analyzed the kinetics of accumulation of H3K27me3 on nascent DNA at the promoters of several repressed genes by re-ChIP assays (Francis et al., 2009; Petruk et al., 2012). Since this type of analysis requires approximately 107 cells per each sample it is not feasible to use purified CD34+CD38− cells in these assays. Thus, we used batches of CD34+ cells which contain significant proportion of CD38− cells. DNA of uninduced CD34+ cells and CD34+ cells induced with EPO was labeled with BrdU for 15 min or for 15 min followed by a chase to 2 hr. Cells were induced with EPO for 24 hrs since induction with this cytokine leads to a longer period of delayed accumulation of H3K27me3 on nascent DNA than after induction with G-CSF (not shown). Chromatin was first immunoprecipitated with antibody to H3K27me3, and in the next step the purified DNA was immunoprecipitated with antibody to BrdU. ChIP assays at the first immunoprecipitation step demonstrate that four examined genes contain similar amounts of H3K27me3 at their promoters before and after EPO induction (Figure 6A), confirming that they remain repressed during erythroid differentiation (Cui et al., 2009). In re-ChIP assays we detected significant accumulation of H3K27me3 on 15 min BrdU-labeled DNA (Figure 6B), suggesting a fast mode of accumulation of H3K27me3 characteristic for early progenitor cells (Figure 5). Accumulation of H3K27me3 is significantly delayed at the promoters of all repressed genes on 15 min of BrdU labeled DNA following induction with EPO (Figure 6B). These results confirm at a gene level that accumulation of H3K27me3 on nascent DNA is delayed following induction of differentiation of HPCs.
Figure 6. Kinetics of H3K27me3 accumulation at nascent DNA of specific genes during erythroid differentiation of CD34+ HPCs.

(A) ChIP assays with the H3K27me3 antibody. Uninduced and 24 hr EPO-induced CD34+ cells were labeled with BrdU for 15 min or 15 min and chased to 60 min. Chromatin was immunoprecipitated with H3K27me3 and IgG antibodies.
(B) ChIP assays with the BrdU antibody. DNA from the first immunoprecipitation step with H3K27me3 antibody (A) was denatured and immunoprecipitated with the BrdU antibody. Q-PCR was performed at genes repressed before and after EPO induction (GPR85, WNT16, HOXB8, HOXB13) and at a non-repressed control gene (CD11B) (Cui et al., 2009). Percent of input for re-ChIP experiments was calculated using the amount of material eluted from the beads following immunoprecipitation with H3K27me3 as 100% input. Data are represented as mean ± SD of technical replicates of one of the independent experiments. p-values were calculated by a one-way ANOVA with Bonferroni’s post-hoc test. The statistical significance was defined as p<0.05.
Cytokine treatment leads to induction of H3K27me3 KDMs UTX and JMJD3 in primitive HPCs
The observed differences in the rate of accumulation of H3K27me3 in CD34+CD38− cells may depend on changes in the amounts or the activities of the H3K27-specific methylating and de-methylating enzymes, such as KMT EZH2 and KDM UTX, respectively. Thus, we examined the presence of these enzymes on DNA following DNA replication. Similar amounts of the KMT EZH2 were detected in uninduced CD34+CD38− cells, and in the same cells induced for 12 hr to myeloid differentiation by G-CSF or M-CSF treatment (Figure 7A). However, UTX was not detected on DNA in untreated CD34+CD38− cells, but was detected on DNA of these cells 12 hr following induction with each cytokine (Figure 7A). These results were confirmed by several independent assays. Western blot analysis revealed an increase in the UTX protein in G-CSF- or M-CSF-treated (12 hr) CD34+CD38− whereas UTX levels did not change after cytokine treatment of the less primitive CD34+CD38+ progenitors (Figure 7B); compared to high amounts of EZH2 in all CD34+ cells, UTX was barely detectable by immunofluorescence (Figure 7C), or by CAA on 1 hr EdU-labeled DNA (Fig. 7D) in the nuclei of more primitive CD90+ cells. Likewise, using CAA we detected low amounts of the homologous H3K27me3 KDM JMJD3 on DNA of the CD90+ cells, and an increase in this protein in the CD90− cells (Figure 7D). Thus, a 12 hr cytokine treatment of primitive HPCs leads to induction of the KDMs UTX and JMJD3 proteins and their further accumulation on DNA. This may explain the switch detected in these cells from the fast to the slow mode of accumulation of post-replicative H3K27me3 after DNA replication (Figure 5B).
Figure 7. Induction of H3K27me3 KDMs UTX and JMJD3 during lineage specification.

(A) Kinetics of EZH2 (left panels) panels and UTX (right panels) accumulation during induction of CD34+CD38− HPCs with G-CSF (upper panels) or M-CSF (lower panels). Purified CD34+CD38− HPCs were induced with G-CSF (left) or M-CSF (right) for 0 and 12 hr. Cells were labeled with EdU for 15 min and chased to 1 hr. CAA was performed between nascent DNA (biotin) and EZH2 (upper panels) or UTX (lower panel) antibodies. PLA, red, EdU (biotin), green, DAPI, blue. Lower panel shows PLA signals only. Graphs represent quantification of the results of CAA experiments shown above by counting the number of PLA signals per EdU-labeled nuclei in 50 cells/each of the three independent experiments. Error bars represent +/− standard deviation. p Values were determined by ANOVA. *, p<0.05.
(B) CD34+CD38− or CD34+CD38+ HPCs were uninduced or induced by treatment with G-CSF or M-CSF. Cells were analyzed by Western blotting with antibody to UTX protein.
(C) CD34+ HPCs were immunostained with antibodies to UTX (red), CD90 (green, indicated by arrows) and EZH2 (blue).
(D) CD34+ HPCs were pulse-labeled with EdU for 1 hr. CAA was performed for UTX and JMJD3 antibodies and EdU (biotin). Cells were then immunostained for CD90 (green, indicated by arrows) and EdU (blue).
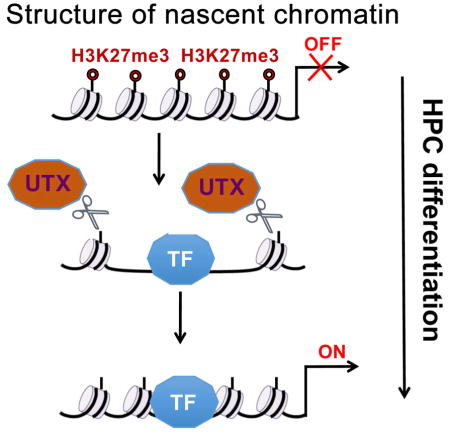
(E) A model for opening of post-replicative chromatin and recruitment of TFs to nascent DNA upon induction of primitive HPCs with lineage-specific cytokines. Primitive (CD34+CD38− or CD34+CD90+) HPCs exhibit the H3K27me3 repressive mark on post-replicative chromatin; treatment with instructive cytokines induces H3K27me3 demethylase activity (UTX in the model) and expression of lineage-determining TFs. H3K27me3-unmodified post-replicative chromatin allows binding of lineage-determining TFs to nascent DNA. Upon commitment to unilineage differentiation, H3K27me3 accumulates fast on nascent DNA preventing binding of additional TFs thus restricting transcriptional program and differentiation potential of these cells.
Discussion
Cytokine-induced lineage specification of CD34+ HPCs involves the recruitment of lineage-determining TFs (e.g., GATA-1 or C/EBPα) to the regulatory regions of differentiation-related genes and is accompanied by complex changes in transcriptional programs and chromatin structure (e.g., patterns of H3K4me3 and H3K27me3 histone marks) (Cui et al., 2009; Lara-Astiaso et al., 2014; Mercer et al., 2011; Wei et al., 2009). However, it is not known whether binding of TFs to new target genes is a regulated process, and whether it is affected by histone modifications and the structure of chromatin. Thus, we have a very limited view of the molecular mechanisms regulating a switch in transcriptional and epigenetic programs in dividing HPCs as these cells undergo lineage specification.
In this study, we uncovered a chromatin-based molecular mechanism of HPCs differentiation towards a specific lineage. The H3K27me3 mark rapidly associates with nascent DNA in primitive human CD34+CD38− cells (see model in Figure 7E) and mouse Lin-Sca-1+Kit+ cells. Upon induction with instructive cytokines, the global accumulation of H3K27me3 on nascent DNA is significantly delayed. In line with this, accumulation of H3K27me3 in nascent chromatin is also globally delayed in the more differentiated CD34+CD38+ HPCs. Our data suggest that transition from a more compact, H3K27me3-modified, to a more de-condensed, H3K27me3-unmodified chromatin structure, may be explained by the induction of H3K27me3 KDMs UTX and JMJD3 that may efficiently de-methylate this residue.
Once established, the H3K27-unmodified chromatin exists for a short period of time at very early stages of differentiation. During this period, cytokines also induce expression of lineage-specific TFs that quickly associate with DNA (Figure 7E). The transient H3K27-unmodified chromatin state is essential for the initial association of these TFs with nascent DNA that occurs at early stages of DNA replication. Remarkably, maintaining the H3K27me3 repressive mark on nascent chromatin of CD34+ HPCs by pharmacological inhibition of the H3K27me3 demethylases blocks the association of lineage-determining TFs to DNA and suppresses cytokine-induced erythroid or myeloid differentiation (Figure 3 and Figure S2). Consistent with these findings, conditional knockout of UTX in female mice induces marked depletion of erythroid progenitors in the bone marrow and of erythrocytes in the peripheral blood, in addition to profound thrombocytopenia and relative leukocytopenia (Thieme et al., 2013).
Fast accumulation of the H3K27me3 mark on nascent DNA resumes once CD34+ HPCs became committed to a single lineage (Figure 7E); this process may be designed to ensure that uni-lineage committed progenitors complete their differentiation program by hindering DNA binding of unwanted TFs that may antagonize the effect of the lineage-determining TF or cooperatively promote differentiation along a closely related lineage (Friedman, 2007).
The lineage-determining TFs analyzed in this work belong to the class of pioneer factors (Iwafuchi-Doi and Zaret, 2014; Zaret and Carroll, 2011) that are able to overcome the barrier of chromatin, and to bind to the so-called L-type chromatin which contains nucleosomes without many activating or repressive marks including H3K27me3 and to facilitate binding of other essential TFs (Iwafuchi-Doi and Zaret, 2016). The pioneer property of these TFs was demonstrated mostly during reprogramming of differentiated cells following ectopic expression of these proteins (Di Stefano et al., 2014; Feng et al., 2008; Heinz et al., 2010; Kulessa et al., 1995; Xie et al., 2004). However, actively repressed chromatin (R-type), which includes genes marked by H3K27me3, is not accessible to any TF, including pioneer factors (Iwafuchi-Doi and Zaret, 2016).
Our work uncovers a chromatin-based mechanism of recruitment to DNA of lineage-determining TFs induced at physiological levels by cytokines during differentiation of hematopoietic progenitor cell subsets. Based on this model, lineage-determining TFs do not need to overcome the chromatin barrier as proposed for pioneer factors on L-type chromatin, but gain access to their specific binding sites on a transiently “open” chromatin of nascent DNA. This mechanism of recruitment to DNA of lineage-determining TFs is not limited to HPCs, as binding to H3K27-unmethylated post-replicative chromatin is also a feature of TFs promoting lineage-specification in mouse and human embryonic stem cells (Petruk, 2017). Thus, these findings of dynamic changes in chromatin conformation may provide a molecular explanation of the biological plasticity of hematopoietic and embryonic stem cells. Future studies may also provide important insights into more mechanism-based approaches in treatment of hematopoietic disorders including leukemia.
Experimental Procedures
Induction of HPCs with different cytokines
Human CD34+CD38+ cells were maintained in SFEM (Stem Cell Technologies) enriched with CC100 cocktail of cytokines (SCF, 100 ng/ml; FLT3L, 100 ng/ml; IL-3, 20 ng/ml; IL-6, 20 ng/ml; Stem Cell Technologies) for 12 hr after thawing. Cells were then washed to eliminate the CC100 cytokines and were plated at a concentration of 106 cells/ml in SFEM with the addition of either 100 ng/ml of G-CSF (Peprotech) or 10,000 U/ml of M-CSF (R&D Systems, USA) to induce myeloid differentiation, or 10 ng/ml of EPO (Peprotech) to induce erythroid differentiation. The cytokines were re-added at every medium-change.
Purification of CD34+CD38− HPCs
Peripheral blood human CD34+ cells mobilized by G-CSF treatment were sorted 12 hr after thawing on the BD FACS Aria machine, according to the expression of CD38 surface marker. Purified CD34+CD38− were induced to myeloid lineage with G-CSF or M-CSF and used in CAA and Western blotting experiments with antibodies to H3K27me3, EZH2 and UTX.
Purification of Lin-Sca-1+Kit+ cells
C57BL/6J mice (The Jackson Laboratory, stock number 000664) were kept in a pathogen free facility and euthanized in accordance with the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University. Femoral, tibial and pelvic bones of 6–8 weeks old mice were dissected from surrounding muscles, and ground in PBS 2% FBS using a mortar and pestle. Bone marrow cells were filtered through a 40 μm Nylon Mesh Cell Strainer (Fisherbrand), and erythrocytes were lysed in 0.8% ammonium chloride (STEMCELLS Technologies). Cells were washed twice before staining with PerCP-Cy 5.5 mouse lineage Antibody Cocktail (BD Pharmigen), PE Rat Anti-Mouse Ly-6A/E (Sca1, BD Pharmigen), FITC Rat Anti-Mouse CD117 (Kit, BD Pharmigen). Lin-Sca1+Kit+ (LSK) cells were sorted using a MoFlo AstriosEQ (Beckman Coulter) and then seeded at 1x106/ml in SFEM (STEMCELLS Technologies) supplemented with SCF (12.5 μg), FLT3L (12.5 μg), IL-3 (5 μg), IL-6 (5 μg), all from Gemini, for 20 hr.
Analysis of protein association with nascent DNA (CAA)
CAA experiments were performed as described before (Petruk et al., 2013; Petruk et al., 2012) with small variations. HPCs were pulse-labeled with 5 μM EdU. Approximately 20,000 cells per each condition were harvested and cytospun (750 rpm for 5 min) on polylysine-coated slides (Polyscience), fixed with 4% formaldehyde, permeabilized in 0.25% Triton, and subjected to Click-iT reaction (Invitrogen) to conjugate biotin to EdU. Cells were incubated with mouse antibody to biotin and rabbit antibodies to the protein of interest. The PLA reaction was performed according to the manufacturer (Olink) and as described before. Following PLA, EdU was detected by immunostaining with Alexa Fluor 488-conjugated anti-biotin mouse antibody.
Re-ChIP with BrdU labeled DNA
For each experiment, 8–10x106 CD34+ peripheral blood G-CSF-mobilized cells were either not induced or induced for 24 hr with EPO. Undifferentiated and induced cells were labeled with 75 μM BrdU (Sigma-Aldrich) for 15 min or 15 min followed by chase for 2 hr and fixed with 1% formaldehyde for 10 min at room temperature. Chromatin was purified and treated with micrococcal nuclease using Simple ChIP Chromatin IP Kit (Cell Signaling). After treatment chromatin was immunoprecipitated first with antibody to H3K27me3 (Millipore) or rabbit IgG (Jackson Immunoresearch). This material was analyzed as conventional ChIP. Following purification, DNA was further immunoprecipitated with antibody to BrdU and used for PCR amplification (re-ChIP). The detailed protocol is described in (Francis et al., 2009; Petruk et al., 2012). All PCR reactions were performed with an Applied Biosystems StepOne Real-Time PCR system. Genes containing H3K27me3 at their promoters (GPR85, WNT16, HOXB8, HOXB13) and a control activated gene (CD11B) before and after cytokine induction were selected according to (Cui et al., 2009).
Treatment with thymidine
To control the efficiency of the thymidine block of DNA replication, CD34+CD38+ cord blood cells were labeled with EdU for 30 min, induced by G-CSF and grown for 24 hr in the presence of 2 mM thymidine. Following release from thymidine block for 2 or 4 hr, cells were labeled with BrdU to monitor resumption of DNA synthesis. Following ‘Click-iT’ reaction to conjugate biotin to EdU (Petruk et al., 2012), cell were fixed in 4% formaldehyde, denaturated in 2N HCL for 30min for detection of BrdU, and immunostained with antibodies to biotin and BrdU. To perform thymidine block analysis for recruitment of C/EBPα to DNA, nascent DNA of CD34+CD38+ cells was labeled for 30 min with EdU, cells were induced with G-CSF and grown for 24 hr in the presence of 2 mM thymidine. Thymidine was removed by three washes with SFEM, cells were grown for the additional 2 or 4 hr and were cytospun on slides, fixed with 4% formaldehyde, and permeabilized in 0.25% Triton. Following ‘Click-iT’ reaction CAA was performed between nascent DNA (biotin) and C/EBPα as described above.
Treatment with GSKJ4
CD34+CD38+ cells (cord blood or peripheral blood G-CSF-mobilized) were induced toward myeloid or erythroid differentiation for 12–72 hr in the presence of 5 μM GSKJ4 (Tocris Bioscience) dissolved in DMSO. In control experiments, cells were incubated with the same concentration of DMSO without GSKJ4. Erythroid differentiation was monitored after 72 hr by flow cytometry analysis detection of CD36 and glycophorin A/GYPA/CD235a double-positive cells. Expression of GATA-1-regulated genes was analyzed at 48 hr by real time PCR with the following primers: GATA-1 forward 5′-GCTACACCAGGTGAACCGGC-3′, reverse 5′-CAGACTGGAGCCCCGTTTCT -3′; ALAS2 forward 5′-TTCAAGACTGTGAACCGCTG -3′, reverse 5′-GCTCATGCCCAGGTAATCAT-3′, GYPA forward 5′-GGGTGATGGCTGGTGTTATTGG-3′, reverse 5′-AAAGGCACGTCTGTGTCAGG-3′; ABCB10 forward 5′-CGGAGGCCCGGAAGCTC-3′, reverse 5′-TTCCCCAGGAAGAAAGGGGC-3′.
Granulocytic differentiation was monitored by flow cytometry analysis of CD33 and CD15 levels (at 72 hr post-G-CSF treatment) and by expression of C/EBPα-regulated genes (at 72 hr post G-CSF treatment) detected by real time PCR with the following primers: CEBPE (C/EBPepsilon) forward 5′-CCAGCCGAGGCAGCTACAAT-3′, reverse 5′-CAAAGGGGCCTTGAGAACGC-3′; MPO (myeloperoxidase) forward 5′-TCTCCTCACCAACCGCTCAG-3′, reverse 5′-CCGGTTGTGCTCCCGAAGTA-3′; ELANE (neutrophilic elastase) forward 5′-CTGGGAGCCCATAACCTCTCG-3′, reverse 5′-CGTTGAGCTGGAGAATCACGA-3′; S100A8 (calgranulin A) forward 5′-ATATCAGGAAAAAGGGTGCAGACG-3′, reverse 5′-GCTGCCACGCCCATCTTTAT-3′. Real time PCR data were normalized for expression of GAPDH detected with the following primers forward 5′-CCCATCCCATCTTCCAGGAG-3′ and reverse 5′-CTTCTCCATGGTGGTGAAGACG-3′.
Antibodies
Anti-human CD34-PE (Miltenyi Biotec #130-081-002) and anti-human CD38-APC (Miltenyi Biotec #130-092-261) were used to analyze the percentage of CD34+ and CD38+ populations. Anti-human CD15-PE-Cy7 (BD Pharmingen #560827), CD33-PE (BD Pharmingen #555450), CD11b-APC (BD Pharmingen #561015) and CD14-PE (BD Pharmingen #555398) were used to test the percentage of cells positive for granulocytic and macrophage differentiation markers; CD235a-Pe-Cy5 (BD Pharmingen #561776), CD36-PE (BD Pharmingen #561821) were used to assess erythroid differentiation. Cells were incubated with PBS + 1% BSA to prevent antibodies’ non-specific binding and then stained with fluorochrome-conjugated antibodies for 15 min at room temperature. Cytofluorimetric analysis was performed on LSRII flow cytometer (BD, USA). For CAA and immunostaining the following antibodies were used: anti-C/EBP (1:200, Abcam); anti-PU.1 (1:100, Santa Cruz); anti-GATA-1 (1:400, Cell Signaling); anti-H3K27me3 (1:2500, Millipore); mouse anti-biotin (1:1000, Jackson Immunoresearch); UTX (1:400, Thermo Fisher Scientific); EZH2 (1:400, Cell Signaling); JMJD3 (1:200, Abgent); BrdU (1:200, BD Bioscience); CD90 (1:100, R&D System).
Statistical Analysis
For CAA experiments and re-ChiP experiment, p-values were calculated by a one-way ANOVA with Bonferroni’s post-hoc test. The statistical significance was defined as p<0.05.
Acknowledgments
This work was supported by the following grants: NIH R01GM075141 to A.M., NIH R01HL127895 to A.M. and B.C., NIH R01AI125650 to A.M and L.I., NIH NS075839 and the Parkinson’s Council and The Strauss Foundation to L.I.
Footnotes
Author contributions
A.M., S.P. and B.C. designed the study and wrote the manuscript. S.P. performed the PLA studies; S.M. and M.D. performed purification and assessed immunophenotype and gene expression of hematopoietic progenitor subsets. P.P. performed differentiation assays of GSKJ4-treated CD34+ cells. V.M and M.D performed purification of mouse mouse HPCs. N.F. provided mobilized CD34+ cells. J.C and L.I. discussed the results and designed experiments. All authors commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bell O, Schwaiger M, Oakeley EJ, Lienert F, Beisel C, Stadler MB, Schubeler D. Accessibility of the Drosophila genome discriminates PcG repression, H4K16 acetylation and replication timing. Nat Struct Mol Biol. 2010;17:894–900. doi: 10.1038/nsmb.1825. [DOI] [PubMed] [Google Scholar]
- Cantor AB, Orkin SH. Hematopoietic development: a balancing act. Curr Opin Genet Dev. 2001;11:513–519. doi: 10.1016/s0959-437x(00)00226-4. [DOI] [PubMed] [Google Scholar]
- Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453:544–547. doi: 10.1038/nature06965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui K, Zang C, Roh TY, Schones DE, Childs RW, Peng W, Zhao K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl R, Walsh JC, Lancki D, Laslo P, Iyer SR, Singh H, Simon MC. Regulation of macrophage and neutrophil cell fates by the PU.1:C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat Immunol. 2003;4:1029–1036. doi: 10.1038/ni973. [DOI] [PubMed] [Google Scholar]
- Di Stefano B, Sardina JL, van Oevelen C, Collombet S, Kallin EM, Vicent GP, Lu J, Thieffry D, Beato M, Graf T. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature. 2014;506:235–239. doi: 10.1038/nature12885. [DOI] [PubMed] [Google Scholar]
- Feng R, Desbordes SC, Xie H, Tillo ES, Pixley F, Stanley ER, Graf T. PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells. Proc Natl Acad Sci U S A. 2008;105:6057–6062. doi: 10.1073/pnas.0711961105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipczyk AA, Laslett AL, Mummery C, Pera MF. Differentiation is coupled to changes in the cell cycle regulatory apparatus of human embryonic stem cells. Stem Cell Res. 2007;1:45–60. doi: 10.1016/j.scr.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Francis NJ, Follmer NE, Simon MD, Aghia G, Butler JD. Polycomb proteins remain bound to chromatin and DNA during DNA replication in vitro. Cell. 2009;137:110–122. doi: 10.1016/j.cell.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AD. Transcriptional control of granulocyte and monocyte development. Oncogene. 2007;26:6816–6828. doi: 10.1038/sj.onc.1210764. [DOI] [PubMed] [Google Scholar]
- Graf T, Enver T. Forcing cells to change lineages. Nature. 2009;462:587–594. doi: 10.1038/nature08533. [DOI] [PubMed] [Google Scholar]
- Grover A, Mancini E, Moore S, Mead AJ, Atkinson D, Rasmussen KD, O’Carroll D, Jacobsen SE, Nerlov C. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J Exp Med. 2014;211:181–188. doi: 10.1084/jem.20131189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida S, Watanabe-Fukunaga R, Nagata S, Fukunaga R. Essential role of C/EBPalpha in G-CSF-induced transcriptional activation and chromatin modification of myeloid-specific genes. Genes Cells. 2008;13:313–327. doi: 10.1111/j.1365-2443.2008.01173.x. [DOI] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014;28:2679–2692. doi: 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, Zaret KS. Cell fate control by pioneer transcription factors. Development. 2016;143:1833–1837. doi: 10.1242/dev.133900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H, Mizuno S, Arinobu Y, Ozawa H, Mori Y, Shigematsu H, Takatsu K, Tenen DG, Akashi K. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006;20:3010–3021. doi: 10.1101/gad.1493506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonk LJ, de Jonge ME, Kruyt FA, Mummery CL, van der Saag PT, Kruijer W. Aggregation and cell cycle dependent retinoic acid receptor mRNA expression in P19 embryonal carcinoma cells. Mech Dev. 1992;36:165–172. doi: 10.1016/0925-4773(92)90067-t. [DOI] [PubMed] [Google Scholar]
- Kapinas K, Grandy R, Ghule P, Medina R, Becker K, Pardee A, Zaidi SK, Lian J, Stein J, van Wijnen A, et al. The abbreviated pluripotent cell cycle. J Cell Physiol. 2013;228:9–20. doi: 10.1002/jcp.24104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012;488:404–408. doi: 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulessa H, Frampton J, Graf T. GATA-1 reprograms avian myelomonocytic cell lines into eosinophils, thromboblasts, and erythroblasts. Genes Dev. 1995;9:1250–1262. doi: 10.1101/gad.9.10.1250. [DOI] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345:943–949. doi: 10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR, Singh H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- Li VC, Ballabeni A, Kirschner MW. Gap 1 phase length and mouse embryonic stem cell self-renewal. Proc Natl Acad Sci U S A. 2012;109:12550–12555. doi: 10.1073/pnas.1206740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer EM, Lin YC, Benner C, Jhunjhunwala S, Dutkowski J, Flores M, Sigvardsson M, Ideker T, Glass CK, Murre C. Multilineage priming of enhancer repertoires precedes commitment to the B and myeloid cell lineages in hematopoietic progenitors. Immunity. 2011;35:413–425. doi: 10.1016/j.immuni.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossadegh-Keller N, Sarrazin S, Kandalla PK, Espinosa L, Stanley ER, Nutt SL, Moore J, Sieweke MH. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature. 2013;497:239–243. doi: 10.1038/nature12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery CL, van den Brink CE, de Laat SW. Commitment to differentiation induced by retinoic acid in P19 embryonal carcinoma cells is cell cycle dependent. Dev Biol. 1987;121:10–19. doi: 10.1016/0012-1606(87)90133-3. [DOI] [PubMed] [Google Scholar]
- Murray AW, Kirschner MW. Dominoes and clocks: the union of two views of the cell cycle. Science. 1989;246:614–621. doi: 10.1126/science.2683077. [DOI] [PubMed] [Google Scholar]
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351:aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet. 2000;1:57–64. doi: 10.1038/35049577. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul F, Arkin Y, Giladi A, Jaitin DA, Kenigsberg E, Keren-Shaul H, Winter D, Lara-Astiaso D, Gury M, Weiner A, et al. Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors. Cell. 2015;163:1663–1677. doi: 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- Perie L, Duffy KR, Kok L, de Boer RJ, Schumacher TN. The Branching Point in Erythro-Myeloid Differentiation. Cell. 2015;163:1655–1662. doi: 10.1016/j.cell.2015.11.059. [DOI] [PubMed] [Google Scholar]
- Petruk S, Black KL, Kovermann SK, Brock HW, Mazo A. Stepwise histone modifications are mediated by multiple enzymes that rapidly associate with nascent DNA during replication. Nat Commun. 2013;4:2841. doi: 10.1038/ncomms3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk S, Cai J, Sussman R, Sun G, Kovermann SK, Mariani SK, Calabretta B, McMahon SB, Brock HW, Iacovitti L, Mazo A. Delayed accumulation of H3K27me3 on nascent DNA is essential for recruitment of transcription factors at early stages of stem cell differentiation. Molecular Cell. 2017 doi: 10.1016/j.molcel.2017.03.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk S, Sedkov Y, Johnston DM, Hodgson JW, Black KL, Kovermann SK, Beck S, Canaani E, Brock HW, Mazo A. TrxG and PcG Proteins but Not Methylated Histones Remain Associated with DNA through Replication. Cell. 2012;150:922–933. doi: 10.1016/j.cell.2012.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science. 2009;325:217–218. doi: 10.1126/science.1171461. [DOI] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- Singh AM, Dalton S. The cell cycle and Myc intersect with mechanisms that regulate pluripotency and reprogramming. Cell Stem Cell. 2009;5:141–149. doi: 10.1016/j.stem.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thieme S, Gyarfas T, Richter C, Ozhan G, Fu J, Alexopoulou D, Muders MH, Michalk I, Jakob C, Dahl A, et al. The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood. 2013;121:2462–2473. doi: 10.1182/blood-2012-08-452003. [DOI] [PubMed] [Google Scholar]
- Till JE, McCulloch EA, Siminovitch L. A Stochastic Model of Stem Cell Proliferation, Based on the Growth of Spleen Colony-Forming Cells. Proc Natl Acad Sci U S A. 1964;51:29–36. doi: 10.1073/pnas.51.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, Blobel GA, Chodosh LA, Weiss MJ. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood. 2004;104:3136–3147. doi: 10.1182/blood-2004-04-1603. [DOI] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, et al. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science. 2012;337:971–975. doi: 10.1126/science.1225237. [DOI] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]