

Abstract
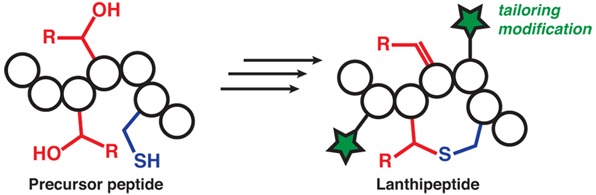
Lanthipeptides are ribosomally synthesized and post-translationally modified peptides (RiPPs) that display a wide variety of biological activities, from antimicrobial to antiallodynic. Lanthipeptides that display antimicrobial activity are called lantibiotics. The post-translational modification reactions of lanthipeptides include dehydration of Ser and Thr residues to dehydroalanine and dehydrobutyrine, a transformation that is carried out in three unique ways in different classes of lanthipeptides. In a cyclization process, Cys residues then attack the dehydrated residues to generate the lanthionine and methyllanthionine thioether cross-linked amino acids from which lanthipeptides derive their name. The resulting polycyclic peptides have constrained conformations that confer their biological activities. After installation of the characteristic thioether cross-links, tailoring enzymes introduce additional post-translational modifications that are unique to each lanthipeptide and that fine-tune their activities and/or stability. This review focuses on studies published over the past decade that have provided much insight into the mechanisms of the enzymes that carry out the post-translational modifications.
1. Introduction
The name lanthipeptide is a short-hand nomenclature for lanthionine-containing peptides,1 with lanthipeptides having antimicrobial activities historically called lantibiotics.2 A lanthionine is a bis-amino-bis acid in which two alanine residues are linked by a thioether group that connects their β-carbons (Figure 1). When incorporated into a peptide chain via both the amino and acid groups, a lanthionine results in a thioether cross-link. Installation of lanthionine residues is accomplished through enzymatic post-translational modifications on peptide substrates. Although genes encoding homologues of lanthipeptide biosynthetic enzymes are also present in some archaea and in higher eukaryotes including mammals,3,4 lanthipeptide detection and isolation is thus far restricted to bacteria. The increase in the number of characterized lanthipeptides as a result of the bacterial genome sequencing projects has led to the realization that their functions are not limited to antimicrobial activities but also include antifungal,5 morphogenetic,6,7 antiviral,8 antinociceptive,9 and antiallodynic functions.10 As a result, lanthipeptide derivatives are undergoing therapeutic evaluation11−25 and have been used for imaging applications.26−34 Lanthipeptide biosynthetic gene clusters are particularly found in the genomes of many genera of Firmicutes, Actinobacteria, Proteobacteria, Bacteroidetes, and Cyanobacteria.35−37
Figure 1.
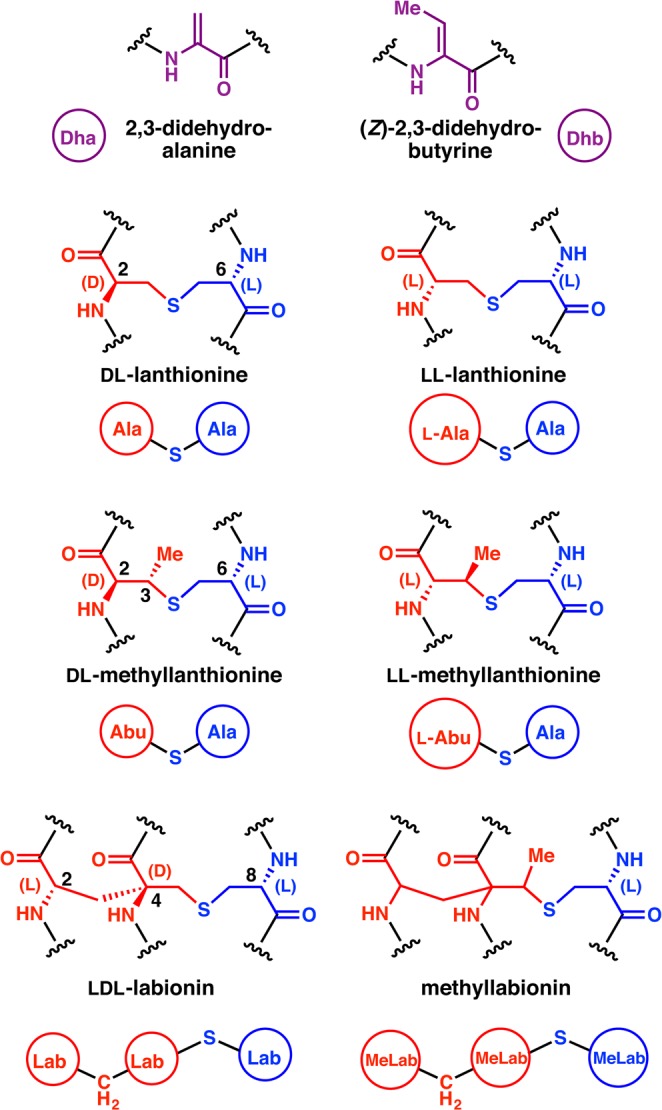
Structures of the thioether cross-links and dehydro amino acids that are characteristic for lanthipeptides. Under each chemical structure is shown a shorthand notation used in this review. At present, two different diastereomers of both Lan and MeLan have been found in natural lanthipeptides, whereas for Dhb and Lab only one diastereomer has been reported thus far. A methyl-substituted Lab (MeLab) has recently been identified and its stereochemistry is unknown. In all drawings of cross-links, atoms originating from Ser/Thr are shown in red, whereas atoms originating from Cys are shown in blue. Abu: α-aminobutyric acid.
The mechanism by which the thioether cross-links are formed is what unifies this family of compounds. For all lanthipeptides, these linkages are introduced by a post-translational modification process that first involves the dehydration of Ser and Thr residues to 2,3-didehydroalanine (Dha) and 2,3-didehydrobutyrine (Dhb) residues, respectively (Figure 2). As discussed in this review, the manner of dehydration can be quite different, which is one of the criteria for dividing lanthipeptides into different classes. The thioether structure is then generated by 1,4-conjugate addition of a Cys onto a dehydro amino acid. The resultant enolate can be protonated to produce either a lanthionine (Lan) from Ser or a methyllanthionine (MeLan) from Thr (Figure 2). Alternatively, the initially formed enolate can attack another dehydro amino acid to produce a carbon–carbon cross-link and a second enolate. Upon protonation, the structure that now introduces two cross-links and contains an α,α-disubstituted amino acid at its center is called a labionin (Lab) when formed from one Cys and two Ser residues,10 or methyllabionin (MeLab) when formed from one Cys, a central Thr, and an N-terminal Ser (Figures 1 and 2).9 At present, no labionin-like structures have been reported in which the electrophile in the second conjugate addition was a Dhb. The lanthipeptide family encompasses any peptide containing a (methyl)lanthionine or a (methyl)labionin provided it is made by dehydration of Ser/Thr and subsequent attack by a Cys residue onto a dehydro amino acid. Thioether cross-links are also found in other peptide natural products, but their biosynthesis does not involve this specific sequence of events and they have therefore not been included in the lanthipeptide family.1
Figure 2.

Post-translational modification reactions leading to the formation of (Me)Lan or (Me)Lab. Color coding as in Figure 1. The stereochemistry for MeLab has not been determined. Xn = peptide of n amino acids. The stereochemistry of (Me)Lan can be different from DL (see Figure 1).
In principle, the conjugate addition process can form two diastereomers for Lan and four diastereomers for MeLan and Lab (when generated from two Dha). In practice, thus far both diastereomers of Lan are indeed found in naturally occurring lanthipeptides, but only two of the four MeLan stereoisomers and one of the four Lab diastereomers have been observed (Figure 1). We emphasize that this situation may change, since for the majority of lanthipeptides the stereochemistry has not been determined, and only very recently was it recognized that more than one stereochemistry can be generated for Lan/MeLan.38
In this review we will use a stereochemical nomenclature that focuses on the stereochemistry at the α-carbons of the former Ser/Thr and Cys residues. We assign as L the stereochemistry that has the original side chain in the same position as in Ser/Thr/Cys and as D the stereochemistry in which the side chain occupies the epimeric position. Thus, (2S,6R)-Lan will be referred to as DL-Lan (D configuration at the former Ser/Thr and L configuration at the former Cys), and (2S,4S,8R)-labionin will be referred to as LDL (Figure 1). This nomenclature has the advantage that it immediately shows the net stereochemistry of the attack of the enolate intermediate onto the electrophile (proton for Lan and MeLan; Dha for Lab). For D stereochemistry this attack is on the opposite face from where the original α-proton was located in Ser/Thr.
The general pathway of lanthipeptide biosynthesis was mostly established by genetic studies in producing organisms that have been comprehensively reviewed.39−45 Since the mid 2000s, advances in biochemical techniques, and more recently the availability of large numbers of genomes, have helped shape a molecular understanding of lanthipeptide biosynthesis that now is increasingly utilized for engineering and synthetic biology applications. In keeping with the theme of this issue, this review will discuss the current knowledge of the details of the molecular mechanisms utilized by the biosynthetic enzymes. We refer the reader to other reviews and primary studies that discuss gene regulation,40,46−62 mode of action,44,55,56,63−67 or bioengineering of lanthipeptides.55,68−111
1.1. Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs)
Lanthipeptides are members of the rapidly expanding RiPP family of natural products. The genome sequencing efforts of the past decade have revealed that many classes of peptide natural products that were initially believed to be made by nonribosomal peptide synthetases are in fact made via a ribosomally synthesized precursor peptide that is then heavily post-translationally modified.1 The direct link between the gene encoding the precursor peptide and the final product renders RiPPs as low-hanging fruits for genome mining exercises, because to a large extent the general shapes of the final structure can be predicted. Based on the frequency of their gene clusters in the currently available genomes, lanthipeptides appear to be the largest group of RiPPs,112−115 although this conclusion may be biased by the genomes that have been sequenced thus far. Whereas comprehensive reviews of lanthipeptides in the mid 2000s discussed compounds that were all discovered via activity-based purification,44,45 many of the compounds discussed in this review were discovered by applying genome mining approaches35,116−123 to lanthipeptide discovery. For these molecules, the biosynthetic gene cluster was known before the compound, and the knowledge of the cluster was often used to obtain the lanthipeptide.124−139 In addition to the high suitability of RiPPs for genome mining, their gene-encoded origin also makes them uniquely accessible to biosynthetic engineering by site-directed mutagenesis.
Although the spectrum of post-translational modifications is diverse,1,140−143 the overall biosynthetic pathways of RiPPs have a key common feature. They almost always involve a precursor peptide that is much larger than the final product, which is generated from the core peptide (Figure 3). The additional sequence can be located at the N-terminus (a leader peptide),144 the C-terminus (a follower peptide),145−148 or at both sides of the core peptide (recognition sequences).149 These additional appendages that do not end up in the final products have proven to be critical recognition sites for many of the biosynthetic enzymes.144,150,151 The separation of substrate recognition from the sites where the post-translational modifications take place have given RiPP biosynthetic systems unique properties of very high substrate tolerance. Indeed, as described in section 4.1, lanthipeptide core peptides can be hypervariable and yet be substrates of a single enzyme.129 These properties provide even more tantalizing opportunities for engineering.
Figure 3.
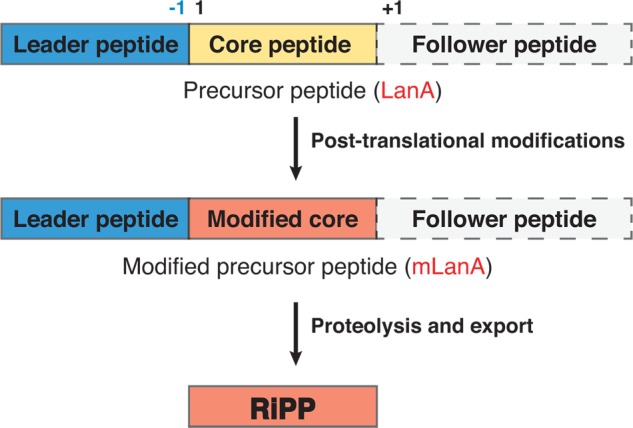
General biosynthetic pathway for RiPPs. For lanthipeptides, thus far no follower peptides have been reported. The general numbering scheme for the precursor peptides is indicated.
For lanthipeptides, thus far only precursors that contain leader peptides have been reported. In this review, we will use the standardized nomenclature recommended by the lanthipeptide community in 2013,1 which is summarized in Figure 3. This nomenclature numbers residues in the precursor peptides starting with the first residue of the core peptide. Residues in the leader peptide are indicated with negative numbers counting back from the junction between the leader peptide and core peptide. In general, biosynthetic enzymes involved in lanthipeptide biosynthesis have been given the generic prefix Lan,152 with a more specific descriptor for specific compounds (e.g., Nis for nisin, Lct for lacticin 481, Cin for cinnamycin). With few historical exceptions for cytolysin (CylL), subtilin (SpaS) and SapB (RamS), the precursor peptides are designated LanA (NisA, LctA, CinA, etc.) and the fully post-translationally modified precursor peptide with the leader peptide still attached is designated mLanA.
1.2. Classification Systems
Many attempts have been undertaken to systematically classify the various lanthipeptides, from efforts that looked at the final structures39 to schemes that focused on the biosynthetic machinery.153 In this review we adopt the latter, updated with classes that were discovered after introduction of the initial classification scheme, but we recognize that already exceptions of currently structurally uncharacterized lanthipeptides can be found in the sequenced genomes. As discussed in this review, at present the distinct mechanisms of the dehydration reaction distinguish class I–IV. As more and more genomes are sequenced, it is highly likely that a single classification scheme may not be possible, as Nature has clearly shuffled biosynthetic genes, not only between lanthipeptide classes, but even across different RiPP families (see sections 2.2 and 4.1). Nevertheless, it is clear that 3–4 quite different strategies are used to biosynthesize the currently known lanthipeptides, and subdividing this very large family of compounds by their biosynthetic logic remains a useful means for classification.
2. Class I Lanthipeptide Biosynthesis
2.1. Overview
The class I lantibiotic nisin produced by Lactococcus lactis, the founding member of the lanthipeptide class of natural products, was discovered in 1928 as a substance that inhibited the growth of Lactobacillus bulgaricus.(154) Although nisin was initially incorrectly suggested to slow acid development in cheese, subsequent demonstration of broad-spectrum bactericidal activity resulted in renewed interest in the 1940s for its use in food preservation. Investigations into its structure elucidation were instigated with the identification of the α,β-unsaturated amino acids Dha and Dhb in nisin155 and in subtilin,156 a structurally closely related lanthipeptide produced by Bacillus subtilis ATCC 6633. Subsequent efforts ultimately culminated in the determination of the chemical structure of nisin by Gross and Morell in 1971 (Figure 4).157 Concurrently, Ingram observed that the levels of incorporation of radioactive Cys into nisin in cultures of L. lactis correlated with the addition of protein synthesis inhibitors,158 and proposed a ribosomal mechanism for lanthipeptide production wherein Dha and Dhb are formed through the dehydration of Ser and Thr, respectively, and subsequent intramolecular 1,4-conjugate addition of Cys onto the dehydro amino acids would afford the characteristic Lan and MeLan rings. Confirmation of a ribosomal origin for lanthipeptides was firmly established in the late 1980s as sequences of several lantibiotic biosynthetic clusters revealed the presence of genes that encode for peptidic precursors of lanthipeptides, such as those for epidermin,2 subtilin,159 nisin,160−162 and gallidermin.163 Importantly, each of these gene sequences codes for peptides that contain an N-terminal leader sequence that is absent in the final products and a C-terminal core sequence that contains codons for Ser, Thr, and Cys at the sites of the post-translational modifications in the final products.
Figure 4.

Structure of the class I lanthipeptide nisin A (A) and its shorthand structure representation (B) that is used throughout this review.
The chemical structure of nisin consists of five thioether linkages that define rings A-E, along with one Dhb and two Dha residues (Figure 4). The configurations of the thioether rings were determined by reductive desulfurization, which yielded D- and L-Ala from rings containing Lan, and D-α-aminobutyric acid (D-Abu) and L-Ala from rings containing MeLan.157 Comparison of the retention times with authentic samples identified the S-configuration at the β-position of the latter.164 These data established the stereochemistry of (2S,6R) for the Lan residue and (2S,3S,6R) for the MeLan residues in nisin (Figures 1 and 4). To date, all known structures of class I lanthipeptides have the same stereochemistry, while other diastereomers have been observed in class II lanthipeptides (section 4.3.3).38 Sequence-specific assignment of the 1H NMR spectrum of subtilin showed that it contains a ring topology similar to that observed for nisin (Figures 4 and 5).165
Figure 5.

Structures of class I lanthipeptides including members containing tailoring modifications (olive green). Shorthand notations used for each post-translational modification (PTM) are indicated. R-stereochemistry has been demonstrated for the lactyl group in epilancin 15X (see section 2.6.1).171
Introduction of the specific patterns of thioether cross-links bestows upon lanthipeptides the ability to recognize specific molecular targets with high specificity and selectivity (e.g., lipid II for nisin,166−168 and phosphatidyl ethanolamine for cinnamycin169,170). Just as medicinal chemists optimize lead structures with synthetic modifications, Nature has optimized the activity and specificity of lanthipeptides within the ecological niche of the producer organism by further tailoring reactions after installation of the characteristic ring structures. Many of these modifications occur at the N- or C-termini to increase the stability of the peptides or to change the overall charge.21,171 Such post-translational modifications in class I lanthipeptides include the installation of a C-terminal S-[(Z)-2-aminovinyl]-d-Cys (AviCys) formed via an enethiol intermediate generated by the LanD oxidative decarboxylase flavoproteins172,173 as found in epidermin (Figure 5), and the reduction of a ketone to an alcohol during installation of an N-terminal 2-hydroxypropionate by LanO dehydrogenases,171 as found in epilancin 15x (Figure 5).174 One of the most potent lantibiotics identified to date is microbisporicin (also known as NAI-107) produced by Microbispora sp. 107891 (Figure 5), which shows potent activity against several clinically relevant pathogenic bacteria.175 The chemical structure of this compound shows two modifications not previously observed in lanthipeptides, specifically, chlorination on a Trp residue and dihydroxylation of a Pro residue (Figure 5). Section 2.6 will discuss the enzymes involved in these tailoring reactions.
The biosynthesis of nisin is encoded by a transcriptional operon of 11 genes (Figure 6) encoding the precursor peptide (nisA), three proteins involved in post-translational modifications (nisB, nisC, and nisP), and an ATP-binding cassette (ABC) type transporter (nisT), along with genes encoding transcriptional regulators (nisR and nisK) and immunity proteins (nisI and nisFEG).55 Comparisons with the gene clusters for subtilin and epidermin, among others, show that four genes (lanABCT) are common across various biosynthetic clusters (Figure 6). Since lanC and lanT have more recently also been found in clusters of class II lanthipeptides, the presence of lanB is what uniquely defines class I lanthipeptides. Notably, the NisA precursor peptide contains 57 residues, while the product nisin is composed of 34 residues. Hence, the precursor peptide comprises a 23-residue N-terminal leader sequence that is excised from the final product and a 34-residue C-terminal core sequence where the enzymatic modifications are installed (Figure 7). The post-translational installation of the (β-methyl)lanthionine rings is carried out by two enzymes: NisB that catalyzes the dehydration of Ser and Thr residues in the core peptide,176 and the NisC cyclase that facilitates the intramolecular addition of Cys onto the resultant dehydro amino acids to generate cyclic NisA with the leader still attached (mNisA).177 The subtilisin-like serine protease NisP removes the leader sequence from the post-translationally modified core178 to yield the bioactive final product. Two partially orthogonal systems confer immunity against nisin to the producing organisms: the extracellularly located NisI lipoprotein that binds and sequesters active nisin,179 and the NisFEG active transporter that is involved in nisin extrusion.180
Figure 6.

Biosynthetic gene clusters of various class I lanthipeptides.2,161,162,181−184 See section 2.1 and Abbreviations section for the general nomenclature of lanthipeptide biosynthetic genes.
Figure 7.
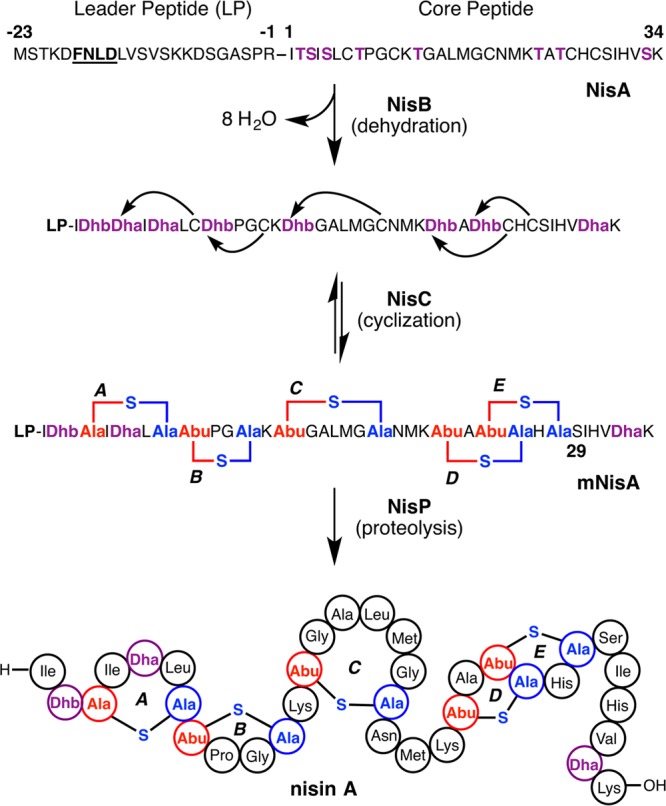
Scheme showing the biosynthetic route to nisin A. For clarity, the process is shown as first completion of dehydration and then cyclization, but this is not necessarily the case. The timing of the different PTMs installed on the NisA precursor peptide is discussed in the text. The FNLD motif in the leader peptide (bolded and underlined) is conserved in other class I lanthipeptide leader peptides and appears important in interactions with both NisB and NisC. The cyclization catalyzed by NisC is reversible as shown by experiments involving resubjection of mNisA to the cyclization conditions (section 2.3).
Genome mining studies have demonstrated that class I lanthipeptide biosynthetic genes are widely distributed across various bacterial phyla.36,37 Phylogenetic analysis of the distribution of the LanC cyclases yields sequences that fall into three distinct groups: group 1 found in Bacteroidetes and Proteobacteria, and groups 2 and 3 found in Actinobacteria and Firmicutes, respectively.36 The latter two groups are related to one another and are distinct from the group 1 sequences. Similar analysis of LanB dehydratases shows that the sequences from Bacteroidetes and Proteobacteria may be derived from Firmicutes, denoting possible differences in the evolutionary origins of the LanBs versus the LanCs.36 While the phylogenetic distribution of LanBs does not completely correlate with that of LanCs, enzymes that are from the same biosynthetic cluster generally group into similar clades.
Class I lanthipeptides may also be distinguished based on similarities of their precursor peptides with members for which experimentally structures have been established. In this classification, the molecules may be demarcated into the nisin-like, epidermin-like, and Pep5-like groups.56 This classification extends only to the lanthipeptide itself and not to the genes in the clusters, as similarities in the structures of the final products do not necessarily reflect an equivalent similarity across the biosynthetic enzymes. For example, although the sequences of the nisin and subtilin precursors and products are similar, their associated LanC enzymes are only distantly related.36 The notion of the precursor sequence possibly directing the final structure is further fortified by experiments in which fusion of a class II core peptide onto the class I NisA leader peptide resulted in processing by the nisin biosynthetic enzymes to yield an antibacterial peptide, which presumably is the natural product.185 Similarly, expression of a chimeric peptide consisting of the subtilin leader and a combination of the nisin and subtilin core peptides with the subtilin biosynthetic machinery in B. subtilis produced a fully processed product.186 Likewise, a chimera of the Staphylococcus epidermidis epilancin 15X core peptide and the NisA leader peptide was processed by the nisin system to afford a product with ring topology and bioactivity similar to epilancin 15X.36
2.2. Dehydration via Glutamylation
Although the genes responsible for nisin182 and subtilin183 production were first identified in 1992, the mechanistic basis for Ser/Thr dehydration by any class I LanB was not elucidated for another 20 years. One of the limiting factors in the characterization of LanB enzymes was the lack of a suitable heterologous expression and purification system for these high molecular weight enzymes; for example, NisB is a 117 kDa protein. In addition, identification of amphipathic α-helices in the primary sequence and immuno-detection of NisB in vesicles of L. lactis lead to the expectation that NisB may be membrane associated,182 which also stalled biochemical efforts. Hence, early experiments were limited in mechanistic explorations, and focused more on genetic characterization. Plasmid-based overexpression of NisB in different nisin producing strains increased the efficiency of dehydration of an engineered NisA variant.187 Additionally, expression of affinity-tagged NisA precursor peptide in a nisB deletion strain yielded unmodified peptide, while expression in a nisC deletion strain yielded a product that had undergone dehydration but was not cyclized.188,189 These data established that NisB carries out the dehydration on the NisA precursor peptide, which is cyclized by NisC.
In the early 2000s, coexpression of SpaB from B. subtilis ATCC 6633 (the subtilin producer) with the GroESL chaperones facilitated purification of the enzyme in milligram quantities.190 However, multiple attempts to reconstitute in vitro activity using the purified protein were unsuccessful. Nearly a decade later, two different methodologies allowed for the production of soluble NisB protein, facilitating functional analysis. First, homologous expression of C-terminally polyhistidine tagged NisB in L. lactis yielded soluble protein that could be purified from the cytosol using affinity chromatography, and showed no propensity for aggregation even after prolonged storage.191 Size-exclusion chromatography, combined with multiangle static light scattering analyses, demonstrated that purified NisB behaved as a homogeneous species with a mass consistent with that of a protein dimer. Surface plasmon resonance experiments documented that purified NisB interacted with the substrate precursor peptide, as well as with the dehydrated NisA with affinities in the micromolar range. In a second approach, coexpression of NisB along with the NisC cyclase and the NisA precursor peptide in Escherichia coli enabled the production of authentic nisin.92 While the dehydratase produced in E. coli using this coexpression strategy was not yet purified, the enzyme had to be functional as the NisA precursor was dehydrated and cyclized in vivo.
Based on the heterologous expression data, recombinant, affinity-tagged NisB was coexpressed with NisA in E. coli, facilitating purification of the enzyme from the soluble fraction. Although in vitro activity could not be demonstrated using purified components, addition of crude E. coli cell extract, along with ATP, MgCl2, and glutamate (the most abundant E. coli metabolite) to reactions of NisB generated up to nine dehydrations on the NisA precursor peptide.192 These studies established the first in vitro biochemically tractable system that allowed monitoring of LanB dehydratase activity, facilitating functional analysis of site-specific LanB variants, and access to possible intermediates in the modification pathway. Ala-scanning mutational analysis was used to identify the role of various polar residues in NisB that are conserved among most LanB enzymes, to assess if any of these residues play a role in the chemistry of dehydration. A group of Ala variants that largely clustered to the N-terminus of NisB (Arg14, Arg83, Arg87, Thr89, Asp121, Asp299, and Arg464) were inactive, suggesting their importance for dehydratase activity. Intriguingly, while Ala variants at residues located at the C-terminus of NisB (Arg786, Arg826, and His961) did not produce a dehydrated product, these variants generated NisA modified by multiple adducts of +129 Da as observed by mass spectrometry.192 The mass shift is consistent with the addition of multiple glutamate residues on the NisA precursor peptide. Polyglutamylation is a post-translational modification in which a free Glu is added via its amino group onto the γ-carboxylate of Glu residues forming an isopeptide bond. Then the added Glu serves as a site for another glutamylation, ultimately leading to polyglutamylation. This modification has previously been observed and extensively studied in eukaryotic tubulin.193
Incubation of glutamylated NisA precursor with wild-type NisB dehydratase resulted in elimination of the glutamate to yield a dehydrated product (Figure 8), and this elimination reaction did not require ATP, MgCl2, or E. coli cell extract.192 Thus, glutamylation could not be on Glu residues but had to be on Ser/Thr residues. These data also demonstrated that the glutamylated NisA is an intermediate in the dehydration pathway, where esterification via the Oγ of Ser/Thr would presumably activate the side chain hydroxyl for subsequent β-elimination. Incubation of glutamylated NisA with Ala variants at the N-terminus of NisB also resulted in glutamate elimination. Collectively, these data suggest that the N-terminal portion of NisB harbors the glutamylation activity, and the glutamate elimination activity resides largely within the C-terminal portion of the polypeptide. However, prior activation of Glu, via either the α- or the side-chain carboxylate, would be required to catalyze glutamate addition onto the side chain Ser/Thr of the precursor substrate, but the NisB dehydratase did not demonstrate any Glu activation activity.
Figure 8.

LanB enzymes achieve dehydration by catalyzing a transesterification reaction from glutamyl-tRNAGlu to the side chain of Ser/Thr followed by β-elimination. Chemoselective activation by glutamate via its α-carboxylate is illustrated here as has been demonstrated for the nonlanthipeptide RiPP goadsporin.195 Conserved LanB residues predicted by mutagenesis studies on NisB to be important for transesterification and elimination are shown in gray. The exact roles of these residues remain to be established.
In order to elucidate the source of activated Glu, E. coli extract was fractionated into high and low molecular weight pools, with activity only present in the high molecular weight fractions.194 Further separation of components using anion exchange chromatography localized the bioactive component to a fraction pool that absorbed strongly at 260 nm and was sensitive to treatment with RNase.194 These data supported glutamyl-tRNAGlu as the source of activated Glu that is necessary for NisB activity. A reconstitution experiment with purified NisB and NisA, along with glutamyl-tRNA synthetase (GluRS) and tRNAGlu illustrated that the latter two are the constituents of E. coli extract that are necessary and sufficient for NisB activity.194 Thus, NisB does not catalyze aminoacylation of tRNA but rather utilizes the glutamyl-tRNAGlu that is available from the cellular pool. These data are consistent with a mechanistic model in which LanB dehydratases activate the side chain hydroxyls of Ser/Thr through the addition of a Glu residue derived from glutamyl-tRNAGlu, and subsequently abstract the α proton of the modified Ser/Thr to facilitate β-elimination to yield the dehydro amino acid (Figure 8). The use of negatively charged glutamyl-tRNAGlu as a cosubstrate may explain the observation that Ser/Thr residues flanked by hydrophobic residues are better substrates for dehydration than those flanked by polar and especially negatively charged residues.74,79
Although NisB could utilize the E. coli glutamyl-tRNAGlu as a source of activated Glu, the LanB from the NAI-107 producing organism Microbispora sp. 107891, MibB, could not do so efficiently.196 Reconstitution of activity for MibB required the addition of the charged cognate CUC anticodon tRNAGlu from the producing organism.196 A comparison of the nucleotide sequence of Microbispora sp. 107891 tRNAGlu with that of E. coli tRNAGlu revealed differences in the acceptor stem. A G73A single mutation and G73A/C72U double mutation were introduced in the E. coli tRNAGlu acceptor stem to reflect the sequence found in Microbispora sp. 107891 tRNAGlu. Notably, reactions using the G73A variant of E. coli tRNAGlu produced up to three dehydrations in the MibA precursor peptide, whereas the G73A/C72U double variant produced up to six dehydrations on the precursor.196 Hence, the tRNA acceptor stem likely serves as a major recognition determinant between the LanB dehydratase and its cognate tRNAGlu.
Further insights into the function of the LanB dehydratases were afforded with the crystal structure of Microbispora sp. 107891 MibB196 and of L. lactis NisB in complex with the NisA precursor peptide.194 Both studies revealed that the LanB enzymes have a didomain architecture consisting of an ∼800 residue N-terminal domain and an ∼350 residue C-terminal domain (Figure 9A). The structures do not show resemblance to any other proteins in the Protein Data Bank,197 and therefore constitute a new fold. Although crystallization of the NisB-NisA complex utilized the entire precursor peptide, and mass spectrometric analysis of dissolved crystals confirmed the integrity of the dehydrated precursor, convincing electron density could only be observed for Lys−20 through Lys−9 of the leader peptide (see Figure 7 for leader peptide numbering scheme), indicating that the remainder of the precursor is conformationally disordered or that the enzyme has low affinity for this region.
Figure 9.

(A) Structure of the NisB homodimer bound to the NisA leader sequence (cyan). The glutamylation domain (brown) and elimination domain (purple) are indicated for one monomer, and the other protomer is colored in gray. (B) Mapping of the electrostatic potential onto the surface of the NisB monomer identified a basic patch (blue) in the vicinity of the NisA leader (green) predicted to engage glutamyl-tRNAGlu. PDB ID 4WD9.
Mapping of electrostatic potentials onto the surface of NisB identified a large basic region in the glutamylation domain, which could engage the tRNAGlu substrate (Figure 9B). Manual docking of tRNAGlu from Thermus thermophilus at this patch positions the aminoacylated CCA terminus in the direct vicinity of residues necessary for glutamylation.194 The NisB structure revealed that residues that were identified via Ala-scanning mutational analysis as important for glutamylation are distributed within a 10 Å radius in the N-terminal domain, and those important for β-elimination are closely distributed within the C-terminal domain (Figure 10).192,194 Analysis of NisB variants generated in a nisin producing strain identified Tyr80 of NisB as a residue that is critical for dehydratase activity, and the Y80F variant could bind the NisA precursor but did not modify the substrate in vivo.198 One possible role for Tyr80 is as a shuttle for glutamate from glutamyl-tRNAGlu to the NisA substrate. The crystal structure of the MibB dehydratase shows an overall conservation of most structural features present in NisB,196 although some local movements near the leader peptide-binding site are evident (see section 2.5).
Figure 10.
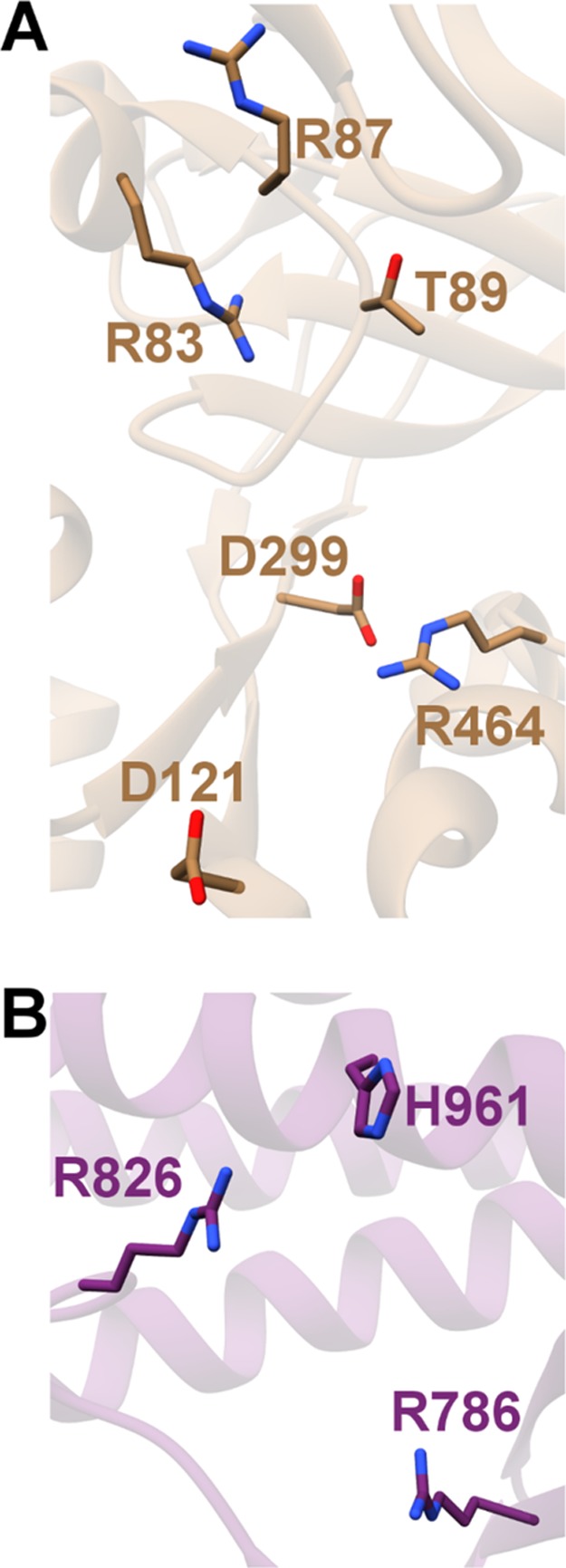
Residues identified by Ala-scanning mutational analysis that are critical for (A) glutamylation and (B) glutamate elimination map to the two different domains of NisB. The exact roles of these residues remain to be established. PDB ID 4WD9.
Confirmation of the attachment of the Glu to the Ser/Thr side chain via its α-carboxylate was provided by a study on the nonlanthipeptide goadsporin. The structure of this RiPP features several dehydro amino acids,199 and its biosynthetic gene cluster contains two open reading frames that would be consistent with one gene that corresponds to a Glu-tRNA dependent glutamylation enzyme and one that corresponds to a Glu elimination enzyme.200 Disruption of the latter in the producing organism resulted in buildup of intermediates that were characterized by NMR spectroscopy.195 These studies unambiguously showed attachment of Glu via its α-carboxylate to the Ser side chain. Similar “split” LanB systems are also present in the biosynthetic gene clusters of thiopeptides (see review by Burkhart et al. in this issue),201 another class of RiPPs, and they have been shown to also utilize Glu-tRNA for dehydration of Ser/Thr in their substrate peptides.202,203 Split LanB enzymes are also found in some lanthipeptide biosynthetic gene clusters35 including one in Chitinophaga pinensis DSM 28390. This cluster produces the pinensins (Figure 5), the first antifungal lantibiotics and the first characterized lanthipeptides from Bacteroidetes.5 With now four LanB and LanB-like enzymes investigated from different genera,194−196,202 the use of Glu-tRNA appears to be a general requirement. These findings also explained a previous report on the in vitro biosynthesis of nisin by expression of nisA, nisB, and nisC in a cell-free translation system.204 This experimental setup contained tRNAs, tRNA transferases, and all amino acids, and hence nisin biosynthesis was observed. We also note that esterification of Ser/Thr, instead of phosphorylation (see sections 4.2 and 5.2), was already suggested back in 1991 as a potential activation mechanism for dehydration.205 Why LanBs use Glu-tRNA and not another amino acyl-tRNA is currently not understood. For an overview of the use of aminoacylated tRNA in natural product biosynthesis, see the review by Gondry and co-workers in this issue.206
To determine the phylogenic distribution of LanB enzymes, a sequence similarity network was constructed for this review with the Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST)207 utilizing all members of the N-terminal Lantibiotic Dehydratase family (Pfam 04738) from the UniProtKB database (version 2016_06; Figure 11). After constructing the network, all sequences that did not contain a C-terminal Lantibiotic Dehydratase family domain (Pfam 14028) were removed, leaving only complete LanB proteins containing both the N-terminal glutamylation and C-terminal elimination domains (i.e., split LanBs and small LanBs194 were removed). The relative similarity between individual LanBs was assessed in Cytoscape208 at an alignment score threshold of 120 (∼30% sequence identity). Actinobacteria constitute the majority of the LanB sequences found in the UniProtKB database and appear to be distributed into two major groups. While the larger cluster is isolated from the clusters of other phyla, the smaller cluster from Actinobacteria appears to be related to LanBs found in Firmicutes. As for the Actinobacteria, LanBs from Bacteroidetes are split into two major clusters. The larger of these two is closely linked to LanBs from Proteobacteria, suggesting a close lineage. Previous phylogenetic analysis of the LanC cyclase enzymes showed that Bacteroidetes and Proteobacteria LanCs are closely related and are distinct from LanCs from Actinobacteria and Firmicutes.36
Figure 11.
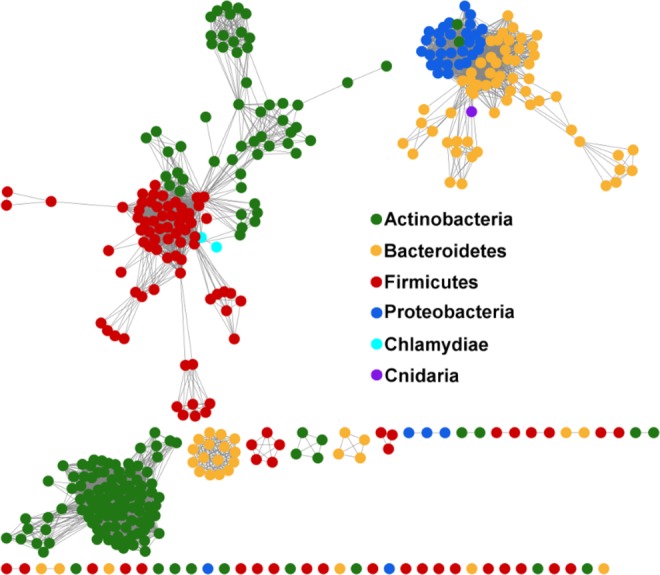
Sequence similarity network generated herein using EFI-EST and visualized in Cytoscape with an alignment score threshold of 120 (∼30% sequence identity) showing the distribution of LanB enzymes across phyla. Each node represents protein sequences sharing >95% sequence identity.
2.3. Cyclization
Formation of the lanthionine ring is completed with the intramolecular attack of a Cys thiolate onto the dehydro amino acid. Conjugate addition of nucleophiles to dehydro amino acids is well characterized and, in principle, formation of lanthionine rings could occur spontaneously at basic pH.209−211 However, attempts to recapitulate the correct ring topology of nisin synthetically yielded a mixture of products. For example, an attempt to form the AB ring system of nisin by biomimetic cyclization of a synthetic model produced multiple products.212 These products were presumed to result from the faster cyclization rate for lanthionines compared to methyllanthionines under nonenzymatic conditions.213 Consequently, enzymatic catalysis is required to overcome this intrinsic reactivity and facilitate proper ring formation.
Initial characterization of the function of a LanC cyclase was carried out via genetic studies of Pep5 biosynthesis, wherein deletion of the C-terminal 231 residues of PepC from a biosynthetic plasmid in the Staphylococcus epidermidis 5 producing strain diminished formation of lanthionine rings.214 Isolation of intermediates from this strain demonstrated that the Ser/Thr residues in the precursor peptide had undergone modification but in most cases the Cys residues were intact. Studies of mutant strains of S. epidermidis Tü3298 that do not produce epidermin revealed an intact biosynthetic pathway but with mutations in the epiC gene, as plasmid complementation with wild-type epiC restored lanthipeptide production.215 These data suggested that formation of the thioether linkages is not spontaneous, and that the LanC cyclase is required for the site-specific addition of Cys onto the dehydro amino acid to achieve the proper ring topology.
The LanC cyclases could be readily purified after overexpression in an E. coli heterologous system, as first demonstrated for EpiC from the epidermin cluster.216 However, in vitro characterization of LanC activity initially proved challenging due to the lack of suitable quantities of the dehydrated precursor substrate and the lack of a means to detect the cyclization reaction, as there is no change in mass of the precursor peptide upon thioether formation. Interestingly, sequencing of the nonproducer mutants of S. epidermidis Tü3298 described above identified G131E or G245E mutations in EpiC (note that numbering is adjusted from ref (216) to correspond with the complete gene), and each of these residues are in motifs conserved throughout LanCs and G245 is in the vicinity of the predicted catalytic acid (His248) in the primary sequence. The authors also noted that two Cys residues (Cys313 and Cys360 in EpiC) are also conserved among LanCs,216 suggesting that these Cys may play a role in catalysis. In 2002, analysis of affinity tagged NisA precursor expressed in mutant strains of L. lactis that lacked NisC showed that the peptide had undergone dehydration but not cyclization, confirming a role for LanC enzymes in the cyclization reaction.188
Chemical analysis of two LanC enzymes (NisC and SpaC) purified from a heterologous E. coli expression system indicated that both enzymes contain stoichiometric amounts of zinc.217 Based on a functional role for zinc in maintaining the reactivity of thiols at neutral pH in other alkylating enzymes,218,219 the zinc in LanC was proposed to function as a Lewis acid to lower the pKa of the Sγ of Cys, enabling the thiolate to participate in conjugate addition to Dha or Dhb residues.217 The in vitro reconstitution of NisC activity was finally achieved using dehydrated NisA purified from a L. lactis strain that encoded the precursor peptide, dehydratase, and transporter but lacked the gene for the cyclase.177 Thioether formation was monitored by mass spectral analysis of the dehydrated NisA that was treated with a thiol-selective modification agent. In the event, the dehydrated peptide was modified five times indicating the presence of five free thiols, whereas peptide that was treated with NisC did not show any mass shift, consistent with a sample devoid of any free Cys as a result of five cyclizations. Proteolytic removal of the leader peptide from the NisC-treated precursor yielded a product that was bioactive against a nisin indicator strain, suggesting that NisC installed the correct ring topology of nisin.177
The crystal structure of NisC revealed an α,α-toroidal fold containing a single zinc ion at the center of the toroid (Figure 12A). Enzyme residues Cys284, Cys330, and His331 coordinate the zinc ion (Figure 12B),177 and these residues are very close in structure to the Gly residue that corresponds to Gly245 in EpiC that was altered to Glu in a nonepidermin-producing strain of S. epidermidis Tü3298. The structure also reveals a small extension adjacent to the zinc ion, which resemble mammalian SH2 domains220 and may analogously serve to bind the peptide substrate. However, there is currently no evidence for a functional role of this SH2-like extension. More recently, mammalian homologues of LanC proteins, termed LanCL (for “LanC-like”) have been identified and characterized to similarly contain an active site zinc (Figure 12C).221
Figure 12.

(A) Overall structure of the NisC cyclase (PDB ID 2G0D) illustrating the α,α-toroid (orange), and the SH2-like extension (blue and purple). (B) Close-up view of NisC showing the zinc ligands and other residues located in the active site. Residues derived from the SH2-like domain are indicated in blue. (C) Active site of LanCL1 showing coordination of enzyme residues (green) and GSH (red) to zinc (PDB ID 3E73).
Site-specific mutations were introduced at residues in the vicinity of the zinc ion, which are conserved between NisC and the subtilin cyclase SpaC, in order to probe their importance in catalysis (Figure 12B). Activity for SpaC was measured by detecting production of subtilin in vivo222 while the activity of NisC variants were monitored by the bioactivity of products formed in vitro in reactions using purified dehydrated NisA.223 Mutation of the zinc-ligands Cys284, Cys330, or His331 in NisC, or the equivalent SpaC residues, diminished zinc binding and eliminated cyclase activity.217,222,223 Additional active site variants of NisC, including H212N, H212F, and D141N, retained affinity for zinc but lacked activity, indicating a possible role for these residues in acid/base catalysis.222,223
The structural and mutational data provide a plausible mechanistic framework for LanC catalysis (Figure 13). Coordination of the Cys residue in the precursor peptide by the catalytically requisite zinc ion lowers the pKa to facilitate deprotonation, which may or may not require a general base. For instance, in protein farnesyltransferase, the pKa of a Cys residue in the peptide substrate is lowered from 8.3 to 6.4 upon binding to the zinc in the enzyme active site.224 A role of the zinc ion in thiol activation, rather than activation of the carbonyl group in the dehydro amino acid electrophile, is supported by both the common use of Zn2+ for thiol activation in other proteins218,219,225 and the observation of a direct interaction between the sulfur of Cys in glutathione (GSH) and the zinc ion in the mammalian protein LanCL1 (Figure 12C). Indeed, activation of thiols for nucleophilic attack by zinc coordination is well supported by model studies with small molecule metal complexes.226−237
Figure 13.
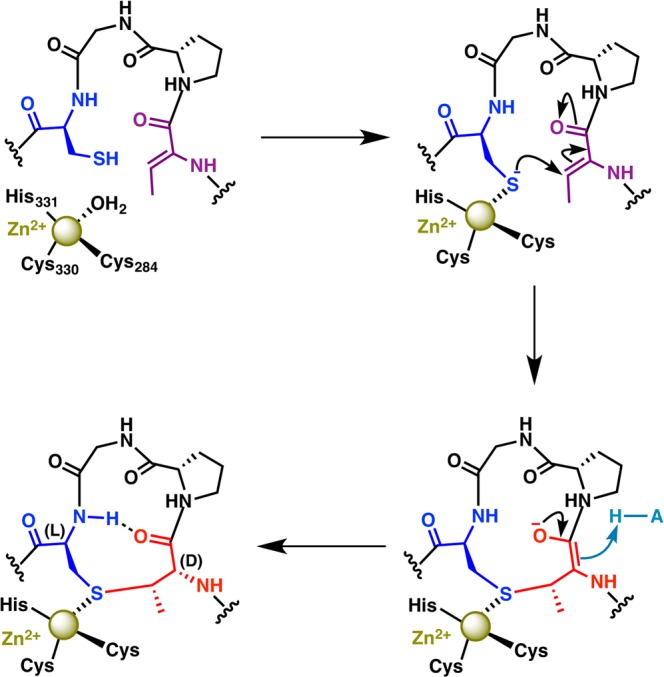
Proposed mechanism of cyclization illustrated for the formation of the B-ring of nisin. The active site acid (H-A) that protonates the enolate is likely His212.
Upon ligation of the Cys thiol, the segment of the peptide containing the dehydrated residue targeted for cyclization would be directed back toward the active site, perhaps through the SH2-like extension, where the thiolate would attack the β-carbon of the dehydro amino acid to generate an enolate. Subsequent protonation provides the D-configuration at the α-carbon via a net anti addition to account for the stereochemistry of MeLan in nisin (Figure 13). The shallow groove formed at the center of the α-toroid may facilitate formation of thioether linkages of different sizes (4–7 amino acids in nisin, Figure 7). The identity of residues that may deprotonate the thiol and/or protonate the enolate has not been equivocally established. Mutation of His212 or its hydrogen-bonding partner Asp141 in NisC abolished activity but the role of these residues is unclear from the mutational analysis.223 More recent studies have shown that cyclization is reversible. In particular, the class I cyclase NisC and the class II lanthipeptide synthetase HalM2 can also catalyze ring opening (i.e., a retro-conjugate addition) following abstraction of the α proton of the (methyl)lanthionine.238 Mutation of His791 in HalM2 (equivalent to His212 in NisC) abolished this proton abstraction suggesting that this residue acts as the base in the retro-conjugate addition. In turn, this suggests that His212 in NisC is the acid that protonates the enolate during ring formation. Mutations of two other conserved amino acids in the active site, Y285F or R280A in NisC223 did not abolish in vitro cyclase activity, suggesting they are not critical for catalysis. A more drastic mutation of the corresponding Tyr in SpaC (Y304A) did result in loss of subtilin production in vivo,222 with similar results observed for the NisC-Y285A mutant.223
At present, it is not understood how the cyclization enzymes control the formation of well-defined ring patterns with widely differing ring sizes and sequences in just a single active site. The dehydrated NisA intermediate shown in Figure 7 contains eight dehydro amino acids and five Cys residues. As noted previously,150 the number of products differing in ring topology that can be generated by a completely nonselective cyclization process would be 6720, with the number going to at least 8.6 × 105 if stereochemistry is included. Yet, NisC makes a single product. As discussed in the next section, one means by which the enzyme may simplify the task is to alternate dehydration and cyclization processes such that the number of dehydro amino acids that each Cys has to differentiate is smaller. Another possibility is that the substrate sequence has an inherent preference for certain ring topologies. This hypothesis is especially supported from data on class II lanthipeptides discussed in section 4.3.3, but some class I systems also provide support. For instance, ericin A and S are made by one set of class I biosynthetic enzymes in Bacillus subtilis A1/3.239 The D ring of ericin S is linked to the E ring similar to the topology found in subtilin whereas the D ring of ericin A is intertwined with the C ring similar to the structure found in microbisporicin (Figure 14). Thus, it appears that depending on the sequence of its substrate, EriC can form products with different topologies.
Figure 14.
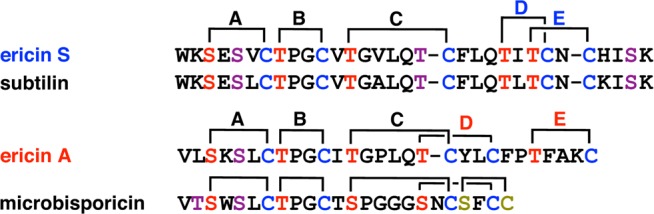
Class I cyclase EriC produces the lanthipeptides ericin S and ericin A with different D and E ring topologies resembling those of subtilin and microbisporicin, respectively.
2.4. Communication between Dehydration and Cyclization Events
Early studies on nisin biosynthesis suggested the existence of a multicomponent enzymatic complex, consisting of the biosynthetic enzymes and the transporter NisT. Co-immunoprecipitation experiments showed that the NisB dehydratase and the NisC cyclase localize together at the cytoplasmic membrane, and likewise yeast two-hybrid studies evidenced interactions between NisB and NisC and between NisC and NisT.240 Similar analysis of the subtilin biosynthetic enzymes indicated that the SpaB dehydratase, SpaC cyclase, and the SpaT transporter may form a multiprotein complex associated with the membrane.241 However, the dehydration and cyclization activities are not mutually contingent, as NisB can dehydrate substrates in the absence of all other modification enzymes,192,194,242 and NisC can catalyze cyclization in the absence of other protein or cellular components.177 Similarly, the leader peptide protease NisP and the transporter NisT can function independently (section 3).243
Although the above data suggest that the individual activities may be decoupled, pulse-labeling experiments (see section 3.1) showed that the kinetics of NisA dehydration by NisB and subsequent secretion by NisT are compromised in the absence of the NisC cyclase.189 The wild type efficiency could be restored by expression of a catalytically inactive NisC mutant (H331A), suggesting that it was interference with complex formation, and not lack of substrate cyclization, that reduced the efficiency of dehydration/transport in the absence of NisC.244 The functional interplay between the dehydration and cyclization reactions was analyzed in vivo using a plasmid-based expression system bearing NisA precursor variants and NisBTC.244 Installation of non-native Ser/Thr residues at positions immediately following the thioether rings did not result in any significant additional dehydration events, suggesting Ser/Thr at these positions are protected; this protection could be removed by preventing formation of the rings by additional mutagenesis.
These studies revealed that for the nisin system the dehydrations are largely independent of each other in the absence of NisC,244 and that additional dehydrations can take place for new Ser/Thr residues that are introduced. However, in the presence of NisC it appears that rings are installed before dehydration has finished such that certain positions become protected from access by the NisB dehydratase. The C-terminal positioning of these protected Ser/Thr residues relative to the rings also suggests a mostly N- to C-directionality of the NisA processing.189 An automated pattern-matching algorithm comparing experimental high-resolution mass spectral data of in vitro generated intermediates against theoretical spectra of hypothetical intermediates is also consistent with a mostly N- to C-terminal directionality for the NisB dehydratase.245
Analogous results suggesting that cyclization and dehydration are at least partly alternating events proceeding with N-to-C-terminal directionality were obtained using an inactive NisC mutant. Specifically, expression of the wild-type NisA precursor with NisB and an inactive NisC cyclase variant yielded a product with one additional Dha at a position directly C-terminal to the E-ring in mNisA that is usually not dehydrated by NisB (i.e., Ser29 in the NisA precursor, see Figure 7).244 These results suggest that E-ring formation prevents dehydration of Ser29 by NisB. The observation that cyclization is interspersed with dehydration is consistent with, but does not prove, a channeling model in which the product from one enzyme is channeled to the next enzyme without dissociation, possibly through a multienzyme complex of NisB and NisC. As discussed in more detail in section 4.4, an alternative to channeling would be a conformational sampling model.
Further details on the mutual influences of dehydrations and cyclizations, as well as on the directionality of processing, were elucidated through in vitro studies of the biosynthesis of NAI-107.196 Processing of the MibA precursor peptide in the presence of only the MibB dehydratase resulted in up to 7 dehydrations. Mass spectral analysis identified the expected dehydrated intermediates, along with masses that corresponded to monoglutamylated MibA, which were formed only after the accumulation of 5-fold dehydrated MibA.196 Notably, upon addition of the MibC cyclase to the reaction, the monoglutamylated intermediates were no longer detected. These observations suggest that glutamate elimination is slower from late stage intermediates than for early intermediates. Use of an inactive MibC mutant did not prevent the formation of glutamylated intermediates, and hence the elimination of these late stage intermediates is facilitated by thioether formation. This observation is consistent with the substrate alternating between modification in the dehydratase and cyclase active sites, which may be facilitated by a multicomplex assembly and, as described above for NisA, may involve channeling or conformational sampling. Surprisingly, pattern-matching algorithmic analysis of MibA processing by MibB suggested that dehydration does not occur with an N- to C-terminal directionality, as observed for nisin, but occurs in a nonlinear fashion.196 This nondirectional order of processing has also been observed for class II246 and class III247 lanthipeptide biosynthetic systems. Hence, there are significant differences between individual lanthipeptide biosynthetic pathways, with some having tightly coupled dehydration and cyclization processes (e.g., NAI-107) and others where such tight coupling is not required for producing the correct final products (e.g., nisin).
2.5. Leader Peptide Dependence and Recognition
As discussed in the Introduction, the sequences of lanthipeptide precursor peptides may be demarcated into an N-terminal leader sequence and a C-terminal core sequence (Figure 3). Post-translational modifications are installed on residues located in the core sequence, after which the leader sequence is removed to afford the final natural product. The initial proposal that the leader peptide may be involved in the biosynthetic process was based on the observation that the leader sequence of the nisin precursor peptide shares similar features with those of subtilin, epidermin, and Pep5.39 Conceptually, the leader sequence may facilitate biosynthesis by interacting with processing enzymes, by assisting in the transport of the processed peptide product, and/or by contributing to immunity within the producing organism.248 Experiments suggest that the leader sequence of the fully modified NisA precursor does not directly interact with the modified core peptide, either in solution or in lipid micelles,249 thus providing no evidence for a direct role of the leader in the chemical transformations in the core peptide.
The leader peptide sequences of most class I lanthipeptides are about 25 residues in length (e.g., 23 for the nisin leader peptide) and generally contain acidic amino acids. Ala-scanning mutational analysis of the leader sequence in the NisA precursor, carried out using a plasmid-encoded gene in a nisin producing strain, identified a four-residue motif, Phe−18/Asn−17/Leu−16/Asp−15 (hereafter FNLD, Figure 7), as critical for nisin production and secretion.248,250,251 Co-immunoprecipitation experiments252 and surface plasmon resonance analysis of the interaction of variants of NisA191 demonstrated the importance of the FNLD sequence for the direct interaction between the NisB dehydratase and the NisA precursor. Isothermal titration calorimetric studies demonstrated that the NisC cyclase also binds to the NisA precursor with a Kd of 2.0 μM, regardless of whether the core is unmodified or processed, and to the leader sequence with a Kd of 3.8 μM.253 Notably, the FNLD sequence in the NisA precursor that is critical for engaging the NisB dehydratase is also essential for binding to NisC, and Ala mutations at either Phe−18 or Leu−16 completely abolished the interaction. Co-immunoprecipitation studies indicated that other regions of the leader may also be necessary for engaging the NisC cyclase.254 Although the leader peptide is sufficient for interactions with the dehydratase and the cyclase individually, the formation of a multienzyme complex that includes NisB and NisC appears to require both the leader and the core sequence, but not necessarily the modified residues.255,256 Interestingly, in vivo NisB can partially dehydrate the NisA core peptide without the leader peptide and more efficiently when the leader peptide is coexpressed in trans.255
The molecular basis for the interaction between the nisin leader peptide and the dehydratase was elucidated by the cocrystal structure of the NisB-NisA complex.194 Electron density could only be observed for residues Lys−9 through Lys−20 of the leader peptide (Figure 15A), which includes the FNLD sequence (Figure 7). Despite expectations based on structure prediction tools that the leader sequence would bind to the dehydratase as an amphipathic α-helix,257 the structural data revealed that the leader forms a β-strand against a small winged helix-turn-helix motif located at the juncture of the glutamylation domain and the glutamate elimination domain.194 A similar winged helix-turn-helix motif is also observed in the structure of unrelated RiPP biosynthetic enzymes, such as the cyanobactin heterocyclases,258,259 PqqD, a peptide-binding protein from the pyrroloquinoline quinone biosynthetic pathway,260,261 and PaaA involved in pantocin biosynthesis.262 Bioinformatic analysis and binding studies established that domains of similar architecture are responsible for leader peptide recognition in a wide array of RiPP biosynthetic systems, including lasso peptides and thiopeptides. The domain was therefore termed the RiPP Recognition Element (RRE).263 Interaction between the domain and the leader peptide has been affirmed in the cocrystal structure of the cyanobactin heterocyclase LynD in complex with the leader peptide of its substrate (Figure 15B),259 and is also supported by mutagenesis studies in a radical epimerase involved in proteusin biosynthesis,264 and binding studies on sactipeptide synthetases.265 Thus, leader peptide binding by the winged helix-turn-helix domain appears to be widespread.
Figure 15.

(A) Close-up view of the interaction between the NisA leader sequence (blue) and the NisB dehydratase (brown). (B) Comparison of the winged helix-turn-helix leader-binding motifs in NisB and the cyanobactin heterocyclase LynD, another RiPP biosynthetic enzyme (PDB ID 4V1T). (C) Superimposition of NisB (brown and gray) with bound NisA leader (blue) and unliganded MibB (red and purple, PDB ID 5EHK), focusing on the winged helix-turn-helix domain. The black arrow illustrates the inward tilt of an ampipathic helix in MibB relative to its counterpart in NisB that interacts with the NisA leader peptide.
The interaction between NisB and the leader sequence of NisA is mediated largely through hydrophobic packing interactions. Within the critical FNLD sequence of NisA, Phe−18 is inserted into a hydrophobic pocket in NisB composed of residues Val176, Val198, Tyr202, Leu209, and Tyr213. Likewise, Leu−16 from this motif is bound in a hydrophobic pocket composed of Ile171, Tyr213, and Leu217.194 Lastly, Asp−15 of the motif is within hydrogen-bonding distance to Arg154 of NisB (Figure 15A). Similar hydrophobic packing interactions are observed in the engagement of the cyanobactin heterocyclase with its cognate leader peptide,259 consistent with prior mutational data that implicated several aliphatic residues in the leader peptide as critical for interactions with this modification enzyme.149 Mutational and deletion studies suggest that the leader peptide of the precursor for mutacin 1140 may have a different recognition motif consisting of the EDLF sequence.266
The FNLD motif is located in the N-terminal half of the leader peptide. The C-terminal half does not appear to be important for specific interactions with NisB as it is not observed in the NisA-NisB cocrystal structure. However, replacing leader peptide residues four-at-a-time with Ala4 did result in less efficient substrates and in some cases to lower amounts of NisB in pull-down experiments.254 Conversely, the amino acids at residues −13 to −8 of the leader peptide could be replaced by six His residues without losing NisB activity.251 Hence, although there may be some interactions with the surface of NisB, generally speaking the C-terminal part of the leader peptide appears to be a spacer251 that is required to bridge the distance of the leader peptide binding site and the glutamylation active site that is visualized by the crystal structure.194 Such a spacer region seems less important for the glutamate elimination activity since a glutamylated core peptide lacking the leader peptide was a substrate for NisB, resulting in glutamate elimination.194 As long as a minimal distance between the leader peptide binding site and the glutamylation active site is satisfied, dehydration can occur, even when the distance is increased significantly as shown by studies where half of a second core peptide fused at the end of NisA was modified by NisB.79 Similarly, Ser/Thr that are sufficiently spaced from the FNLD motif do not need to be in register with the motif since the intervening distance can be changed without losing the ability of NisB to perform the correct dehydrations.267 Similar findings have also been reported for the class I lanthipeptide mutacin 1140,268 as well as for class II lanthipeptides269,270 and class III systems.271
The structure of the MibB dehydratase from Microbispora sp. 107891 has been determined in the absence of the leader peptide. A comparison of the MibB structure with that of the NisB-NisA complex shows differences in domain orientations near the winged helix-turn-helix motif, which may reflect conformational changes that accompany binding of the leader sequence (Figure 15C). In the NisB-NisA cocrystal structure, portions of the leader peptide bind to a hydrophobic region containing several residues from an amphipathic helix, while in the MibB structure the equivalent helix is tilted inward such that the hydrophobic residues are oriented toward the interior of the protein (Figure 15C). This shift results in compensatory movements of the elimination domain to form a more compact overall structure. However, the lack of a MibB-MibA cocrystal structure precludes any conclusion at present regarding the importance of these conformational movements.
2.6. Tailoring Enzymes
In addition to the characteristic thioether rings that define the lanthipeptides, several class I molecules also contain additional tailoring modifications installed by enzymes that are not generally found across biosynthetic clusters. Biochemical studies of these tailoring modifications have not received as much focus but continue to provide examples of novel enzymology. Most tailoring reactions do not require the presence of the leader peptide or take place after leader peptide removal.
2.6.1. Dehydrogenation and Acetylation at the N-Terminus
A subset of class I lanthipeptides, including epilancin 15X,171 epicidin 280,272 and epilancin K7 (Figure 5 and 16A),273 are characterized by the presence of a lactyl group (Lac) on the N-terminus of the processed product. The corresponding precursor peptides all contain a Ser residue immediately following the leader sequence, which is converted to Dha by the cognate LanB dehydratase. Upon removal of the leader peptide, the resultant N-terminal Dha1 presumably spontaneously tautomerizes to an unstable imine, which then undergoes nonenzymatic hydrolysis to generate a 2-oxopropionyl (pyruvyl) moiety, as has been observed in the structure of lactocin S (see section 4.6.1)274,275 and the pinensins (Figure 5).5 Likewise, leader excision, and nonenzymatic hydrolysis of the N-terminal Dhb in Pep5 leads to the formation of a 2-oxobutyryl group (Figure 5).276 Enzymatic reduction of the 2-oxopropionate yields the 2-hydroxypropionate (lactyl) cap observed in the structures of epilancin 15X,174 epilancin K7277 and epicidin 280.272 Elucidation of the biosynthetic cluster of epicidin 280 identified a putative oxidoreductase (eciO) within the gene cluster, which may catalyze this enzymatic reduction.272 Likewise, the biosynthetic cluster of epilancin 15x evidenced a putative oxidoreductase (elxO).171
Figure 16.
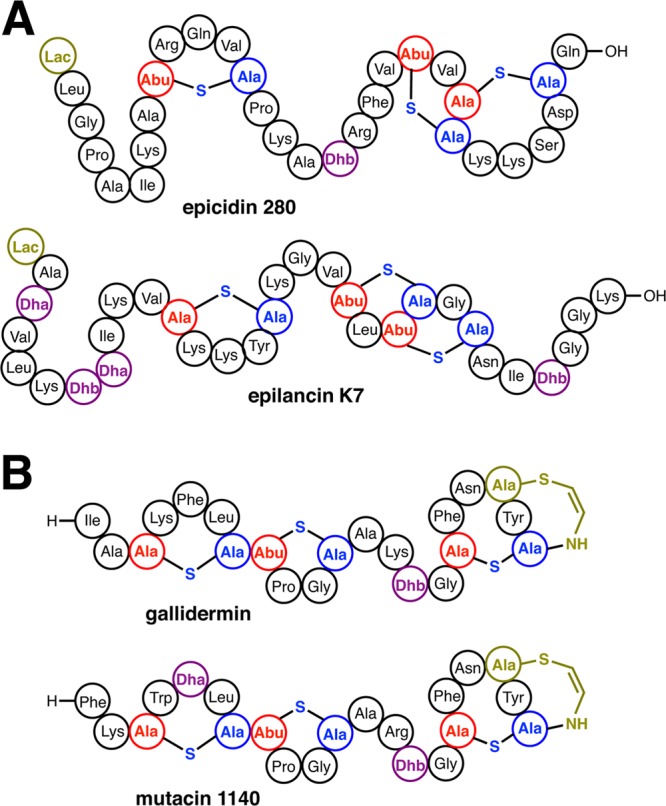
Tailoring modifications in class I lanthipeptide biosynthesis include installation of an N-terminal lactate (Lac) (A) and installation of a C-terminal aminovinyl Cys (B). The structure shown for epicidin 280 is hypothetical and based on analogy to Pep5 but is known to contain an N-terminal Lac.272
Heterologous expression and purification of the putative oxidoreductase ElxO from the epilancin 15X biosynthetic cluster in Staphylococcus epidermidis 15X154 facilitated reconstitution of enzymatic activity.171 Although the physiological substrate (dehydroepilancin 15X) could not be accessed, recombinant ElxO catalyzed the reduction of a synthetic analog containing an N-terminal pyruvyl group (Pyr) followed by five residues resembling the N-terminus of the ElxA core peptide (Pyr-AAIVK; Figure 5). Reduction of this synthetic substrate to Lac required an NADPH cofactor, and LC-MS analysis using synthetic standards assigned the stereochemical configuration of Lac as R.171 Characterization of the substrate scope of ElxO, using a library of peptides, demonstrated the enzyme as generally tolerant of amino acids that are appended to the N-terminal Pyr, but some trends in scope were observed.278 First, the kcat/Km values increased with substrate length, as peptides with 1–3 residues were generally poor substrates and a 13-residue peptide had a higher kcat/Km compared to a peptide of 5 residues. Furthermore, peptides with a longer N-terminal oxobutyryl modification were accepted as substrates, but those with a shorter glyoxyl group were not.278 As ElxO tolerates amino acid substitutions at residues following Pyr, the enzyme may have potential as a biotechnological tool for the installation of N-terminal alcohols on other lanthipeptides. For instance, incubation of ElxO with lactocin S, which contains a native N-terminal pyruvyl moiety (see section 4.6.1),274 resulted in reduction to the alcohol product.278
The crystal structure of ElxO in complex with NADP(H) affirmed its classification as a member of the short chain dehydrogenase/reductase (SDR) family of oxidoreductases.278 As with other members of the SDR family, the architecture of ElxO is built around a Rossmann-fold dinucleotide-binding motif (Figure 17A). The NADP(H) cofactor binds in an extended conformation. Other SDR enzymes utilize a catalytic Tyr that acts as both a general acid and base, flanked by an adjacent Lys that lowers the pKa of the Tyr, and a Ser that polarizes the substrate carbonyl.279 The active site of ElxO indicates that Ser139, Tyr153, and Lys157 could function in these roles (Figure 17B), and mutations of any of these residues resulted in a strong reduction of the reaction rate, reflecting their importance in catalysis. In the proposed catalytic mechanism, Tyr153 and Ser139 activate the N-terminal carbonyl of the substrate through hydrogen bonding, creating an environment that facilitates hydride transfer from NADPH to the Si-face of the carbonyl to produce the R-Lac product. A long groove is present along the enzyme surface adjacent to the NADP(H)-binding site and likely harbors the peptide-binding site (Figure 17C).
Figure 17.

(A) Structure of the ElxO oxidoreductase involved in N-terminal Lac installation. (B) Close-up view of the active site showing critical residues and bound NADP(H) (yellow). (C) Electrostatic surface view of the structure illustrating an obvious groove that may be involved in binding to the peptide substrate. PDB ID 4QEC.
Another novel tailoring modification that was recently discovered in the natural product paenibacillin is N-terminal acetylation.280,281 The putative paenibacillin biosynthetic cluster from Paenibacillus polymyxa OSY-DF contains genes that encode for canonical class I lanthipeptide proteins, including LanBCT, along with a precursor peptide bearing the FNLD motif. Inactivation of the lanB gene by targeted mutagenesis eliminated production of paenibacillin, confirming a role in biosynthesis. The gene cluster also encodes a 29-kDa polypeptide (PaeN) that bears sequence similarity with the TraX enzymes involved in N-acetylation of Ala in the F-pilin in E. coli.282 However, biochemical verification of PaeN activity has not yet been carried out. While the order of installation of the modifications has not been determined, the acetylation must occur after thioether formation as the target N-Ala residue is only exposed upon excision of the leader peptide.
2.6.2. Oxidative Decarboxylation at the C-Terminus
Several lanthipeptides, including epidermin, gallidermin, NAI-107, and mutacin 1140 contain the amino acid S-[(Z)-2-aminovinyl]-d-cysteine (AviCys) at their C-termini (Figures 5 and 16B). This modification was first identified through structural elucidation of epidermin from S. epidermidis Tü3298,173 and subsequent heterologous expression of the epiD gene from the corresponding biosynthetic cluster produced a yellow flavoprotein that was presumed to function in AviCys formation.172 Reconstitution studies of EpiD, using either the precursor peptide EpiA283 or a synthetic peptide corresponding to the epidermin core,284 indicated a loss of 46 Da from the substrate corresponding to the loss of CO2 and two H atoms. These studies confirmed that EpiD carries out oxidative decarboxylation of the C-terminal Cys, to produce a (Z)-enethiol product. As the mechanism of LanD enzymes has been previously reviewed,44 it will only briefly be discussed.
Detailed analysis of the substrate specificity for EpiD demonstrates that the decarboxylation reaction does not require the leader sequence,285 and studies using several truncated variants of the epidermin core sequence localized the terminal four residues of EpiA (SYCC) as the minimal substrate for EpiD.284 Modifications to the peptide C-terminus, such as ethylation, prevented oxidative decarboxylation, indicating that a free carboxy terminus is required.284 Mutational analysis of EpiD,286 along with the cocrystal structure with a peptide substrate (Figure 18),287 provides the basis for a plausible mechanism for AviCys installation. The flavin cofactor (FMN in EpiD) first oxidizes the terminal Cys to a thioaldehyde, followed by spontaneous decarboxylation to form the thioenolate.287,288 Attack of this enethiol onto the Dha residue, possibly catalyzed by the EpiC cyclase, would yield the AviCys product. EpiD as well as the homologous enzymes MrsD and MibD involved in mersacidin and NAI-107 biosynthesis,289−291 respectively, have sequence homology with enzymes that carry out two reactions during the biosynthesis of 4′-phosphopantetheine in coenzyme A biosynthesis. These latter enzymes first catalyze flavin-dependent oxidative decarboxylation of a Cys similarly to the LanD proteins, and subsequently reduce the resulting aminoethenethiol group to the corresponding 2-mercaptoethylamine. This class of proteins, including LanD, has been named the homo-oligomeric flavin-containing Cys decarboxylase (HFCD) family.286,292 Mechanistic studies on the enzymes involved in biosynthesis of 4′-phosphopantetheine are consistent with the proposed mechanism of LanD proteins.293−296
Figure 18.

(A) Structure of the EpiD flavoprotein homotrimer involved in AviCys installation. (B) Close-up view of the FMN (yellow) cofactor in the vicinity of a bound short peptide substrate (purple). PDB ID 1G5Q.
It should be noted that AviCys residues are also observed in two other classes of RiPP natural products, namely linaridins297,298 and thioviridamides.299 Although linaridins such as cypemycin297 and grisemycin298 contain the dehydro amino acid Dhb, the biosynthetic gene clusters for these molecules do not encode obvious homologues of either the lanthipeptide dehydratase that is necessary for installation of the Dha/Dhb or the lanthipeptide cyclase implicated in AviCys formation. CypD from the cypemycin cluster is also a member of the HFCD family and catalyzes oxidative decarboxylation of the terminal Cys to yield the enethiolate.297 However, subsequent attack of the enethiolate to a Dha to yield the AviCys occurs through a mechanism that has yet to be established. The thioviridamide biosynthetic cluster300 likewise contains a HFCD family gene (tvaF) but lacks any homologues of the LanB and LanC enzymes.
2.6.3. Trp Halogenation and Pro Hydroxylation
The lanthipeptide NAI-107 exhibits potent antibacterial activity against many Gram-positive pathogens175 and has demonstrated efficacy in experimental models of infection.16 The compound consists of a complex of related molecules produced by Microbispora sp. 107891, and these all contain two modifications not previously reported in lanthipeptides, namely halogenation on Trp and (di)hydroxylation on Pro (Figure 5).175 The two predominant congeners differ in the presence of one or two hydroxyl groups on Pro14 of the final product. The biosynthetic gene cluster of microbisporicin encodes the expected cadre of class I lanthipeptide biosynthetic enzymes, including the MibB dehydratase, the MibC cyclase, and the MibT transporter in addition to the substrate MibA.301 The cluster also contains additional genes that are presumed to install the two novel modifications, based on sequence similarities with enzymes that catalyze similar reactions. Specifically, the MibH and MibS proteins are similar to flavin-dependent halogenases and their corresponding flavin reductases that together catalyze halogenation in an FADH2-dependent manner.302 Recent in vitro studies confirmed that MibH catalyzes the chlorination at Trp4 of MibA and that MibS provides reduced FAD.291 MibH is the first halogenase that acts on Trp embedded in a peptide sequence as all prior Trp halogenases acted either on free tryptophan or tryptophan (or its analogs) tethered to coenzyme A or a carrier protein.302 The crystal structure of MibH was determined, revealing a much larger binding cavity than previously characterized Trp halogenases.291 This finding explains the very high substrate specificity of MibH, which requires preinstallation of the thioether rings in the substrate MibA for chlorination activity. Interestingly, inclusion of KBr in the production medium of the NAI-107 producer yielded bromination (rather than chlorination) on Trp4 of the mature lanthipeptide, suggesting some flexibility in the halogenase.303 The MibO polypeptide shows similarities to cytochrome P450 enzymes, and is likely responsible for hydroxylation of Pro14.301 Biochemical characterization of the hydroxylase has yet to be reported.
2.7. Immunity Proteins
Organisms that produce natural products that are bioactive against other closely related strains must protect themselves against toxicity from the final product. As most lanthipeptides are not active until removal of the leader sequence,248,304 coupling of leader excision to active transport provides a level of innate defense. Two other strategies have been adopted by strains that produce class I lanthipeptides to afford immunity, and these include active transport to avert unwanted build-up and sequestering of the mature lanthipeptide in the extracellular environment. Lipid II is the target of many lanthipeptides including nisin,166,167 mersacidin,305 lacticin 481/nukacin ISK-1,94,306 and many two-component lantibiotics307−309 (and their structural analogs). Lipid II is present both on the cytoplasmic side of the membrane and, after translocation, on the extracellular side. Thus, immunity needs to be provided both intracellularly and extracellularly. Lanthipeptide efflux from producing organisms is mediated by the LanFEG complex, an integral membrane ABC type transport system. Disruption of either nisE and/or nisF in L. lactis does not affect the ability to produce nisin but results in an increase in susceptibility, confirming their role in immunity.310,311 However, the lanFEG locus is not associated with some class I lantibiotic clusters, such as the Pep5214 and epicidin272 clusters. Hence, the LanFEG transporter system is not a universal mechanism of immunity.
The LanI proteins are thought to provide immunity to producing strains by sequestering the extracellularly secreted product. Some LanI proteins contain a consensus signal sequence for export, followed by an N-terminal Cys,312 which is modified by a diacylglycerol membrane anchor313 to localize the polypeptide on the cell surface.314,315 Expression of NisI in L. lactis ΔlanI enabled a significant increase in resistance to nisin.312 Conversely, knockout of nisI in L. lactis producing strains rendered them much more susceptible to nisin.310 For more discussion of genetic experiments aimed to understand lantibiotic immunity, we refer the reader to several reviews on this topic316−318 and focus here only on the systems for which structural information has recently been reported.
The solution NMR structures of SpaI,319 NisI,320 and MlbQ321 provide some rationale for the mode of actions of these proteins. The structure of the 16.8 kDa SpaI protein from B. subtilis showed a new fold consisting of a six-stranded antiparallel twisted β sheet architecture (Figure 19A). However, the construct used for structural work lacked the N-terminal 17 residues and did not confer immunity.319 The deleted region consists largely of basic Lys residues, suggesting that these N-terminal 17 residues play a role in stabilizing the interaction between SpaI and the acidic head groups of membrane phospholipids. The subsequent NMR structure of the 25.8 kDa NisI from L. lactis revealed a two-domain architecture consisting of tandem repeats of a seven-stranded β sheet fold resembling the fold in SpaI (Figure 19A). Each domain could be individually aligned with the structure of SpaI despite a lack of any notable similarity in sequence. Binding of NisI to nisin was observed by titrating in nisin and monitoring changes in the chemical shifts of NisI, which showed that nisin binds to the C-terminal domain of NisI (Kd of 22 μM), presumably along an extended hydrophobic patch. Structure-based mutational analysis suggested a role for Tyr153 and Asp155 of NisI in mediating binding to nisin.320
Figure 19.
(A) Solution NMR structures showing the overall folds of the SpaI (PDB ID 2LVL), NisI (PDB ID 2N2E and 2N32) and MlbQ resistance proteins (PDB ID 2MVO). The fold of MlbQ is distinct from those of SpaI and NisI. (B) Crystal structure of the S. agalactiae nisin resistance protein (SaNSR). PDB ID 4Y68.
The MlbQ polypeptide is a small lipoprotein from the NAI-107 biosynthetic gene cluster in Microbispora ATCC PTA-5024 that confers specific resistance to NAI-107-like lantibiotics.321 Like the coordinated immunity provided by LanI and LanEFG proteins, protection against NAI-107 like lantibiotics is conferred by several proteins, MlbJQYX.321 Although MlbQ sequesters NAI-107 and functions analogously to NisI and SpaI, its structure shows a novel topology consisting of four β strands topped by two short helices (Figure 19A). Despite the lack of structural similarities, MlbQ also contains an extended hydrophobic patch where the lanthipeptide may bind. However, the molecular details as to how these proteins recognize their cognate lantibiotics are still undetermined. Similarly, the molecular mechanism of immunity is not understood in systems where nisEFG and nisI homologues are missing (e.g. ref (171)).
Several strains of L. lactis that do not produce nisin have been shown to encode for a different type of resistance system.322,323 The resistance protein NSR is a protease that cleaves the C-terminal six residues from nisin, resulting in a significant reduction in bactericidal activity.324 More recently this system has also been found in pathogens. An operon in Streptococcus agalactiae that confers 20-fold resistance against nisin when expressed in L. lactis contains genes for an ABC-type transporter and a membrane-associated NSR (SaNSR).325 The crystal structure of SaNSR consists of an S41 C-terminal serine endopeptidase fold326 with an appended N-terminal helical bundle and protease capping domains (Figure 19B).327 Modeling studies suggest that the C-terminal residues of the nisin substrate could be positioned in a hydrophobic tunnel formed between the capping and protease domains. Enzymatic activity was contingent on the D and E rings of nisin because SaNSR could not process nisin variants that lacked these last two rings.
3. Proteases and Export in Class I and II Lanthipeptides
The late stages of lanthipeptide biosynthesis involve proteolytic cleavage of the leader peptide and export. The leader peptide is often necessary to direct the secretion of the modified peptide out of the producing organism, after which the leader peptide is removed to furnish the mature peptide, but sometimes proteolysis precedes secretion (vide infra). Two general strategies have been employed to accomplish these two tasks. The first discussed in section 3.1 was long thought to be characteristic for class I lanthipeptides and involves dedicated LanT transporters and LanP proteases. However, more recently, genomes especially in Proteobacteria36 and Bacteroidetes5 have been sequenced that contain class I lanthipeptide biosynthetic gene clusters with a bifunctional transporter/protease. For many years such bifunctional transporter/proteases were thought to be characteristic of class II lanthipeptides. Conversely, LanP proteins were long confined to class I lanthipeptides. The only exception was lactocin S, a class II lanthipeptide encoded by a gene cluster that contains genes for a dedicated LanT transporter and LanP protease.50 However, lanP genes are now regularly detected in class II gene clusters (section 3.2). Since distinction between class I and class II lanthipeptides based on proteolysis and transport no longer holds, both classses are discussed here in one section. The class II lantibiotic cinnamycin produced by Streptomyces cinnamoneus cinnamoneus DSM 40005 is unusual as its leader peptide appears to be removed by a protease of the general secretory (Sec) system.328 In fact, at present, lanthipeptide-specific proteases are very frequently absent from biosynthetic gene clusters in streptomycetes.35
3.1. Leader Peptide Removal and Export by Independent Proteases and Transporters
As noted in section 2.1, sequencing of the subtilin183 and nisin182 biosynthetic clusters identified genes for LanT that showed sequence similarity to integral membrane transporters, suggesting that the polypeptide was a dedicated transporter for export of the mLanA. More detailed analysis of the primary sequence showed strong conservation of the SpaT sequence with multidrug transporters and the transporter that when mutated causes cystic-fibrosis.329 Especially characteristic are the two Walker-motif sequences involved in ATP-binding, indicating that LanTs fall within the ABC family of transporters.330 Genetic deletion of the spaT gene in the subtilin-producing B. subtilis ATCC 6633 strain resulted in cell agglutination and a loss of viability, presumably as the result of intracellular accumulation of subtilin.183 Likewise, deletion of pepT from a plasmid directing the production of Pep5 in a S. epidermidis host reduced production of the lantibiotic to about 10% of that produced by a strain containing the intact plasmid.214 Lanthipeptides could also be detected in other lanT deletion strains, as evidenced in variant-producing strains of mutacin 1140268 and epidermin,181 (as well the class II lanthipeptide lacticin 481331). It is currently unclear how these compounds are exported when their LanTs are missing. Conversely, NisT is essential for nisin export as disruption of nisT prevented nisin transport.332
Immuno-detection using antibodies against the leader peptide, coupled with mass spectrometric analysis of culture supernatants, was used to determine the substrate tolerance of the NisT transporter in L. lactis strains bearing plasmids with various combinations of nisin biosynthetic enzymes.243 An expression strain with a plasmid containing only the genes encoding the transporter and the precursor peptide was capable of exporting unmodified precursor, indicating that the lanthionine rings were not required for export. Likewise, the strain also exported chimeras of the leader sequence fused to various biologically active peptides, demonstrating that the NisT transporter is broadly tolerant of substrates, as long as the leader peptide is present.243 However, kinetic analysis of peptide extrusion using expression strains pulse-labeled with [35S]-Met indicated that deletion of the NisB dehydratase significantly reduces the precursor export, and a substrate-channeling mechanism between the biosynthetic enzymes and the transporter has been invoked to explain these observations.189 Biochemical evidence for such a multiprotein complex with the transporter has not yet been established, but, as noted in section 2.4, yeast two-hybrid studies have suggested a physical interaction of NisC and NisT, and NisC and NisB, and co-immunoprecipitation studies indicated interactions between NisB and NisC and suggested a membrane localization for both.240 Similar results were also reported for subtilin biosynthesis in B. subtilis.(241)
The leader peptide-dependence of the transporter is consistent with removal of the leader peptide after export of the modified peptide for most class I lanthipeptides. There are two different groups of lanthipeptide proteases that are found in the corresponding biosynthetic clusters. The nisin leader protease NisP is the first characterized class I leader protease,178 and is a member of the S8 clade of subtilisin-like proteases.333 Sequence analysis identified four regions in the NisP polypeptide including an N-terminal 22-residue signal peptide, a prodomain consisting of residues Glu23 through Arg195, a serine protease catalytic domain encompassing Ser224 through Arg566, and a C-terminal ∼110-residue peptide that contains a canonical LPxTG sorting signal to anchor the polypeptide to the cell wall of the producer L. lactis via amidation of Thr655 with a pentaglycyl murein peptidoglycan.334 The C-terminal anchoring peptide can also be excised from NisP by autoproteolysis, likely freeing it from the cell wall.335
The crystal structure of the catalytic domain of the NisP protease has been determined to 1.1 Å resolution, revealing a fold that is similar to that observed for other S8 proteases (Figure 20A).335 Although the sequence of the prodomain was included in the protein construct used for crystallization, the reported structure consists only of the serine protease catalytic domain composed of Ser224 through Arg566. SDS-PAGE analysis of dissolved crystals revealed that the protein had undergone proteolytic degradation in situ, facilitating crystallization of the resultant fragment consisting of residues 224–566. The crystal structure revealed a canonical catalytic triad consisting of Asp259, His306, and Ser512 positioned along a heart-shaped open cleft on the protein surface.335 The NisP active site is flanked by electron density consistent with the sequence of the C-terminal region comprising Ala635 through Arg647, suggesting that autocatalytic processing may separate the protein from the membrane-anchoring C-terminal region to liberate NisP from the cell wall.335 A common feature among canonical S8 proteases is a rearrangement of a high-affinity calcium-binding loop in the N-terminal prodomain to displace it away from the active site following autoprocessing, resulting in a stable structure and explaining the calcium-dependent activity of these enzymes.336 Yet, the prodomain is absent in the reported NisP structure, suggesting that perhaps the catalytic domain is stable in its absence. However, at present no additional details are available that may rationalize this observation.335
Figure 20.

(A) Overall structure of the NisP catalytic domain. PDB ID 4MZD. (B) Docking model of the unmodified NisA peptide (green) onto the NisP active site (pink) evidencing steric clashes that may explain the substrate preference of NisP for mNisA. (C) Structure of the LicP protease including the prodomain. PDB ID 4ZOQ.
Sequence alignments of NisA with other nisin-like precursor peptides identified a conserved Gly-Ala-(Xxx)2-Arg-Ile motif, wherein NisP cleaves between the Arg and Ile residues.248 Mutational analysis of NisA variants demonstrated that precursors in which Ala had been altered to Asp resulted in products that had undergone post-translational modifications but from which the leader sequence was not excised.248 Likewise, mutation of the Arg in this motif to a Gln yielded a modified peptide product with an intact leader. Tryptic removal of the leader peptide from the product of the Ala variant strain afforded a peptide with bioactivity comparable to that of authentic nisin. These experiments confirmed that leader removal must occur only after export of the modified peptide, and is the last step in the maturation of fully bioactive nisin.
The substrate tolerance of NisP was further explored through mass-spectrometric analysis of in vitro activity of NisP-expressing cells on a series of modified NisA peptides.243 Treatment of fully modified NisA (mNisA) with cells displaying NisP resulted in facile removal of the leader sequence. However, NisP was unable to cleave the leader sequence from either unmodified NisA or a precursor containing dehydro amino acids but lacking the lanthionine rings. These experiments demonstrate that fully modified nisin with the thioether linkages is the true substrate for the NisP protease. Similar substrate specificity has been reported for the LanP involved in production of mutacin 1140.268 A manual docking model of the unmodified NisA peptide into the NisP active site suggests that the installation of ring A in nisin may be necessary to induce peptide curvature that avoids steric clashes with the enzyme (Figure 20B).337 Studies monitoring the timing of secretion by NisT and leader peptide removal by NisP indicate that these two processes are not tightly coupled.189
A second type of class I lanthipeptide LanP proteases was identified as part of the cluster responsible for the production of epilancin 15X.171 Like NisP, the leader protease from this cluster, ElxP, is a canonical S8-peptidase family serine protease. However, unlike NisP that functions only on substrates with the lanthionine bridges installed, recombinant ElxP can catalyze leader peptide removal from linear, unmodified ElxA precursor peptide171,278 (although it remains to be established if the modified peptide is a better substrate than the precursor). Residues that likely comprise the catalytic triad of ElxP (Asp27, His62, and Ser240) were identified based on sequence similarities with other characterized LanP proteases and subtilisin family members.171
Two distinguishing characteristics differentiate the ElxP structure from the NisP group of class I lanthipeptide leader proteases. First, the primary sequence lacks an N-terminal secretion signal and the C-terminal LPxTG sorting signal suggesting that ElxP is located in the cytoplasm. The cytoplasmic localization of ElxP suggests that the ElxA leader is removed prior to extrusion of the mature lanthipeptide outside of the cell, although this has not yet been definitively established. If so, the transporter ElxT would not require a leader peptide (see section 3.2 for discussion of transporters). Second, the recognition sequence in ElxA flanking the cleavage site is distinct from that for NisP and related signal proteases. An alignment of the ElxA leader sequence with those of other homologous precursor peptides showed a conserved Asp/Glu-Leu/Val-(Xxx)2-Gln-Ser/Thr sequence at the C-terminus of the leader sequence that is distinct from the Gly-Ala-(Xxx)2-Arg-Ile motif found in NisA and related precursors. The Ser/Thr residue in the ElxA motif is dehydrated by the corresponding LanB dehydratase and ElxP cleaves after the Gln residue.278 Ala-scanning mutational analysis of the ElxA leader peptide revealed that mutations at Asp, Leu, or Gln of the motif resulted in a significant reduction in cleavage efficiency by ElxP as determined by mass spectrometric analysis.278 Michaelis–Menten kinetic constants determined for the Gln and Leu ElxA variants indicated that kcat was decreased by more than 10-fold compared to native ElxA, while the Km was largely unchanged.278 Conversely, Ala mutations at the two intervening residues within this motif had no effect on proteolytic efficiency. Notably, although the NisA precursor is not a substrate for ElxP, insertion of the Asp-Leu-(Xxx)2-Gln-Ala motif present in ElxA (where the Ala mimics the Dha found in processed ElxA precursor) into the NisA precursor yielded a chimeric peptide that was readily cleaved by ElxP.278
3.2. Leader Peptide Removal and Export by Bifunctional Enzymes
While independent proteases and transporters carry out the processing of most class I lanthipeptides, some class I lanthipeptide and most class II lanthipeptide clusters contain a single polypeptide that harbors both protease and transport activities. These bifunctional LanT proteins are members of the ABC-transporter maturation and secretion (AMS) protein family.338,339 For instance, the biosynthetic cluster of the class II lanthipeptide lactococcin DR (identical to lacticin 481, section 4.2) from L. lactis subsp. lactis ADRIA 85LO30 encodes a protein featuring a high level of sequence similarity to ATP-dependent transporters340 but that also encompasses an N-terminal domain with homology to members of the C39 family of papain-like Cys proteases.341 Historically, these bifunctional transporters with a protease domain have been given the same LanT designation as the transporters without a protease domain, which is somewhat unfortunate since it does not provide immediate indication about the type of protein. Analogously to how the multifunctional class III lanthipeptide synthetases have been named LanKC (for kinase and cyclase), we suggest calling the transporters that contain a protease domain LanTp (for transporter and protease). We use the subscript p rather than capital P (i.e., LanTP) to avoid confusion with LanT + LanP, and to follow the guidelines of the American Society of Microbiology. In the remainder of the review we will use this nomenclature.
The leader sequences of lactococcin DR, as well as those of streptococcin A-FF22,342 salivaricin A,343 and enterococcal cytolysin,344 show strong similarities with other nonlanthipeptide bacteriocins that are generally cleaved after two conserved glycine residues.338 Mutations at either of the two Gly in the MutA precursor peptide abolish both proteolytic processing and export of mutacin II (Figure 21).345 This cleavage motif, which also includes GA and GS sequences as variations, has been named the double-Gly motif.
Figure 21.
Structures of bovicin HJ50, nukacin ISK-1, and mutacin II. For the first two compounds, the protease activity of the cognate bifunctional LanTps has been reconstituted. Bovicin HJ50 incorporates a disulfide tailoring modification (section 4.6).
Characterization of the processing of the nonlantibiotic bacteriocin lactococcin G revealed that the LagD AMS transporter also harbored the leader peptide excision activity, which was mapped to the N-terminal 150 residues of the polypeptide.338 Subsequent characterization of the isolated protease domains of the ComA AMS transporter involved in quorum sensing,346 and of the CvaB AMS transporter of the colicin V secretion system,347 as well as genetic experiments on the LcnC transporter involved in the biosynthesis of other lactococcins,339 established the general architecture for this class of ABC-type bacteriocin transporters. The crystal structure of the protease domain of ComA revealed an overall fold that resembled the papain-like C39 family of Cys proteases, as predicted from the primary sequence.348 Docking studies with a model ComC leader peptide are consistent with the leader peptide binding as an amphipathic α-helix in a hydrophobic groove adjacent to the active site, thus positioning the double Gly motif in the enzyme’s active site.
Full reconstitution of in vitro protease activity for a lanthipeptide AMS protein was established through the characterization of the N-terminal 150-residue protease domain of the LctT transporter (= LctTp in the nomenclature used in this review) involved in the processing of the class II lanthipeptide lacticin 481.349 Unlike the NisP-type class I stand-alone proteases, leader cleavage from the LctA precursor peptide was not contingent on installation of the lanthionine rings. Although the LctTp protease domain could remove the leader sequence from full-length LctA precursor peptide, a truncated 10-residue substrate, corresponding to Asp−6 through Ser4 of the substrate, was not processed. Likewise, replacement of residues in the leader sequence with Pro also compromised activity.349 These studies are consistent with the expectation that a helical conformation of the leader sequence is critical for substrate recognition by the protease. Protease activity has also been established for the N-terminal 150 amino acids of BovTp, which removed the leader peptide from mBovA to generate the lantibiotic bovicin HJ50 (Figure 21).350
Based on the sequence similarity to other AMS transporters and to papain-like proteases, the catalytic triad of the protease domain of LctTp was proposed to consist of Asp106, His90, and Cys12. Both the C12A and C12S variants were devoid of activity but mutations at Asp106 had no effect on activity, suggesting that this residue is not essential for catalysis.349 Surprisingly, although the H90A variant was active, this mutant cleaved the substrate peptide at a different site, suggesting that this residue may play a role in site specificity of amide bond hydrolysis. A mutation at the equivalent His in the protease domain of ComC346 or CvaB347 resulted in loss of catalytic activity.
AMS proteins are quite tolerant of the cargo they transport and proteolyze. Peptide chimeras, in which the LctA leader peptide was appended to different core peptides, were processed at varying rates by the LctTp protease domain suggesting some role of the core sequence on substrate processing.349 Similarly, the LtnTp AMS transporter involved in biosynthesis of lacticin 3147 has been shown to secrete various noncognate peptides attached to the LtnA2 leader peptide and to remove the leader peptide in the process.351 The presence of the class II lanthionine synthetase LtnM2 (section 4) was not required for transport by LtnTp. Substrate tolerance is also demonstrated by AMS proteins that have more than one substrate such as the transporters HalTp124,125 and LicTp103 that each transport two different modified peptides (mHalA1 and mHalA2 for HalTp and mLicA1 and mLicA2 for LicTp, see Figure 22), and the transporter ProcTp that presumably secretes all 30 of its substrates (section 4.1).129 Not only are these transporters tolerant of the various cargo peptides, they also demonstrate relaxed specificity with regard to the leader peptides since the leader peptides of the individual precursors of two-component peptides are quite diverse,103,124 and these transporters can even act on noncognate leader peptides.77 Based on sequence homology and site-directed mutagenesis experiments, an ELXnBXG-G/A/S (B is a hydrophobic residue, n = 2–8) at the end of the leader peptide appears important for proteolytic cleavage.103,349
Figure 22.

Biosynthesis of the two-component lantibiotic lichenicidin. The structures of Licα and Licβ were assigned by NMR and MS analyses.363 LicTp acts on both peptides to remove the entire leader peptide of Licα and all but six C-terminal residues, NDVNPE (bolded and underlined), of Licβ. The latter are recognized by LicP in the final proteolysis step. The stereochemistry of the Licβ A-ring is unknown but predicted to be LL as it derives from a Dhx-Dhx-Xxx-Xxx-Cys motif (Dhx = Dha/Dhb; section 4.3.3). For discussion of the LanM proteins, see section 4. For clarity, the process is shown as first completion of dehydration and then cyclization, but this is not necessarily the case. For instance, for the related two-component lantibiotic haloduracin, cyclization commences before dehydration is completed.364,365
The topology of AMS proteins has been investigated showing that the protease domain is located at the cytoplasmic side of the membrane.339,352 Yeast two-hybrid studies demonstrated a physical interaction between NukTp and NukM involved in the biosynthesis of the class II lantibiotic nukacin ISK-1 (Figure 21).353 This study also suggested that like for class I lanthipeptides, the biosynthetic enzymes NukM and NukTp can function independently in vitro and in heterologous expression systems but physiologically may be in a membrane-localized multienzyme complex. Expression of full-length NukTp in inside-out membrane vesicles showed that mNukA, but not unmodified NukA, was a substrate for proteolysis.354 Interestingly, this study also showed that adenosine triphosphate (ATP) was required for proteolysis, suggesting a tight coupling between the transport domain and the protease domain of NukTp. Indeed, mutations in the conserved Walker motifs and H-loop of the ATP-binding domain of NukTp resulted not only in accumulation of mNukA inside the cells, indicating disruption of transport activity, but also in abolishment of leader peptide removal.352 Similar observations were also made with inside-out vesicles. Analogously, mutations to the catalytic residues in the C39 protease domain interfered with both proteolysis and transport.352 Thus, although isolated C39 protease domains of AMS proteins have proteolytic activity and do not need ATP, for intact AMS proteins, proteolysis and transport appear to be cooperative.
The crystal structures of a putative AMS protein provide insights into the transport cycle of LanTp and other transporters in this family.355 At present, the function of this peptidase-containing ATP-binding cassette transporter from Clostridium thermocellum that was identified by genome mining is not known, but it seems to be associated with a 90-residue precursor peptide containing a 24 amino acid leader peptide ending in GG. The architecture of the transporter consists of three domains, an N-terminal peptidase domain, a six-helical transmembrane domain (TMD), and a C-terminal nucleotide-binding domain (NBD) (Figure 23). Structures were obtained both in the absence and presence of an inert nucleotide analog, revealing two conformations of the transporter (Figure 23A,B). In the absence of bound nucleotide, the structure reveals an inward-facing conformation wherein the transmembrane helices of the homodimer form an open tunnel that is oriented toward the active site of the peptidase domain. This state would position the core peptide within the tunnel and orient the double Gly sequence of the leader toward the peptidase domain. Nucleotide binding induces a rotation of the C-terminal nucleotide-binding domains to close the transmembrane tunnel. This conformational reorganization displaces the peptidase domains, which may help facilitate translocation, and subsequent release, of the cleaved core peptide across the membrane.355 In the nucleotide-bound structure, the transmembrane tunnel is empty and not open to the outside, but if a core peptide were in the cavity, it might induce formation of an extracellular gate. The authors also noted that unlike the tunnel of the general Sec system, which transports unfolded extended polypeptide chains or small helices, the transmembrane cavity of the AMS protein is large enough to accommodate a small folded protein.355 Presumably, this also extends to a post-translationally modified core peptide in the case of RiPPs.
Figure 23.

Homodimeric structure of the AMS family protease/transporter from C. thermocellum determined in the absence (A) and presence (B) of an inert nucleotide analog. The transmembrane domain is colored in dark and light green and the nucleotide-binding domain is colored in dark and light orange. The protease domain shown in dark and light blue is not ordered in the nucleotide bound structure, and is presumably directed away from the core of the transporter. In the absence of nucleotide, the entrance to the transmembrane tunnel is situated at the interface with the protease domain and positions the active site residues right at the gateway. A close-up view of the active site residues of the protease domain is shown in the inset box. PDB ID 4S0F and 4RY2.
This model explains the general tolerance of transporters like NisT and ProcTp with respect to the cargo peptides (engineered in the case of NisT243 and 30 different natural peptides for ProcTp,129 see section 4.1). The model in which the core peptide first backs into the tunnel can also explain more substrate-restricted systems that require the post-translationally modified cognate peptide if the tunnel has shape and/or charge complementarity to the post-translationally modified peptide. Unlike the observations mentioned above for NukTp,354 the protease domain of the AMS protein from C. thermocellum was active in the absence of ATP.355 Instead, proteolysis was actually inhibited by ATP binding, because the cytoplasmic opening is closed in the ATP bound state. In this ATP-bound state the protease domains are displaced from the membrane-spanning and nucleotide binding domains and in this conformation the protease domains exhibit similar low level activity as observed with excised C39 protease domains. Indeed, in trans addition of the protease domain to the TMD-NBD fragment of the C. thermocellum protein increased the proteolytic activity, presumably by reconstituting a complete protein.355 Thus, like the NukTp study, the crystallographic and biochemical characterization of the AMS protein from C. thermocellum shows strong cooperativity between the nucleotide-binding domain and the protease domain. One possible explanation for the observed dependence of ATP for proteolysis in the NukTp study is that during the first turnover perhaps the cleaved nukacin ISK-1 may be trapped in the transmembrane tunnel and ATP hydrolysis is required to reset the system that recognizes the full length mNukA. Indeed, the system from C. thermocellum was only inhibited by ATP in the absence of Mg2+.355
With the discovery of lanthipeptide gene clusters in genomes of Gram-negative bacteria, another variation of the multifunctional transporter-protease has come to light. Several of the lanthipeptide gene clusters in Cyanobacteria, Bacteroidetes, and Proteobacteria contain in addition to a LanTp a series of proteins that appear to resemble type 1 protein secretion systems.356 These clusters encode a protein from the major facilitator superfamily (MFS) that would be localized in the inner membrane,357−359 a HlyD-type protein that would span the periplasm, and a TolC-dependent efflux pump that would be localized in the outer membrane. At present it has not yet been investigated how this secretion system would interact with the LanTp, but a model has been proposed.5
A few class II lanthipeptide biosynthetic clusters contain genes that encode for both an AMS protease-transporter and a stand-alone S8 subtilisin-like LanP protease. Examples include the clusters encoding the enterococcal cytolysin,360 cerecidin,133 carnolysin,361 and lichenicidin.91 In these clusters, the modified precursor peptide is first partially processed by the LanTp to remove the leader peptide at the double Gly motif, and subsequently processed by the LanP protease to trim additional remaining residues to elaborate the final product,360 as illustrated in Figure 22 for lichenicidin β.91 The crystal structure of LicP, the LanP involved in the final processing of lichenicidin β, shows an overall structure similar to other S8 type subtilisin-like Ser proteases (Figure 20C).362 LicP is autocatalytically processed to a prodomain and a catalytic domain as in the case of NisP, but the crystal structure of LicP contains both domains and thus provides insights into the protease activation mechanism. Maturation of subtilisin and related proteases proceeds by the displacement and subsequent stabilization of the N-terminus of the catalytic domain through interactions with a requisite calcium ion. In LicP, the equivalent calcium-binding site is replaced with a hydrophobic pocket, and stabilization is achieved by the displacement of Trp111 of the catalytic domain into this pocket.362
Processing of the mLicA2 precursor peptide by the LicTp C39 protease-transporter after the double Gly sequence of the leader generates an NDVNPE sequence appended to the core, and the LicP S8 protease then removes these six residues to produce the mature product (Figure 22).91 Reconstitution of LicP activity in vitro demonstrated that the protease is fairly tolerant to alterations with regard to the substrate peptide, and can excise the leader from full-length linear LicA2 precursor peptide, a partly proteolyzed precursor peptide (containing the DVNPE sequence), and a processed full-length precursor containing the (methyl)lanthionine rings.362 The activity toward unmodified precursor peptides is reminiscent of that observed for ElxP but is notably distinct from the substrate requirements of NisP, which only cleaves the leader sequences from fully modified precursors. However, LicP processed the lanthionine-bearing peptide much faster than the unmodified peptide.362 LicP could also process a number of synthetic peptide substrates of variable sequences incorporating the NDVNPE recognition sequence, and its tolerance toward peptide substrates with divergent amino acids in the P′ sites may be exploited for biotechnology.362
4. Class II Lanthipeptide Biosynthesis
4.1. Overview
In the mid 1990s, the gene clusters of lacticin 481 (then also called lactococcin DR) and the enterococcal cytolysin were sequenced and it was discovered that they did not encode LanB and LanC proteins.340,344 Instead, apparent fusion proteins were present that contained a C-terminal domain that had homology with the LanC proteins,344 but N-terminal domains that did not show any obvious sequence homology with known proteins in the database. These proteins were designated the general term LanM,40 and they form the basis for classifying lanthipeptides in class II. For many years, another distinguishing feature of biosynthetic gene clusters of class II lanthipeptides was the presence of a member of the AMS protein family.338 These LanTp proteins contain an N-terminal papain-like C39 protease domain discussed in section 3.2 that typically cleaves after a double Gly motif. But as more and more genomes have become available, genes encoding AMS proteins are also found in gene clusters of class I lanthipeptides and the manner in which the leader peptide is removed is becoming less uniform. Although some mixing of class I and II biosynthetic genes is starting to appear, for instance clusters that contain both lanM and lanC genes (see Figure 24),35 at present it is still possible to use the presence of a LanB protein as the defining feature of class I lanthipeptides and the presence of a LanM protein for inclusion with class II lanthipeptides. Like the LanB proteins, which show up in gene clusters of other RiPPs,195,202,366 LanM proteins also make appearances in other RiPP clusters, most notably the polytheonamides,367 and also in clusters that appear to encode a new class of RiPPs that contain D-amino acids (see section 4.6.1).
Figure 24.

Sequence similarity network of LanMs generated herein using EFI-EST and visualized in Cytoscape with an alignment score threshold of 110 (∼35% sequence identity). Each node represents protein sequences sharing 100% sequence identity. LanMs discussed throughout this review are indicated. The enzymes containing three Cys as Zn2+ ligands (“CCG” group) are mostly found within the dashed circle. Enzymes from clusters that contain both LanM and LanC enzymes (e.g., a cluster from Streptomyces globisporus C-1027) group together.
Phylogenetic analysis of LanM enzymes was completed for this review using a sequence similarity network (SSN) (Figure 24). Sequences for the network were accessed by a 5000 sequence return, two-iteration Position-Specific Iterated BLAST (PSI-BLAST) of the lanthipeptide synthetase CylM (accession number AAK67266.1). The SSN was built using the online Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST)207 using the 1811 sequences obtained from PSI-BLAST that had both an E-value of ≤e–18 and were ≥500 amino acids in length. The 100% identity representative node network, which contains 1687 nodes, was visualized in Cytoscape208 with an alignment score threshold of 110 (∼35% sequence identity).
As described in more detail in the following sections, LanM proteins are bifunctional enzymes that carry out both dehydration and cyclization of the substrate. The currently known LanM proteins can be divided into clusters and the main cluster is clearly subdivided into different groups (Figure 24).36 Although the differences between these groups are throughout the protein sequence, the most eye-catching differences can be found in the cyclization domain and will be discussed in section 4.3. One group comprises the majority of LanMs from Cyanobacteria368 and many of its members are encoded near multiple putative LanA substrates and thus predicted to have high substrate tolerance as was first demonstrated for ProcM, encoded in Prochlorococcus MIT9313.129 The genome of this organism codes for 30 substrate peptides (ProcAs) with strongly conserved, long leader peptides (∼70 amino acids), but relatively short and highly diverse core peptides (Figure 25). One precursor gene is truncated and may be a pseudogene.368 ProcM dehydrates and cyclizes all ProcA peptides tested thus far (18)129,368 and turns them into products with single ring topologies.369 Remarkably, these ring topologies are highly diverse (Figure 26),129,370 raising the intriguing question of how one enzyme can make so many different products. Section 4.3 will provide a potential answer to this question. The leader peptides of the ProcA substrates belong to a large family of peptides resembling Nif11 nitrogen-fixing proteins (N11P) that was first identified in a bioinformatic study.371 These Nif11 peptides are not only found as leader peptides on lanthipeptide precursor peptides but also for other classes of RiPPs.372 Another widespread family of RiPP precursors from Cyanobacteria,368 which includes putative lanthipeptides, have unusually long nitrile hydratase-like leader peptide (NHLP) sequences.367,371 The evolutionary and functional significance, if any, of these sequence homologies is currently not clear.
Figure 25.

ProcM has 30 putative substrates, 29 of which contain Cys residues in the core peptide. One substrate, ProcAT.1, has a truncated leader peptide. Fully conserved identical residues in the leader peptide are shown in dark orange and fully conserved similar residues are shown in green. Cys residues in the core peptide are shown in blue and Ser/Thr residues in purple.
Figure 26.

Six structurally characterized prochlorosins demonstrate the diverse ring topologies installed by ProcM.
4.2. Dehydration via Phosphorylation
Early gene disruption experiments in the clusters encoding lacticin 481 and the enterococcal cytolysin suggested the LanM proteins were essential for lanthipeptide maturation.340,344 The C-terminal domain that displays sequence homology with LanC proteins (section 2.3) was postulated to be responsible for the cyclization step. However, the manner in which the dehydration was catalyzed, presumably by the N-terminal domain, was not clear. In 2004, in vitro experiments demonstrated that the LctM protein was indeed capable of converting the precursor peptide LctA into a fully dehydrated and cyclized intermediate, which after leader peptide removal provided lacticin 481 (Figure 27),304 the founding member of a large group of class II lanthipeptides with similar structures.373 The process required ATP and Mg2+, and follow-up experiments with inefficient substrate analogs resulted in buildup of intermediates that were phosphorylated on the Ser and Thr residues destined for dehydration.374 Experiments with synthetic phosphorylated LctA peptides demonstrated that the enzyme can eliminate the phosphate group to convert phosphoSer (pSer) and phosphoThr (pThr) into Dha and Dhb, respectively. Thus, class II LanM enzymes utilize a very different mechanism for activation of the side chain hydroxyl groups of Ser/Thr from the glutamylation strategy discussed in section 2.2 for class I LanB dehydratases (Figure 28).
Figure 27.
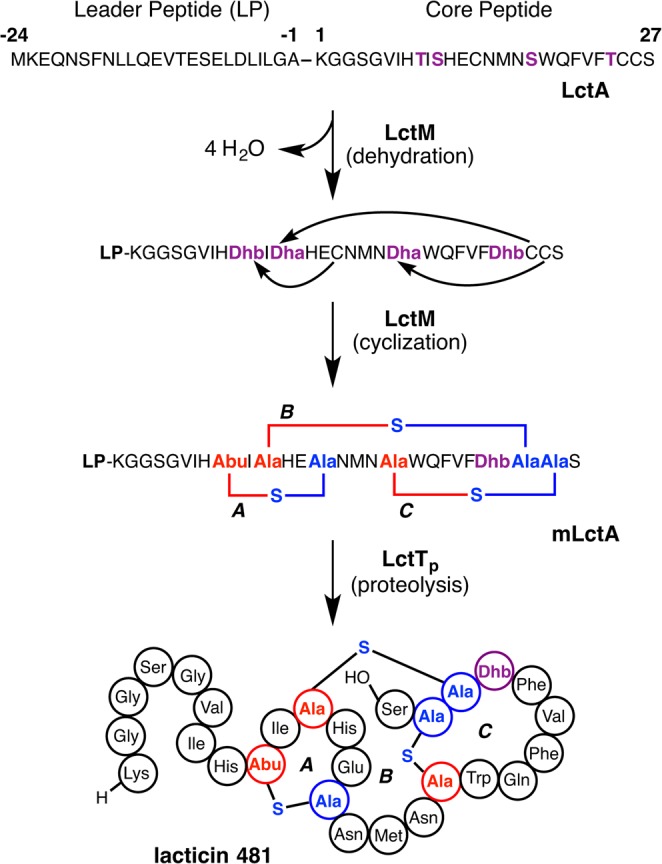
Biosynthesis of lacticin 481. For clarity, the process is shown as first completion of dehydration and then cyclization, but this is not actually the case since cyclization commences before dehydration is completed.375 See section 4.5.
Figure 28.
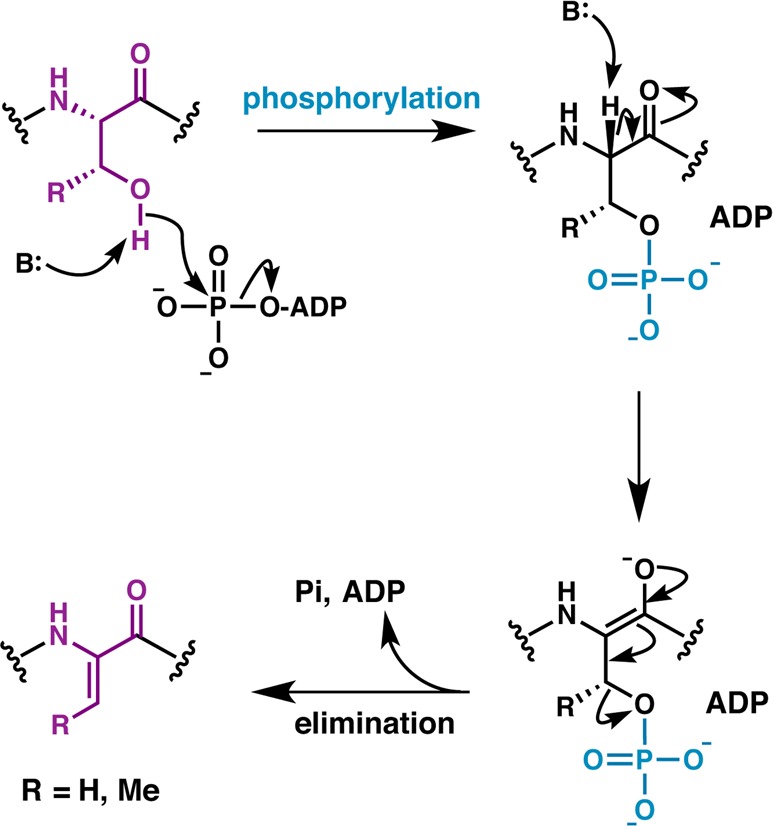
Dehydration during biosynthesis of class II lanthipeptides involves phosphorylation followed by phosphate elimination with anti stereoselectivity in all cases for which the stereochemistry has been investigated.
Interestingly, the elimination of the phosphate group from pSer and pThr required ADP, suggesting that the phosphorylation and elimination steps might take place in the same active site. In this model, elimination of the phosphate might require ADP to bind deeper in the active site pocket to set up the required environment for binding the phosphorylated peptide. Also noteworthy, phosphorylated peptides are typically not observed during in vitro reactions with class II enzymes, suggesting that phosphate elimination is faster than dissociation of the intermediate phosphopeptide.
Although these studies revealed the molecular mechanism of dehydration, how the enzyme catalyzes these chemical transformations remained mostly enigmatic. Mutagenesis experiments on LctM in which all conserved amino acids with side chains that could be involved in catalysis376 were mutated revealed several amino acids that appeared to be important for the phosphorylation step, including Asp242 and Asp259.377 Two other amino acid residues, Arg399 and Thr405, were shown to be important for elimination of the phosphate, and one residue, Lys159, appeared to be involved in both phosphorylation and phosphate elimination.377,378 A model was proposed that made analogies to the mechanism of Ser/Thr protein kinases, but at the sequence level no homology with such proteins was detected. The importance of these same residues was later also confirmed by mutagenesis of BovM involved in bovicin HJ50 biosynthesis379 and ProcM involved in the production of prochlorosins.380 More recently, the mutagenesis experiments were placed in a structural context with the first X-ray structure of a LanM protein, the CylM protein that generates both peptides of the enterococcal cytolysin (Figure 29). It was fitting for this protein to be the first structurally characterized LanM since it was also one of the first family members for which the sequence was determined in 1994.344
Figure 29.

Biosynthesis of both peptides that make up cytolysin. For clarity, the process is shown as first completion of dehydration and then cyclization, but the order of the PTMs is currently not known.
Cytolysin is a member of the two-component lantibiotics. This group of peptides displays maximum activity when two different post-translationally modified peptides are administered together.381 For most two-component lantibiotics, one peptide, called the α-peptide, binds to lipid II and the second β-peptide is believed to bind to this complex to form pores in the bacterial membrane.270,307,309,382,383 Thus far, two-component lantibiotics are confined to class II lanthipeptides, although some systems are present in actinobacterial genomes that could possibly be two-component lanthipeptides spanning two different classes.35 For most two-component lantibiotics, two different LanM enzymes carry out the dehydrations and cyclizations of two different LanA substrates that have divergent leader and core peptides, with little or no crossover (e.g., Figure 22 for lichenicidin).124,137,381,384,385 CylM is an exception in that it modifies both substrate peptides, CylLS and CylLL (Figure 29).38,386 Two other unusual aspects of the enterococcal cytolysin related to its bioactivity are that it functions as a virulence peptide that lyses mammalian cells including immune cells during infection,387 and that it has neither sequence nor structural homology with the two-component lanthipeptides that bind lipid II (e.g., compare Figures 22 and 29).
As anticipated based on sequence analysis, the structure of CylM contains two domains, an N-terminal dehydration domain and a C-terminal cyclization domain (Figure 30A).388 The two domains do not interact much with each other, explaining the observation by several laboratories that dehydration and cyclization activity of LanMs can be achieved independently by expressing the two domains individually.369,375,388−390 As expected, the cyclization domain has a structure similar to that of NisC described in section 2.3 and will be discussed further in the next section. Unexpectedly, however, the overall fold of the dehydration domain bears strong resemblance to that of eukaryotic lipid kinases and lipid-kinase like protein kinases.388 Although the overall fold has clear structural homology, a structure-based alignment showed that the secondary structural elements are connected in a unique manner. Nevertheless, homology detection and structure prediction by profile HMM-HMM (hidden Markov model) alignments391 performed for this review does identify the structural homology between LanM enzymes and lipid kinases. The CylM structure contains the typical N- and C-terminal lobes of Ser/Thr protein kinases, but also the characteristic kα10 and kα11 helices usually found in lipid kinases and lipid kinase-like protein kinases (Figure 30). Compared with canonical lipid kinases like phosphoinositide 3-kinase (PI3K), the CylM dehydration domain is significantly larger and contains several additional subdomains. One of those is the kinase-activation (KA) domain that plays at least two important roles, providing catalytic residues to the active site and ordering the activation loop.
Figure 30.

(A) Structure of the CylM class II lanthipeptide synthetase showing the dehydration domain (green) and cyclization domain (orange). (B) Close-up view of the dehydration active site showing critical active site residues and bound nucleotide. (C) and (D) Color-coded diagrams highlighting in (C) the N- and C-terminal kinase lobes (blue and red) as well as the kα10 and kα11 helices (yellow), and in (D) the LanM specific KA-domain (purple), activation loop (red), and capping helices (brown). PDB ID 5DZT.
The two residues mentioned above that were identified by site-directed mutagenesis in other LanM proteins as being critical for phosphate elimination, Arg506 and Thr512 in CylM numbering (Figure 30B), are located in the KA domain. Indeed mutagenesis of these two residues in CylM again resulted in mutant enzymes that still carried out phosphorylation but were strongly impaired in phosphate elimination. The side chains of these two residues insert into an active site that otherwise looks strikingly similar to typical kinases, with Asp252 and His254 on the P-loop that activate the phosphate to be transferred from ATP to the substrate, and Asp347 and His349 on the C-lobe that accept a proton from the Ser/Thr nucleophile of the substrate. The active site also contains Lys274, which in other kinases binds to the γ-phosphate of ATP to guide it to the phosphoryl acceptor. As noted above, mutagenesis of this residue in LctM slowed down both phosphorylation and phosphate elimination, suggesting the Lys still engages the phosphate group after phosphoryl transfer and facilitates elimination by stabilizing the leaving group. The active site is completed with a series of conserved residues including Asn352 and Asp364 that likely interact with the nucleotide, possibly mediated by a Mg2+ ion. Since the structure only contained AMP and not the peptide substrate, details of the active site organization in the transition states of the two chemical transformations are not available, but a possible model is shown in Figure 31. The CylM structure confirmed that phosphorylation and elimination take place in overlapping active sites, and explains why ADP needs to be present for elimination of the phosphate from exogenously supplied phosphorylated peptides.365,374
Figure 31.
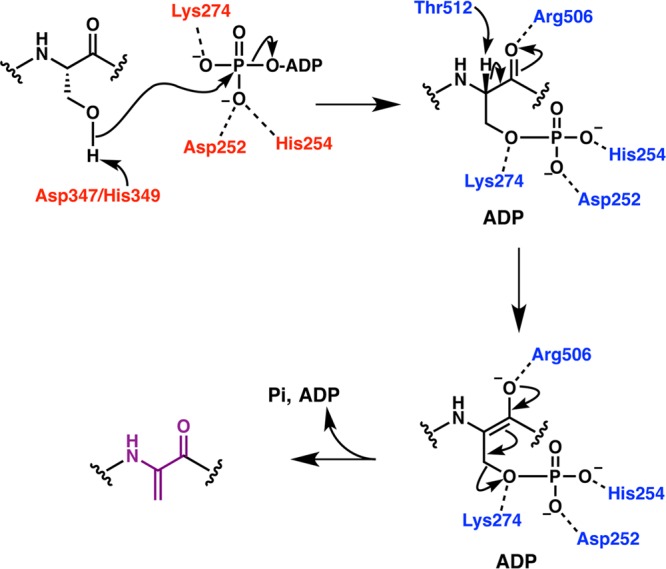
Proposed mechanism of phosphorylation and phosphate elimination by CylM based on mutagenesis studies. In the absence of a cocrystal structure, many details are still missing. For instance, the interactions of Asp252/His254 with the nucleotide phosphate could be mediated via a Mg2+. Not shown are Asn352, Asp364, and Glu366, which also appear to engage the nucleotide phosphates.
The second role of the KA domain is to orient the activation loop. In canonical lipid kinases this loop is disordered in the absence of substrate or other specific interacting proteins. Phosphorylation or autophosphorylation of residues in the activation loop often results in an ordering of the loop to adopt a productive conformation for substrate binding and catalysis. In CylM, the activation loop is well ordered via multiple interactions between residues in the KA domain and the loop (Figure 30D). The KA domain is, in turn, held in place by interactions with a set of helices termed the capping helices. Both the KA domain and these capping helices are currently unique to LanM proteins, and the overall topology that results in a well-ordered activation loop may explain why LanM enzymes essentially have constitutive kinase activity. At present there is no evidence for autophosphorylation as an activation mechanism.392 One question that remained unanswered with the structure of CylM is where the leader peptide binds. The enzyme does not contain an obvious winged helix-turn-helix motif that engages the leader peptide in class I lanthipeptides and many other RiPP biosynthetic enzymes (section 2.5), nor does it have the SH2-like domain identified in NisC that may interact with the substrate (section 2.3). CylM does contain a subdomain inserted in the cyclization domain that may be important for substrate binding (see section 4.3.1).
Several other residues have been reported to be important for LanM activity based on mutagenesis studies. These include Tyr230, Glu266, Glu446, and Gln495 in BovM (corresponding to Tyr330, Glu366, Glu555, and Gln611 in CylM).379 Inspection of the CylM structure only suggests a clear role for Glu366, which may stabilize ATP. The other residues are not close to the phosphorylation/elimination active site. Furthermore, mutation of Ile254, Leu290, and Leu597 in BovM resulted in an 5 to 7-fold decrease in affinity for the substrate BovA.379 The basis for the loss of affinity is currently not known, and mapping these residues on the CylM structure does not provide clear clues as to where substrate leader peptide binding occurs. The N-terminal dehydratase domain has been expressed individually and carries out dehydration of the CylL substrates, suggesting that leader peptide binding for the dehydration reaction would take place in the N-terminal domain.388
4.3. Cyclization
The cyclization events in lanthipeptide biosynthesis require remarkable control over regio- and stereoselectivity. Studies geared toward understanding the cyclization mechanism and selectivity started with class I LanC enzymes (section 2.3) but have recently become an intense focus in class II lanthipeptide research in light of several unanticipated developments. First, the class II lanthipeptide synthetase ProcM was shown to have 29 predicted substrates that appear to undergo selective cyclization to afford a single ring topology per substrate, with the individual topologies being highly diverse (Figure 25 and 26; ProcM has 30 substrates but one does not have Cys residues and hence cannot cyclize).129 Second, the two-component class II lanthipeptide cytolysin was reported to contain (Me)Lan with both DL- and LL stereochemistry (Figure 29), constituting the first characterized example of non-DL-stereochemistry in a lanthipeptide.38 A single enzyme, CylM, catalyzes the formation of both DL and LL (Me)Lan in the same peptide with high stereoselectivity.393 Subsequently, similar observations were made for the class II lanthipeptides carnolysin,361 haloduracin,38 geobacillin II,394 and the flavecins.137 These observations and the remarkable substrate tolerance of ProcM-like proteins imply an even greater plasticity of the cyclization catalysts than the already impressive control discussed in section 2.3.
While the various solutions for the dehydration step in lanthipeptide biosynthesis are interesting for their evolutionary origins, chemically, the dehydration reaction is not too challenging. However, control over a set of cyclizations to give one final lanthipeptide product of defined ring topology and stereochemistry catalyzed by a single enzyme with one active site is highly impressive, and that level of control currently cannot be achieved in synthetic organic chemistry. Consider the number of potential products that could arise from cyclization of dehydrated CylLL, the precursor to the large peptide of cytolysin (Figure 29). Dehydrated CylLL contains three cysteines and 7 dehydro amino acids. If all cysteines form rings, cyclization could lead to 210 possible constitutional isomers without considering stereochemistry. If DL- and LL-stereochemistries are both possible, and both configurations at the β-carbon of MeLan are considered, then over 6000 potential isomeric products can be formed. Out of over 6000 possibilities, CylM generates only one isomer. This remarkable level of control begs the question of how selectivity is governed. Do the enzymes or the substrates determine selectivity or is it a combination of both? Is selectivity under kinetic or thermodynamic control? How does ProcM take control over cyclization to another level by doing this not for one, but 29 different substrates? These questions have fueled much of the recent research on cyclization in class II lanthipeptide biosynthesis. The current understanding regarding regioselectivity and stereoselectivity of cyclization will be discussed in sections 4.3.2 and 4.3.3, respectively.
4.3.1. Structure and Phylogeny of LanM Cyclization Domains
As described in section 4.2, the dehydratase domain in LanM proteins differs entirely from that of class I LanB dehydratases, but the cyclase domain is analogous to class I LanCs. The cyclase domain in CylM shares with NisC the α,α-barrel fold that encapsulates a Zn2+ ion ligated by one His and two Cys residues; in the case of CylM these residues are Cys875, Cys911, and His912 (Figure 32).388 For NisC, the peptide substrate is expected to bind at an SH2-like substructure that resembles eukaryotic peptide-binding motifs (section 2.3), but this feature is absent in CylM. Instead two putative substrate-binding grooves, one in the dehydratase domain and one in the cyclase domain near the Zn2+ ion, were observed. Interestingly, an antiparallel β-sheet is present in the cyclase domain (Ile666-Leu690; purple in Figure 32) near the interface with the dehydratase domain that provides a potential binding surface that has similarities to the leader peptide binding region observed in NisB (section 2.5). However, incubation of CylLS with CylM(1–625) lacking this β-sheet did not significantly alter dehydration activity,388 suggesting that this putative binding site is not critical for dehydration, but it might yet be important for cyclization. Although it has not been confirmed that the precursor peptides CylLL and CylLS have a binding site on the cyclase domain, substrate affinity for the cyclase domain is supported by several other studies on LanMs. In particular, cyclase activity of isolated cyclization domains in the absence of a dehydration domain has been observed for ProcM369 and BovM,390 implying that these domains have substrate affinity. Although nonenzymatic cyclization in these experiments cannot be ruled out (see section 4.3.2), the correct regioselectivity of ring formation observed with the isolated ProcM cyclase domain suggests that the cyclization is enzymatic.369 A role of the leader peptide in cyclization is also suggested by investigations on the haloduracin β synthetase HalM2.238 In this study, the enzyme was shown to require the leader peptide to catalyze the reverse of cyclization, a retro-conjugate addition that opened up each of the four rings of haloduracin β attached to its leader peptide (i.e., mHalA2, Figure 33). Binding interactions of LanA substrates in both dehydration and cyclization domains were also implicated by early yeast two-hybrid studies that suggested binding interactions of the LctA substrate throughout the LctM protein.376 However, affinity of class II lanthipeptide precursors for their LanM cyclase domains does not appear to be universal. NukA, the precursor to the lanthipeptide nukacin ISK-1 (Figure 21), has much lower affinity for the NukM cyclase domain (NukMC) than for its dehydratase domain and no binding was detected between the leader peptide of NukA and NukMC.389
Figure 32.
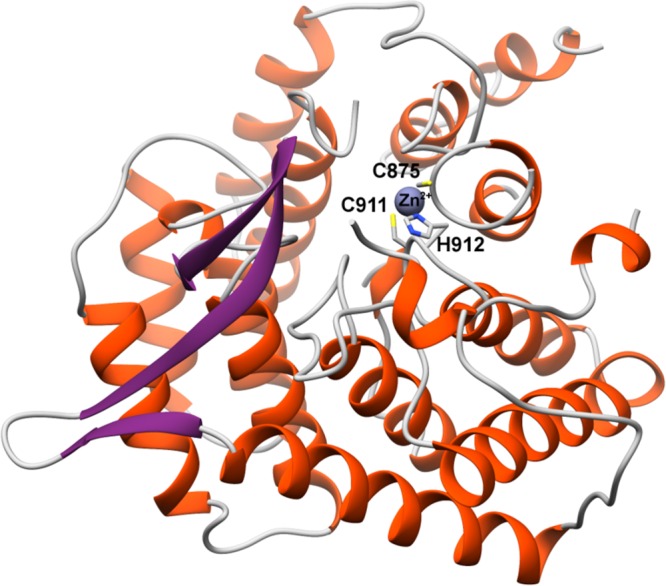
Structure of the CylM cyclization domain shown in the same orientation as NisC in Figure 12A. The antiparallel β-sheet that may constitute a substrate-binding platform is shown in purple.
Figure 33.
Biosynthesis of haloduracin β. BH1491 is an uncharacterized protease that has been suggested to remove residues −1 through −6 after cleavage by HalTp at the GS motif.91 Although for clarity the process is drawn as dehydration and then cyclization, the cyclization process commences before dehydration is completed.364,365 See section 4.4.
Although LanM and LanC proteins share certain structural features in the cyclization domain (Figures 12A and 32), a phylogenetic analysis of these enzyme classes showed that LanM cyclase domains and LanC enzymes fall into two different clades with deep roots, suggesting that the cyclase domain in LanMs was not obtained by horizontal gene transfer from LanC, unless it happened very early.36 The LanM sequence similarity network (Figure 24) shows one major cluster and many small clusters. Much of the clustering is based on phyla. For example, in the main cluster proteins from Firmicutes and Actinobacteria primarily group among themselves. More interesting is the observation that the phylogeny is reflective of the Zn2+-coordinating ligands. Many LanMs that contain three Cys ligands for zinc instead of two Cys ligands and one His ligand cluster together.36 The prediction of a Cys-Cys-Cys ligand environment is based on the presence of a “CCG” instead of a “CHG” motif370 (e.g., Cys911, His912, Gly913 for CylM), hence this subcluster is referred to as the “CCG” group (Figure 24). The significance of this difference in zinc ligands as it affects reactivity is discussed in section 4.3.2. Although the importance of the “CCG” motif was first investigated for ProcM and is found in many LanMs from Cyanobacteria, members of various phyla contain this “CCG” motif and cluster within this group, including Actinobacteria, Firmicutes, Proteobacteria, and Euryarchaeota. Euryarchaeota, albeit a minor source of LanMs, cluster almost entirely within the “CCG” group.
The chemical mechanism of cyclization catalyzed by LanMs is thought to mirror that of LanCs (section 2.3). Cyclization is predicted to involve activation of a cysteine thiol by coordination to the Zn2+ ion. The zinc dependence of lanthipeptide cyclases was first illustrated for NisC and SpaC217 and then for LanMs in 2007 for the case of LctM,395 the synthetase involved in lacticin 481 production (Figure 27). Two of the predicted zinc ligands of LctM, Cys781 and Cys836, were demonstrated to be important for metal binding as the zinc content was reduced in LctM-C781A and C836A mutants and even further with a double mutant. Notably, mutation of either Cys781 or Cys836 also resulted in reduced efficiency of cyclization in vitro. The efficiency of dehydration was not affected suggesting that these residues are only important for cyclization.
Additional molecular complexities of the cyclization event have recently come to light; in particular, cyclization to form (Me)Lan rings was shown to be reversible. This reversibility was demonstrated for the class II enzyme HalM2 with its fully modified substrate mHalA2 (Figure 33), and for the class I cyclase NisC with its fully modified substrate peptide mNisA (section 2.3).238 mHalA2 was generated by in vitro modification in D2O so that the intermediate enolate would pick up deuterium instead of a proton to afford α-deuterated (Me)Lan (Figure 34). Reexposure of α-D-labeled mHalA2 to HalM2 in H2O resulted in deuterium/protium exchange for all rings and incubation of mHalA2 and mNisA in the presence of the requisite synthetase and the thiol-selective electrophile N-ethylmaleimide (NEM) resulted in alkylation of all cysteines in each peptide (Figure 34). Because these studies show that the cyclization is reversible, the possibility of thermodynamic control cannot be ruled out. A more definitive test would be to expose synthetic products or intermediates with incorrect rings to these enzymes to monitor possible correction of the incorrect rings, but the difficulty with such a study is identifying which incorrect ring system to prepare. As noted in section 2.3, more than 6700 different ring topologies are possible for mNisA, and 840 for mHalA2 (Figure 33).
Figure 34.

Cyclization catalyzed by both HalM2 and NisC is reversible, as shown with α-D-labeled mHalA2 and mNisA. D/H exchange in α-D-labeled mHalA2 occurs in the presence of HalM2 but could occur either by a deprotonation/reprotonation mechanism or by a reversible cyclization mechanism. The reversibility of cyclization was confirmed for α-D-labeled mHalA2 and mNisA by trapping of the ring-opened species by conjugate addition to NEM.238
The active sites of LanC cyclases and LanM cyclase domains contain a conserved His residue (e.g., His212 for NisC; His725 for LctM and His791 for HalM2) that has been proposed to function either as a base that deprotonates the thiol to facilitate attack on the dehydro amino acid or as an acid that protonates the enolate to complete cyclization. It is unlikely that the His performs both functions given that conjugate attack and protonation occur on opposite faces of the alkene based on the stereochemistry of both DL- and LL-MeLan that demonstrates a net anti addition. The ability to study the retro-conjugate addition provided indirect insights into the role of the active site His in HalM2 (His791). Specifically, exposure of modified deuterium-labeled HalA2 to HalM2-H791A did not result in any deuterium exchange or ring opening, implicating His791 in the deprotonation step of the retro-conjugate addition, and hence protonation of the enolate during cyclization. If His791 were responsible for thiol protonation/deprotonation then deuterium exchange might have been observed. These findings are in agreement with examples of related Zn-dependent enzymes that activate thiols but do not contain an active site base for thiol deprotonation,225 and studies showing that the pKa of a thiol can be dramatically lowered on binding to Zn (e.g., 3 orders of magnitude).219,224
4.3.2. Regio- and Chemoselectivity of Cyclization of Class II Lanthipeptides
As described above, cyclization of lanthipeptides typically proceeds selectively to afford a single ring topology for each precursor peptide despite the myriad of isomeric possibilities. Studies aimed at distinguishing the roles of the enzyme and the substrate in this selectivity have relied on controlling the enzyme state (inactive vs active) so that enzymatic and nonenzymatic activity could be compared. Formation of the same products from enzymatic and nonenzymatic cyclizations would support a model in which the substrate controls selectivity. Two methods of controlling the enzyme state have been introduced: (1) separating the activity of the two domains of the LanM, which is a recent development that allows analysis of the dehydration and cyclization events separately,369,375,388−390,396 and (2) mutagenesis of the cyclase domain to generate a cyclization-deficient mutant.395
Early results suggesting that the enzyme plays a role in the chemoselectivity of cyclization came out of mutational analysis of the LctM cyclase domain. Lacticin 481 was not produced by mutant enzymes in which the Cys ligands to the zinc were replaced by Ala,395 but some cyclization nevertheless did occur, suggesting that the cyclization could be nonenzymatic. Exposure of LctA(−24–20, S11A/Q20A), which contains Cys14, Thr9, and Ser18 to LctM gave the correct A ring topology (Figure 35), but exposure to LctM-C781A resulted in preference for a different ring topology resulting from Cys14 attack on Dha18 instead of Dhb9.397 Thus, the substrate inherently prefers to form Lan rings (i.e., cysteine thiols react faster with Dha than with Dhb residues) and the enzyme is necessary to favor MeLan formation. A similar result was obtained using truncated LctM containing only the dehydratase domain (LctMΔcyc). Treating LctA with the leader and core peptides in trans with LctMΔcyc resulted in a non-native Dha11-Cys14 cross-link.375 An analogous nonenzymatic cyclization is predicted for the closely related nukacin synthetase truncant NukMΔcyc where a single ring formed,389 and similar observations have been made for class I lanthipeptides (section 2.3). These results suggest that nonenzymatically Dha reacts faster than Dhb to form an incorrect ring and the enzyme is required to overcome this intrinsic reactivity and facilitate proper ring formation.
Figure 35.
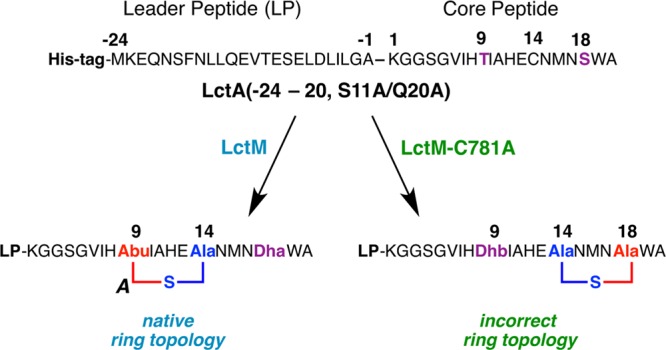
Modification of LctA(−24–20, S11A/Q20A) by LctM and by a cyclization-deficient mutant showed that LctM enforces the chemoselectivity of cyclization. The major products are shown but the native ring topology was observed as a minor product using the cyclization-deficient mutant.
The studies on LctM suggest that the enzyme is required for cyclization. However, LctM like most LanM enzymes, modifies only one substrate, LctA. As discussed above, ProcM has 30 predicted substrates. It is more difficult to imagine how one enzyme could selectively catalyze ring formation, which rekindled the notion that some of the cyclizations might be nonenzymatic. Intriguingly, ProcM is distinct from LanCs, LctM, and other characterized LanMs in that all three zinc-binding ligands are Cys.370 Based on model studies with low molecular weight metal complexes,226−237 the replacement of a His ligand with a Cys should increase thiolate reactivity compared to other LanC/LanM proteins. In turn, this increased reactivity might assist ProcM in processing so many diverse substrates.
The possible involvement of nonenzymatic cyclization during ProcM catalysis was recently studied in detail for two ProcA substrates with different ring topologies, ProcA2.8 and ProcA3.3.246 The prochlorosin products of both peptides contain two rings but one forms nonoverlapping rings (ProcA2.8) and the other overlapping rings (ProcA3.3) (Figure 26). A new strategy for controlling enzyme activity was developed by generating semisynthetic ProcA2.8 and 3.3 substrate analogs in which one Cys was free to react and the other Cys was blocked with an ortho-nitrophenyl protecting group that can be removed by photolysis. This strategy allowed investigation of nonenzymatic cyclization after preorganization of the peptide by enzymatic installation of another ring. Thus, two different monoprotected variants of both ProcA2.8 and ProcA3.3 were first subjected to enzymatic cyclization to form one ring. Then ProcM was removed, the protected thiol was uncaged, and nonenzymatic cyclization was monitored at pH 8.0 or pH 8.5 (Figure 36). In all four peptides investigated, nonenzymatic cyclization was much slower than enzymatic cyclization, suggesting that ring formation in prochlorosins 2.8 and 3.3 is enzymatic. One advantage of this approach is that the individual cyclization events can be monitored one at a time, which is not possible in the previously discussed approaches that rely on mutagenesis and separation of the dehydration and cyclization domains.
Figure 36.

Nonenzymatic and enzymatic cyclization of ProcA substrates containing a single ring installed by ProcM-catalyzed modification of a linear substrate with one Cys protected. In all cases, nonenzymatic cyclization was much slower (A and B). When B ring formation was blocked by Cys protection of ProcA3.3, an incorrect ring topology was favored as characterized for the enzymatic cyclization (B). LP = leader peptide.
Interestingly, the order of ProcM dehydration and cyclization are distinct. While both processes are ordered, dehydration occurs with C-to-N directionality (see section 4.4) but cyclization does not appear to be directional. Cyclization is ordered but the order depends on the substrate and it appears the smaller rings form faster.246 Notably, in the ProcA3.3 substrate described above where B ring formation was blocked, cyclization in the presence of ProcM afforded primarily an incorrect, smaller ring, ultimately resulting in a derivative with two nonoverlapping small rings (Figure 36B). The importance of the unique ligand set of ProcM was shown by the formation of the same incorrect ring topology upon exposure of the ProcA3.3 precursor peptide to ProcM-C971H, in which the Cys-Cys-Cys ligand set was mutated to the more common Cys-Cys-His coordination environment. The cyclization reactions catalyzed by the mutant were also significantly slower than with wild-type ProcM,369 suggesting that ProcM uses the competitive advantage of a zinc site with increased reactivity offered by a third Cys ligand to control the regioselectivity of cyclization.
At present, all studies on both class I and class II lanthipeptides suggest that the enzyme catalyzes all cyclizations and is responsible for facilitating the correct ring formation. However, several investigations have demonstrated that both the enzyme and the substrate are critical to the observed cyclization selectivity, and, for some systems, the ring topology may be encoded in the substrate sequence itself. Phylogenetic analysis of LanMs shows that enzymes that produce lanthipeptides with similar structure do not necessarily cluster together,36 suggesting that the ring topology may be defined by the substrate sequence, not by the LanM. In support of this hypothesis, several class II lanthipeptides have been prepared correctly using the synthetic machinery of completely different lanthipeptides. For example, ProcM modified the core peptide of LctA fused to the ProcA3.2 leader peptide to afford the correct ring topology of lacticin 481.36 This finding is remarkable since the LctA core peptide and the resulting ring topology are very different from any of the 30 ProcA substrates of ProcM. Furthermore, as discussed in section 4.4, the LctM-catalyzed post-translational modifications that lead to lacticin 481 are a highly coordinated set of reactions. Interestingly, the enzyme and the peptide substrate do not even need to be from the same lanthipeptide class to facilitate cyclization of a dehydrated peptide to a bioactive product. The nisin maturation enzymes were used to modify two putative class II lanthipeptide sequences from one gene cluster for which the natural products had never before been accessed.185 The core peptides of the putative class II LanAs were fused to the NisA leader and the hybrid peptides were expressed with the NisBC enzymes, which resulted in antibacterial peptides after leader peptide removal. It would be interesting to determine the structures of both the natural products and the products formed by NisBC to find out if they are the same. In addition to these studies that suggest the lanthipeptide substrates have an inherent preference for the observed regioselectivity of cyclization, investigations discussed in section 4.3.3 provide support for a role of the substrate in controlling the stereochemical outcome of cyclization. Thus, lanthipeptide cyclization may have commonalities with protein folding. The substrate peptide may already have a propensity to form a certain ring topology and stereochemistry because of its conformational energy landscape. Lanthipeptide cyclases could serve like folding chaperones to steer the peptide into the desired conformation or to let it adopt its preferred conformation and to then lock this conformation by catalyzing covalent cross-link formation.
The chemo- and regioselectivity of cyclization could be governed via either thermodynamic or kinetic control based on the experiments described above evidencing the reversibility of cyclization (section 4.3.1). In the thermodynamic case, intermediates would ultimately funnel predominantly to the product with the lowest free energy and this would not be determined by the enzyme. Under kinetic control, the transition state energies for formation of the correct rings would be lower than those for formation of incorrect rings. Kinetic control could be exerted entirely by the enzyme by only allowing certain transition states to be attainable through the active site geometry. Alternatively, kinetic control could be entirely governed by the conformational energy landscape of the peptide when bound to the protein, where the enzyme would only increase the reactivity of the Cys nucleophile and dehydro amino acid (Dhx) electrophile, but which Cys reacts with which Dhx would be determined by the substrate sequence. The latter situation is likely the case for ProcM. Finally, and probably the most likely scenario for most lanthipeptide biosynthetic enzymes (see section 4.4), it could be a combination of both, where the enzyme alters the conformational energy landscape of the substrate through hydrophobic or electrostatic interactions with the peptide, thereby achieving lower transition state energies leading to defined ring topologies.
Thermodynamic control was first suggested as an explanation for how ProcM could form so many ring topologies selectively.246,369 As in the experiments that determined the reversibility of cyclization for NisC (section 2.3) and HalM2 (section 4.3.1), fully modified ProcA2.8 and 3.3 substrates were produced by ProcM in D2O, resulting in deuterium incorporation in every ring. These mProcA peptides were then exposed to ProcM in H2O. In contrast to the observations with NisC and HalM2, D/H exchange generally did not occur or was inefficient, and free Cys could not be trapped with an electrophile such as NEM.246 These observations suggest that the first step of a retro-conjugate addition reaction, deprotonation at the α-carbon of Lan/MeLan, may still be possible, but that ring opening did not occur. One likely explanation for the different observations is that NisC and HalM2 have coevolved with their respective substrates to make a single product whereas ProcM makes 30 different products. Thus, NisC and HalM2 will have a certain amount of shape and charge complementarity with their products, and therefore have the necessary affinity for the final product and intermediates to catalyze retro-conjugate addition reactions. On the other hand, ProcM cannot be complementary in shape and/or electrostatics with its 30 products and their intermediates. Hence, the fully modified core peptides likely do not have much if any affinity for the cyclization active site. Another piece of evidence against thermodynamic control for ProcM was obtained by exposure of incorrectly cyclized ProcA3.3 or an incorrectly cyclized intermediate to the enzyme, which did not result in conversion to the correct ring topology.369 As noted above, the reversibility observed for HalM2 and NisC does not necessarily mean that their reactions are under thermodynamic control because it has not been demonstrated whether HalM2 and NisC could convert incorrectly cyclized products to the natural products.
4.3.3. Stereoselectivity in Class II Lanthipeptide Cyclization
As discussed in the previous section, the ring topology in class II lanthipeptide biosynthesis is at least in part determined by the substrate sequence but the enzyme is required to enforce selectivity. Interestingly, a similar explanation has been offered for stereoselectivity in which the substrate sequence is largely responsible for the stereochemical outcome. Until very recently it was thought that all rings in lanthipeptides had DL stereochemistry, which results from attack of cysteine onto the Si-face of the dehydro amino acid and subsequent enolate protonation to result in a net anti addition (note, only for Dhb are the two faces at the β-carbon stereogenic and hence only for Dhb is the Si-face defined). Alternatively, LL stereochemistry would arise from an anti addition via attack of Cys onto the Re-face of the alkene. In 2013, unusual results were reported for the two-component class II lanthipeptide cytolysin consisting of CylLL″ and CylLS″ (Figure 29).38 Cytolysin was demonstrated to contain both DL- and LL-(Me)Lan structures by peptide hydrolysis and subsequent GC-MS analysis. CylLS″ contains an LL-MeLan A-ring and a DL-Lan B-ring, and CylLL″ contains an LL-MeLan A-ring, an LL-Lan B-ring, and a DL-Lan C-ring (Figure 29). For each of the LL rings, the precursor sequence is Dhx-Dhx-Xxx-Xxx-Cys (Xxx = any amino acid except Cys) providing the first hint that the substrate might be responsible for the stereoselectivity. Although LL stereochemistry had not been seen until the report on cytolysin in 2013, a careful analysis of other lanthipeptides revealed the presence of the Dhx-Dhx-Xxx-Xxx-Cys sequence as likely intermediates for the biosynthesis of several other compounds, and indeed they all contain LL-(Me)Lan.137,361,394 Three other lanthipeptides, cerecidins A1 and A7, isolated from a Bacillus cereus gene cluster,133 and lichenicidin β isolated from various Bacillus licheniformis strains contain a putative Dhx-Dhx-Xxx-Xxx-Cys motif (Figure 22) and are therefore predicted to have LL stereochemistry. The dehydrated precursor peptide to prochlorosin 1.6 (Figure 25) also potentially contains this motif but the sites of dehydration, and its ring topology and stereochemistry, have not yet been determined. At present LL stereochemistry has only been reported for class II lanthipeptides.
Given that LL stereochemistry is only observed upon cyclization of the Dhx-Dhx-Xxx-Xxx-Cys motif, the substrate is clearly important for determining stereoselectivity. Furthermore, the enzymes that contain the motif in their substrates generate both DL and LL stereochemistry in their products (e.g., see Figures 29 and 33 for cytolysin and haloduracin). The reasons for the observed selectivity have been the focus of both experimental and computational investigations. First, the role of the enzyme was explored by replacing the leader peptide of CylLS with a different class II leader peptide and exposing this hybrid substrate to the enzyme associated with the leader peptide. This approach was used with leader peptides of HalA2, ProcA3.2, and LtnA2 and their respective cognate modification enzymes HalM2, ProcM, and LtnM2, respectively. ProcM and LtnM2 do not natively form rings with LL stereochemistry in their physiological substrates. Nevertheless, the major product with all three hybrid peptides that differed in their leader peptides but contained the CylLS core peptide contained an A ring with LL stereochemistry.393 In a follow up experiment, dehydrated CylLS was generated and cyclized nonenzymatically,396 again affording the LL-Lan A-ring as the major product. Remarkably, the nonenzymatic cyclization occurred rapidly at pH 7.5,396 whereas other nonenzymatic cyclizations for lanthionine formation require pH 8 or higher and are very slow.246 Thus, the Dhx-Dhx-Xxx-Xxx-Cys sequence appears to not only preferentially form LL stereochemistry but to also greatly facilitate the cyclization reaction.
To better understand the substrate requirements for LL stereoselectivity, both CylM-catalyzed and nonenzymatic cyclizations were attempted on substrates where the underlined Dhx in the Dhx-Dhx-Xxx-Xxx-Cys motif was mutated to Ala.396 For CylLS-T2A, which after dehydration resulted in a Dhb-Ala-Pro-Ala-Cys sequence, nonenzymatic cyclization was extremely sluggish but the CylM-catalyzed reaction still favored LL stereochemistry although with reduced diastereoselectivity. To facilitate nonenzymatic cyclization, the Dhb of the mutant was replaced with Dha (A-ring sequence: Dha-Ala-Pro-Ala-Cys);396 nonenzymatic cyclization of this substrate took place but with the canonical DL stereoselectivity. These results suggest that CylM is enforcing the LL stereoselectivity even in the absence of the Dhx-Dhx-Xxx-Xxx-Cys sequence and against the inherent preference of the substrate. Similar findings were also reported for the biosynthesis of geobacillin II.394 On the other hand, exposure of HalA2-T2A (A-ring sequence: Dhb-Ala-Trp-Pro-Cys) to HalM2 resulted in preferential DL-ring formation.393 Thus, whether or not the enzyme overrides the inherent selectivity of a mutant substrate is system-dependent.
The molecular origins of the stereoselectivity and reactivity induced by the Dhx-Dhx-Xxx-Xxx-Cys motif have been illuminated through computational analysis.393 The A rings for CylLS and HalA2 derive from Dhb-Dhb-Pro-Ala-Cys and Dhb-Dhb-Trp-Pro-Cys, respectively (Figure 29 and 33). For both, computational analysis showed that Re-face attack to give the LL stereochemistry is favored. For the CylLS A ring Re-face attack is favored over Si-face attack by 3.2 kcal mol–1, whereas for the HalA2 A-ring the transition state difference is 1.9 kcal mol–1. Interestingly, although both favor Re-face attack, the conformations of the lowest energy transition states differ greatly. For CylLS, the negative charge buildup on the reactive Dhb carbonyl is stabilized by a hydrogen bonding network from Ala and Cys backbone amide hydrogens. On the contrary, the HalA2 transition state is much less ordered. For HalA2, the T2A mutation shown experimentally to result in DL selectivity for the A ring even in the presence of its synthetase (which installs LL stereochemistry for the native sequence), was also modeled computationally. This mutation results in a minor conformational change, but it is significant enough that Si-face attack is now favored, consistent with the substrate determining the stereochemical outcome in this case.
Additional efforts have been undertaken to understand how and why LL stereochemistry is incorporated in select lanthipeptides. Notably, many of the LL-rings are located at the N-terminus of the core peptide such as for geobacillin II,394 haloduracin β,38 CylLS″,38 CrnA2′,361 and the flavecins.137 The effect of ring size on LL stereoselectivity was also explored. Precursors were designed for CylLL that increased or decreased the ring size (i.e., Dhx-Dhx-Xxx-Xxx-Xxx-Cys or Dhx-Dhx-Xxx-Cys), and for both the stereoselectivity switched to DL.396 All of these results suggest that the Dhx-Dhx-Xxx-Xxx-Cys motif is important for the LL-stereochemical preference and that perhaps the substrates evolved to form LL-rings at very precise positions.
At this point, it is mostly unknown how important the LL stereochemistry is for the bioactivities of these peptides but it seems likely that there is a reason why they evolved this stereochemistry, just as in other natural products where the stereochemistry is generally critical and altering it has drastic effects on the bioactivity. Before the realization that these LL-rings existed, stereochemistry had not been extensively explored in lanthipeptide biosynthesis. In a recent effort to understand the role of stereochemistry for lanthipeptides, LL stereochemistry was incorporated synthetically into lacticin 481, which naturally only contains DL-rings. Three mutants were prepared each with one of the rings changed to LL stereochemistry.398 Interestingly, all three diastereomers had retention times by HPLC distinct from that of wild-type lacticin 481 prepared in the same manner, suggesting that the LL diastereomers adopt quite different conformations. Growth inhibition assays showed that all three diastereomers did not display any antimicrobial activity. In these examples, native DL stereochemistry was changed to LL stereochemistry. Conversely, at present little is known whether the LL stereochemistry is important for the activity of the molecules where it is found naturally. The only information currently available is that changing the LL-stereochemistry in the small subunit of cytolysin (CylLS″) to DL using synthetic chemistry did not affect the hemolytic activity of cytolysin but did decrease its antimicrobial activity 10-fold.399
Although these studies predict that stereochemistry in lanthipeptides is generally important, an unusual example of a lack of stereochemical control was reported in the biosynthesis of the two-component lanthipeptide bicereucin. Bicereucin is the first example of a two-component lanthipeptide where one of the components, bicereucin α, does not contain (Me)Lan as there are no cysteines in the precursor peptide (Figure 37).400 The second component, bicereucin β, contains a single C-terminal Lan. Oddly, unlike what has been observed for other lanthipeptides, cyclization of this ring by BsjM is not stereoselective in vitro, but the cross-link in bicereucin β is important to the bioactivity. It is possible that the stereochemistry is not important in this case but the independent bioactivity of the two diastereomers has not been evaluated. The lack of selectivity may be explained by the fact that the bicereucin lanthipeptide synthetase BsjM lacks two of the Cys-Cys-His/Cys zinc ligands and active site His catalytic acid conserved in other LanCs and LanM cyclase domains. In other words, its cyclization active site appears disrupted. Recent genome mining has shown that several LanMs lack some or all of these conserved active site residues, and many of their substrates lack Cys suggesting coevolution of the LanA and LanM cyclization domain.35,368 These LanMs lacking conserved active site residues do not appear to cluster in the SSN (Figure 24). For example, both BsjM and NpnM, a LanM that also lacks the active site His acid and processes cysteine-free LanAs (see section 4.6.1), cluster separately from each other but with LanMs featuring fully conserved cyclization active sites. It is important to note that the class III LanKC enzymes also lack these active site residues and yet catalyze cyclization (see section 5). Interestingly, a bioinformatics genome mining study identified clusters that are very similar to that of bicereucin.35 These clusters also contain two substrate peptides with one lacking Cys, and a dehydrogenase that likely reduces Dhx to the corresponding D-amino acid (see section 4.6.1). But instead of only a LanM they contain both a LanM and a LanKC.35 It remains to be determined whether these are class III two-component lanthipeptides.
Figure 37.

Class II two-component natural product bicereucin comprises a lanthipeptide and a linear peptide. The Lan ring was produced as a mixture of stereoisomers in the in vitro reconstitution of bicereucin biosynthesis.400
4.4. Communication between Dehydration and Cyclization Events
Class II lanthipeptide synthetases catalyze a remarkable number of reactions on a substrate that changes shape with each consecutive transformation. As noted, these enzymes typically carry out their iterative reactions with defined order.238,246,364,375 Some operate with clear directionality (N-to-C terminus or vice versa) whereas others follow a process that is not directional but is still ordered; for some enzymes like ProcM, the directionality/order is dependent on what substrate the enzyme acts on.246 Although many questions still remain, in recent years the mechanisms by which the enzymes control the order in which they catalyze these multiple chemical processes have slowly been coming into focus. One very useful tool developed in recent years is the use of synthetic isotopically labeled substrates in which a single Ser or Thr residue is labeled at the α-carbon with deuterium.401 This label allows determination of the timing of dehydration of the labeled residue since it involves loss of 19 Da whereas the dehydration of all other Ser/Thr in the peptide involve loss of 18 Da (Figure 38).401 By using a series of synthetic peptides in which the labeled Ser/Thr is at different positions, the order of dehydration can be established even in cases where cyclization prevents use of tandem MS approaches to follow the timing of each dehydration. This technique has been used to study both class II and class III dehydration, including the assignment of C-to-N directionality for ProcM dehydration.246,247,375,401 Another important development in advancing our understanding has been the establishment of quantitative methods to measure the kinetics of individual dehydration and cyclization reactions by mass spectrometry and the use of computer programs to evaluate various kinetic models. This approach was first used for HalM2, the enzyme that makes the β-peptide of the two-component lantibiotic haloduracin (Figure 33) and ProcM, the single enzyme that makes 30 different prochlorosins (section 4.1).365 With HalM2, a series of interesting observations was made. The enzyme had already been shown to function with N-to-C terminal directionality.238,364 The kinetic analysis demonstrated that the enzyme alternates between dehydrating and cyclizing its HalA2 substrate peptide.365 After a certain number of dehydrations has occurred, the first cyclization takes place. Subsequent dehydrations do not happen until after the first cyclization. One possible explanation for the latter observation is that each formation of a ring brings the more C-terminally located Ser/Thr closer to the leader peptide, and possibly to the dehydration active site. Another important observation was that after a relatively slow initiation process, subsequent cyclization reactions are progressively faster as if each ring formation either improves the productive interaction with the enzyme or facilitates the subsequent ring formation by adopting a conformation that favors cyclization. In the former explanation, the enzyme and final product could potentially be complementary in shape and/or electrostatics.365 The complete reversibility of the cyclization process with HalM2 (section 4.3.1) is an indirect demonstration that the intermediates and final cyclized product can bind reasonably well to the cyclization active site.238
Figure 38.
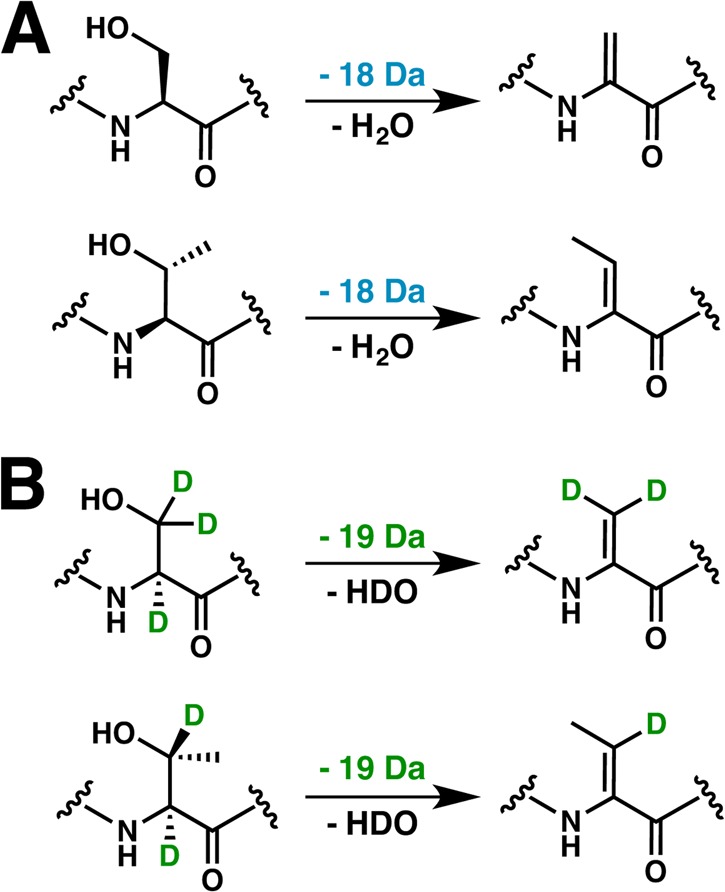
Residue-specific incorporation of deuterium at the α-carbon of Ser/Thr enables monitoring of the directionality of dehydration. Dehydration of unlabeled Ser/Thr involves a loss of 18 Da (A), whereas dehydration of α-deuterated Ser/Thr involves a loss of 19 Da (B).
A final interesting observation is that in general the rate constants of the various dehydration and cyclization steps performed by HalM2 are quite similar, such that many intermediates are observed (no step is strongly rate limiting) and several parallel pathways are operational. Parallel pathways have also been observed for NisB dehydration245 and catalysis by class III synthetases.247,401 This observation is not too surprising since a very tightly ordered process in which only a specific mandatory order of events leads to the final product seems difficult to achieve with a single iterative enzyme. In addition, such a scenario would likely render these enzymes much less substrate tolerant than they are. On the other hand, given that dehydration and cyclization steps are very different chemical reactions and that the substrate is constantly changing, the similarity in rate constants is surprising. It suggests that perhaps the overall rate of each reaction may not be governed by chemistry but perhaps by a physical step. One possibility would be that the overall rates of reaction are determined by conformational sampling, in which after leader peptide binding, the core peptide samples the dehydration and cyclization active sites.365 For some intermediates, a dehydration might be the energetically best next reaction, and for other intermediates a cyclization step may be favored, either because of preferred binding of the intermediate in one of the active sites (Km-like effect), or because of faster chemistry (kcat-like effect). Although the details remain to be elucidated and likely will require molecular dynamic simulations, collectively, these findings paint a picture of coevolution of the substrate sequence with the LanM enzyme to arrive at the present-day well-orchestrated post-translational modification process that results in a powerful antibiotic with nanomolar minimal inhibitory concentrations against select bacteria.
Kinetic analysis of ProcM provided a completely different picture.365 As noted in section 4.1, this enzyme has 30 different substrates with highly diverse core peptides that are converted into a library of cyclic peptides with very different ring topologies (Figures 25 and 26). Thus, it is difficult to imagine an active site that could be shape and/or charge complementary with all 30 of its products. Indeed, the kinetics of processing of the ProcA substrates investigated thus far suggests that this is not the case. Unlike HalM2, which alternates dehydration and cyclization steps, ProcM first dehydrates all Ser and Thr in the core peptides of those ProcA substrates that have been analyzed.246,365,369 The kinetics of dehydration are not very different from those observed for dehydration of HalA2 by HalM2, but the cyclizations by ProcM are significantly slower, and contrary to HalM2, ProcM slows down with each successive cyclization reaction. This observation suggests that, once a ring has been installed, the intermediate becomes a less good substrate for subsequent cyclizations, presumably because, unlike HalM2, ProcM has not evolved for optimal catalysis to form just one product. Thus, increased substrate tolerance comes at the expense of slower kinetics. Why the cyanobacteria that utilize this group of ProcM-like enzymes put a larger premium on a diversity of products over optimizing one product is at present not clear. The limited resources available to many of these organisms living in the nutrient-poor open ocean and the associated small genomes402,403 may offer an explanation. The advantages provided by a system that very efficiently uses minimal genetic information for the production of a library of compounds would likely be of more value under these circumstances. A different question is what the functions are of these lanthipeptides and how the diversity is maintained, preventing the system to evolve to make one particular compound. The former question is still entirely unclear, whereas one possible answer to the latter question is that these molecules act together, in which case there would be evolutionary pressure on keeping ProcM substrate tolerant.404 While on the surface the sequences of the 30 core peptides and their associated products appear very diverse or even random, mutagenesis experiments suggest that the ring topologies made from these core peptides have some inherently favorable attributes that make them particularly tolerant to further sequence diversification within the constraints of an existing ring topology.405 Similarly, these experiments show that ProcM cannot turn just any sequence into a well-defined cyclic product. It appears that the sequences, diverse as they are, evolved because they are prone to make a single product of defined topology. These results fit well with the proposal described in section 4.3 that the outcome of the cyclization process is determined by the substrate peptide.
A third system for which the kinetics of dehydration and cyclization has been investigated is the lacticin 481 synthetase LctM.375 This study focused on the role of the leader peptide in the overall kinetics and order of the various steps that convert LctA to mLctA. The results of this study illustrate that the leader peptide is critical for ensuring the efficient and correct order of events and will be discussed in the next section.
4.5. Leader Peptide Dependence
Similar to class I lanthipeptides, the leader peptide in the biosynthesis of class II lanthipeptides is required for the efficient installation of the (Me)Lan cross-links144,150,151 but is often not required for tailoring modifications as discussed in section 4.6. Studies showing that non-natural substrates can be processed by LanMs if they are fused to the cognate leader peptide confirmed an important role for the leader peptide in facilitating PTMs. For example, as described in section 4.3.2, the class II synthetase ProcM can modify the LctA core fused to the ProcA3.2 leader peptide to afford lacticin 481.36 Extensive studies on the lacticin 481 synthetase LctM both in vitro and in vivo have shown that this enzyme can modify a variety of peptides fused to the LctA leader.81,269,304,397,406−408 Some examples include a truncated LctA core lacking 7397 or 14304 C-terminal residues, core peptides of the class II lanthipeptides nukacin ISK-1 and mutacin II,406,407 and completely unrelated peptides.81 One limitation to LctM processing also evidenced for class I and III lanthionine synthases is that the dehydratable residues must be a certain distance from the leader peptide, presumably in order for those residues to access the active site.269 Although LanM proteins clearly recognize the leader peptide, it is important to note that LanM recognition of the core peptide is also generally predicted. For example, although an LctA leader-NukA core fusion can be processed by LctM to afford nukacin ISK-1 both in vivo407 and in vitro,406 in vivo experiments have shown that NukM, the LanM involved in nukacin ISK-1 production (Figure 21), does not convert a NukA leader-LctA core fusion to lacticin 481,407 suggesting that NukM recognizes the core peptide of NukA.
Unlike in class I biosynthesis where a cocrystal structure of NisA and NisB witnessed interactions between the leader peptide and enzyme as described in section 2.5, the precise molecular interactions involved in leader peptide binding have not been elucidated for any class II lanthipeptide synthetases. No crystal structures of substrate-bound LanM synthetases have been solved but the first crystal structure of a LanM (CylM) was recently reported.388 Interestingly, the leader peptide binding domain found in class I dehydratases is not conserved in CylM, suggesting an alternative mechanism of leader peptide binding to the enzyme. This hypothesis of an alternative mechanism is further substantiated by differences in the leader peptide sequences. Class II lanthipeptide leaders lack the conserved FNLD motif found in class I substrates and are instead characterized by the incorporation of many negatively charged residues as well as a conserved C-terminal ELXXBX motif (e.g., positions −3 to −8 for LctA and NukA), where B is a hydrophobic residue (Val, Leu, or Ile). Truncants of LctA,304 NukA,407 and ProcA2.8 precursor peptides368 containing this motif but shortened at the N-terminus (by 8, 9, and 40 residues, respectively) were fully processed by their respective LanM enzymes, illustrating that unlike class I, the leader peptide recognition site is located in the C-terminal half of the leader peptide.
The importance of the conserved leader peptide residues for the efficiency of PTMs has been explored in vitro for lacticin 481406 and bovicin HJ50,379 and in vivo for nukacin ISK-1407 and mutacin II.345 Most point mutations did not dramatically reduce production of the natural products and the effects of mutations were similar among the three systems. Notably, incorporation of a charged residue at positions −4 or −7 reduced processing efficiency.345,379,406,407 Incorporation of proline within the ELXXBX motif as evaluated for lacticin 481 and nukacin ISK-1 resulted in reduced LctM efficiency406 or little or no lanthipeptide production,407 respectively. The detrimental effect of Pro incorporation suggests that the leader peptide may adopt a helical conformation. Indeed, structural prediction tools have predicted helical regions encompassing the ELXXBX motif for the LctA406 and NukA leader peptides407 and the NukA peptide adopts a partial helical structure in trifluoroethanol as determined by CD.407 However, more definitive evidence is required as similar predictions were made for the NisA leader peptide,257 but the cocrystal structure with NisB subsequently revealed that the leader peptide engaged the enzyme in an extended conformation. Interestingly, the in vitro mutagenesis studies also hint that the binding interactions with LanM proteins during dehydration and cyclization may differ.406 Although the point mutations E–8P, D–6P, and I–4P in LctA all result in much reduced dehydration efficiency, the effects of these mutations on cyclization are distinct. Cyclization of fully dehydrated mLctA goes to completion for the D–6P and I–4P mutants but not for the E–8P mutant. The requirement of the leader peptide for retro-conjugate addition238 and the independent activities of separately expressed dehydration and cyclization domains369,396 also suggest potentially separate interactions of these domains with the leader peptide.
In addition to how the leader peptide is recognized by the enzyme, the mechanism of how this interaction facilitates catalysis has been under considerable investigation. One possible mechanism proposed early on is that binding of the leader peptide to the enzyme induces a conformational change to expose the enzyme active site and promote post-translational modification. However, in 2007 it was reported that LctM does not absolutely require the leader peptide for dehydration although the efficiency was dramatically reduced409 and the same observations have since been reported for HalM2.385 In trans supplementation of the LctA core peptide with the LctA leader peptide partially restored LctM processing,409 indicating that the leader and core do not need to be fused as has also been described for class I lanthipeptides in section 2.5 and some class III enzymes in section 5.3. As the leader peptide is not required for catalysis, a mechanism in which the leader peptide induces a conformational change seems unlikely. Instead, this data suggests a conformational selection model150 in which the enzyme exists in the absence of the precursor peptide in an equilibrium of inactive and active forms, favoring the inactive form (Figure 39). Binding of the leader peptide stabilizes the active form, shifting the equilibrium to increase the concentration of active enzyme. The core peptide brought in proximity by the leader is then able to bind at the enzyme active site. Support for this synergistic leader and core peptide binding model has been reported for some enzymes as discussed below. Further support for the conformational selection model came from the development of a constitutively active fusion (ConFusion) enzyme where LctM is connected through its N-terminus by a short linker to the LctA leader peptide (LctCE-GS15).408 The model predicts that since LctCE-GS15 is constantly in proximity to the leader peptide, the enzyme population should favor the active form resulting in efficient processing. In the event, treatment of the LctA core with LctCE-GS15 gave 4-fold dehydrated LctA as the major species after 3 h whereas the otherwise identical in trans experiment with a 1:1 ratio of LctA leader and LctM mimicking the ConFusion stoichiometry gave 3-fold dehydrated LctA as the major species.408
Figure 39.
Conformational selection model whereby leader peptide binding shifts an equilibrium to an active enzyme state that promotes core peptide binding to the enzyme and facilitates catalysis. The enzyme, leader peptide, and core peptide are shown in blue, pink, and green, respectively. The leader peptide in class II LanAs is predicted to form a helix but this conformation has not been directly proven in the context of interactions with the modifying enzymes and is not generalizable to class I lanthipeptide biosynthesis, as the precursor NisA does not adopt a helical conformation in binding to its modifying enzyme NisB (section 2.5).
Although affinity of the leader peptide for the enzyme is clearly important for catalysis, the core peptide can also independently bind to the enzyme as is evident from the in trans409 and ConFusion408 experiments. To investigate the relative roles of the leader and core peptide in LanM binding, modification of haloduracin precursor peptide chimeras (HalA1-A2, containing the A1 leader and A2 core, and HalA2-A1) was explored.385 Exposure of HalA1-A2 to HalM1 and HalA2-A1 to HalM2 resulted in 5 and 2 dehydrations as the major species, respectively, compared to 7 for wild-type HalA2 and 3 for wild-type HalA1. Modification of these chimeras containing matched leader peptides and synthetases supports the importance of leader peptide binding. Alternatively, exposure of HalA1-A2 to HalM2 and HalA2-A1 to HalM1 resulted in 6 and 1 dehydrations, respectively. These results suggested that the mismatched leader peptides and enzymes might have affinity for each other or, alternatively, that the core peptides might also have affinity for their cognate enzymes. Fluorescence polarization assays have confirmed both hypotheses.385 The HalA1 leader proved a competent partner for HalM2, binding with a Kd of 4.1 μM, comparable to that of the HalA2 leader and HalM2 (Kd = 3.7 μM). The HalA2 core peptide has low affinity for HalM2 with a Kd > 500 μM but interestingly, a HalM2 ConFusion enzyme has much higher affinity for the core peptide (Kd = 85 μM). This result demonstrates for the first time synergistic binding of the leader and core peptides and supports a mechanistic model in which binding of the leader peptide not only favors an active enzyme conformation but facilitates core peptide binding to enable catalysis. Notably, the dissociation constants reflect a much lower binding affinity for HalM2 and its substrate than has been demonstrated for two other characterized class II LanMs. As determined by surface plasmon resonance studies, the nukacin ISK-1 synthetase NukM and its substrate NukA bind with a Kd of 60 nM353,389 and the bovicin HJ50 synthetase BovM and its substrate BovA bind with a Kd of 16.4 nM.379 The reasons for these large differences in affinity are not yet clear.
The chimeric haloduracin precursor peptides described above were never fully processed suggesting that the native leader peptide may be required for proper positioning of the core peptide.385 Indeed, kinetic investigation into the processing of LctA by LctM has revealed a role for the LctA leader peptide in spatial positioning.375 To evaluate the kinetic importance of the covalent linkage of leader and core peptides, several systems were explored using LctA mutants incorporating deuterium at dehydratable Ser and Thr residues to monitor the directionality of dehydration (Figure 38). Wild-type LctM was shown to process deuterated LctA-T24S with N-to-C directionality; Thr9 and Ser11 were dehydrated first followed by Ser18 and Ser24. Notably, exposure of deuterated LctA to LctM lacking the cyclase domain (LctMΔcyc) resulted in much slower processing but the same directionality. Surprisingly, very different results were obtained when the same kinetics experiments were conducted with in trans provided LctA leader and core peptides for LctM and LctMΔcyc, or for the LctM ConFusion enzyme with the core peptide. The rate of both dehydration and cyclization was reduced in all three cases. Intriguingly, the directionality of dehydration was also changed; Ser11/Thr24 was dehydrated first, then Thr9/Ser11/Thr24, then Thr9/Thr24, and finally Ser18. The regioselectivity of cyclization was also disrupted as analyzed by tandem MS. Specifically, a non-native Dha11-Cys14 ring formed, likely due to a nonenzymatic cyclization event that becomes competitive in these less efficient systems. Thus, the leader peptide not only increases the efficiency of catalysis through allostery, but is also important for the correct order of dehydration presumably by proper positioning of the core peptide. In turn the correct order prevents incorrect nonenzymatic cyclization.375
4.6. Tailoring Enzymes
Like the class I lanthipeptides described in section 2.6, many class II lanthipeptides contain PTMs in addition to the characteristic thioether cross-links and dehydro amino acids. These additional modifications contribute to the broad structural diversity of lanthipeptides and also often appear to increase their bioactivities. The presumably nonenzymatic hydrolysis of N-terminal Dha or Dhb that occurs upon leader peptide proteolysis to afford N-terminal 2-oxopropionyl and 2-oxobutyryl groups, respectively, is seen in both class I and II lanthipeptides (e.g., Pep5 and pinensin from class I;276 lichenicidin, lacticin 3147, and lactocin S from class II; Figures 5, 23, and 40).410 In addition, disulfide formation is observed in class II and class III, with examples in class II including haloduracin α and bovicin HJ50 (Figure 21). As these gene clusters either lack a dedicated thiol–disulfide oxidoreductase124 or it is not required for bioactivity as shown for bovicin HJ50,411 this tailoring modification will not be discussed further. Other than these two modifications, tailoring PTMs tend not to be shared between classes of lanthipeptides based on the compounds characterized thus far. The general lack of PTM crossover between classes could be explained by different evolutionary origins or it may be that as more lanthipeptide gene clusters are sequenced PTMs will emerge in class II that were already seen in other classes and vice versa. It is only within the past few years that most of the enzymes responsible for additional PTMs in class II have been studied. Generally, these enzymes can be divided into two groups with respect to their substrate selectivity, although for some the selectivity is not fully characterized. One group is very permissive and can process both linear and cyclic peptides. These enzymes have much potential for bioengineering. The second group is highly selective and only acts on cyclized peptides.
Figure 40.
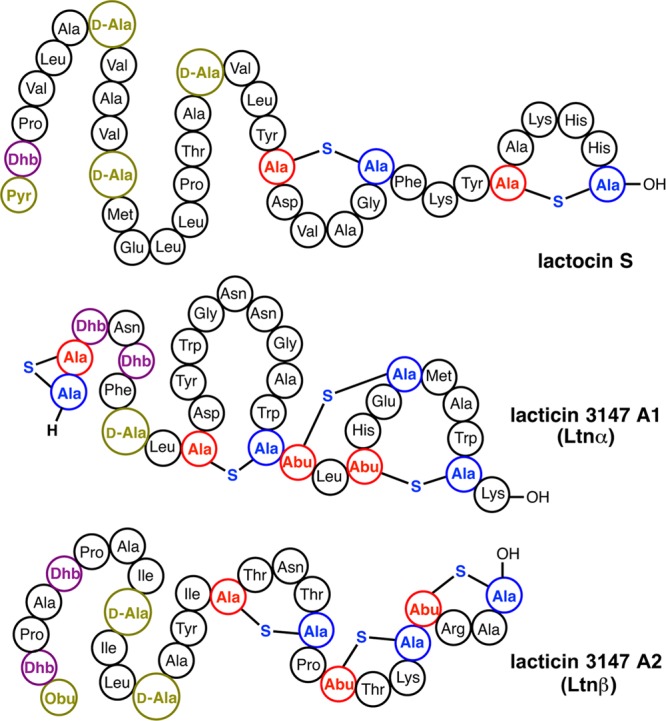
Examples of class II lanthipeptides containing d-Ala residues and a 2-oxobutyryl (Obu) or 2-oxopropionyl (pyruvyl, Pyr) tailoring modification at the N-terminus.
4.6.1. Incorporation of D-Amino Acids
D-Amino acids have been found in ribosomally synthesized natural products of both eukaryotic and prokaryotic origin.367,412−416 This unusual stereochemistry is thought to contribute to proteolytic stability,87,417−419 structural conformation,420 and bioactivity.413,414,421 As only L-amino acids are genetically encoded, access to the D stereochemistry requires post-translational modification and several unique mechanisms for this transformation have been uncovered. For nonlanthipeptide natural products, D-amino acids are incorporated by epimerization of the corresponding L-amino acid, either by a deprotonation-protonation mechanism422,423 or by a radical mechanism.424 When the first lanthipeptide containing D-amino acids was identified in 1994,274 it was immediately clear that a different mechanism must be operative. Certain serines encoded in the precursor gene for lactocin S were not found in the natural product; instead, alanine residues were found in their place. Incorporation of nongenetically encoded Ala was confirmed by amino acid composition analysis, and the D-stereochemistry of these Ala residues was shown by GC-MS analysis with a chiral stationary phase (Figure 40). Based on the mechanism of (Me)Lan formation in lanthipeptides, the conversion of l-serine to d-alanine was suggested to occur by initial LanM-catalyzed dehydration of l-Ser to afford Dha.274 Instead of lanthionine formation, diastereoselective hydrogenation of Dha by a hypothetical enzyme (later generically named LanJ361,421,425) could then give d-Ala.
A few years later the same observation was made during the partial structural characterization of the two-component lanthipeptide lacticin 3147, composed of Ltnα and Ltnβ and produced by L. lactis (Figure 40).426 Chiral GC-MS analysis of the hydrolyzed peptides showed that Ltnα contains one d-Ala at residue 7426 and Ltnβ contains two d-Ala (later shown to be residues 9 and 12).410 Subsequent studies identified the biological role of the d-Ala residues and the substrate specificity of dehydrogenases that convert Dha to d-Ala.421
The hypothetical dehydrogenase for converting Dha to d-Ala was first characterized in the lacticin 3147 gene cluster,95,421 confirming that indeed d-Ala is incorporated by dehydration of Ser followed by diastereoselective hydrogenation. Specifically, it was shown that d-Ala was not formed in the absence of the enzyme LtnJ.421 Ltnα and Ltnβ produced in vivo in a ΔltnJ strain contained Dha residues in place of d-Ala residues. In addition, the Ltnα mutant from the ΔltnJ strain featured 4-fold lower activity than wild-type Ltnα when applied in an equimolar ratio with wild-type Ltnβ to susceptible L. lactis; the bioactivity of the Ltnβ d-Ala to Dha mutant could not be evaluated due to poor expression yields. Together with experiments showing reduced activity for mutants where d-Ala was replaced with l-Ala,421 these studies showed that the incorporation of alanine as the D stereoisomer is important to the bioactivity of lacticin 3147. Interestingly, site-directed mutagenesis to incorporate Thr at d-Ala precursor sites in the LtnA1 and LtnA2 precursor peptides resulted in products that had undergone dehydration of Thr to afford Dhb but not hydrogenation to afford D-Abu;421 these results suggested that LtnJ can reduce Dha but not Dhb.
LtnJ belongs to the family of zinc-dependent alcohol dehydrogenases. Genome mining for orthologs of LtnJ uncovered three other putative dehydrogenases;421 SacJ, which is located in the Staphylococcus aureus C55 operon for the biosynthesis of staphylococcin C55,427 a lanthipeptide predicted to be very similar in structure to lacticin 3147; PenN from Pediococcus pentosaceus FBB61, which is required to access bioactive pediocin A;428 and NpnJ (originally named NstJ),421 which is from a cyanobacterium Nostoc punctiforme PCC73102 cluster that contains a LanM but does not produce lanthipeptides as the precursor peptides (NpnA) do not contain cysteines.368 LtnJ, NpnJ, PenN, and SacJ share many conserved residues including three or four conserved Cys residues predicted to bind zinc (NpnJ has three of the conserved Cys, LtnJ, SacJ, and PenN have all four).429 These four enzymes have been classified as LanJA-type dehydrogenases based on their zinc and NADPH-dependence.425 The dehydrogenase activity for PenN and SacJ was demonstrated in vivo with the lacticin 3147 substrate, confirming their function and suggesting that these enzymes do not require a leader peptide as a non-native substrate was accepted.429 Specifically, supplementing the ΔltnJ strain described above with penN restored d-Ala formation in both Ltnα and Ltnβ, whereas supplementation with sacJ restored d-Ala formation in Ltnα but not in Ltnβ. No effect was observed with npnJ, likely due to poor expression (vide infra). The activity of penN is particularly intriguing; although penN has proven necessary for accessing bioactive pediocin A428 and PenN reduced Dha in LtnA1 and LtnA2, surprisingly the gene cluster containing PenN does not contain a LanM or LanB. To our knowledge, the structure and the encoding gene of pediocin A remain unknown, thus whether or not pediocin A incorporates D-amino acids has not been established.
The important residues in LtnJ were identified by in vivo expression studies429 using the ΔltnJ strain supplemented with ltnJ mutants where 15 residues conserved among LtnJ, SacJ, PenN, and NpnJ were individually replaced. Leu54, Gly99, Thr166, Ala189, and Leu317 were found to be critical for LtnJ activity but Asn63, Ser89, Gly127, Ala157, Leu185, Thr213, and Gly308 were not, as wild-type lacticin 3147 was obtained without significant change in bioactivity. Gly272 and Asp335 were important for efficient reduction in LtnA2. The most surprising effect was observed with the LtnJ-K359A mutant; lacticin 3147 of the correct mass was still produced but the bioactivity was 1.5 fold lower than for the ΔltnJ strain and >20-fold lower than for wild-type lacticin 3147. Remarkably, the bioactivity is reduced because a single mutation to Lys359 results in an enzyme that is no longer stereoselective. For a completely diastereoselective hydrogenation, the ratio of d-Ala to l-Ala should be 1:2 for both Ltnα and Ltnβ (Figure 40), but mutating Lys359 resulted in ratios of 1:4.4 and 1:6.6, respectively. These studies offer intriguing insights into how the chirality is transferred from the enzyme to the substrate. Although the precise roles of the important LtnJ residues have not been assigned and would be greatly facilitated by an X-ray structure, one possible function of Lys359 is to stabilize negative charge buildup on the carbonyl group resulting from initial hydride conjugate addition to the Dha.
Introduction of many PTMs in lanthipeptide biosynthesis is highly dependent on the substrate sequence, including the stereochemical outcome of cyclizations as discussed in section 4.3.3. In contrast, a high degree of flexibility has been reported for LanJA enzymes with no apparent loss of stereoselectivity. For example, LtnJ has been used to incorporate d-Ala into nisin, once again suggesting that LtnJ does not require a leader peptide.430,431 These studies used an in vivo expression system in L. lactis of LtnJ, NisA, and NisBCT, which suggests that LtnJ can act on cyclic peptides given the coupled activity predicted for NisBC (section 2.4). Additionally, LtnJ was shown to modify a mutant of NisA lacking all cysteines, showing that LtnJ can act on both linear and cyclized substrates. A variety of flanking hydrophobic residues were well accepted by LtnJ. However, flanking charged residues including Lys and Glu as well as an adjacent Dhx were not tolerated. Finally, with this system it was once again shown that LtnJ can reduce Dha but not Dhb. The substrate scope of LanJA is also substantiated by the fact that there are no remaining Dha in Ltnα, Ltnβ, and lactocin S (Figure 40) and that all of the d-Ala are flanked by hydrophobic residues. It is also intriguing that the three Dha that react to form Lan in Ltnα and Ltnβ are adjacent to polar, charged, or Dhx residues; it seems likely that LtnM completes its function first, but it is interesting that the Dha residues involved in ring formation are predicted to be poor substrates for LtnJ.
The broad substrate scope of LanJA enzymes has also been illustrated for the activity of NpnJ in what was the first in vitro reconstitution of a LanJ enzyme.425 In prior in vivo studies, NpnJ was not functional,429 but use of an MBP-tag provided soluble, active NpnJ. As for LtnJ, the leader peptide was not required for activity and only Dha could be hydrogenated, not Dhb. A variety of flanking residues were tolerated although C-terminal charged residues greatly reduced efficiency, and Trp and Pro were also poorly tolerated. NpnJ was also shown to act on non-native substrates, including dehydrated LctA in vitro and mLtnA1 and mLtnA2 in E. coli. These experiments provided in vitro support for the substrate scope predicted from in vivo studies on LanJAs and also inspired a straightforward method for analyzing d-Ala incorporation. Traditionally, this analysis is carried out either by GC-MS of the hydrolyzed peptide or by LC-MS analysis of the hydrolyzed peptides modified by Marfey’s reagent. However, these methods are complicated when the hydrogenation does not go to full conversion or when the peptides also contain many gene-encoded l-Ala residues. By carrying out the reduction of the peptides in vitro in D2O with NpnJ and NADP2H generated in situ from NADP+ by phosphite dehydrogenase, two 2H atoms are incorporated into every d-Ala. Subsequent treatment with Marfey’s reagent results in d-Ala and l-Ala-bearing molecules with distinct LC retention times and distinct masses, thus facilitating analysis.425
More recently, a new class of LanJs was characterized. This enzyme class, termed LanJB,425 are flavin-dependent oxidoreductases. Notably, although the LanJ involved in lactocin S biosynthesis has not been characterized it is also predicted to be a LanJB based on sequence homology425 (the protein of unknown function was originally annotated as LasN).50 CrnJ, involved in the biosynthesis of carnolysin, was the first of two recently explored LanJBs.361 Carnolysin is a two-component lanthipeptide composed of CrnA1′ and CrnA2′ and interestingly it contains both unusual ring stereochemistry (LL-Lan and LL-MeLan, see section 4.3.3) and D-amino acids. Another unusual feature is that both d-Ala and d-Abu are incorporated, resulting from hydrogenation of Dha and Dhb, respectively. CrnA1′ contains four d-Ala and CrnA2′ contains one d-Ala and one d-Abu. Coexpression in E. coli of the precursor peptides CrnA1 or CrnA2 with CrnM in the presence or absence of CrnJ showed that CrnJ is responsible for both Dha and Dhb hydrogenation.361
The second of two LanJBs explored was BsjJB,400 the enzyme responsible for hydrogenation in the recently discovered two-component lanthipeptide bicereucin (Figure 37). In addition to the D-amino acids, another unusual feature of bicereucin is that only one component is a lanthipeptide and its lanthionine cross-link is formed as a mixture of stereoisomers (see section 4.3.3). The activity of BsjJB was reconstituted both in a heterologous coexpression system in E. coli and in vitro using dehydrated BsjA1 as the substrate in the presence of FMN and NADH. As observed for CrnJ, BsjJB reduces both Dha and Dhb. Specifically, Bsjα contains four d-Ala while Bsjβ contains one d-Abu and three d-Ala (Figure 37). At this point it is unclear why the LanJB class of enzymes can hydrogenate both Dha and Dhb whereas the LanJA class can only reduce Dha.
Although the sequence specificity of LanJB enzymes has not been as extensively explored by mutagenesis as for LanJA enzymes, some trends can be gleaned from the structures of carnolysin and bicereucin. LanJA and LanJB enzymes feature similarities in their requirements for flanking residues, which are commonly hydrophobic and no hydrogenation occurs adjacent to Pro. In addition, Dhx flanked by thioether cross-links are not hydrogenated likely due to steric hindrance or possibly the presence of a D-stereocenter in (Me)Lan. Spatial positioning of the Dhx residues may be more important for the LanJB class than for the LanJA class, which reduces Dha at many different positions. For instance, BsjJB does not act on Dhb33 near the C-terminus of Bsjα. Second, when ring formation is blocked in Bsjβ by mutating Cys40 (see Figure 37 for numbering) neither Dha35 nor Dhb39 were reduced; however, another possible explanation is that these residues are not hydrogenated due to flanking polar/charged residues.
Now that it has been demonstrated that there are two independent enzyme families that convert dehydro amino acids to the corresponding D-amino acids, we propose that for clarity and consistency the gene/protein designation of LanJ proteins always indicate whether they are members of the Zn2+-dependent dehydrogenase family (i.e., LtnJA, SacJA, PenJA, and NpnJA) or the flavin dependent dehydrogenase family (LasJB, CrnJB, and BsjJB).
4.6.2. Lysinoalanine Formation and Aspartate β-Hydroxylation
Cinnamycin (formerly also named Ro 09-0198 and lanthiopeptin),432−435 cinnamycin B,436 duramycin (formerly also named leucopeptin),437 and duramycins B and C438,439 form a small group of globular class II lanthipeptides that share high sequence identity and a fully conserved set of PTMs as determined by extensive NMR analysis.433,440−443 Each lanthipeptide comprises 19 amino acids (Figure 41); duramycin and duramycin B differ from cinnamycin by a single residue, and cinnamycin B and duramycin C differ by five and six residues, respectively. These peptides are an unusual example, analogous to the prochlorosin natural products, in which Lan and (Me)Lan are formed by both C-to-N directional cyclization, and by N-to-C directional cyclization wherein the nucleophilic Cys is N-terminal to the reactive Dhx. These duramycin-type lanthipeptides contain three DL-(Me)Lan, an erythro-3-hydroxy-l-aspartic acid at position 15, and a lysinoalanine (Lal) cross-link that forms between Ser6 and Lys19 with LL-stereoselectivity.441 Although Lal formation and β-Asp hydroxylation are otherwise unknown for lanthipeptides, a mammalian Asp β-hydroxylase has been characterized and is an α-ketoglutarate/iron(II)-dependent hydroxylase.444 Additionally, nonenzymatically generated Lal is prevalent in aging and interestingly forms in a manner reminiscent of lanthipeptide biosynthesis by Lys attack on Dha.445
Figure 41.
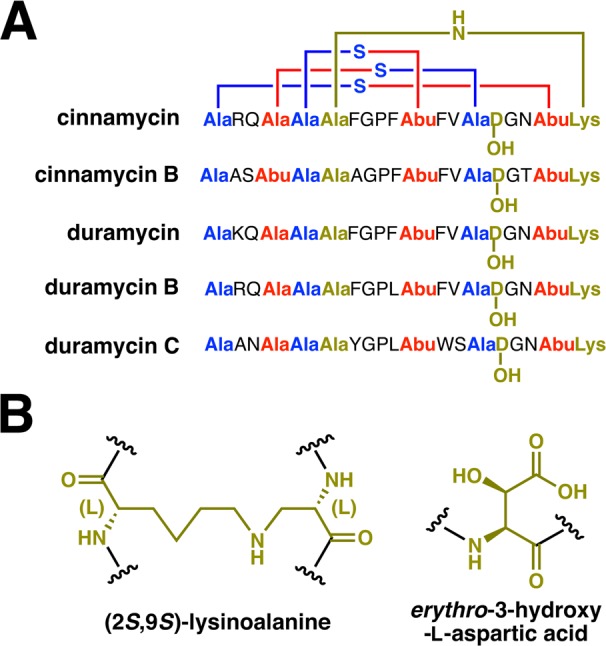
(A) Cinnamycin and duramycin peptides share identical post-translational modifications including Lal formation and Asp hydroxylation, for which the structures are shown in (B).
In these molecules, the well-defined pocket set up by the four cyclization events recognizes the target phosphatidyl ethanolamine (PE) with high affinity and selectivity. As a result, various derivatives of duramycin have been used to image PE in cells and inside organisms,26−34,65,446,447 the compound has been shown to inhibit viral entry,448 and duramycin has been used to effect chloride secretion in lung epithelial cells11−15 and calcium release in cancer cells.449
In groundbreaking studies on cinnamycin biosynthesis, its biosynthetic gene cluster was cloned from Streptomyces cinnamoneus cinnamoneus DSM 40005 and heterologously expressed in Streptomyces lividans.328 Subsequent genetic experiments identified four out of 21 predicted open reading frames (orfs) as critical to cinnamycin production: genes encoding the precursor peptide CinA, the lanthionine synthetase CinM, an α-ketoglutarate/iron(II)-dependent hydroxylase homologue CinX, and Cinorf7, for which no homologues with assigned function had been found. The respective roles of cinM, cinX, and cinorf7 were determined by evaluating each orf individually.450 CinM and CinX functions were reconstituted in vitro and as a coexpression system in E. coli. As predicted by sequence homology, CinX was confirmed to be an α-ketoglutarate (α-KG)/iron(II)-dependent hydroxylase responsible for Asp15 hydroxylation. CinX could process both linear CinA and CinM-modified CinA and also did not require the leader peptide,450 as is common for tailoring enzymes.
CinM was shown to dehydrate all Ser/Thr in the CinA core peptide, including the Ser involved in Lal formation (Ser6), thus suggesting that Lal formation results from Lys attack on Dha (Figure 41).450 However, CinM does not catalyze Lys attack, serving only to catalyze Cys conjugate addition that results in the three (Me)Lan. Instead, Cinorf7 was found to promote Lal formation when coexpressed with CinAMX in E. coli. Unfortunately, the activity of Cinorf7, a small protein of just 13 kDa, could not been reconstituted in vitro; alternatively, exposure of CinM and CinX-modified CinA to pH 9.5 in vitro resulted in nonenzymatic cyclization to afford Lal. The stereoselectivity of this cyclization was not confirmed due to complicating epimerization of Lal groups upon the peptide hydrolysis necessary for chiral GC-MS analysis. However, the substrate-controlled stereoselectivity observed in (Me)Lan formation (section 4.3.3) suggests that nonenzymatic Lys attack to form Lal may give the native LL-cross-link, especially given the very compact and well-ordered structure of the final product, which may not allow formation of the DL-Lal isomer.443
Very recently, the biosynthetic gene cluster of duramycin was also reported.451 The overall gene organization and the biochemical activities of the enzymes DurM and DurX were very similar to those reported for the cinnamycin orthologs. A Cinorf7 homologue was required for lysinoalanine formation and was designated the name DurN (with LanN instead of Lanorf7 as a general designation for orthologous lysinoalanine synthases). The mechanistic details of lysinoalanine formation await further investigation but the reported findings suggest that it will be a unique mechanism despite the deceptive similarities to (Me)Lan formation.
4.6.3. Sulfoxide Formation
Another unusual tailoring modification in class II lanthipeptides is the installation of a single sulfoxide by oxidation of a MeLan thioether. This modification has only been found in the 19-amino acid globular lanthipeptide actagardine (also known as gardimycin)452 and Ala(0)-actagardine, which differs from actagardine by a single additional Ala residue at the N-terminus (Figure 42).453 The structure of actagardine was determined by a combination of Edman sequencing, MS, and NMR analysis,454−456 revealing that oxidation occurs site-selectivity in the D-ring.454 The genetic origin of sulfoxide formation was uncovered through a comparison of the gene clusters of actagardine and another lanthipeptide michiganin A,86 which is similar in structure to actagardine but lacks the sulfoxide.457 Annotation of the actagardine gene cluster from Actinoplanes garbadinensis ATCC 31049 revealed the unique presence of a gene encoding a luciferase-like flavin-dependent monoxygenase termed garO. A ΔgarO mutant of A. garbadinensis ATCC 31049 produced only deoxyactagardine and Ala(0)-deoxyactagardine. The actagardine gene cluster also encodes GarA, the lanthipeptide precursor peptide, and GarM, the lanthipeptide synthetase.
Figure 42.
Structures of actagardine and Ala(0)-actagardine.
Actagardine has been heterologously produced in S. lividans(86) and in E. coli.458 Similar to the gene deletion studies, coexpressing only GarA and GarM in E. coli gave deoxyactagardine, whereas coexpression with GarO resulted in actagardine. The activity of GarO was reconstituted in vitro with FMN and NADH as cofactors and shown to not require the leader peptide.458 As with other luciferase-like flavin-dependent monooxygenases,459,460 the proposed mechanism involves oxidation of FMN to a hydroperoxide species that serves as the oxygen donor to the substrate, followed by reduction of the oxidized FMN by NADH. The molecular explanation for regioselective oxidation of the D-ring remains to be determined but in vitro results suggest that the selectivity is under catalyst control. Multiple oxidations were sometimes observed in vitro, likely resulting from precedented nonenzymatic oxidation by a hydrogen peroxide side product rather than direct transfer of oxygen from FMN.461 The stereochemistry of the sulfoxide in actagardine is currently not known.
4.7. Immunity Against Class II Lanthipeptides
Many class II lanthipeptide gene clusters also encode LanEFG transporters that were discussed in section 2.7 for class I lanthipeptides, and some encode LanI-like proteins.317,462,463 In some cases, a different type of immunity protein cooperates with LanEFG secretion systems such as the NukH protein involved in self-resistance of the nukacin ISK-1 producer.464−466
A unique immunity mechanism is found in the producer of cinnamycin, Streptomyces cinnamoneus DSM 40646.467 Cinnamycin and similar compounds (section 4.6.2) exert their antimicrobial activity by binding to phosphatidyl ethanolamine (PE),170,468,469 a target that is a major membrane lipid in streptomycetes. To protect the producing strain, the cinorf10 gene encodes a PE monomethyltransferase.467 Transcription of cinorf10 is initiated upon production of a low concentration of cinnamycin, leading to PE methylation in the producer prior to high-level production of cinnamycin. Based on the structure of cinnamycin bound to PE,468,469 monomethylated PE is unlikely to be accommodated in the binding pocket. Immunity to cinnamycin or the closely related duramycin was conferred by expressing cinorf10 in a previously sensitive Streptomyces lividans strain, which was shown to result in accumulation of monomethylated PE.467 Interestingly, this immunity mechanism does not appear to be universal among the producers of the cinnamycin-group of lanthipeptides because a homologue of cinorf10 was not found in the genome of Streptomyces cinnamoneus ATCC 12686 (previously also known as Streptoverticillium cinnamoneum forma azacoluta),451 a producer of duramycin.470 How the organism then defends itself against the duramycin it produces is currently not known.
5. Class III and IV Lanthipeptide Biosynthesis
5.1. Discovery of Class III and IV Pathways
The two most recently discovered classes of lanthipeptides have a common mechanism of dehydration that is distinct from class I and II, but they differ in their cyclization strategies. Because their shared dehydration reactions are much better understood than the cyclization processes, these classes will be discussed together in this review. Class III lanthipeptides were first established when Willey and co-workers reported that the small morphogenetic peptide SapB was a lanthipeptide.6 SapB was known to be important for formation of the aerial mycelium at the start of the sporulation process of Streptomyces coelicolor.(471) Its structure had been unknown, but genetic studies had identified the locus for rapid aerial mycelium (ram) formation.472,473 This gene cluster had no similarities with known lanthipeptide gene clusters, but it did encode a 42-amino acid peptide (RamS) as well as a protein RamC with homology to Ser/Thr protein kinases.474 A similar cluster (aerial mycelium formation, amf) was found in Streptomyces griseus and encodes the AmfS peptide.475,476 A sequence analysis of RamC suggested that the protein, in addition to a Ser/Thr protein kinase domain, contained a C-terminal domain with homology to the C-terminal cyclization domain of the class II lanthipeptide synthetases CinM and MrsM that are involved in the biosynthesis of cinnamycin and mersacidin, respectively.6 Even though RamC lacks the catalytic residues and Zn2+-binding residues that are conserved in the cyclase domains of LanM and in LanC proteins (sections 4.3.1 and 2.3, respectively), the authors showed that the mass of SapB was consistent with the C-terminal 21 residues of RamS after four dehydrations.6 Chemical derivatization studies followed by mass spectrometry, Edman degradation, and amino acid analyses provided strong evidence for the structure shown in Figure 43. A decade later, analysis of the available Actinobacteria genomes showed that the ram/amf-like gene clusters are the most common lanthipeptide gene cluster family in this phylum.35 Another morphogenetic peptide, SapT, is also involved in aerial mycelium formation in Streptomyces tendae and was also shown to be a lanthipeptide.7 Whereas SapB was too hydrophobic to obtain an NMR structure because of aggregation, SapT was amenable to NMR structure elucidation (Figure 44A).7 Although the gene cluster encoding SapT has not yet been reported, a BLAST search performed for this review with its putative precursor core peptide sequence returns a 50-amino acid peptide in Streptomyces reticuli (GenBank accession number CUW32634.1) with a potential core peptide that differs in just two residues (Figure 44B). Interestingly, immediately adjacent to the putative substrate gene is a lanB gene. The contig does not provide additional information, but another 56-amino acid peptide from Streptomyces albulus CCRC11814 (GenBank accession number EPY92828.1) differs in nine residues from SapT in its putative core region and is predicted to contain the same 4-ring topology of SapT but also to have a fifth ring at its C-terminus (Figure 44B). Similarly, this gene is flanked by a lanB gene, but the cluster also clearly contains genes for a LanC and a standalone LanB C_terminal domain protein (Pfam 14028; homologous to LanB glutamate elimination domain, section 2.2), with the function of the latter unclear. Thus, it is very likely that these gene clusters encode class I lanthipeptides and that SapT also belongs in this class and not class III as previously assigned based on bioactivity.54
Figure 43.
Biosynthesis of SapB. For clarity the process is drawn as first complete dehydration and then cyclization, but the order of the PTMs for SapB is not actually known.
Figure 44.

SapT (A) is predicted to be a class I lanthipeptide based on sequence alignment (B) with putative LanAs from S. reticuli and S. albulus that cluster with genes encoding putative LanB enzymes. Asterisks indicate fully conserved identical residues whereas colons indicate conserved similar residues.
A few years after the recognition that SapB was a lanthipeptide and that there was a third biosynthetic pathway to this family of RiPPs, two studies in 2010 provided key new information on this pathway. The first study reported the structure of the labyrinthopeptins and the associated gene cluster.10 Structure elucidation of a series of congeners of these compounds had proven unusually challenging, hence their name, and only when a crystal structure of one of them was obtained did it become clear why: the compound contains two entirely unprecedented and unanticipated cross-links in which new carbon–carbon bonds are made between the α-carbon of what used to be a Ser and the β-carbon of what used to be another Ser (Figures 1 and 45A).10 This new type of cross-link was termed labionin. The presumed mechanism of its formation involves initial attack of a Cys thiol onto a Dha. Instead of protonation of the resulting enolate to form a lanthionine, another conjugate addition is thought to take place in which the enolate attacks a different Dha to form a second enolate, which upon protonation forms the labionin (Figure 2).10 The gene cluster that encodes for labyrinthopeptins showed the presence of a protein that has clear homology with RamC comprising a kinase domain and a C-terminal cyclase domain. In vitro studies confirmed that this protein carries out both dehydration and labionin formation.477 These studies also observed phosphorylated intermediates that demonstrated that the Ser/Thr kinase domain indeed carries out the presumed reaction and resulted in the designation LanKC for this class of proteins (for lanthipeptide kinase and cyclase). As noted above, the C-terminal domain of RamC/LanKC shows sequence homology with LanC enzymes and the C-termini of LanM proteins but lacks the catalytic residues and zinc-binding ligands of the latter two protein families (Figure 46).477
Figure 45.
(A) X-ray crystal structure of labyrinthopeptin A2 (CCDC number 721326). The color coding uses standard atom-type colors (red, oxygen; blue, nitrogen; yellow, sulfur). For Lab, the carbons originating from Cys are shown in light blue and those originating from Ser are in magenta. (B) Biosynthesis of labyrinthopeptin A2. For clarity the process is drawn as complete dehydration and then cyclization, but the order of the PTMs can be more complicated; e.g. see text and Figure 49 for curvopeptin.
Figure 46.
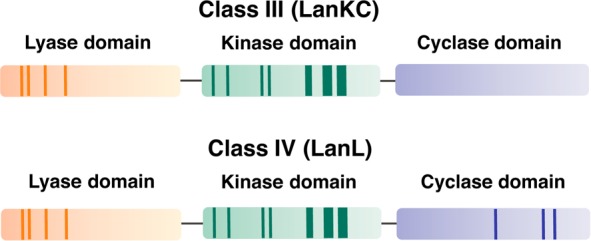
Class III and class IV lanthipeptide synthetases are distinguished by their cyclase domains. LanL (class IV) contains zinc-binding ligands conserved in class I and II but these ligands are absent in the class III LanKC.
A second independent study reported in 2010 provided additional insights into RamC/LanKC catalysis.128 Genome mining of Actinobacteria uncovered examples of proteins with a C-terminal LanC-like domain (this time including the zinc ligands and catalytic residues), a central Ser/Thr kinase domain, and an N-terminal domain that resembled type III effector proteins of Gram-negative pathogens including Salmonella, Pseudomonas, and Shigella (Figure 46). These effector proteins recognize a phosphorylated Thr on the activation loop of mitogen activated protein kinases (MAPK) and eliminate the phosphate to inactivate these proteins that are a key part of the immune response.478−481 In vitro studies with the putative lanthipeptide biosynthetic protein discovered in Streptomyces venezuelae showed that it indeed catalyzed three independent reactions (phosphorylation, phosphate elimination, and cyclization) on its substrate peptide (VenA) as discussed in the next section.128 Homologous phospholyase domains that presumably also catalyze phosphate elimination were also detected at the N-termini of the RamC/LanKC family (Figure 46).128 However, the clear differences in the cyclization domains required separation of the enzymes into two classes of lanthipeptide synthetases (class III and IV); the enzymes with the zinc-binding ligands were given the generic name LanL (Figure 46).128
Interestingly, the biological activities of class III lanthipeptides are very diverse and many do not display antimicrobial activities against the organisms tested. As a result, following the lead of the prototypical labyrinthopeptin, class III peptides have been designated with the suffix–peptin,1,132 rather than the often used suffix -cin or -cidin, which implies a toxic activity. Based on the currently available genomes, class III lanthipeptide gene clusters significantly outnumber class IV clusters.35 To date, all class IV lanthipeptides are close analogs of the original prototype from S. venezuelae and they have been generically called venezuelins (Figure 47).35 Whereas the original venezuelin was not produced under a variety of growth conditions, sequence variants with the same ring topology are produced by Streptomyces collinus Tü 365 (streptocollin),134S. katrae ISP5550, S. lavendulae subsp. lavendulae NRRL B-2508, and Streptomyces sp. strain NRRL B-2375.35 Despite several attempts to determine the biological activity of the venezuelins, thus far their function(s) are not clear.128,134
Figure 47.
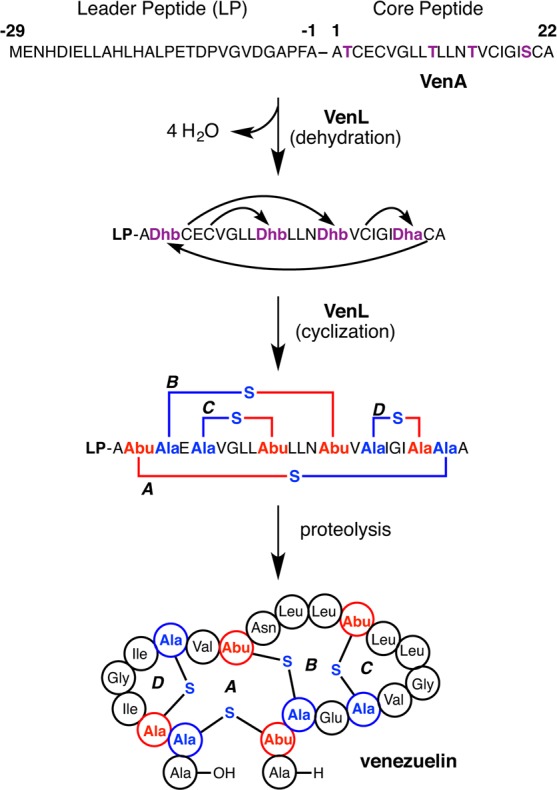
Biosynthesis of venezuelin. For clarity the process is drawn as completion of dehydration before cyclization, but the order of the PTMs for venezuelin is not currently known.
5.2. Mechanism of Phosphorylation, Phosphate Elimination, and Cyclization
Class III and IV lanthipeptide synthetases consist of three different domains, each presumably with its own active site. Hence, the mechanism and timing of each consecutive reaction is more complicated than in the case of class II LanM enzymes for which phosphorylation and phosphate elimination take place in one active site and are usually tightly coupled (section 4.2). Although X-ray structures are available for orthologs of each of the three individual domains,177,480,482,483 thus far LanKC and LanL proteins have not yielded crystal structures, possibly because some of the proteins that have been characterized in vitro have needed fusions with solubilizing proteins such as SUMO132 or coexpression with folding chaperones484 to achieve reasonable quantities of soluble protein. The three proteins and their substrates that are the best behaved and did not need additional measures to obtain soluble enzyme, LabKC,271,477 CurKC,131,247 and VenL,128,485 have been investigated in more detail with respect to the three activities catalyzed by these trifunctional enzymes. Notably, CurKC is encoded in the genome of the thermophile Thermomonospora curvata,131 which may provide improved thermostability of both the final product and the biosynthetic enzymes as seen for the class II lanthipeptide geobacilin II.394,486
5.2.1. Dehydration via Phosphorylation
Although RamC was the first discovered class III lanthipeptide synthetase, the extreme insolubility of its substrate RamS precluded any in vitro studies. LabKC, the enzyme responsible for the biosynthesis of the labyrinthopeptins, could be studied in vitro and was shown to convert synthetic LabA2 into mLabA2 (Figure 45B), confirming that the enzyme catalyzes both dehydration and cyclization, despite the absence of zinc-ligands.477 Metal analysis of the AciKC ortholog involved in catenulipeptin biosynthesis (Figure 48) corroborated that the protein did not bind zinc nor any other metal.130 The in vitro reconstitution of LabKC demonstrated another unusual feature, the requirement of guanosine triphosphate (GTP) or deoxy-GTP for dehydration instead of ATP that is utilized by class II LanM enzymes.477 Subsequent studies on a series of LanKC enzymes showed that the nucleotide specificity is not uniform: EryKC involved in erythreapeptin biosynthesis is ATP dependent,132 AciKC that generates catenulipeptin accepts ATP, GTP, CTP, or TTP as a cosubstrate,130 and StaKC484 and CurKC131 responsible for stackepeptin and curvopeptin biosynthesis, respectively, can utilize all NTPs/dNTPs but prefer purine nucleoside triphosphates as the phosphoryl donor for the central kinase domain (for structures of these class III lanthipeptides, see Figure 48).
Figure 48.

Structures of class III lanthipeptides. Labyrinthopeptin A3 differs from labyrinthopeptin A1 by a single additional Asp residue at the N-terminus thought to derive from incomplete leader removal (section 5.4).
The order of dehydration has been investigated for two enzymes, LabKC and CurKC, using the Ser/Thr deuterium-labeling strategy described in section 4.4 but that was originally developed for studies on LabKC (Figure 38).401 Both enzymes are distributive, and unlike class II lanthipeptide synthetases, phosphorylated intermediates are detected with wild-type substrates. LabKC and CurKC catalyze dehydration with net C-to-N terminal directionality.247,401 Although both enzymes clearly display overall directionality, the differentiation between two consecutively dehydrated residues is not always complete and hence several parallel pathways are operational,247,401 similar to what is observed for class I and class II dehydration.245,364,365,375 Thus, it may be that the model of conformational sampling discussed in section 4.4 is a general feature of dehydration in all three enzyme classes; the order and directionality for class IV LanL enzymes has thus far not been investigated.
As noted above, the dehydration reaction involves two separate active sites with phosphorylation taking place in the central kinase domain and phosphate elimination in the N-terminal pSer/pThr lyase domain. The timing of these two processes has been most extensively investigated for CurKC. Surprisingly, phosphorylation and elimination are not always consecutive reactions. During processing of CurA by CurKC, an initially phosphorylated Ser1 remains phosphorylated as the enzyme dehydrates Ser12 and Ser15, leading to bisphosphorylated intermediates (Figure 49).247 Only after Ser12 and Ser15 have been converted into Dha is the phosphate at pSer1 eliminated, and it appears that only then is cyclization initiated. Interestingly, when Ser1 was mutated to Ala, Ser2 was phosphorylated first.247 Thus, it appears that leader peptide binding juxtaposes the core peptide in such a way with respect to the kinase active site that a residue at its N-terminus is the preferred substrate for the first phosphorylation. However, in the ensuing competition between the lyase and kinase domain to either eliminate the phosphate from pSer1 or phosphorylate a second time at Ser12/15, the kinase wins out possibly because pSer1 is a particularly unfavorable substrate for the lyase domain. After Dha12/15 are both installed, a new competition is set up between elimination at pSer1 and phosphorylation at Ser4, which apparently now favors the lyase reaction. Interestingly, it is proposed that immediately after introduction of the Dha1, the cyclase domain catalyzes Lan formation between Cys19 and Dha12.247 Thus, it appears that the phosphate at Ser1 prevented cyclization. Once the Lan is introduced, Ser4 and subsequently Ser2 become improved substrates of the kinase. For all lanthipeptide synthetases, such models can likely only be tested with cocrystal structures of enzymes bound to intermediates and/or by kinetic studies with (semi)synthetic intermediates of defined structures. Given the size of the substrates and the complexity of the overall process, both of these approaches are highly challenging.
Figure 49.
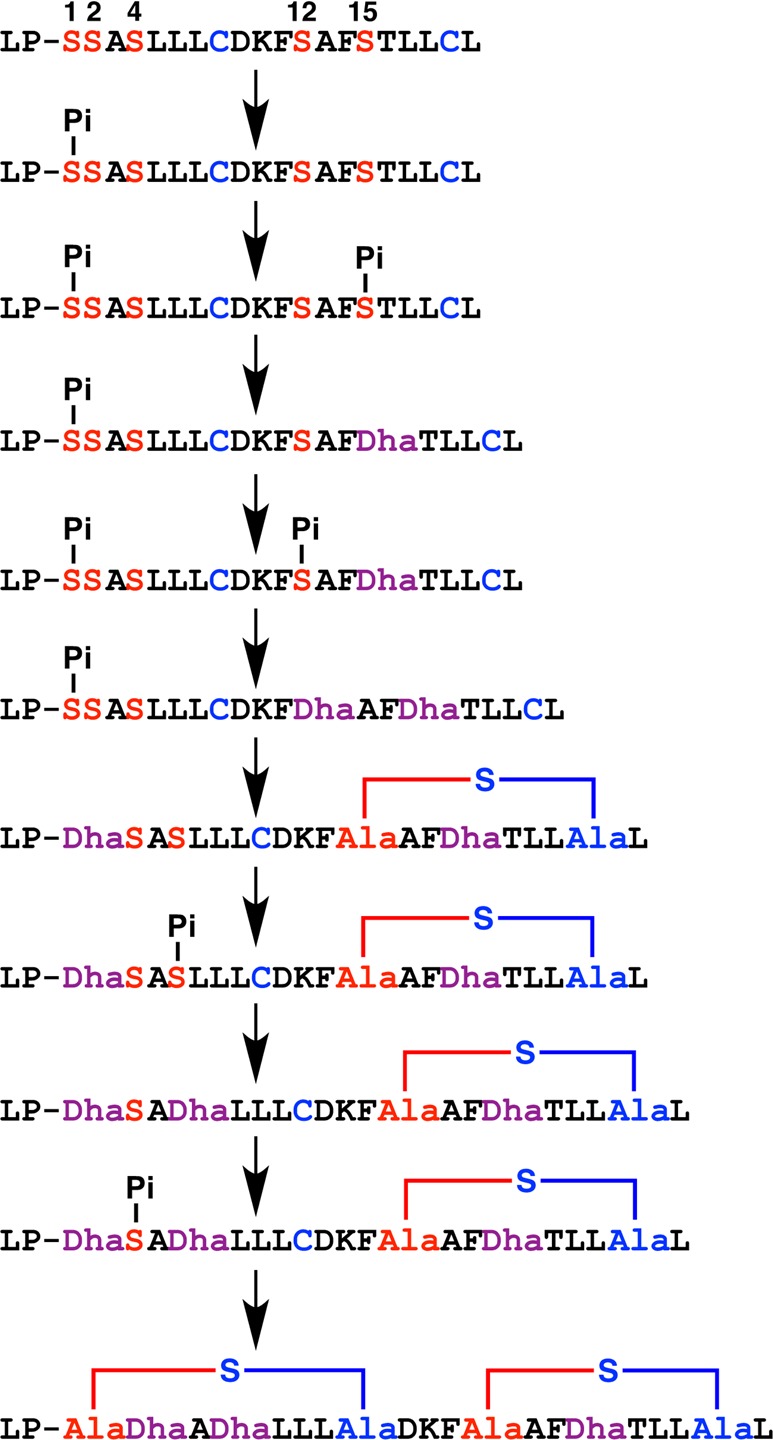
CurKC catalyzes phosphorylation, phosphate elimination, and cyclization of CurA in a distributive fashion and unusual order. Only the main pathway is shown, with minor parallel pathways also observed.247 Pi = phosphate; LP = leader peptide.
The phosphate elimination activity in the N-terminal lyase site follows a very similar mechanism as in the homologous type III effector proteins of the OspF family that catalyze phosphate elimination from a very specific pThr in MAP kinases.478−481 It is interesting that these effector proteins are found in Gram-negative pathogens,487 whereas the LanKC and LanL enzymes are phylogenetically more diverse and are found in Actinobacteria, Firmicutes, and Proteobacteria.36 As expected from the structure of the effector proteins and from the fact that elimination takes place in a separate active site from phosphorylation, unlike for class II LanM enzymes phosphate elimination for class III/IV does not require the presence of an NDP as shown for LabKC.401 At present no structures are available for class III and IV lanthipeptide synthetases nor for their three individual domains. However, mutagenesis studies on VenL illustrate that several of the conserved catalytic residues in OspF proteins are also important for catalysis by class III/IV lanthipeptide synthetases (vide infra).
A truncated form of VenL containing only the lyase and kinase domains, termed VenL-Δcyc, was able to fully dehydrate the VenA substrate, illustrating that cyclization is not required for full dehydration.128 However, this finding does not rule out a mechanism whereby full length VenL processes VenA by alternating dehydration and cyclization events, as has been observed for several lanthipeptide synthetases. Expression and purification of the individual kinase and lyase domains of VenL and incubation of these proteins with VenA resulted in incomplete conversion, indicating that the domains can operate separately, albeit less efficiently than when present in one polypeptide.128 Whether the decreased efficiency signifies some level of communication between the kinase and lyase domains, for instance by sharing a leader peptide binding site, or whether it simply results from less well-folded proteins, is currently not known. Indeed, many questions remain that in principle can be investigated such as whether or not the leader peptide is required for all three activities of LanL/LanKC proteins.
In SpvC, a type III effector protein from Salmonella, Lys136 is responsible for deprotonation at the α-carbon of the pThr targeted for phosphate elimination (Figure 50),480 and accordingly mutation of the corresponding Lys80 in VenL resulted in phosphorylated VenA products, indicating interference with the elimination step.485 His106 in SpvC is thought to act as the catalytic acid that protonates the bridging oxygen in the phosphate leaving group, and indeed mutation of the corresponding His53 in VenL interfered with phosphate elimination. A third critical residue in SpvC, Lys104, is thought to decrease the pKa of the α-proton of the pThr residue by electrostatic activation of its carbonyl group.481 The corresponding residue in VenL, Lys51, was also shown to be important for phosphate elimination. However, two other residues that are fully conserved in OspF, LanL, and RamC/LanKC family members,485 Thr106 and Tyr108 in VenL (Thr156 and Tyr158 in SpvC), were not critical for VenL catalyzed dehydration of its VenA substrate.485 Also different are the interactions with the nonbridging phosphate oxygens. In SpvC, three Arg residues (Arg213, Arg220, and Arg148) stabilize the negatively charged phosphate group. A homology model predicts that in VenL these roles may be fulfilled by residues that are not conserved in the OspF family but are in the LanL/LanKC families: Arg156 and Lys103. A possible model for the elimination reaction in VenL is shown in Figure 51.485
Figure 50.

(A) Structure of SpvC type III effector protein K136A mutant (purple) bound to a model pThr-bearing substrate (brown). (B) Close-up view of the SpvC active site. PDB ID 2Z8P.
Figure 51.
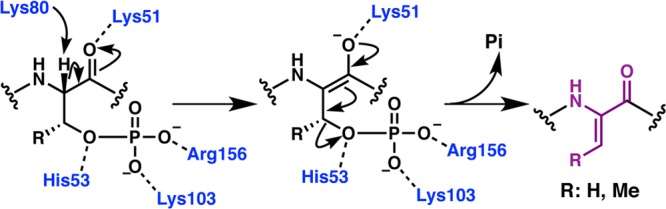
Proposed mechanism of VenL-catalyzed phosphate elimination to afford Dhx.
5.2.2. Cyclization
The most intriguing aspect of class III lanthipeptides is that they can contain either lanthionine or labionin residues. Comparison of the core peptide sequences of RamS (leading to SapB) and LabA2 (leading to labyrinthopeptin A2) demonstrates very similar SX2SX3C motifs but one leads to lanthionine and one to labionin (Figure 52). Phylogenetic analysis of class III enzymes that make Lan or Lab did not provide any clear indications that could be predictive of the outcome.36 Even more interestingly, some LanKC enzymes such as EryKC132 and StaKC484 can form both Lan and Lab in the same product (e.g., Figure 48 for the product of EryKC). Although the molecular mechanism of how the enzymes achieve the remarkable tandem cross-linking reaction resulting in labionin is still mostly unclear, several insightful observations have been reported. It is notable that whereas some of the enzymes appear to have strong preference to turn a given substrate sequence into either Lab or Lan, the selectivity is not complete since small amounts of Lab have been detected by GC-MS after acid hydrolysis of SapB132 and curvopeptin,131 two Lan-containing class III lanthipeptides (Figure 48). In addition to GC-MS analysis, which can detect small amounts of minor products, Lan and Lab have been distinguished by taking advantage of the reactivity of a Dha that will remain upon formation of Lan but not Lab (Figure 52). This noncyclized Dha will react selectively with either a thiol nucleophile or NaBH4.6,484
Figure 52.
Class III lanthipeptides labyrinthopeptin A2 and SapB contain different PTMs (Lab and Lan, respectively) despite high similarity of their precursor sequences.
A Lab introduces two cross-links when incorporated into a peptide. Of these, the carbacyclic ring is N-terminal and the thioether ring is C-terminal. A common nomenclature labels these two rings as A and A′, respectively, if they are the first Lab, B and B′ if they are the second Lab, etc. (e.g., Figure 52 for labyrinthopeptin A2). Whereas the majority of Lab structures in the currently characterized class III lanthipeptides are formed from a SX2SX3C motif, other sequences have also been shown to result in Lab formation. Labyrinthopeptins A1 and A3 contain a C-terminal labionin formed from an SX2SX5C sequence that has a larger thioether ring made up of seven residues (Figure 48),10 and the C-terminal labionin in catenulipeptin contains a smaller thioether ring formed from an SX2SX2C sequence (Figure 48).130 Variations in the ring size are not limited to the C-terminal ring; the N-terminal Lab residue in NAI-112 incorporates a six-residue thioether ring generated from an SX2SX4C motif (Figure 48).9 Indeed, in vivo and in vitro engineering studies have also demonstrated the ability of LabKC to generate thioether rings with expanded or contracted rings.488 Conversely, the carbacyclic ring in (Me)Lab appears more restricted since all such rings reported thus far in natural products consist of four residues, and attempts to increase or decrease this size by engineering were not successful.488 Similarly, it has been challenging to change a Lab into a Lan,488 suggesting that the conformation of the substrate peptide and the active site geometry of the enzymes appear to be quite specific for one product or the other for a given precursor peptide. An intriguing group of LanKCs were recently found by genome mining that have putative precursor peptides that do not contain the canonical labionin motif but that do contain a conserved CXSX2S motif. Whether this motif also encodes a labionin structure formed in the opposite direction (Dha residues localized C-terminal to the Cys) remains to be determined.35 The discussion below focuses on two Lab-forming class III enzymes (LabKC and AciKC), one Lan-forming class III enzyme (CurKC), and one Lan-forming class IV enzyme (VenL) that have been investigated in more detail with respect to the cyclization process.
Similarly to the directionality of phosphorylation and elimination discussed in section 5.2.1, the cyclization appears to take place with C-to-N directionality based on mutagenesis studies. Mutation of serines that prevent formation of C-terminal rings, whether Lab in catenulipeptin,130 Lan in curvopeptin,247 or either Lab or Lan in stackepeptin,484 negatively affected ring formation involving N-terminal residues. The timing of cyclization has also been determined by tandem MS analysis of intermediates formed during the biosynthesis of curvopeptin by CurKC, with the results again pointing at C-to-N directionality of cyclization.247 Formation of the C-terminal Lan in curvopeptin appears to take place only after dehydration of Ser1, Ser12, and Ser15 (Figure 49). This means that the dehydro amino acids required for labionin installation are available, but yet the enzyme preferentially forms a Lan. The N-terminal Lan is proposed to be generated only after completion of dehydration, including introduction of Dha2, and it has been suggested that this order of events may facilitate proper positioning of the intermediate peptide for formation of the Lan A-ring in curvopeptin.247 A strict requirement of the leader peptide for the cyclization reaction has been determined for AciKC,130 and hence leader peptide binding and the resulting juxtaposition with respect to the cyclase active site may also be important for the observed order and specificity of cyclization. Whether the leader peptide utilizes a single binding site or whether all three domains have their own individual binding sites is currently unknown. Sequence comparison does not locate a winged helix-turn-helix domain like that found in LanB in either LanKC or LanL. Another intriguing and currently unresolved question is what the functional significance is of a clade of class IV enzymes that have a zinc site with a Cys-Cys-Cys ligand set.36 As discussed in section 4.3, this ligand environment in LanM enzymes is associated with higher reactivity and substrate tolerance.
The stereochemistry of the venezuelin family of class IV lanthipeptides has been determined to be the canonical DL,35 but for class III lanthipeptides stereochemistry has not yet been explored in much detail. The stereochemistry of the founding member of labionin-containing peptides, labyrinthopeptin A2, was firmly established by X-ray crystallography.10 However, the stereochemistry of the MeLab present in NAI-112 (Figure 48) is currently unknown.9 Similarly, the stereochemistry of Lab in many other family members is inferred and has not been experimentally demonstrated. Given the unexpected findings of noncanonical stereochemistry in class II lanthipeptides that turned out to be sequence-dependent,38 it could be that the stereochemistry of labionin formed from sequences other than the SX2SX3C motif in labyrinthopeptin A2 is also different. A GC-MS method has been developed that can detect labionins,489 but the use of this method to confirm the stereochemistry of other labionin-containing lanthipeptides has not yet been reported. Unambiguous determination of stereochemistry would require synthesis of all four diastereomers of Lab, as was done for Lan and MeLan,490 or alternatively use of the diastereomers of α-methyl-2,4-diaminoglutaric acid, the product formed upon application of a desulfurization protocol with NiCl2/NaBH4 to labionin-containing peptides.489
Similarly, the stereochemistry of lanthionines formed in class III lanthipeptides has not been reported. We note that the L stereochemistry at carbon 2 of labionin requires protonation of the enolate from the opposite face of the canonical protonation at carbon 2 in DL-Lan (as seen in Figure 2). Although the stereochemical nomenclature is S in both cases, the actual stereochemistry is different because of the change in Cahn-Ingold-Prelog priorities upon sulfur substitution at the β-carbon of Lan compared to carbon substitution in Lab. If protonation occurs from the same face during formation of both Lab and Lan, then SapB and other lanthionine-containing class III lanthipeptides would form LL-Lan. Conversely, it is possible that the enzyme presents different faces of the enolate to the active site acid for protonation during Lab or Lan formation. This would be analogous to the different faces of protonation induced by a Dhx-Dhx-Xxx-Xxx-Cys motif discussed for class II lanthipeptide synthetases in section 4.3.3.
5.3. Substrate Recognition
The features of the substrate peptides that are important for biosynthesis of class III and IV lanthipeptides have been most extensively investigated for labyrinthopeptin A2.271,488 Truncation experiments on the LabA2 precursor peptide showed that the LabKC enzyme requires its leader peptide. Addition of the leader peptide in trans did not result in any activity.271 Systematic truncation of residues at the N- or C-terminus of the leader peptide as well as site-specific mutagenesis showed that the first three amino acids of the leader peptide are dispensable, but that a subsequent I-L-E-L-Q motif is very important. Substitution of individual amino acids suggested that the second and fourth residues in this five-amino acid motif (both Leu) may be most important, whereas the Glu and Gln could be replaced with Ala without losing processing by LabKC. Truncations and replacement of residues at the C-terminus of the leader peptide showed that residues located C-terminal to the above-mentioned motif are not important for catalysis but are critical to position Ser/Thr residues that are destined for dehydration at a sufficient distance from the leader peptide binding region. If a Ser/Thr is too close to the binding site, it is not dehydrated.271 This observation is very similar to that in class I and class II lanthipeptide synthetases,194,251,269 and in those proteins likely reflects the distance from the leader peptide binding site on the protein to the phosphorylation/glutamylation active sites. Interestingly, when the RamS leader peptide was fused to the N-terminus of a truncated LabA2 core peptide, LabKC catalyzed dehydration of both Ser residues albeit more slowly than with the wild type leader peptide.271 Indeed, the leader peptide of RamS contains an LFDLQ motif at its N-terminus that is similar to the ILELQ motif of LabA2, and this motif is widely found in class III precursor peptides (Figure 53). Hence, leader peptide recognition may be a conserved feature. Whether cyclization of the RamS-LabA2 hybrid also led to labionin formation (as in labyrinthopeptin), or resulted in lanthionine generation (as in SapB), was not reported and constitutes an interesting question.
Figure 53.

Alignment of class III lanthipeptide precursors. Lanthipeptides used: labyrinthopeptins A1-A3 (LabA1-A3), SapB (RamS), curvopeptin (CurA), griseopeptin (Streptomyces griseus AmfS), avermipeptin (Streptomyces avermitilis AmfS), erythreapeptin (EryS), flavipeptin (FlaA), catenulipeptin (AciA), stackepeptin (StaA), and NAI-112 (LabA). Fully conserved identical residues in the leader region are shown in dark orange and conserved similar residues are shown in green. Ser/Thr residues that undergo dehydration are shown in purple, and Cys residues are shown in blue.
Although LabKC did not show any activity in the absence of the leader peptide or when the leader peptide was presented in trans, the StaKC enzyme involved in stackepeptin biosynthesis did show a low level basal activity on its core peptide, and the activity was enhanced by addition of the leader peptide in trans.484 Thus, it may be that the leader peptide also functions as an allosteric activator as in many other RiPP biosynthetic enzymes.150 Like the substrates for class I and II lanthipeptides,257 and those for cyanobactins491 and microcin B17492 (two other RiPP families), the LabA2 leader peptide has a propensity to form an α-helical secondary structure.271 However, for class I lanthipeptides, cyanobactins, and microcin B17-like RiPPs, it has been shown that, despite their propensity to form helices, the leader peptides bind to a conserved domain in an extended conformation.194,259,263
5.4. Leader Peptide Removal
The removal of the leader peptide of class III and IV lanthipeptides is quite different from that of class I/II compounds. First, most clusters lack a gene encoding a dedicated protease. Second, for many family members, a series of congeners have been observed that contain differing numbers of amino acids originating from the leader peptide at their N-termini. This observation was first made for the labyrinthopeptins; the A1 and A3 congeners differ only in that A3 contains an additional Asp at its N-terminus (Figure 48).10 Subsequently, similar observations have been made for erythreapeptin (five congeners with different numbers of remaining leader peptide residues), avermipeptin (seven congeners), griseopeptin (two congeners), curvopeptin (five congeners), stackepeptin (two congeners), and even SapB (two congeners).131,132,484 Similar observations have been made for the class IV compounds venezuelin35 and streptocollin134 for which two variants of each were observed. Thus, it appears that the class III and IV lanthipeptides are generated by stepwise trimming of the leader peptides by currently unidentified amino peptidases. It is possible that for some or all members, an N-terminal segment of the leader peptide is first removed in one step by a specific endopeptidase. Indeed, this scenario has been experimentally demonstrated for flavipeptin from Kribbella flavida. Its biosynthetic gene cluster contains a prolyl oligopeptidase (POP) designated FlaP (note that it is not a member of the LanP Ser proteases of the subtilisin family) that is predicted to be cytoplasmic.493 In vitro reconstitution of FlaP activity showed that the enzyme is specific for cyclized FlaA. The enzyme cleaves mFlaA after Pro−12 but did not act on linear FlaA or the leader peptide, and very slowly processed dehydrated FlaA. Hence, the rings are important for substrate recognition. POPs were shown to be encoded in a small subset of class III gene clusters,493 but quite a few class III LanA substrates have Pro in their leader peptides, and hence cleavage by a POP and subsequent trimming by aminopeptidases could be a more general pathway. A POP has also been shown to be involved in the biosynthesis of the RiPP class of amanitins produced by mushrooms.494 Rather than the POP encoded in the flavipeptin biosynthetic gene cluster, a different type of protease appears to be encoded in the cluster responsible for the biosynthesis of NAI-112 (Figure 48).9 Thus far, the activity of this protein has not been reported.
5.5. Tailoring Reactions: Disulfide Bond Formation and Glycosylation
Class III and IV lanthipeptides are not as richly decorated with PTMs resulting from tailoring reactions as class I and II lanthipeptides. Only two types of modification have been reported thus far. The prototype class III lanthipeptide labyrinthopeptin A2 contains a disulfide linkage made by Cys9 and Cys18 that spans the C-terminal labionin (Figure 45), and labyrinthopeptin A1/A3 contain a similarly positioned disulfide formed from Cys9 and Cys20 (Figure 48).10 Like other disulfide containing class II lanthipeptides such as haloduracin α and bovicin HJ50,124,411 the gene cluster encoding the labyrinthopeptins does not appear to contain a disulfide isomerase that is required for disulfide formation.10 A second tailoring PTM was discovered in NAI-112 produced by Actinoplanes DSM 24059. This compound, which also contains the first example of a MeLab, is unusual in that it is the first example of a glycosylated lanthipeptide (Figure 48).9 Furthermore, the glycosylation is unusual as a 6-deoxyhexose is attached to the indole nitrogen of Trp. At present the identity of the 6-deoxyhexose is not known. Interestingly, a putative glycosyltransferase is encoded within the gene cluster just downstream of the lanA gene.
6. Concluding Remarks and Outlook
As discussed in this review, lanthipeptide biosynthesis is very widely distributed, and based on the number of lanthipeptide gene clusters, this family is currently the most abundant of all RiPPs. The origin of this widespread occurrence may lie in the ease by which lanthipeptides are fashioned from a linear peptide. Both the dehydration of Ser/Thr and the conjugate addition of thiol nucleophiles to dehydro amino acids are chemically relatively straightforward reactions to turn a linear peptide into a polycyclic, conformationally constrained peptide. Cyclization offers important evolutionary advantages of higher stability87,176,178,495,496 and improved target recognition.468,497 The notion that at least four different routes to lanthipeptides have evolved may be a further demonstration of both the ease of evolution of these pathways, and the privileged nature of the thioether cross-link to access biological activity space.
At least three very different solutions have evolved for the dehydration step. Glutamylation in class I lanthipeptides is entirely novel and the evolutionary origins are at present still enigmatic. Classes II–IV all involve Ser/Thr activation via phosphorylation by a kinase-like protein followed by phosphate elimination, but different solutions have evolved for the latter step. For class II lanthipeptides, the elimination activity evolved out of the kinase active site, whereas for class III and IV a separate standalone elimination domain was recruited. It is possible that there are other dehydration mechanisms that remain to be discovered for which the associated biosynthetic gene clusters have not yet been recognized as lanthipeptide gene clusters. Indeed, most genome mining efforts in the lanthipeptides field have used knowledge of existing biosynthetic strategies as queries, which would only return members of known classes. Even when limiting genome mining efforts to the four known classes of lanthipeptides, the amount of diversity and biosynthetic novelty remaining to be discovered is staggering. For instance, an analysis of 830 actinobacterial genomes illustrated that known lanthipeptides from this phylum cover only a very small fraction of biosynthetic space.35 Many gene cluster families that are quite common in these genomes at present do not have a member with a characterized structure of the final product, and many clusters contain putative biosynthetic enzymes for which the function currently is unclear, such as radical-SAM proteins and methyltransferases.35,37 It is also noteworthy that the utilization of the lanthipeptide biosynthetic enzymes, or domains thereof, is also observed in other classes of RiPPs, such as thiopeptides202 and proteusins,367 and RiPPs that are formed from precursor peptides lacking Cys,368 and they are even found in gene clusters for natural products that are not RiPPs such as NRPS and PKS clusters.35
Despite the variety of ways by which dehydration is achieved, remarkable similarities have started to emerge with respect to the role of the leader peptide and the determination of ring topology. In three of the four classes, a low basal level of dehydration activity has been observed in the absence of a leader peptide, and addition of the leader peptide in trans has increased this activity. These observations are consistent with a model in which the leader peptide functions as an allosteric activator.150 Another similarity is the existence of multiple parallel modification pathways in an overall ordered process, suggesting that the energy differences governing site selectivity can be small for residues that are close in space. A third commonality is the observation that dehydration and cyclization are often coupled, such that a later dehydration may require a prior cyclization. This is not observed for all systems, however, since in all classes there are some dehydratases or dehydration domains that can complete dehydration of all targeted sites even when no cyclization has occurred (e.g., NisB,243 ProcM,246,365 NukM,498 and CurKC131).
Although the classification scheme for lanthipeptides highlights their differences with regards to the dehydration reaction, the cyclization processes also have distinct features despite being catalyzed by enzymes or domains with clear sequence similarities. Phylogenetic analyses of the LanC enzymes with the cyclization domains of class II–IV lanthipeptide synthetases show that they have evolved separately with perhaps a common ancient ancestor.35,36,404
Phylogenetic analysis also supports coevolution of the biosynthetic enzymes and their substrates. This has been seen in the tRNA and leader peptide specificity of class I dehydratases, and in the observation of clusters containing substrates that lack Cys and synthetases that have lost the conserved catalytic residues for cyclization.196,368 With respect to the regio- and stereoselectivity of cyclization, several studies have demonstrated that the final outcome may be at least in part determined by the substrate. The enzyme is still required for efficient catalysis, as indicated by comparisons with nonenzymatic processes, but the ultimate ring topology and stereochemistry is in some of the cases that have been investigated determined by the substrate sequence. As such, similarities exist with protein folding in that lanthipeptide cyclases could serve like folding chaperones to let the substrate adopt its preferred conformation on the enzyme and to then lock this conformation by catalyzing covalent cross-link formation.
Even though the enzymology of lanthipeptide biosynthesis is much better understood than a decade ago,44 many questions remain and new questions have been raised by the discovery of two new pathways and the mechanistic, crystallographic, and bioinformatics studies that have been conducted on the previously known pathways. Further advancement of our understanding will likely require studies on the dynamic interactions of the enzymes and their substrates and investigation of the makeup of the lanthipeptide synthetase complexes. While challenging to study, the lanthipeptide biosynthetic enzymes offer so much promise with respect to bioengineering and synthetic biology applications that continuing investigations are warranted.
Acknowledgments
The authors thank Dr. Brian San Francisco in the laboratory of Professor John A. Gerlt (UIUC) and Dr. Mark Walker (van der Donk group) for help with constructing the SSN figures. The authors also thank Dr. Mark Walker for performing profile HMM alignments for this review. This work was supported by grants from the National Institutes of Health (R37 GM 058822 to W.A.v.d.D., R01 GM 079038 to S.K.N., and F32 GM108275 to L.M.R.).
Glossary
Abbreviations
- α-KG
α-ketoglutarate
- Δcyc
lacking the cyclase domain
- ABC
ATP-binding cassette
- Abu
α-aminobutyric acid
- ADP
adenosine diphosphate
- amf
aerial mycelium formation
- AMP
adenosine monophosphate
- AMS
ABC-transporter maturation and secretion
- ATCC
American Type Culture Collection
- ATP
adenosine triphosphate
- AviCys
S-[(Z)-2-aminovinyl]-d-cysteine
- BLAST
Basic Local Alignment Search Tool
- CD
circular dichroism
- CTP
cytidine triphosphate
- ConFusion
constitutively active fusion
- Dha
2,3-didehydroalanine
- Dhb
(Z)-2,3-didehydrobutyrine
- Dhx
dehydro amino acid
- dNTP
deoxynucleoside triphosphate
- DSM
Deutsche Sammlung von Mikroorganismen
- EFI-EST
Enzyme Function Initiative-Enzyme Similarity Tool
- FADH2
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- GC
gas chromatography
- GluRS
glutamyl-tRNA synthetase
- GSH
glutathione
- GTP
guanosine triphosphate
- HFCD
homo-oligomeric flavin-containing Cys decarboxylase
- HMM
hidden Markov model
- HPLC
high performance liquid chromatography
- KA
kinase-activation
- kcat
catalytic rate constant
- Kd
dissociation constant
- Km
Michaelis constant
- Lac
lactyl
- Lab
labionin
- Lal
lysinoalanine
- Lan
lanthionine
- LanA
generic designation for precursor peptides in lanthipeptide biosynthesis
- LanB
generic designation for lanthipeptide dehydratases
- LanC
generic designation for lanthipeptide cyclases
- LanCL
LanC-like protein
- LanD
generic designation for lanthipeptide oxidative decarboxylases belonging to the HFCD family
- LanE
generic designation for component of the ABC transport complex involved in lanthipeptide self-immunity
- LanF
generic designation for component of the ABC transport complex involved in lanthipeptide self-immunity
- LanG
generic designation for component of the ABC transport complex involved in lanthipeptide self-immunity
- LanI
generic designation for lanthipeptide immunity proteins
- LanJ
generic designation for lanthipeptide dehydrogenases
- LanJA
generic designation for zinc and NADPH-dependent lanthipeptide dehydrogenases
- LanJB
generic designation for flavin-dependent lanthipeptide dehydrogenases
- LanK
generic designation for lanthipeptide regulatory histidine kinases
- LanKC
generic designation of class III bifunctional enzymes catalyzing both dehydration and cyclization reactions
- LanL
generic designation of class IV bifunctional enzymes catalyzing both dehydration and cyclization reactions
- LanM
generic designation of class II bifunctional enzymes catalyzing both dehydration and cyclization reactions
- LanO
generic designation for lanthipeptide oxidoreductases that convert Pyr to Lac
- LanP
generic designation of proteases that remove lanthipeptide leader peptides
- LanR
generic designation for lanthipeptide response regulator protein
- LanT
generic designation of ABC transporters that excrete lanthipeptides after biosynthesis
- LanTp
generic designation of bifunctional ABC transporters that excrete lanthipeptides after biosynthesis and remove the leader peptide
- LC
liquid chromatography
- LP
leader peptide
- MAPK
mitogen activated protein kinase
- MBP
maltose binding protein
- MeLan
methyllanthionine
- MeLab
methyllabionin
- MFS
major facilitator superfamily
- mLanA
generic designation for modified precursor peptides in lanthipeptide biosynthesis
- MS
mass spectrometry
- N11P
Nif11 nitrogen-fixing protein
- NADH
nicotinamide adenine dinucleotide
- NADPH
nicotinamide adenine dinucleotide phosphate
- NBD
nucleotide binding domain
- NDP
nucleoside diphosphate
- NEM
N-ethylmaleimide
- NHLP
nitrile hydratase-like leader peptide
- NMR
nuclear magnetic resonance
- NRPS
nonribosomal peptide synthetase
- NSR
nisin resistance protein
- NTP
nucleoside triphosphate
- NukMC
NukM cyclase domain
- Obu
2-oxobutyryl
- orf
open reading frame
- Pfam
protein family
- PE
phosphatidyl ethanolamine
- Pi
phosphate
- PI3K
phosphoinositide 3-kinase
- PKS
polyketide synthase
- POP
prolyl oligopeptidase
- pSer
phosphoSer
- pThr
phosphoThr
- PSI-BLAST
Position-Specific Iterated BLAST
- PTM
post-translational modification
- Pyr
2-oxopropionyl (pyruvyl)
- ram
rapid aerial mycelium
- RiPPs
ribosomally synthesized and post-translationally modified peptides
- RNase
ribonuclease
- RRE
RiPP Recognition Element
- SaNSR
Streptococcus agalactiae nisin resistance protein
- SAM
S-adenosyl methionine
- SDR
short chain dehydrogenase/reductase
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- Sec
secretory pathway
- SH2
Src homology 2
- SSN
sequence similarity network
- SUMO
small ubiquitin-related modifier
- TMD
transmembrane domain
- TTP
thymidine triphosphate
- UniProtKB
UniProt Knowledgebase
Biographies
Lindsay M. Repka received her B.A. in Chemistry from Barnard College in 2008. She completed her Ph.D. in Chemistry in 2013 under the direction of Prof. Sarah E. Reisman at the California Institute of Technology. Lindsay is currently a postdoctoral researcher in the group of Prof. Wilfred A. van der Donk at the University of Illinois. Her research focuses on investigating the mechanistic details of lanthipeptide biosynthetic enzymes. In 2017, she will begin her independent career as an Assistant Professor of Chemistry at Middlebury College.
Jonathan R. Chekan received his B.S. degree in Microbiology from the Pennsylvania State University in 2011. Under Prof. Satish K. Nair, he obtained his Ph.D. in Biochemistry from the University of Illinois in 2016. He is currently a postdoctoral researcher under Prof. Bradley S. Moore at the Scripps Institution of Oceanography. His research has primarily focused on structural and biochemical studies of enzymes involved in the biosynthesis of RiPP natural products.
Satish K. Nair obtained his Sc.B. (with Honors) in Chemistry from Brown University in 1989. He obtained his Ph.D. in Chemistry (1994) with Prof. David W. Christianson at the University of Pennsylvania. Following postdoctoral studies with Prof. Stephen K. Burley at the Rockefeller University, he began his independent career at the University of Illinois in 2001, where he currently holds the I.C. Gunsalus chair in the Department of Biochemistry. His research focus aims to utilize physical chemical methods, especially X-ray crystallography, to study the function of biosynthetic enzymes and bacterial receptor signaling.
Wilfred A. van der Donk obtained his B.S. and M.S. at Leiden University in The Netherlands under the direction of Jan Reedijk and Willem Driessen. In 1989 he moved to Rice University where he completed his Ph.D. in organic chemistry with Kevin Burgess. After postdoctoral studies with JoAnne Stubbe at MIT, he started his independent career in 1997 at the University of Illinois, where he currently holds the Richard E. Heckert chair in the Department of Chemistry. Since 2008, he is an Investigator of the Howard Hughes Medical Institute.
Author Contributions
∥ L.M.R. and J.R.C. contributed equally.
The authors declare no competing financial interest.
References
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30 (1), 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell N.; Entian K. D.; Schneider U.; Götz F.; Zahner H.; Kellner R.; Jung G. Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature 1988, 333 (6170), 276–278. 10.1038/333276a0. [DOI] [PubMed] [Google Scholar]
- Mayer H.; Bauer H.; Breuss J.; Ziegler S.; Prohaska R. Characterization of rat LANCL1, a novel member of the lanthionine synthetase C-like protein family, highly expressed in testis and brain. Gene 2001, 269 (1–2), 73–80. 10.1016/S0378-1119(01)00463-2. [DOI] [PubMed] [Google Scholar]
- Park S.; James C. D. Lanthionine synthetase components C-like 2 increases cellular sensitivity to adriamycin by decreasing the expression of P-glycoprotein through a transcription-mediated mechanism. Cancer Res. 2003, 63 (3), 723–727. [PubMed] [Google Scholar]
- Mohr K. I.; Volz C.; Jansen R.; Wray V.; Hoffmann J.; Bernecker S.; Wink J.; Gerth K.; Stadler M.; Müller R. Pinensins: the first antifungal lantibiotics. Angew. Chem., Int. Ed. 2015, 54 (38), 11254–11258. 10.1002/anie.201500927. [DOI] [PubMed] [Google Scholar]
- Kodani S.; Hudson M. E.; Durrant M. C.; Buttner M. J.; Nodwell J. R.; Willey J. M. The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (31), 11448–11453. 10.1073/pnas.0404220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodani S.; Lodato M. A.; Durrant M. C.; Picart F.; Willey J. M. SapT, a lanthionine-containing peptide involved in aerial hyphae formation in the streptomycetes. Mol. Microbiol. 2005, 58, 1368–1380. 10.1111/j.1365-2958.2005.04921.x. [DOI] [PubMed] [Google Scholar]
- Férir G.; Petrova M. I.; Andrei G.; Huskens D.; Hoorelbeke B.; Snoeck R.; Vanderleyden J.; Balzarini J.; Bartoschek S.; Brönstrup M.; et al. The lantibiotic peptide labyrinthopeptin A1 demonstrates broad anti-HIV and anti-HSV activity with potential for microbicidal applications. PLoS One 2013, 8 (5), e64010. 10.1371/journal.pone.0064010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio M.; Sasso O.; Maffioli S. I.; Bertorelli R.; Monciardini P.; Sosio M.; Bonezzi F.; Summa M.; Brunati C.; Bordoni R.; et al. A glycosylated, labionin-containing lanthipeptide with marked antinociceptive activity. ACS Chem. Biol. 2014, 9 (2), 398–404. 10.1021/cb400692w. [DOI] [PubMed] [Google Scholar]
- Meindl K.; Schmiederer T.; Schneider K.; Reicke A.; Butz D.; Keller S.; Guhring H.; Vertesy L.; Wink J.; Hoffmann H.; et al. Labyrinthopeptins: a new class of carbacyclic lantibiotics. Angew. Chem., Int. Ed. 2010, 49 (6), 1151–1154. 10.1002/anie.200905773. [DOI] [PubMed] [Google Scholar]
- Grasemann H.; Stehling F.; Brunar H.; Widmann R.; Laliberte T. W.; Molina L.; Doring G.; Ratjen F. Inhalation of Moli1901 in patients with cystic fibrosis. Chest 2007, 131 (5), 1461–1466. 10.1378/chest.06-2085. [DOI] [PubMed] [Google Scholar]
- McNulty M. J.; Hutabarat R. H.; Findlay J. W.; Devereux K.; Knick V. C.; Harvey R. J.; Molina L. Pharmacokinetics and tissue distribution of the nonadecapeptide Moli1901 in rats and mice. Xenobiotica 2003, 33 (2), 197–210. 10.1080/0049825021000022320. [DOI] [PubMed] [Google Scholar]
- Steiner I.; Errhalt P.; Kubesch K.; Hubner M.; Holy M.; Bauer M.; Muller M.; Hinterberger S.; Widmann R.; Mascher D.; et al. Pulmonary pharmacokinetics and safety of nebulized duramycin in healthy male volunteers. Naunyn-Schmiedeberg's Arch. Pharmacol. 2008, 378 (3), 323–333. 10.1007/s00210-008-0293-8. [DOI] [PubMed] [Google Scholar]
- Jones A. M.; Helm J. M. Emerging treatments in cystic fibrosis. Drugs 2009, 69 (14), 1903–1910. 10.2165/11318500-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Oliynyk I.; Varelogianni G.; Roomans G. M.; Johannesson M. Effect of duramycin on chloride transport and intracellular calcium concentration in cystic fibrosis and non-cystic fibrosis epithelia. APMIS 2010, 118 (12), 982–990. 10.1111/j.1600-0463.2010.02680.x. [DOI] [PubMed] [Google Scholar]
- Jabés D.; Brunati C.; Candiani G.; Riva S.; Romanó G.; Donadio S. Efficacy of the new lantibiotic NAI-107 in experimental infections induced by multidrug-resistant Gram-positive pathogens. Antimicrob. Agents Chemother. 2011, 55 (4), 1671–1676. 10.1128/AAC.01288-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther G. S.; Baines S. D.; Todhunter S. L.; Freeman J.; Chilton C. H.; Wilcox M. H. Evaluation of NVB302 versus vancomycin activity in an in vitro human gut model of Clostridium difficile infection. J. Antimicrob. Chemother. 2013, 68 (1), 168–176. 10.1093/jac/dks359. [DOI] [PubMed] [Google Scholar]
- Lepak A. J.; Marchillo K.; Craig W. A.; Andes D. R. In vivo pharmacokinetics and pharmacodynamics of the lantibiotic NAI-107 in a neutropenic murine thigh infection model. Antimicrob. Agents Chemother. 2015, 59 (2), 1258–1264. 10.1128/AAC.04444-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boakes S.; Weiss W. J.; Vinson M.; Wadman S.; Dawson M. J. Antibacterial activity of the novel semisynthetic lantibiotic NVB333 in vitro and in experimental infection models. J. Antibiot. 2016, 69 (12), 850–857. 10.1038/ja.2016.47. [DOI] [PubMed] [Google Scholar]
- Thomsen T. T.; Mojsoska B.; Cruz J. C.; Donadio S.; Jenssen H.; Lobner-Olesen A.; Rewitz K. The lantibiotic NAI-107 efficiently rescues Drosophila melanogaster from infection with methicillin-resistant Staphylococcus aureus USA300. Antimicrob. Agents Chemother. 2016, 60 (9), 5427–5436. 10.1128/AAC.02965-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffioli S. I.; Cruz J. C.; Monciardini P.; Sosio M.; Donadio S. Advancing cell wall inhibitors towards clinical applications. J. Ind. Microbiol. Biotechnol. 2016, 43 (2–3), 177–184. 10.1007/s10295-015-1703-9. [DOI] [PubMed] [Google Scholar]
- Ghobrial O. G.; Derendorf H.; Hillman J. D. Pharmacodynamic activity of the lantibiotic MU1140. Int. J. Antimicrob. Agents 2009, 33 (1), 70–74. 10.1016/j.ijantimicag.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghobrial O. G.; Derendorf H.; Hillman J. D. Development and validation of a LC-MS quantification method for the lantibiotic MU1140 in rat plasma. J. Pharm. Biomed. Anal. 2009, 49 (4), 970–975. 10.1016/j.jpba.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Ghobrial O.; Derendorf H.; Hillman J. D. Human serum binding and its effect on the pharmacodynamics of the lantibiotic MU1140. Eur. J. Pharm. Sci. 2010, 41 (5), 658–664. 10.1016/j.ejps.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Ghobrial O.; Derendorf H.; Hillman J. D. Pharmacokinetic and pharmacodynamic evaluation of the lantibiotic MU1140. J. Pharm. Sci. 2010, 99 (5), 2521–2528. 10.1002/jps.22015. [DOI] [PubMed] [Google Scholar]
- Johnson S. E.; Li Z.; Liu Y.; Moulder J. E.; Zhao M. Whole-body imaging of high-dose ionizing irradiation-induced tissue injuries using 99mTc-duramycin. J. Nucl. Med. 2013, 54 (8), 1397–1403. 10.2967/jnumed.112.112490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S.; Hu K.; Tang G.; Liang X.; Du K.; Nie D.; Jiang S.; Zang L. Positron emission tomography imaging of cell death with [(18)F]FPDuramycin. Apoptosis 2014, 19 (5), 841–850. 10.1007/s10495-013-0964-x. [DOI] [PubMed] [Google Scholar]
- Audi S. H.; Jacobs E. R.; Zhao M.; Roerig D. L.; Haworth S. T.; Clough A. V. In vivo detection of hyperoxia-induced pulmonary endothelial cell death using (99m)Tc-duramycin. Nucl. Med. Biol. 2015, 42 (1), 46–52. 10.1016/j.nucmedbio.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Wang F.; Fang W.; Johnson S. E.; Audi S.; Zimmer M.; Holly T. A.; Lee D. C.; Zhu B.; Zhu H.; et al. The feasibility of imaging myocardial ischemic/reperfusion injury using (99m)Tc-labeled duramycin in a porcine model. Nucl. Med. Biol. 2015, 42 (2), 198–204. 10.1016/j.nucmedbio.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvas F.; Vangestel C.; Rapic S.; Verhaeghe J.; Gray B.; Pak K.; Stroobants S.; Staelens S.; Wyffels L. Characterization of [(99m)Tc]duramycin as a SPECT imaging agent for early assessment of tumor apoptosis. Mol. Imaging Biol. 2015, 17 (6), 838–847. 10.1007/s11307-015-0852-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elvas F.; Vangestel C.; Pak K.; Vermeulen P.; Gray B.; Stroobants S.; Staelens S.; Wyffels L. Early prediction of tumor response to treatment: preclinical validation of 99mTc-duramycin. J. Nucl. Med. 2016, 57 (5), 805–811. 10.2967/jnumed.115.168344. [DOI] [PubMed] [Google Scholar]
- Luo R.; Niu L.; Qiu F.; Fang W.; Fu T.; Zhao M.; Zhang Y. J.; Hua Z. C.; Li X. F.; Wang F.. Monitoring apoptosis of breast cancer xenograft after paclitaxel treatment with 99mTc-Labeled duramycin SPECT/CT. Mol. Imaging 2016, 15, DOI: 10.1177/1536012115624918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medhora M.; Haworth S.; Liu Y.; Narayanan J.; Gao F.; Zhao M.; Audi S.; Jacobs E. R.; Fish B. L.; Clough A. V. Biomarkers for radiation pneumonitis using noninvasive molecular imaging. J. Nucl. Med. 2016, 57 (8), 1296–1301. 10.2967/jnumed.115.160291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Larsen B. T.; Lerman L. O.; Gray B. D.; Barber C.; Hedayat A. F.; Zhao M.; Furenlid L. R.; Pak K. Y.; Woolfenden J. M. Detection of atherosclerotic plaques in ApoE-deficient mice using (99m)Tc-duramycin. Nucl. Med. Biol. 2016, 43 (8), 496–505. 10.1016/j.nucmedbio.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Doroghazi J. R.; Zhao X.; Walker M. C.; van der Donk W. A. Expanded natural product diversity revealed by analysis of lanthipeptide-like gene clusters in Actinobacteria. Appl. Environ. Microbiol. 2015, 81 (13), 4339–4350. 10.1128/AEM.00635-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Yu Y.; Velásquez J. E.; van der Donk W. A. Evolution of lanthipeptide synthetases. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (45), 18361–18366. 10.1073/pnas.1210393109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh A. J.; O’Sullivan O.; Ross R. P.; Cotter P. D.; Hill C. In silico analysis highlights the frequency and diversity of type 1 lantibiotic gene clusters in genome sequenced bacteria. BMC Genomics 2010, 11, 679. 10.1186/1471-2164-11-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.; van der Donk W. A. The sequence of the enterococcal cytolysin imparts unusual lanthionine stereochemistry. Nat. Chem. Biol. 2013, 9 (3), 157–159. 10.1038/nchembio.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G. Lantibiotics-Ribosomally synthesized biologically active polypeptides containing sulfide bridges and α,β-dehydroamino acids. Angew. Chem., Int. Ed. Engl. 1991, 30, 1051–1068. 10.1002/anie.199110513. [DOI] [Google Scholar]
- Siezen R. J.; Kuipers O. P.; de Vos W. M. Comparison of lantibiotic gene clusters and encoded proteins. Antonie van Leeuwenhoek 1996, 69 (2), 171–184. 10.1007/BF00399422. [DOI] [PubMed] [Google Scholar]
- Sahl H. G.; Bierbaum G. Lantibiotics: biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu. Rev. Microbiol. 1998, 52, 41–79. 10.1146/annurev.micro.52.1.41. [DOI] [PubMed] [Google Scholar]
- van Kraaij C.; de Vos W. M.; Siezen R. J.; Kuipers O. P. Lantibiotics: biosynthesis, mode of action and applications. Nat. Prod. Rep. 1999, 16 (5), 575–587. 10.1039/a804531c. [DOI] [PubMed] [Google Scholar]
- McAuliffe O.; Ross R. P.; Hill C. Lantibiotics: structure, biosynthesis and mode of action. FEMS Microbiol. Rev. 2001, 25 (3), 285–308. 10.1111/j.1574-6976.2001.tb00579.x. [DOI] [PubMed] [Google Scholar]
- Chatterjee C.; Paul M.; Xie L.; van der Donk W. A. Biosynthesis and mode of action of lantibiotics. Chem. Rev. 2005, 105, 633–684. 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- Cotter P. D.; Hill C.; Ross R. P. Bacterial lantibiotics: strategies to improve therapeutic potential. Curr. Protein Pept. Sci. 2005, 6 (1), 61–75. 10.2174/1389203053027584. [DOI] [PubMed] [Google Scholar]
- Chandrapati S.; O’Sullivan D. J. Nisin independent induction of the nisA promoter in Lactococcus lactis during growth in lactose or galactose. FEMS Microbiol. Lett. 1999, 170 (1), 191–198. 10.1111/j.1574-6968.1999.tb13374.x. [DOI] [PubMed] [Google Scholar]
- Jack R.; Bierbaum G.; Heidrich C.; Sahl H.-G. The genetics of lantibiotic biosynthesis. BioEssays 1995, 17, 793–802. 10.1002/bies.950170909. [DOI] [PubMed] [Google Scholar]
- Chandrapati S.; O’Sullivan D. J. Characterization of the promoter regions involved in galactose- and nisin-mediated induction of the nisA gene in Lactococcus lactis ATCC 11454. Mol. Microbiol. 2002, 46 (2), 467–477. 10.1046/j.1365-2958.2002.03163.x. [DOI] [PubMed] [Google Scholar]
- Li H.; O’Sullivan D. J. Heterologous expression of the Lactococcus lactis bacteriocin, nisin, in a dairy Enterococcus strain. Appl. Environ. Microbiol. 2002, 68 (7), 3392–3400. 10.1128/AEM.68.7.3392-3400.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaugen M.; Andersen E. L.; Christie V. H.; Nes I. F. Identification, characterization, and expression of a second, bicistronic, operon involved in the production of lactocin S in Lactobacillus sakei L45. Appl. Environ. Microbiol. 2002, 68 (2), 720–727. 10.1128/AEM.68.2.720-727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein T.; Heinzmann S.; Kiesau P.; Himmel B.; Entian K. D. The spa-box for transcriptional activation of subtilin biosynthesis and immunity in Bacillus subtilis. Mol. Microbiol. 2003, 47 (6), 1627–1636. 10.1046/j.1365-2958.2003.03374.x. [DOI] [PubMed] [Google Scholar]
- Schmitz S.; Hoffmann A.; Szekat C.; Rudd B.; Bierbaum G. The lantibiotic mersacidin is an autoinducing peptide. Appl. Environ. Microbiol. 2006, 72 (11), 7270–7277. 10.1128/AEM.00723-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; O’Sullivan D. J. Identification of a nisI promoter within the nisABCTIP operon that may enable establishment of nisin immunity prior to induction of the operon via signal transduction. J. Bacteriol. 2006, 188 (24), 8496–8503. 10.1128/JB.00946-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willey J. M.; van der Donk W. A. Lantibiotics: peptides of diverse structure and function. Annu. Rev. Microbiol. 2007, 61, 477–501. 10.1146/annurev.micro.61.080706.093501. [DOI] [PubMed] [Google Scholar]
- Lubelski J.; Rink R.; Khusainov R.; Moll G. N.; Kuipers O. P. Biosynthesis, immunity, regulation, mode of action and engineering of the model lantibiotic nisin. Cell. Mol. Life Sci. 2008, 65, 455–476. 10.1007/s00018-007-7171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierbaum G.; Sahl H. G. Lantibiotics: mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol. 2009, 10 (1), 2–18. 10.2174/138920109787048616. [DOI] [PubMed] [Google Scholar]
- Foulston L.; Bibb M. Feed-forward regulation of microbisporicin biosynthesis in Microbispora corallina. J. Bacteriol. 2011, 193 (12), 3064–3071. 10.1128/JB.00250-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J.; Teng K.; Liu G.; Qiao C.; Huan L.; Zhong J. Autoregulation of lantibiotic bovicin HJ50 biosynthesis by the BovK-BovR two-component signal transduction system in Streptococcus bovis HJ50. Appl. Environ. Microbiol. 2011, 77 (2), 407–415. 10.1128/AEM.01278-10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lee J. H.; Li X.; O’Sullivan D. J. Transcription analysis of a lantibiotic gene cluster from Bifidobacterium longum DJO10A. Appl. Environ. Microbiol. 2011, 77 (17), 5879–5887. 10.1128/AEM.00571-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaskell A. A.; Giovinazzo J. A.; Fonte V.; Willey J. M. Multi-tier regulation of the streptomycete morphogenetic peptide SapB. Mol. Microbiol. 2012, 84 (3), 501–515. 10.1111/j.1365-2958.2012.08041.x. [DOI] [PubMed] [Google Scholar]
- Sherwood E. J.; Bibb M. J. The antibiotic planosporicin coordinates its own production in the actinomycete. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (27), E2500–2509. 10.1073/pnas.1305392110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Martínez L. T.; Gomez-Escribano J. P.; Bibb M. J. A relA-dependent regulatory cascade for auto-induction of microbisporicin production in Microbispora corallina. Mol. Microbiol. 2015, 97 (3), 502–514. 10.1111/mmi.13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breukink E.; de Kruijff B. Lipid II as a target for antibiotics. Nat. Rev. Drug Discovery 2006, 5 (4), 321–332. 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- Schneider T.; Sahl H. G. Lipid II and other bactoprenol-bound cell wall precursors as drug targets. Curr. Opin. Investig. Drugs 2010, 11 (2), 157–164. [PubMed] [Google Scholar]
- Zhao M. Lantibiotics as probes for phosphatidylethanolamine. Amino Acids 2011, 41 (5), 1071–1079. 10.1007/s00726-009-0386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. R.; Nagao J.; Zendo T.; Sonomoto K. Antimicrobial mechanism of lantibiotics. Biochem. Soc. Trans. 2012, 40 (6), 1528–1533. 10.1042/BST20120190. [DOI] [PubMed] [Google Scholar]
- Barbour A.; Tagg J.; Abou-Zied O. K.; Philip K. New insights into the mode of action of the lantibiotic salivaricin B. Sci. Rep. 2016, 6, 31749. 10.1038/srep31749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers O. P.; Rollema H. S.; Yap W. M.; Boot H. J.; Siezen R. J.; de Vos W. M. Engineering dehydrated amino acid residues in the antimicrobial peptide nisin. J. Biol. Chem. 1992, 267 (34), 24340–24346. [PubMed] [Google Scholar]
- Liu W.; Hansen J. N. Enhancement of the chemical and antimicrobial properties of subtilin by site-directed mutagenesis. J. Biol. Chem. 1992, 267 (35), 25078–25085. [PubMed] [Google Scholar]
- Rollema H. S.; Kuipers O. P.; Both P.; de Vos W. M.; Siezen R. J. Improvement of solubility and stability of the antimicrobial peptide nisin by protein engineering. Appl. Environ. Microbiol. 1995, 61 (8), 2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierbaum G.; Szekat C.; Josten M.; Heidrich C.; Kempter C.; Jung G.; Sahl H. G. Engineering of a novel thioether bridge and role of modified residues in the lantibiotic Pep5. Appl. Environ. Microbiol. 1996, 62 (2), 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers O. P.; Bierbaum G.; Ottenwälder B.; Dodd H. M.; Horn N.; Metzger J.; Kupke T.; Gnau V.; Bongers R.; van den Bogaard P.; et al. Protein engineering of lantibiotics. Antonie van Leeuwenhoek 1996, 69 (2), 161–169. 10.1007/BF00399421. [DOI] [PubMed] [Google Scholar]
- Szekat C.; Jack R. W.; Skutlarek D.; Farber H.; Bierbaum G. Construction of an expression system for site-directed mutagenesis of the lantibiotic mersacidin. Appl. Environ. Microbiol. 2003, 69 (7), 3777–3783. 10.1128/AEM.69.7.3777-3783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink R.; Kuipers A.; de Boef E.; Leenhouts K. J.; Driessen A. J.; Moll G. N.; Kuipers O. P. Lantibiotic structures as guidelines for the design of peptides that can be modified by lantibiotic enzymes. Biochemistry 2005, 44 (24), 8873–8882. 10.1021/bi050081h. [DOI] [PubMed] [Google Scholar]
- Nagao J.; Asaduzzaman S. M.; Aso Y.; Okuda K.; Nakayama J.; Sonomoto K. Lantibiotics: insight and foresight for new paradigm. J. Biosci. Bioeng. 2006, 102 (3), 139–149. 10.1263/jbb.102.139. [DOI] [PubMed] [Google Scholar]
- Cotter P. D.; Deegan L. H.; Lawton E. M.; Draper L. A.; O’Connor P. M.; Hill C.; Ross R. P. Complete alanine scanning of the two-component lantibiotic lacticin 3147: generating a blueprint for rational drug design. Mol. Microbiol. 2006, 62 (3), 735–747. 10.1111/j.1365-2958.2006.05398.x. [DOI] [PubMed] [Google Scholar]
- Nagao J.; Aso Y.; Shioya K.; Nakayama J.; Sonomoto K. Lantibiotic engineering: molecular characterization and exploitation of lantibiotic-synthesizing enzymes for peptide engineering. J. Mol. Microbiol. Biotechnol. 2007, 13 (4), 235–242. 10.1159/000104749. [DOI] [PubMed] [Google Scholar]
- Rink R.; Wierenga J.; Kuipers A.; Kluskens L. D.; Driessen A. J. M.; Kuipers O. P.; Moll G. N. Dissection and modulation of the four distinct activities of nisin by mutagenesis of rings A and B and by C-terminal truncation. Appl. Environ. Microbiol. 2007, 73 (18), 5809–5816. 10.1128/AEM.01104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink R.; Wierenga J.; Kuipers A.; Kluskens L. D.; Driessen A. J. M.; Kuipers O. P.; Moll G. N. Production of dehydroamino acid-containing peptides by Lactococcus lactis. Appl. Environ. Microbiol. 2007, 73 (6), 1792–1796. 10.1128/AEM.02350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink R.; Kluskens L. D.; Kuipers A.; Driessen A. J.; Kuipers O. P.; Moll G. N. NisC, the cyclase of the lantibiotic nisin, can catalyze cyclization of designed nonlantibiotic peptides. Biochemistry 2007, 46, 13179–13189. 10.1021/bi700106z. [DOI] [PubMed] [Google Scholar]
- Levengood M. R.; van der Donk W. A. Use of lantibiotic synthetases for the preparation of bioactive constrained peptides. Bioorg. Med. Chem. Lett. 2008, 18, 3025–3028. 10.1016/j.bmcl.2008.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleyard A. N.; Choi S.; Read D. M.; Lightfoot A.; Boakes S.; Hoffmann A.; Chopra I.; Bierbaum G.; Rudd B. A.; Dawson M. J.; et al. Dissecting structural and functional diversity of the lantibiotic mersacidin. Chem. Biol. 2009, 16 (5), 490–498. 10.1016/j.chembiol.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortés J.; Appleyard A. N.; Dawson M. J. Whole-cell generation of lantibiotic variants. Methods Enzymol. 2009, 458, 559–574. 10.1016/S0076-6879(09)04822-8. [DOI] [PubMed] [Google Scholar]
- Islam M. R.; Shioya K.; Nagao J.; Nishie M.; Jikuya H.; Zendo T.; Nakayama J.; Sonomoto K. Evaluation of essential and variable residues of nukacin ISK-1 by NNK scanning. Mol. Microbiol. 2009, 72 (6), 1438–1447. 10.1111/j.1365-2958.2009.06733.x. [DOI] [PubMed] [Google Scholar]
- Levengood M. R.; Knerr P. J.; Oman T. J.; van der Donk W. A. In vitro mutasynthesis of lantibiotic analogues containing nonproteinogenic amino acids. J. Am. Chem. Soc. 2009, 131 (34), 12024–12025. 10.1021/ja903239s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boakes S.; Cortés J.; Appleyard A. N.; Rudd B. A.; Dawson M. J. Organization of the genes encoding the biosynthesis of actagardine and engineering of a variant generation system. Mol. Microbiol. 2009, 72 (5), 1126–1136. 10.1111/j.1365-2958.2009.06708.x. [DOI] [PubMed] [Google Scholar]
- Rink R.; Arkema-Meter A.; Baudoin I.; Post E.; Kuipers A.; Nelemans S. A.; Akanbi M. H.; Moll G. N. To protect peptide pharmaceuticals against peptidases. J. Pharmacol. Toxicol. Methods 2010, 61 (2), 210–218. 10.1016/j.vascn.2010.02.010. [DOI] [PubMed] [Google Scholar]
- Field D.; Hill C.; Cotter P. D.; Ross R. P. The dawning of a ’Golden era’ in lantibiotic bioengineering. Mol. Microbiol. 2010, 78, 1077–1087. 10.1111/j.1365-2958.2010.07406.x. [DOI] [PubMed] [Google Scholar]
- Moll G. N.; Kuipers A.; Rink R. Microbial engineering of dehydro-amino acids and lanthionines in non-lantibiotic peptides. Antonie van Leeuwenhoek 2010, 97 (4), 319–333. 10.1007/s10482-010-9418-4. [DOI] [PubMed] [Google Scholar]
- Ross A. C.; Vederas J. C. Fundamental functionality: recent developments in understanding the structure-activity relationships of lantibiotic peptides. J. Antibiot. 2011, 64 (1), 27–34. 10.1038/ja.2010.136. [DOI] [PubMed] [Google Scholar]
- Caetano T.; Krawczyk J. M.; Mosker E.; Süssmuth R. D.; Mendo S. Heterologous expression, biosynthesis, and mutagenesis of type II lantibiotics from Bacillus licheniformis in Escherichia coli. Chem. Biol. 2011, 18 (1), 90–100. 10.1016/j.chembiol.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Yang X.; Garg N.; van der Donk W. A. Production of lantipeptides in Escherichia coli. J. Am. Chem. Soc. 2011, 133 (8), 2338–2341. 10.1021/ja109044r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao J.; Shioya K.; Harada Y.; Okuda K.; Zendo T.; Nakayama J.; Sonomoto K. Engineering unusual amino acids into peptides using lantibiotic synthetase. Methods Mol. Biol. 2011, 705, 225–236. 10.1007/978-1-61737-967-3_13. [DOI] [PubMed] [Google Scholar]
- Knerr P. J.; Oman T. J.; Garcia de Gonzalo C.; Lupoli T. J.; Walker S.; van der Donk W. A. Non-proteinogenic amino acids in lacticin 481 analogues result in more potent inhibition of peptidoglycan transglycosylation. ACS Chem. Biol. 2012, 7, 1791–1795. 10.1021/cb300372b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda S.; Cotter P. D.; Hill C.; Ross R. P. Lacticin 3147-biosynthesis, molecular analysis, immunity, bioengineering and applications. Curr. Protein Pept. Sci. 2012, 13 (3), 193–204. 10.2174/138920312800785021. [DOI] [PubMed] [Google Scholar]
- Oldach F.; Al Toma R.; Kuthning A.; Caetano T.; Mendo S.; Budisa N.; Süssmuth R. D. Congeneric lantibiotics from ribosomal in vivo peptide synthesis with noncanonical amino acids. Angew. Chem., Int. Ed. 2012, 51, 415–418. 10.1002/anie.201106154. [DOI] [PubMed] [Google Scholar]
- Boakes S.; Ayala T.; Herman M.; Appleyard A. N.; Dawson M. J.; Cortés J. Generation of an actagardine A variant library through saturation mutagenesis. Appl. Microbiol. Biotechnol. 2012, 15, 1509–1517. 10.1007/s00253-012-4041-0. [DOI] [PubMed] [Google Scholar]
- Montalbán-López M.; Zhou L.; Buivydas A.; van Heel A. J.; Kuipers O. P. Increasing the success rate of lantibiotic drug discovery by synthetic biology. Expert Opin. Drug Discovery 2012, 7 (8), 695–709. 10.1517/17460441.2012.693476. [DOI] [PubMed] [Google Scholar]
- Field D.; Begley M.; O’Connor P. M.; Daly K. M.; Hugenholtz F.; Cotter P. D.; Hill C.; Ross R. P. Bioengineered nisin A derivatives with enhanced activity against both Gram positive and Gram negative pathogens. PLoS One 2012, 7 (10), e46884. 10.1371/journal.pone.0046884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy E. M.; Ross R. P.; Hill C. ’Bac’ to the future: bioengineering lantibiotics for designer purposes. Biochem. Soc. Trans. 2012, 40 (6), 1492–1497. 10.1042/BST20120193. [DOI] [PubMed] [Google Scholar]
- Chen S.; Wilson-Stanford S.; Cromwell W.; Hillman J. D.; Guerrero A.; Allen C. A.; Sorg J. A.; Smith L. Site-directed mutations in the lanthipeptide mutacin 1140. Appl. Environ. Microbiol. 2013, 79 (13), 4015–4023. 10.1128/AEM.00704-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy E. M.; Field D.; PM O. C.; Cotter P. D.; Hill C.; Ross R. P. Saturation mutagenesis of lysine 12 leads to the identification of derivatives of nisin A with enhanced antimicrobial activity. PLoS One 2013, 8 (3), e58530. 10.1371/journal.pone.0058530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano T.; Barbosa J.; Moesker E.; Süssmuth R. D.; Mendo S. Bioengineering of lanthipeptides in Escherichia coli: assessing the specificity of lichenicidin and haloduracin biosynthetic machinery. Res. Microbiol. 2014, 165 (7), 600–604. 10.1016/j.resmic.2014.07.006. [DOI] [PubMed] [Google Scholar]
- Kuthning A.; Mösker E.; Süssmuth R. D. Engineering the heterologous expression of lanthipeptides in Escherichia coli by multigene assembly. Appl. Microbiol. Biotechnol. 2015, 99 (15), 6351–6361. 10.1007/s00253-015-6557-6. [DOI] [PubMed] [Google Scholar]
- Escano J.; Smith L. Multipronged approach for engineering novel peptide analogues of existing lantibiotics. Expert Opin. Drug Discovery 2015, 10 (8), 857–870. 10.1517/17460441.2015.1049527. [DOI] [PubMed] [Google Scholar]
- Kong W.; Kapuganti V. S.; Lu T. A gene network engineering platform for lactic acid bacteria. Nucleic Acids Res. 2016, 44 (4), e37. 10.1093/nar/gkv1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W.; Lu T. Cloning and optimization of a nisin biosynthesis pathway for bacteriocin harvest. ACS Synth. Biol. 2014, 3 (7), 439–445. 10.1021/sb500225r. [DOI] [PubMed] [Google Scholar]
- Kuthning A.; Durkin P.; Oehm S.; Hoesl M. G.; Budisa N.; Süssmuth R. D. Towards biocontained cell factories: an evolutionarily adapted Escherichia coli strain produces a new-to-nature bioactive lantibiotic containing thienopyrrole-alanine. Sci. Rep. 2016, 6, 33447. 10.1038/srep33447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalbán-López M.; van Heel A. J.; Kuipers O. P.. Employing the promiscuity of lantibiotic biosynthetic machineries to produce novel antimicrobials. FEMS Microbiol. Rev. 2016, DOI: 10.1093/femsre/fuw034. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Shao J.; Li Q.; van Heel A. J.; de Vries M. P.; Broos J.; Kuipers O. P. Incorporation of tryptophan analogues into the lantibiotic nisin. Amino Acids 2016, 48 (5), 1309–1318. 10.1007/s00726-016-2186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; van Heel A. J.; Montalbán-López M.; Kuipers O. P. Potentiating the activity of nisin against Escherichia coli. Front. Cell Dev. Biol. 2016, 4, 7. 10.3389/fcell.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroghazi J. R.; Albright J. C.; Goering A. W.; Ju K. S.; Haines R. R.; Tchalukov K. A.; Labeda D. P.; Kelleher N. L.; Metcalf W. W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014, 10 (11), 963–968. 10.1038/nchembio.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimermancic P.; Medema M. H.; Claesen J.; Kurita K.; Wieland Brown L. C.; Mavrommatis K.; Pati A.; Godfrey P. A.; Koehrsen M.; Clardy J.; et al. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 2014, 158 (2), 412–421. 10.1016/j.cell.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. J.; Guinane C. M.; Hill C.; Ross R. P.; O’Toole P. W.; Cotter P. D. In silico identification of bacteriocin gene clusters in the gastrointestinal tract, based on the Human Microbiome Project’s reference genome database. BMC Microbiol. 2015, 15, 183. 10.1186/s12866-015-0515-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinnider M. A.; Johnston C. W.; Edgar R. E.; Dejong C. A.; Merwin N. J.; Rees P. N.; Magarvey N. A. Genomic charting of ribosomally synthesized natural product chemical space facilitates targeted mining. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (42), E6343–E6351. 10.1073/pnas.1609014113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velásquez J. E.; van der Donk W. A. Genome mining for ribosomally synthesized natural products. Curr. Opin. Chem. Biol. 2011, 15 (1), 11–21. 10.1016/j.cbpa.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong A.; van Heel A. J.; Kok J.; Kuipers O. P. BAGEL2: mining for bacteriocins in genomic data. Nucleic Acids Res. 2010, 38, W647–651. 10.1093/nar/gkq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M. H.; Blin K.; Cimermancic P.; de Jager V.; Zakrzewski P.; Fischbach M. A.; Weber T.; Takano E.; Breitling R. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339. 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten R. D.; Yang Y.-L.; Xu Y.; Cimermancic P.; Nam S.-J.; Fenical W.; Fischbach M. A.; Moore B. S.; Dorrestein P. C. A mass spectrometry–guided genome mining approach for natural product peptidogenomics. Nat. Chem. Biol. 2011, 7, 794–802. 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel A. J.; de Jong A.; Montalbán-López M.; Kok J.; Kuipers O. P. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013, 41, W448–453. 10.1093/nar/gkt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H.; Kersten R. D.; Liu W. T.; Wang M.; Purvine S. O.; Wu S.; Brewer H. M.; Pasa-Tolic L.; Bandeira N.; Moore B. S.; et al. Automated genome mining of ribosomal peptide natural products. ACS Chem. Biol. 2014, 9 (7), 1545–1551. 10.1021/cb500199h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M.; Sareen D. Novel LanT associated lantibiotic clusters identified by genome database mining. PLoS One 2014, 9 (3), e91352. 10.1371/journal.pone.0091352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T.; Blin K.; Duddela S.; Krug D.; Kim H. U.; Bruccoleri R.; Lee S. Y.; Fischbach M. A.; Müller R.; Wohlleben W.; et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43 (W1), W237–243. 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClerren A. L.; Cooper L. E.; Quan C.; Thomas P. M.; Kelleher N. L.; van der Donk W. A. Discovery and in vitro biosynthesis of haloduracin, a two-component lantibiotic. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (46), 17243–17248. 10.1073/pnas.0606088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton E. M.; Cotter P. D.; Hill C.; Ross R. P. Identification of a novel two-peptide lantibiotic, Haloduracin, produced by the alkaliphile Bacillus halodurans C-125. FEMS Microbiol. Lett. 2007, 267, 64–71. 10.1111/j.1574-6968.2006.00539.x. [DOI] [PubMed] [Google Scholar]
- Begley M.; Cotter P. D.; Hill C.; Ross R. P. Identification of a novel two-peptide lantibiotic, lichenicidin, following rational genome mining for LanM proteins. Appl. Environ. Microbiol. 2009, 75 (17), 5451–5460. 10.1128/AEM.00730-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dischinger J.; Josten M.; Szekat C.; Sahl H. G.; Bierbaum G. Production of the novel two-peptide lantibiotic lichenicidin by Bacillus licheniformis DSM 13. PLoS One 2009, 4 (8), e6788. 10.1371/journal.pone.0006788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y.; Li B.; Claesen J.; Shi Y.; Bibb M. J.; van der Donk W. A. Discovery of unique lanthionine synthetases reveals new mechanistic and evolutionary insights. PLoS Biol. 2010, 8 (3), e1000339. 10.1371/journal.pbio.1000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Sher D.; Kelly L.; Shi Y.; Huang K.; Knerr P. J.; Joewono I.; Rusch D.; Chisholm S. W.; van der Donk W. A. Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (23), 10430–10435. 10.1073/pnas.0913677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; van der Donk W. A. Biosynthesis of the class III lantipeptide catenulipeptin. ACS Chem. Biol. 2012, 7, 1529–1535. 10.1021/cb3002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk B.; Völler G. H.; Völler J.; Ensle P.; Süssmuth R. D. Curvopeptin: a new lanthionine-containing class III lantibiotic and its co-substrate promiscuous synthetase. ChemBioChem 2012, 13 (14), 2065–2071. 10.1002/cbic.201200417. [DOI] [PubMed] [Google Scholar]
- Völler G. H.; Krawczyk J. M.; Pesic A.; Krawczyk B.; Nachtigall J.; Süssmuth R. D. Characterization of new class III lantibiotics-erythreapeptin, avermipeptin and griseopeptin from Saccharopolyspora erythraea, Streptomyces avermitilis and Streptomyces griseus demonstrates stepwise N-terminal leader processing. ChemBioChem 2012, 13, 1174–1183. 10.1002/cbic.201200118. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zhang L.; Teng K.; Sun S.; Sun Z.; Zhong J. Cerecidins, novel lantibiotics from Bacillus cereus with potent antimicrobial activity. Appl. Environ. Microbiol. 2014, 80 (8), 2633–2643. 10.1128/AEM.03751-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iftime D.; Jasyk M.; Kulik A.; Imhoff J. F.; Stegmann E.; Wohlleben W.; Süssmuth R. D.; Weber T. Streptocollin, a type IV lanthipeptide produced by Streptomyces collinus Tü 365. ChemBioChem 2015, 16 (18), 2615–2623. 10.1002/cbic.201500377. [DOI] [PubMed] [Google Scholar]
- Wang J.; Ma H.; Ge X.; Zhang J.; Teng K.; Sun Z.; Zhong J. Bovicin HJ50-like lantibiotics, a novel subgroup of lantibiotics featured by an indispensable disulfide bridge. PLoS One 2014, 9 (5), e97121. 10.1371/journal.pone.0097121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel A. J.; Kloosterman T. G.; Montalbán-López M.; Deng J.; Plat A.; Baudu B.; Hendriks D.; Moll G. N.; Kuipers O. P. Discovery, production and modification of five novel lantibiotics using the promiscuous nisin modification machinery. ACS Synth. Biol. 2016, 5, 1146–1154. 10.1021/acssynbio.6b00033. [DOI] [PubMed] [Google Scholar]
- Zhao X.; van der Donk W. A. Structural characterization and bioactivity analysis of the two-component lantibiotic Flv system from a ruminant bacterium. Cell Chem. Biol. 2016, 23 (2), 246–256. 10.1016/j.chembiol.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Zhang L.; Zhang J.; Ma H.; Sun S.; Zhong J. Semi-in vitro biosynthesis of cryptic lanthipeptide CLA 124 from Streptomyces clavuligerus. Wei Sheng Wu Xue Bao 2015, 55 (11), 1402–1408. [PubMed] [Google Scholar]
- Basi-Chipalu S.; Dischinger J.; Josten M.; Szekat C.; Zweynert A.; Sahl H. G.; Bierbaum G. Pseudomycoicidin, a class II lantibiotic from Bacillus pseudomycoides. Appl. Environ. Microbiol. 2015, 81 (10), 3419–3429. 10.1128/AEM.00299-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh J. A.; Donia M. S.; Schmidt E. W. Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Nat. Prod. Rep. 2009, 26, 537–559. 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega M. A.; van der Donk W. A. New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products. Cell Chem. Biol. 2016, 23 (1), 31–44. 10.1016/j.chembiol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar K. L.; Mitchell D. A. Revealing nature’s synthetic potential through the study of ribosomal natural product biosynthesis. ACS Chem. Biol. 2013, 8 (3), 473–487. 10.1021/cb3005325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. T. Blurring the lines between ribosomal and nonribosomal peptide scaffolds. ACS Chem. Biol. 2014, 9 (8), 1653–1661. 10.1021/cb5003587. [DOI] [PubMed] [Google Scholar]
- Oman T. J.; van der Donk W. A. Follow the leader: the use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 2010, 6, 9–18. 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crone W. J. K.; Leeper F. J.; Truman A. W. Identification and characterisation of the gene cluster for the anti-MRSA antibiotic bottromycin:expanding the biosynthetic diversity of ribosomal peptides. Chem. Sci. 2012, 3, 3516–3521. 10.1039/c2sc21190d. [DOI] [Google Scholar]
- Gomez-Escribano J. P.; Song L.; Bibb M. J.; Challis G. L. Posttranslational β-methylation and macrolactamidination in the biosynthesis of the bottromycin complex of ribosomal peptide antibiotics. Chem. Sci. 2012, 3, 3522–3525. 10.1039/c2sc21183a. [DOI] [Google Scholar]
- Hou Y.; Tianero M. D.; Kwan J. C.; Wyche T. P.; Michel C. R.; Ellis G. A.; Vazquez-Rivera E.; Braun D. R.; Rose W. E.; Schmidt E. W.; et al. Structure and biosynthesis of the antibiotic bottromycin D. Org. Lett. 2012, 14, 5050–5053. 10.1021/ol3022758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L.; Rachid S.; Stadler M.; Wenzel S. C.; Müller R. Synthetic biotechnology to study and engineer ribosomal bottromycin biosynthesis. Chem. Biol. 2012, 19, 1278–1287. 10.1016/j.chembiol.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Sardar D.; Pierce E.; McIntosh J. A.; Schmidt E. W. Recognition sequences and substrate evolution in cyanobactin biosynthesis. ACS Synth. Biol. 2015, 4 (2), 167–176. 10.1021/sb500019b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; van der Donk W. A. Ribosomally synthesized and post-translationally modified peptide natural products: new insights into the role of leader and core peptides during biosynthesis. Chem. - Eur. J. 2013, 19, 7662–7677. 10.1002/chem.201300401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plat A.; Kuipers A.; Rink R.; Moll G. N. Mechanistic aspects of lanthipeptide leaders. Curr. Protein Pept. Sci. 2013, 14, 85–96. 10.2174/1389203711314020001. [DOI] [PubMed] [Google Scholar]
- de Vos W. M.; Jung G.; Sahl H.-G. In Nisin and Novel Lantibiotics; Jung G., Sahl H.-G., Eds.; ESCOM: Leiden, 1991; pp 457–464. [Google Scholar]
- Pag U.; Sahl H. G. Multiple activities in lantibiotics--models for the design of novel antibiotics?. Curr. Pharm. Des. 2002, 8 (9), 815–833. 10.2174/1381612023395439. [DOI] [PubMed] [Google Scholar]
- Rogers L. A. The inhibiting effect of Streptococcus lactis on Lactobacillus bulgaricus. J. Bacteriol. 1928, 16, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E.; Morell J. L. The presence of dehydroalanine in the antibiotic nisin and its relationship to activity. J. Am. Chem. Soc. 1967, 89 (11), 2791–2792. 10.1021/ja00987a084. [DOI] [PubMed] [Google Scholar]
- Gross E.; Morell J. L.; Craig L. C. Dehydroalanyllysine: identical COOH-terminal structures in the peptide antibiotics nisin and subtilin. Proc. Natl. Acad. Sci. U. S. A. 1969, 62 (3), 952–956. 10.1073/pnas.62.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E.; Morell J. L. The structure of nisin. J. Am. Chem. Soc. 1971, 93, 4634–4635. 10.1021/ja00747a073. [DOI] [PubMed] [Google Scholar]
- Ingram L. A ribosomal mechanism for synthesis of peptides related to nisin. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1970, 224 (1), 263–265. 10.1016/0005-2787(70)90642-8. [DOI] [PubMed] [Google Scholar]
- Banerjee S.; Hansen J. N. Structure and expression of a gene encoding the precursor of subtilin, a small protein antibiotic. J. Biol. Chem. 1988, 262, 9508–9514. [PubMed] [Google Scholar]
- Buchman G. W.; Banerjee S.; Hansen J. N. Structure, expression, and evolution of a gene encoding the precursor of nisin, a small protein antibiotic. J. Biol. Chem. 1988, 263 (31), 16260–16266. [PubMed] [Google Scholar]
- Kaletta C.; Entian K. D. Nisin, a peptide antibiotic: cloning and sequencing of the nisA gene and posttranslational processing of its peptide product. J. Bacteriol. 1989, 171 (3), 1597–1601. 10.1128/jb.171.3.1597-1601.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd H. M.; Horn N.; Gasson M. J. Analysis of the genetic determinant for production of the peptide antibiotic nisin. J. Gen. Microbiol. 1990, 136 (3), 555–566. 10.1099/00221287-136-3-555. [DOI] [PubMed] [Google Scholar]
- Schnell N.; Entian K. D.; Götz F.; Horner T.; Kellner R.; Jung G. Structural gene isolation and prepeptide sequence of gallidermin, a new lanthionine containing antibiotic. FEMS Microbiol. Lett. 1989, 58 (2–3), 263–267. 10.1111/j.1574-6968.1989.tb03056.x. [DOI] [PubMed] [Google Scholar]
- Morell J. L.; Gross E. Configuration of the beta-carbon atoms of the beta-methyllanthionine residues in nisin. J. Am. Chem. Soc. 1973, 95 (19), 6480–6481. 10.1021/ja00800a069. [DOI] [PubMed] [Google Scholar]
- Chan W. C.; Bycroft B. W.; Leyland M. L.; Lian L. Y.; Yang J. C.; Roberts G. C. Sequence-specific resonance assignment and conformational analysis of subtilin by 2D NMR. FEBS Lett. 1992, 300 (1), 56–62. 10.1016/0014-5793(92)80163-B. [DOI] [PubMed] [Google Scholar]
- Brötz H.; Josten M.; Wiedemann I.; Schneider U.; Götz F.; Bierbaum G.; Sahl H.-G. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol. Microbiol. 1998, 30 (2), 317–327. 10.1046/j.1365-2958.1998.01065.x. [DOI] [PubMed] [Google Scholar]
- Breukink E.; Wiedemann I.; van Kraaij C.; Kuipers O. P.; Sahl H. G.; de Kruijff B. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 1999, 286 (5448), 2361–2364. 10.1126/science.286.5448.2361. [DOI] [PubMed] [Google Scholar]
- Hasper H. E.; Kramer N. E.; Smith J. L.; Hillman J. D.; Zachariah C.; Kuipers O. P.; de Kruijff B.; Breukink E. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 2006, 313 (5793), 1636–1637. 10.1126/science.1129818. [DOI] [PubMed] [Google Scholar]
- Wakamatsu K.; Choung S. Y.; Kobayashi T.; Inoue K.; Higashijima T.; Miyazawa T. Complex formation of peptide antibiotic Ro09–0198 with lysophosphatidylethanolamine: 1H NMR analyses in dimethyl sulfoxide solution. Biochemistry 1990, 29 (1), 113–118. 10.1021/bi00453a013. [DOI] [PubMed] [Google Scholar]
- Märki F.; Hanni E.; Fredenhagen A.; van Oostrum J. Mode of action of the lanthionine-containing peptide antibiotics duramycin, duramycin B and C, and cinnamycin as indirect inhibitors of phospholipase A2. Biochem. Pharmacol. 1991, 42 (10), 2027–2035. 10.1016/0006-2952(91)90604-4. [DOI] [PubMed] [Google Scholar]
- Velásquez J. E.; Zhang X.; van der Donk W. A. Biosynthesis of the antimicrobial peptide epilancin 15X and its unusual N-terminal lactate moiety. Chem. Biol. 2011, 18, 857–867. 10.1016/j.chembiol.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupke T.; Stevanovic S.; Sahl H. G.; Götz F. Purification and characterization of EpiD, a flavoprotein involved in the biosynthesis of the lantibiotic epidermin. J. Bacteriol. 1992, 174 (16), 5354–5361. 10.1128/jb.174.16.5354-5361.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allgaier H.; Jung G.; Werner R. G.; Schneider U. Elucidation of the structure of epidermin, a ribosomally synthesized, tetracyclic heterodetic polypeptide antibiotic. Angew. Chem., Int. Ed. Engl. 1985, 24, 1051–1053. 10.1002/anie.198510511. [DOI] [Google Scholar]
- Ekkelenkamp M. B.; Hanssen M.; Danny Hsu S. T.; de Jong A.; Milatovic D.; Verhoef J.; van Nuland N. A. Isolation and structural characterization of epilancin 15X, a novel lantibiotic from a clinical strain of Staphylococcus epidermidis. FEBS Lett. 2005, 579 (9), 1917–1922. 10.1016/j.febslet.2005.01.083. [DOI] [PubMed] [Google Scholar]
- Castiglione F.; Lazzarini A.; Carrano L.; Corti E.; Ciciliato I.; Gastaldo L.; Candiani P.; Losi D.; Marinelli F.; Selva E.; et al. Determining the structure and mode of action of microbisporicin, a potent lantibiotic active against multiresistant pathogens. Chem. Biol. 2008, 15 (1), 22–31. 10.1016/j.chembiol.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Kluskens L. D.; Kuipers A.; Rink R.; de Boef E.; Fekken S.; Driessen A. J.; Kuipers O. P.; Moll G. N. Post-translational modification of therapeutic peptides by NisB, the dehydratase of the lantibiotic nisin. Biochemistry 2005, 44 (38), 12827–12834. 10.1021/bi050805p. [DOI] [PubMed] [Google Scholar]
- Li B.; Yu J. P.; Brunzelle J. S.; Moll G. N.; van der Donk W. A.; Nair S. K. Structure and mechanism of the lantibiotic cyclase involved in nisin biosynthesis. Science 2006, 311 (5766), 1464–1467. 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- van der Meer J. R.; Polman J.; Beerthuyzen M. M.; Siezen R. J.; Kuipers O. P.; de Vos W. M. Characterization of the Lactococcus lactis nisin A operon genes nisP, encoding a subtilisin-like serine protease involved in precursor processing, and nisR, encoding a regulatory protein involved in nisin biosynthesis. J. Bacteriol. 1993, 175 (9), 2578–2588. 10.1128/jb.175.9.2578-2588.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koponen O.; Takala T. M.; Saarela U.; Qiao M.; Saris P. E. J. Distribution of the NisI immunity protein and enhancement of nisin activity by the lipid-free NisI. FEMS Microbiol. Lett. 2004, 231 (1), 85–90. 10.1016/S0378-1097(03)00934-0. [DOI] [PubMed] [Google Scholar]
- Stein T.; Heinzmann S.; Solovieva I.; Entian K. D. Function of Lactococcus lactis nisin immunity genes nisI and nisFEG after coordinated expression in the surrogate host Bacillus subtilis. J. Biol. Chem. 2002, 278 (1), 89–94. 10.1074/jbc.M207237200. [DOI] [PubMed] [Google Scholar]
- Schnell N.; Engelke G.; Augustin J.; Rosenstein R.; Ungermann V.; Götz F.; Entian K.-D. Analysis of genes involved in the biosynthesis of the lantibiotic epidermin. Eur. J. Biochem. 1992, 204 (1), 57–68. 10.1111/j.1432-1033.1992.tb16605.x. [DOI] [PubMed] [Google Scholar]
- Engelke G.; Gutowski-Eckel Z.; Hammelmann M.; Entian K.-D. Biosynthesis of the lantibiotic nisin: genomic organization and membrane localization of the NisB protein. Appl. Environ. Microbiol. 1992, 58 (11), 3730–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C.; Kaletta C.; Schnell N.; Entian K.-D. Analysis of genes involved in biosynthesis of the lantibiotic subtilin. Appl. Environ. Microbiol. 1992, 58 (1), 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y. J.; Steen M. T.; Hansen J. N. The subtilin gene of Bacillus subtilis ATCC 6633 is encoded in an operon that contains a homolog of the hemolysin B transport protein. J. Bacteriol. 1992, 174 (4), 1417–1422. 10.1128/jb.174.4.1417-1422.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majchrzykiewicz J. A.; Lubelski J.; Moll G. N.; Kuipers A.; Bijlsma J. J.; Kuipers O. P.; Rink R. Production of a class II two-component lantibiotic of Streptococcus pneumoniae using the class I nisin synthetic machinery and leader sequence. Antimicrob. Agents Chemother. 2010, 54 (4), 1498–1505. 10.1128/AAC.00883-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakicherla A.; Hansen J. N. Role of the leader and structural regions of prelantibiotic peptides as assessed by expressing nisin-subtilin chimeras in Bacillus subtilis 168, and characterization of their physical, chemical, and antimicrobial properties. J. Biol. Chem. 1995, 270 (40), 23533–23539. 10.1074/jbc.270.40.23533. [DOI] [PubMed] [Google Scholar]
- Sen A. K.; Narbad A.; Horn N.; Dodd H. M.; Parr A. J.; Colquhoun I.; Gasson M. J. Post-translational modification of nisin. The involvement of NisB in the dehydration process. Eur. J. Biochem. 1999, 261 (2), 524–532. 10.1046/j.1432-1327.1999.00303.x. [DOI] [PubMed] [Google Scholar]
- Koponen O.; Tolonen M.; Qiao M.; Wahlstrom G.; Helin J.; Saris P. E. J. NisB is required for the dehydration and NisC for the lanthionine formation in the post-translational modification of nisin. Microbiology 2002, 148 (11), 3561–3568. 10.1099/00221287-148-11-3561. [DOI] [PubMed] [Google Scholar]
- van den Berg van Saparoea H. B.; Bakkes P. J.; Moll G. N.; Driessen A. J. Distinct contributions of the nisin biosynthesis enzymes NisB and NisC and transporter NisT to prenisin production by Lactococcus lactis. Appl. Environ. Microbiol. 2008, 74 (17), 5541–5548. 10.1128/AEM.00342-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L.; Chatterjee C.; Balsara R.; Okeley N. M.; van der Donk W. A. Heterologous expression and purification of SpaB involved in subtilin biosynthesis. Biochem. Biophys. Res. Commun. 2002, 295 (4), 952–957. 10.1016/S0006-291X(02)00783-0. [DOI] [PubMed] [Google Scholar]
- Mavaro A.; Abts A.; Bakkes P. J.; Moll G. N.; Driessen A. J.; Smits S. H.; Schmitt L. Substrate recognition and specificity of the NisB protein, the lantibiotic dehydratase involved in nisin biosynthesis. J. Biol. Chem. 2011, 286 (35), 30552–30560. 10.1074/jbc.M111.263210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg N.; Salazar-Ocampo L. M.; van der Donk W. A. In vitro activity of the nisin dehydratase NisB. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (18), 7258–7263. 10.1073/pnas.1222488110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edde B.; Rossier J.; Le Caer J. P.; Desbruyeres E.; Gros F.; Denoulet P. Posttranslational glutamylation of alpha-tubulin. Science 1990, 247 (4938), 83–85. 10.1126/science.1967194. [DOI] [PubMed] [Google Scholar]
- Ortega M. A.; Hao Y.; Zhang Q.; Walker M. C.; van der Donk W. A.; Nair S. K. Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB. Nature 2014, 517 (7535), 509–512. 10.1038/nature13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki T.; Kurokawa Y.; Hayashi S.; Oku N.; Asamizu S.; Igarashi Y.; Onaka H. Insights into the biosynthesis of dehydroalanines in goadsporin. ChemBioChem 2016, 17 (3), 218–223. 10.1002/cbic.201500541. [DOI] [PubMed] [Google Scholar]
- Ortega M. A.; Hao Y.; Walker M. C.; Donadio S.; Sosio M.; Nair S. K.; van der Donk W. A. Structure and tRNA specificity of MibB, a lantibiotic dehydratase from Actinobacteria involved in NAI-107 biosynthesis. Cell Chem. Biol. 2016, 23 (3), 370–380. 10.1016/j.chembiol.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook J.; Feng Z.; Jain S.; Bhat T. N.; Thanki N.; Ravichandran V.; Gilliland G. L.; Bluhm W.; Weissig H.; Greer D. S.; et al. The Protein Data Bank: unifying the archive. Nucleic Acids Res. 2002, 30 (1), 245–248. 10.1093/nar/30.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusainov R.; van Heel A. J.; Lubelski J.; Moll G. N.; Kuipers O. P. Identification of essential amino acid residues in the nisin dehydratase NisB. Front. Microbiol. 2015, 6, 102. 10.3389/fmicb.2015.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi Y.; Kan Y.; Fujii K.; Fujita T.; Harada K.; Naoki H.; Tabata H.; Onaka H.; Furumai T. Goadsporin, a chemical substance which promotes secondary metabolism and morphogenesis in streptomycetes. II. Structure determination. J. Antibiot. 2001, 54 (12), 1045–1053. 10.7164/antibiotics.54.1045. [DOI] [PubMed] [Google Scholar]
- Onaka H.; Nakaho M.; Hayashi K.; Igarashi Y.; Furumai T. Cloning and characterization of the goadsporin biosynthetic gene cluster from Streptomyces sp. TP-A0584. Microbiology 2005, 151 (12), 3923–3933. 10.1099/mic.0.28420-0. [DOI] [PubMed] [Google Scholar]
- Burkhart B. J.; Schwalen C.; Mann G.; Naismith J. H.; Mitchell D. A.. YcaO-dependent posttranslational amide activation: biosynthesis, structure, and function. Chem. Rev. 2017, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson G. A.; Zhang Z.; Tietz J. I.; Mitchell D. A.; van der Donk W. A. In vitro biosynthesis of the core scaffold of the thiopeptide thiomuracin. J. Am. Chem. Soc. 2015, 137 (51), 16012–16015. 10.1021/jacs.5b10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Hudson G. A.; Mahanta N.; Tietz J. I.; van der Donk W. A.; Mitchell D. A. Biosynthetic timing and substrate specificity for the thiopeptide thiomuracin. J. Am. Chem. Soc. 2016, 138, 15511–15514. 10.1021/jacs.6b08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F.; Takala T. M.; Saris P. E. Nisin biosynthesis in vitro. J. Mol. Microbiol. Biotechnol. 2007, 13 (4), 248–254. 10.1159/000104754. [DOI] [PubMed] [Google Scholar]
- Jung G. In Nisin and Novel Lantibiotics; Jung G., Sahl H. G., Eds.; ESCOM: Leiden, The Netherlands, 1991; pp 1–34. [Google Scholar]
- Moutiez M.; Belin P.; Gondry M.. Aminoacyl-tRNA-utilizing enzymes in natural product biosynthesis. Chem. Rev. 2017, 117, DOI: 10.1021/acs.chemrev.6b00523. [DOI] [PubMed] [Google Scholar]
- Gerlt J. A.; Bouvier J. T.; Davidson D. B.; Imker H. J.; Sadkhin B.; Slater D. R.; Whalen K. L. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854 (8), 1019–1037. 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P.; Markiel A.; Ozier O.; Baliga N. S.; Wang J. T.; Ramage D.; Amin N.; Schwikowski B.; Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13 (11), 2498–2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toogood P. L. Model studies of lantibiotic biogenesis. Tetrahedron Lett. 1993, 34, 7833–7836. 10.1016/S0040-4039(00)61488-0. [DOI] [Google Scholar]
- Burrage S.; Raynham T.; Williams G.; Essex J. W.; Allen C.; Cardno M.; Swali V.; Bradley M. Biomimetic synthesis of lantibiotics. Chem. - Eur. J. 2000, 6 (8), 1455–1466. 10.1002/(SICI)1521-3765(20000417)6:8<1455::AID-CHEM1455>3.3.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Okeley N. M.; Zhu Y.; van der Donk W. A. Facile chemoselective synthesis of dehydroalanine-containing peptides. Org. Lett. 2000, 2, 3603–3606. 10.1021/ol006485d. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Gieselman M.; Zhou H.; Averin O.; van der Donk W. A. Biomimetic studies on the mechanism of stereoselective lanthionine formation. Org. Biomol. Chem. 2003, 1, 3304–3315. 10.1039/b304945k. [DOI] [PubMed] [Google Scholar]
- Zhou H.; van der Donk W. A. Biomimetic stereoselective formation of methyllanthionine. Org. Lett. 2002, 4, 1335–1338. 10.1021/ol025629g. [DOI] [PubMed] [Google Scholar]
- Meyer C.; Bierbaum G.; Heidrich C.; Reis M.; Suling J.; Iglesias-Wind M. I.; Kempter C.; Molitor E.; Sahl H. G. Nucleotide sequence of the lantibiotic Pep5 biosynthetic gene cluster and functional analysis of PepP and PepC. Evidence for a role of PepC in thioether formation. Eur. J. Biochem. 1995, 232 (2), 478–489. 10.1111/j.1432-1033.1995.tb20834.x. [DOI] [PubMed] [Google Scholar]
- Augustin J.; Rosenstein R.; Wieland B.; Schneider U.; Schnell N.; Engelke G.; Entian K. D.; Götz F. Genetic analysis of epidermin biosynthetic genes and epidermin-negative mutants of Staphylococcus epidermidis. Eur. J. Biochem. 1992, 204 (3), 1149–1154. 10.1111/j.1432-1033.1992.tb16740.x. [DOI] [PubMed] [Google Scholar]
- Kupke T.; Götz F. Expression, purification, and characterization of EpiC, an enzyme involved in the biosynthesis of the lantibiotic epidermin, and sequence analysis of Staphylococcus epidermidis epiC mutants. J. Bacteriol. 1996, 178, 1335–1340. 10.1128/jb.178.5.1335-1340.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeley N. M.; Paul M.; Stasser J. P.; Blackburn N.; van der Donk W. A. SpaC and NisC, the cyclases involved in subtilin and nisin biosynthesis, are zinc proteins. Biochemistry 2003, 42, 13613–13624. 10.1021/bi0354942. [DOI] [PubMed] [Google Scholar]
- Matthews R. G.; Goulding C. W. Enzyme-catalyzed methyl transfers to thiols: the role of zinc. Curr. Opin. Chem. Biol. 1997, 1 (3), 332–339. 10.1016/S1367-5931(97)80070-1. [DOI] [PubMed] [Google Scholar]
- Rozema D. B.; Poulter C. D. Yeast protein farnesyltransferase. pKas of peptide substrates bound as zinc thiolates. Biochemistry 1999, 38 (40), 13138–13146. 10.1021/bi990794y. [DOI] [PubMed] [Google Scholar]
- Bradshaw J. M.; Waksman G. Molecular recognition by SH2 domains. Adv. Protein Chem. 2002, 61, 161–210. 10.1016/S0065-3233(02)61005-8. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Wang L.; Liu Y.; Xu J.; Zhu G.; Cang H.; Li X.; Bartlam M.; Hensley K.; Li G.; et al. Structure of human lanthionine synthetase C-like protein 1 and its interaction with Eps8 and glutathione. Genes Dev. 2009, 23 (12), 1387–1392. 10.1101/gad.1789209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich M.; Entian K. D.; Stein T. Structure-function relationships of the lanthionine cyclase SpaC involved in biosynthesis of the Bacillus subtilis peptide antibiotic subtilin. Biochemistry 2007, 46 (11), 3224–3233. 10.1021/bi062124f. [DOI] [PubMed] [Google Scholar]
- Li B.; van der Donk W. A. Identification of essential catalytic residues of the cyclase NisC involved in the biosynthesis of nisin. J. Biol. Chem. 2007, 282, 21169–21175. 10.1074/jbc.M701802200. [DOI] [PubMed] [Google Scholar]
- Hightower K. E.; Huang C. C.; Casey P. J.; Fierke C. A. H-Ras peptide and protein substrates bind protein farnesyltransferase as an ionized thiolate. Biochemistry 1998, 37 (44), 15555–15562. 10.1021/bi981525v. [DOI] [PubMed] [Google Scholar]
- Hightower K. E.; Fierke C. A. Zinc-catalyzed sulfur alkylation: insights from protein farnesyltransferase. Curr. Opin. Chem. Biol. 1999, 3 (2), 176–181. 10.1016/S1367-5931(99)80030-1. [DOI] [PubMed] [Google Scholar]
- Wilker J. J.; Lippard S. J. Alkyl transfer to metal thiolates: kinetics, active species identification, and relevance to the DNA methyl phosphotriester repair center of Escherichia coli Ada. Inorg. Chem. 1997, 36 (6), 969–978. 10.1021/ic961082o. [DOI] [PubMed] [Google Scholar]
- Warthen C. R.; Hammes B. S.; Carrano C. J.; Crans D. C. Methylation of neutral pseudotetrahedral zinc thiolate complexes: model reactions for alkyl group transfer to sulfur by zinc-containing enzymes. JBIC, J. Biol. Inorg. Chem. 2001, 6 (1), 82–90. 10.1007/s007750000171. [DOI] [PubMed] [Google Scholar]
- Brand U.; Rombach M.; Seebacher J.; Vahrenkamp H. Functional modeling of cobalamine-independent methionine synthase with pyrazolylborate-zinc-thiolate complexes. Inorg. Chem. 2001, 40 (24), 6151–6157. 10.1021/ic0105112. [DOI] [PubMed] [Google Scholar]
- Chiou S. J.; Riordan C. G.; Rheingold A. L. Synthetic modeling of zinc thiolates: Quantitative assessment of hydrogen bonding in modulating sulfur alkylation rates. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (7), 3695–3700. 10.1073/pnas.0637221100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilker J. J.; Lippard S. J. Modeling the DNA methylphosphotriester repair site in Escherichia coli Ada. Why zinc and four cysteines?. J. Am. Chem. Soc. 1995, 117 (33), 8682–8683. 10.1021/ja00138a031. [DOI] [Google Scholar]
- Chiou S.-J.; Innocent J.; Riordan C. G.; Lam K.-C.; Liable-Sands L.; Rheingold A. L. Synthetic models for the zinc sites in the methionine synthases. Inorg. Chem. 2000, 39 (19), 4347–4353. 10.1021/ic000505q. [DOI] [PubMed] [Google Scholar]
- Parkin G. Synthetic analogues relevant to the structure and function of zinc enzymes. Chem. Rev. 2004, 104 (2), 699–767. 10.1021/cr0206263. [DOI] [PubMed] [Google Scholar]
- Morlok M. M.; Janak K. E.; Zhu G.; Quarless D. A.; Parkin G. Intramolecular N-H···S hydrogen bonding in the zinc thiolate complex [Tm(Ph)]ZnSCH2C(O)NHPh: a mechanistic investigation of thiolate alkylation as probed by kinetics studies and by kinetic isotope effects. J. Am. Chem. Soc. 2005, 127 (40), 14039–14050. 10.1021/ja0536670. [DOI] [PubMed] [Google Scholar]
- Ibrahim M. M.; Seebacher J.; Steinfeld G.; Vahrenkamp H. Tris(thioimidazolyl)borate-zinc-thiolate complexes for the modeling of biological thiolate alkylations. Inorg. Chem. 2005, 44 (23), 8531–8538. 10.1021/ic0508270. [DOI] [PubMed] [Google Scholar]
- Ji M.; Benkmil B.; Vahrenkamp H. Zinc-thiolate complexes of the bis(pyrazolyl) (thioimidazolyl)hydroborate tripods for the modeling of thiolate alkylating enzymes. Inorg. Chem. 2005, 44 (10), 3518–3523. 10.1021/ic0484147. [DOI] [PubMed] [Google Scholar]
- Melnick J. G.; Zhu G.; Buccella D.; Parkin G. Thiolate exchange in [TmR]ZnSR’ complexes and relevance to the mechanisms of thiolate alkylation reactions involving zinc enzymes and proteins. J. Inorg. Biochem. 2006, 100 (5–6), 1147–1154. 10.1016/j.jinorgbio.2005.12.023. [DOI] [PubMed] [Google Scholar]
- Rombach M.; Seebacher J.; Ji M.; Zhang G.; He G.; Ibrahim M. M.; Benkmil B.; Vahrenkamp H. Thiolate alkylation in tripod zinc complexes: a comparative kinetic study. Inorg. Chem. 2006, 45 (11), 4571–4575. 10.1021/ic060301v. [DOI] [PubMed] [Google Scholar]
- Yang X.; van der Donk W. A. Michael-type cyclizations in lantibiotic biosynthesis are reversible. ACS Chem. Biol. 2015, 10 (5), 1234–1238. 10.1021/acschembio.5b00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein T.; Borchert S.; Conrad B.; Feesche J.; Hofemeister B.; Hofemeister J.; Entian K.-D. Two different lantibiotic-like peptides originate from the ericin gene cluster of Bacillus subtilis A1/3. J. Bacteriol. 2002, 184 (6), 1703–1711. 10.1128/JB.184.6.1703-1711.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegers K.; Heinzmann S.; Entian K.-D. Biosynthesis of lantibiotic nisin. Posttranslational modification of its prepeptide occurs at a multimeric membrane-associated lanthionine synthetase complex. J. Biol. Chem. 1996, 271, 12294–12301. 10.1074/jbc.271.21.12294. [DOI] [PubMed] [Google Scholar]
- Kiesau P.; Eikmanns U.; Gutowski-Eckel Z.; Weber S.; Hammelmann M.; Entian K.-D. Evidence for a multimeric subtilin synthetase complex. J. Bacteriol. 1997, 179 (5), 1475–1481. 10.1128/jb.179.5.1475-1481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers A.; Wierenga J.; Rink R.; Kluskens L. D.; Driessen A. J.; Kuipers O. P.; Moll G. N. Sec-mediated transport of post-translationally dehydrated peptides in Lactococcus lactis. Appl. Environ. Microbiol. 2006, 72, 7626–7633. 10.1128/AEM.01802-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers A.; De Boef E.; Rink R.; Fekken S.; Kluskens L. D.; Driessen A. J.; Leenhouts K.; Kuipers O. P.; Moll G. N. NisT, the transporter of the lantibiotic nisin, can transport fully modified, dehydrated and unmodified prenisin and fusions of the leader peptide with non-lantibiotic peptides. J. Biol. Chem. 2004, 279, 22176–22182. 10.1074/jbc.M312789200. [DOI] [PubMed] [Google Scholar]
- Lubelski J.; Khusainov R.; Kuipers O. P. Directionality and coordination of dehydration and ring formation during biosynthesis of the lantibiotic nisin. J. Biol. Chem. 2009, 284 (38), 25962–25972. 10.1074/jbc.M109.026690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Ortega M.; Shi Y.; Wang H.; Melby J. O.; Tang W.; Mitchell D. A.; van der Donk W. A. Structural investigation of ribosomally synthesized natural products by hypothetical structure enumeration and evaluation using tandem MS. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (33), 12031–12036. 10.1073/pnas.1406418111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; van der Donk W. A. Mechanistic studies on the substrate-tolerant lanthipeptide synthetase ProcM. J. Am. Chem. Soc. 2014, 136 (29), 10450–10459. 10.1021/ja504692v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann N. A.; Krawczyk B.; Tietzmann M.; Ensle P.; Süssmuth R. D. Dissecting reactions of nonlinear precursor peptide processing of the class III lanthipeptide curvopeptin. J. Am. Chem. Soc. 2014, 136 (43), 15222–15228. 10.1021/ja5062054. [DOI] [PubMed] [Google Scholar]
- van der Meer J. R.; Rollema H. S.; Siezen R. J.; Beerthuyzen M. M.; Kuipers O. P.; de Vos W. M. Influence of amino acid substitutions in the nisin leader peptide on biosynthesis and secretion of nisin by Lactococcus lactis. J. Biol. Chem. 1994, 269 (5), 3555–3562. [PubMed] [Google Scholar]
- van den Hooven H. W.; Rollema H. S.; Siezen R. J.; Hilbers C. W.; Kuipers O. P. Structural features of the final intermediate in the biosynthesis of the lantibiotic nisin. Influence of the leader peptide. Biochemistry 1997, 36 (46), 14137–14145. 10.1021/bi9713106. [DOI] [PubMed] [Google Scholar]
- Kuipers O. P.; Rollema H. S.; de Vos W. M.; Siezen R. J. Biosynthesis and secretion of a precursor of nisin Z by Lactococcus lactis, directed by the leader peptide of the homologous lantibiotic subtilin from Bacillus subtilis. FEBS Lett. 1993, 330 (1), 23–27. 10.1016/0014-5793(93)80911-D. [DOI] [PubMed] [Google Scholar]
- Plat A.; Kluskens L. D.; Kuipers A.; Rink R.; Moll G. N. Requirements of the engineered leader peptide of nisin for inducing modification, export, and cleavage. Appl. Environ. Microbiol. 2011, 77 (2), 604–611. 10.1128/AEM.01503-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusainov R.; Heils R.; Lubelski J.; Moll G. N.; Kuipers O. P. Determining sites of interaction between prenisin and its modification enzymes NisB and NisC. Mol. Microbiol. 2011, 82, 706–718. 10.1111/j.1365-2958.2011.07846.x. [DOI] [PubMed] [Google Scholar]
- Abts A.; Montalbán-Lopez M.; Kuipers O. P.; Smits S. H.; Schmitt L. NisC binds the FxLx motif of the nisin leader peptide. Biochemistry 2013, 52 (32), 5387–5395. 10.1021/bi4008116. [DOI] [PubMed] [Google Scholar]
- Khusainov R.; Moll G. N.; Kuipers O. P. Identification of distinct nisin leader peptide regions that determine interactions with the modification enzymes NisB and NisC. FEBS Open Bio. 2013, 3, 237–242. 10.1016/j.fob.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khusainov R.; Kuipers O. P. When the leader gets loose: in vivo biosynthesis of a leaderless prenisin is stimulated by a trans-acting leader peptide. ChemBioChem 2012, 13, 2433–2438. 10.1002/cbic.201200437. [DOI] [PubMed] [Google Scholar]
- Khusainov R.; Kuipers O. P. The presence of modifiable residues in the core peptide part of precursor nisin is not crucial for precursor nisin interactions with NisB- and NisC. PLoS One 2013, 8 (9), e74890. 10.1371/journal.pone.0074890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck-Sickinger A. G.; Jung G. In Nisin and Novel Lantibiotics; Jung G., Sahl H.-G., Eds.; ESCOM: Leiden, 1991; pp 218–230. [Google Scholar]
- Koehnke J.; Bent A. F.; Zollman D.; Smith K.; Houssen W. E.; Zhu X.; Mann G.; Lebl T.; Scharff R.; Shirran S.; et al. The cyanobactin heterocyclase enzyme: a processive adenylase that operates with a defined order of reaction. Angew. Chem., Int. Ed. 2013, 52 (52), 13991–13996. 10.1002/anie.201306302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J.; Mann G.; Bent A. F.; Ludewig H.; Shirran S.; Botting C.; Lebl T.; Houssen W. E.; Jaspars M.; Naismith J. H. Structural analysis of leader peptide binding enables leader-free cyanobactin processing. Nat. Chem. Biol. 2015, 11 (8), 558–563. 10.1038/nchembio.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai T. Y.; Yang C. Y.; Shih H. L.; Wang A. H.; Chou S. H. Xanthomonas campestris PqqD in the pyrroloquinoline quinone biosynthesis operon adopts a novel saddle-like fold that possibly serves as a PQQ carrier. Proteins: Struct., Funct., Genet. 2009, 76 (4), 1042–1048. 10.1002/prot.22461. [DOI] [PubMed] [Google Scholar]
- Latham J. A.; Iavarone A. T.; Barr I.; Juthani P. V.; Klinman J. P. PqqD is a novel peptide chaperone that forms a ternary complex with the radical S-adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway. J. Biol. Chem. 2015, 290 (20), 12908–12918. 10.1074/jbc.M115.646521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodge S. V.; Biernat K. A.; Bassett S. J.; Redinbo M. R.; Bowers A. A. Post-translational Claisen condensation and decarboxylation en route to the bicyclic core of pantocin A. J. Am. Chem. Soc. 2016, 138 (17), 5487–5490. 10.1021/jacs.5b13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart B. J.; Hudson G. A.; Dunbar K. L.; Mitchell D. A. A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat. Chem. Biol. 2015, 11 (8), 564–570. 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S. W.; Lackner G.; Morinaka B. I.; Morishita Y.; Asai T.; Riniker S.; Piel J. A lanthipeptide-like N-terminal leader region guides peptide epimerization by radical SAM epimerases: implications for RiPP evolution. Angew. Chem., Int. Ed. 2016, 55 (40), 12330–12333. 10.1002/anie.201602863. [DOI] [PubMed] [Google Scholar]
- Wieckowski B. M.; Hegemann J. D.; Mielcarek A.; Boss L.; Burghaus O.; Marahiel M. A. The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation. FEBS Lett. 2015, 589 (15), 1802–1806. 10.1016/j.febslet.2015.05.032. [DOI] [PubMed] [Google Scholar]
- Escano J.; Stauffer B.; Brennan J.; Bullock M.; Smith L. The leader peptide of mutacin 1140 has distinct structural components compared to related class I lantibiotics. MicrobiologyOpen 2014, 3 (6), 961–972. 10.1002/mbo3.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelski J.; Overkamp W.; Kluskens L. D.; Moll G. N.; Kuipers O. P. Influence of shifting positions of Ser, Thr, and Cys residues in prenisin on the efficiency of modification reactions and on the antimicrobial activities of the modified prepeptides. Appl. Environ. Microbiol. 2008, 74 (15), 4680–4685. 10.1128/AEM.00112-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escano J.; Stauffer B.; Brennan J.; Bullock M.; Smith L. Biosynthesis and transport of the lantibiotic mutacin 1140 produced by Streptococcus mutans. J. Bacteriol. 2015, 197 (7), 1173–1184. 10.1128/JB.02531-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee C.; Patton G. C.; Cooper L.; Paul M.; van der Donk W. A. Engineering dehydro amino acids and thioethers into peptides using lacticin 481 synthetase. Chem. Biol. 2006, 13, 1109–1117. 10.1016/j.chembiol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Bindman N. A.; van der Donk W. A. A general method for fluorescent labeling of the N-termini of lanthipeptides and its application to visualize their cellular localization. J. Am. Chem. Soc. 2013, 135 (28), 10362–10371. 10.1021/ja4010706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller W. M.; Ensle P.; Krawczyk B.; Süssmuth R. D. Leader peptide-directed processing of labyrinthopeptin A2 precursor peptide by the modifying enzyme LabKC. Biochemistry 2011, 50 (39), 8362–8373. 10.1021/bi200526q. [DOI] [PubMed] [Google Scholar]
- Heidrich C.; Pag U.; Josten M.; Metzger J.; Jack R. W.; Bierbaum G.; Jung G.; Sahl H. G. Isolation, characterization, and heterologous expression of the novel lantibiotic epicidin 280 and analysis of its biosynthetic gene cluster. Appl. Environ. Microbiol. 1998, 64 (9), 3140–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Kamp M.; van den Hooven H. W.; Konings R. N. H.; Bierbaum G.; Sahl H.-G.; Kuipers O. P.; Siezen R. J.; de Vos W.; Hilbers C. W.; van de Ven F. J. Elucidation of the primary structure of the lantibiotic epilancin K7 from Staphylococcus epidermis K7. Cloning and characterization of the epilancin-K7-encoding gene and NMR analysis of mature epilancin K7. Eur. J. Biochem. 1995, 230 (2), 587–600. 10.1111/j.1432-1033.1995.tb20600.x. [DOI] [PubMed] [Google Scholar]
- Skaugen M.; Nissenmeyer J.; Jung G.; Stevanovic S.; Sletten K.; Abildgaard C. I. M.; Nes I. F. In vivo conversion of L-serine to D-alanine in a ribosomally synthesized polypeptide. J. Biol. Chem. 1994, 269 (44), 27183–27185. [PubMed] [Google Scholar]
- Mortvedt C. I.; Nissen-Meyer J.; Sletten K.; Nes I. F. Purification and amino acid sequence of lactocin S, a bacteriocin produced by Lactobacillus sake L45. Appl. Environ. Microbiol. 1991, 57 (6), 1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner R.; Jung G.; Josten M.; Kaletta C.; Entian K. D.; Sahl H. G. Pep5: structure elucidation of a large lantibiotic. Angew. Chem. 1989, 101 (5), 618–621. 10.1002/ange.19891010514. [DOI] [Google Scholar]
- van de Kamp M.; van den Hooven H. W.; Konings R. N.; Bierbaum G.; Sahl H. G.; Kuipers O. P.; Siezen R. J.; de Vos W. M.; Hilbers C. W.; van de Ven F. J. Elucidation of the primary structure of the lantibiotic epilancin K7 from Staphylococcus epidermidis K7. Cloning and characterisation of the epilancin-K7-encoding gene and NMR analysis of mature epilancin K7. Eur. J. Biochem. 1995, 230 (2), 587–600. 10.1111/j.1432-1033.1995.tb20600.x. [DOI] [PubMed] [Google Scholar]
- Ortega M. A.; Velásquez J. E.; Garg N.; Zhang Q.; Joyce R. E.; Nair S. K.; van der Donk W. A. Substrate specificity of the lanthipeptide peptidase ElxP and the oxidoreductase ElxO. ACS Chem. Biol. 2014, 9 (8), 1718–1725. 10.1021/cb5002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh K. L.; Jörnvall H.; Persson B.; Oppermann U. Medium- and short-chain dehydrogenase/reductase gene and protein families: the SDR superfamily: functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 2008, 65 (24), 3895–3906. 10.1007/s00018-008-8588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E.; Yousef A. E. Biosynthesis of paenibacillin, a lantibiotic with N-terminal acetylation, by Paenibacillus polymyxa. Microbiol. Res. 2015, 181, 15–21. 10.1016/j.micres.2015.08.001. [DOI] [PubMed] [Google Scholar]
- He Z.; Yuan C.; Zhang L.; Yousef A. E. N-Terminal acetylation in paenibacillin, a novel lantibiotic. FEBS Lett. 2008, 582 (18), 2787–2792. 10.1016/j.febslet.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Moore D.; Hamilton C. M.; Maneewannakul K.; Mintz Y.; Frost L. S.; Ippen-Ihler K. The Escherichia coli K-12 F plasmid gene traX is required for acetylation of F pilin. J. Bacteriol. 1993, 175 (5), 1375–1383. 10.1128/jb.175.5.1375-1383.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupke T.; Götz F. In vivo reaction of affinity-tag-labelled epidermin precursor peptide with flavoenzyme EpiD. FEMS Microbiol. Lett. 1997, 153 (1), 25–32. 10.1111/j.1574-6968.1997.tb10459.x. [DOI] [PubMed] [Google Scholar]
- Kupke T.; Kempter C.; Jung G.; Götz F. Oxidative decarboxylation of peptides catalyzed by flavoprotein EpiD. Determination of substrate specificity using peptide libraries and neutral loss mass spectrometry. J. Biol. Chem. 1995, 270 (19), 11282–11289. 10.1074/jbc.270.19.11282. [DOI] [PubMed] [Google Scholar]
- Kupke T.; Kempter C.; Gnau V.; Jung G.; Götz F. Mass spectroscopic analysis of a novel enzymatic reaction. Oxidative decarboxylation of the lantibiotic precursor peptide EpiA catalyzed by the flavoprotein EpiD. J. Biol. Chem. 1994, 269 (8), 5653–5659. [PubMed] [Google Scholar]
- Kupke T.; Uebele M.; Schmid D.; Jung G.; Blaesse M.; Steinbacher S. Molecular characterization of lantibiotic-synthesizing enzyme EpiD reveals a function for bacterial Dfp proteins in coenzyme A biosynthesis. J. Biol. Chem. 2000, 275 (41), 31838–31846. 10.1074/jbc.M004273200. [DOI] [PubMed] [Google Scholar]
- Blaesse M.; Kupke T.; Huber R.; Steinbacher S. Crystal structure of the peptidyl-cysteine decarboxylase EpiD complexed with a pentapeptide substrate. EMBO J. 2000, 19 (23), 6299–6310. 10.1093/emboj/19.23.6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupke T.; Götz F. The enethiolate anion reaction products of EpiD. pKa value of the enethiol side chain is lower than that of the thiol side chain of peptides. J. Biol. Chem. 1997, 272 (8), 4759–4762. 10.1074/jbc.272.8.4759. [DOI] [PubMed] [Google Scholar]
- Majer F.; Schmid D. G.; Altena K.; Bierbaum G.; Kupke T. The flavoprotein MrsD catalyzes the oxidative decarboxylation reaction involved in formation of the peptidoglycan biosynthesis inhibitor mersacidin. J. Bacteriol. 2002, 184 (5), 1234–1243. 10.1128/JB.184.5.1234-1243.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse M.; Kupke T.; Huber R.; Steinbacher S. Structure of MrsD, an FAD-binding protein of the HFCD family. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2003, 59 (8), 1414–1421. 10.1107/S0907444903011831. [DOI] [PubMed] [Google Scholar]
- Ortega M. A.; Cogan D. P.; Mukherjee S.; Garg N.; Li B.; Thibodeaux G. N.; Maffioli S.; Donadio S.; Sosio M.; Escano J.. et al. Two flavoenzymes catalyze the post-translational generation of 5-chlorotryptophan and 2-aminovinyl-cysteine during NAI-107 biosynthesis. ACS Chem. Biol. 2017, DOI: 10.1021/acschembio.6b01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbacher S.; Hernandez-Acosta P.; Bieseler B.; Blaesse M.; Huber R.; Culianez-Macia F. A.; Kupke T. Crystal structure of the plant PPC decarboxylase AtHAL3a complexed with an ene-thiol reaction intermediate. J. Mol. Biol. 2003, 327 (1), 193–202. 10.1016/S0022-2836(03)00092-5. [DOI] [PubMed] [Google Scholar]
- Strauss E.; Begley T. P. Stereochemical studies on phosphopantothenoylcysteine decarboxylase from Escherichia coli. Bioorg. Med. Chem. Lett. 2003, 13 (3), 339–342. 10.1016/S0960-894X(02)01018-1. [DOI] [PubMed] [Google Scholar]
- Strauss E.; Begley T. P. Mechanistic studies on phosphopantothenoylcysteine decarboxylase. J. Am. Chem. Soc. 2001, 123 (26), 6449–6450. 10.1021/ja016020y. [DOI] [PubMed] [Google Scholar]
- Hernandez-Acosta P.; Schmid D. G.; Jung G.; Culianez-Macia F. A.; Kupke T. Molecular characterization of the Arabidopsis thaliana flavoprotein AtHAL3a reveals the general reaction mechanism of 4′-phosphopantothenoylcysteine decarboxylases. J. Biol. Chem. 2002, 277 (23), 20490–20498. 10.1074/jbc.M201557200. [DOI] [PubMed] [Google Scholar]
- Strauss E.; Zhai H.; Brand L. A.; McLafferty F. W.; Begley T. P. Mechanistic studies on phosphopantothenoylcysteine decarboxylase: trapping of an enethiolate intermediate with a mechanism-based inactivating agent. Biochemistry 2004, 43 (49), 15520–15533. 10.1021/bi048340a. [DOI] [PubMed] [Google Scholar]
- Claesen J.; Bibb M. J. Genome mining and genetic analysis of cypemycin biosynthesis reveal an unusual class of posttranslationally modified peptides. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (37), 16297–16302. 10.1073/pnas.1008608107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesen J.; Bibb M. J. Biosynthesis and regulation of grisemycin, a new member of the linaridin family of ribosomally synthesized peptides produced by Streptomyces griseus IFO 13350. J. Bacteriol. 2011, 193 (10), 2510–2516. 10.1128/JB.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y.; Sasaki K.; Nagai K.; Shin-ya K.; Furihata K. Structure of thioviridamide, a novel apoptosis inducer from Streptomyces olivoviridis. J. Antibiot. 2006, 59 (1), 6–10. 10.1038/ja.2006.2. [DOI] [PubMed] [Google Scholar]
- Izawa M.; Kawasaki T.; Hayakawa Y. Cloning and heterologous expression of the thioviridamide biosynthesis gene cluster from Streptomyces olivoviridis. Appl. Environ. Microbiol. 2013, 79 (22), 7110–7113. 10.1128/AEM.01978-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulston L. C.; Bibb M. J. Microbisporicin gene cluster reveals unusual features of lantibiotic biosynthesis in actinomycetes. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (30), 13461–13466. 10.1073/pnas.1008285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Peé K. H.; Patallo E. P. Flavin-dependent halogenases involved in secondary metabolism in bacteria. Appl. Microbiol. Biotechnol. 2006, 70 (6), 631–641. 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- Cruz J. C.; Iorio M.; Monciardini P.; Simone M.; Brunati C.; Gaspari E.; Maffioli S. I.; Wellington E.; Sosio M.; Donadio S. Brominated variant of the lantibiotic NAI-107 with enhanced antibacterial potency. J. Nat. Prod. 2015, 78 (11), 2642–2647. 10.1021/acs.jnatprod.5b00576. [DOI] [PubMed] [Google Scholar]
- Xie L.; Miller L. M.; Chatterjee C.; Averin O.; Kelleher N. L.; van der Donk W. A. Lacticin 481: in vitro reconstitution of lantibiotic synthetase activity. Science 2004, 303 (5658), 679–681. 10.1126/science.1092600. [DOI] [PubMed] [Google Scholar]
- Brötz H.; Bierbaum G.; Leopold K.; Reynolds P. E.; Sahl H. G. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 1998, 42 (1), 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M. R.; Nishie M.; Nagao J.; Zendo T.; Keller S.; Nakayama J.; Kohda D.; Sahl H. G.; Sonomoto K. Ring A of nukacin ISK-1: a lipid II-binding motif for type-A(II) lantibiotic. J. Am. Chem. Soc. 2012, 134 (8), 3687–3690. 10.1021/ja300007h. [DOI] [PubMed] [Google Scholar]
- Wiedemann I.; Bottiger T.; Bonelli R. R.; Wiese A.; Hagge S. O.; Gutsmann T.; Seydel U.; Deegan L.; Hill C.; Ross P.; et al. The mode of action of the lantibiotic lacticin 3147--a complex mechanism involving specific interaction of two peptides and the cell wall precursor lipid II. Mol. Microbiol. 2006, 61 (2), 285–296. 10.1111/j.1365-2958.2006.05223.x. [DOI] [PubMed] [Google Scholar]
- Breukink E. A lesson in efficient killing from two-component lantibiotics. Mol. Microbiol. 2006, 61 (2), 271–273. 10.1111/j.1365-2958.2006.05239.x. [DOI] [PubMed] [Google Scholar]
- Oman T. J.; Lupoli T. J.; Wang T.-S. A.; Kahne D.; Walker S.; van der Donk W. A. Haloduracin α binds the peptidoglycan precursor lipid II with 2:1 stoichiometry. J. Am. Chem. Soc. 2011, 133, 17544–17547. 10.1021/ja206281k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegers K.; Entian K. D. Genes involved in immunity to the lantibiotic nisin produced by Lactococcus lactis 6F3. Appl. Environ. Microbiol. 1995, 61 (3), 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immonen T.; Saris P. E. Characterization of the nisFEG operon of the nisin Z producing Lactococcus lactis subsp. lactis N8 strain. DNA Sequence 1998, 9 (5–6), 263–274. 10.3109/10425179809008466. [DOI] [PubMed] [Google Scholar]
- Kuipers O. P.; Beerthuyzen M. M.; Siezen R. J.; de Vos W. M. Characterization of the nisin gene cluster nisABTCIPR of Lactococcus lactis. Requirement of expression of the nisA and nisI genes for development of immunity. Eur. J. Biochem. 1993, 216 (1), 281–291. 10.1111/j.1432-1033.1993.tb18143.x. [DOI] [PubMed] [Google Scholar]
- Halami P. M.; Stein T.; Chandrashekar A.; Entian K. D. Maturation and processing of SpaI, the lipoprotein involved in subtilin immunity in Bacillus subtilis ATCC 6633. Microbiol. Res. 2010, 165 (3), 183–189. 10.1016/j.micres.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Reis M.; Eschbach-Bludau M.; Iglesias-Wind M. I.; Kupke T.; Sahl H. G. Producer immunity towards the lantibiotic Pep5: identification of the immunity gene pepI and localization and functional analysis of its gene product. Appl. Environ. Microbiol. 1994, 60 (8), 2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao M.; Immonen T.; Koponen O.; Saris P. E. J. The cellular location and effect on nisin immunity of the NisI protein from Lactococcus lactis N8 expressed in Escherichia coli and L. lactis. FEMS Microbiol. Lett. 1995, 131 (1), 75–80. 10.1111/j.1574-6968.1995.tb07757.x. [DOI] [PubMed] [Google Scholar]
- Saris P. E.; Immonen T.; Reis M.; Sahl H. G. Immunity to lantibiotics. Antonie van Leeuwenhoek 1996, 69 (2), 151–159. 10.1007/BF00399420. [DOI] [PubMed] [Google Scholar]
- Draper L. A.; Ross R. P.; Hill C.; Cotter P. D. Lantibiotic immunity. Curr. Protein Pept. Sci. 2008, 9 (1), 39–49. 10.2174/138920308783565750. [DOI] [PubMed] [Google Scholar]
- Okuda K.; Sonomoto K. Structural and functional diversity of lantibiotic immunity proteins. Curr. Pharm. Biotechnol. 2011, 12 (8), 1231–1239. 10.2174/138920111796117274. [DOI] [PubMed] [Google Scholar]
- Christ N. A.; Bochmann S.; Gottstein D.; Duchardt-Ferner E.; Hellmich U. A.; Dusterhus S.; Kotter P.; Guntert P.; Entian K. D.; Wohnert J. The first structure of a lantibiotic immunity protein, SpaI from Bacillus subtilis, reveals a novel fold. J. Biol. Chem. 2012, 287 (42), 35286–35298. 10.1074/jbc.M112.401620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker C.; Christ N. A.; Duchardt-Ferner E.; Korn S.; Gobl C.; Berninger L.; Dusterhus S.; Hellmich U. A.; Madl T.; Kotter P.; et al. The solution structure of the lantibiotic immunity protein NisI and its interactions with nisin. J. Biol. Chem. 2015, 290 (48), 28869–28886. 10.1074/jbc.M115.679969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi R.; Coles M.; Linke D.; Kulik A.; Nega M.; Wohlleben W.; Stegmann E. Distinct mechanisms contribute to immunity in the lantibiotic NAI-107 producer strain Microbispora ATCC PTA-5024. Environ. Microbiol. 2016, 18 (1), 118–132. 10.1111/1462-2920.12892. [DOI] [PubMed] [Google Scholar]
- Froseth B. R.; McKay L. L. Molecular characterization of the nisin resistance region of Lactococcus lactis subsp. lactis biovar diacetylactis DRC3. Appl. Environ. Microbiol. 1991, 57 (3), 804–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S.; Chen X.; Yang W.; Chen M.; Huan L. Isolation and characterization of a plasmid pTS50, which encodes nisin resistance determinant in Lactococcus lactis TS1640. Wei Sheng Wu Xue Bao 2001, 41 (5), 536–541. [PubMed] [Google Scholar]
- Sun Z.; Zhong J.; Liang X.; Liu J.; Chen X.; Huan L. Novel mechanism for nisin resistance via proteolytic degradation of nisin by the nisin resistance protein NSR. Antimicrob. Agents Chemother. 2009, 53 (5), 1964–1973. 10.1128/AAC.01382-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosa S.; Alkhatib Z.; Smits S. H. NSR from Streptococcus agalactiae confers resistance against nisin and is encoded by a conserved nsr operon. Biol. Chem. 2013, 394 (11), 1543–1549. 10.1515/hsz-2013-0167. [DOI] [PubMed] [Google Scholar]
- Liao D. I.; Qian J.; Chisholm D. A.; Jordan D. B.; Diner B. A. Crystal structures of the photosystem II D1 C-terminal processing protease. Nat. Struct. Biol. 2000, 7 (9), 749–753. 10.1038/78973. [DOI] [PubMed] [Google Scholar]
- Khosa S.; Frieg B.; Mulnaes D.; Kleinschrodt D.; Hoeppner A.; Gohlke H.; Smits S. H. Structural basis of lantibiotic recognition by the nisin resistance protein from Streptococcus agalactiae. Sci. Rep. 2016, 6, 18679. 10.1038/srep18679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widdick D. A.; Dodd H. M.; Barraille P.; White J.; Stein T. H.; Chater K. F.; Gasson M. J.; Bibb M. J. Cloning and engineering of the cinnamycin biosynthetic gene cluster from Streptomyces cinnamoneus cinnamoneus DSM 40005. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (7), 4316–4321. 10.1073/pnas.0230516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doige C. A.; Ames G. F. ATP-dependent transport systems in bacteria and humans: relevance to cystic fibrosis and multidrug resistance. Annu. Rev. Microbiol. 1993, 47, 291–319. 10.1146/annurev.mi.47.100193.001451. [DOI] [PubMed] [Google Scholar]
- Hollenstein K.; Dawson R. J.; Locher K. P. Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007, 17 (4), 412–418. 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Uguen P.; Hindré T.; Didelot S.; Marty C.; Haras D.; Le Pennec J. P.; Vallee-Rehel K.; Dufour A. Maturation by LctT is required for biosynthesis of full-length lantibiotic lacticin 481. Appl. Environ. Microbiol. 2005, 71 (1), 562–565. 10.1128/AEM.71.1.562-565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao M.; Saris P. E. J. Evidence for a role of NisT in transport of the lantibiotic nisin produced by Lactococcus lactis N8. FEMS Microbiol. Lett. 1996, 144 (1), 89–93. 10.1111/j.1574-6968.1996.tb08513.x. [DOI] [PubMed] [Google Scholar]
- Page M. J.; Di Cera E. Serine peptidases: classification, structure and function. Cell. Mol. Life Sci. 2008, 65 (7–8), 1220–1236. 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian S. K.; Ton-That H.; Schneewind O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 2001, 40 (5), 1049–1057. 10.1046/j.1365-2958.2001.02411.x. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Li X.; Li R.; Li S.; Ni H.; Wang H.; Xu H.; Zhou W.; Saris P. E.; Yang W.; et al. Structure of the nisin leader peptidase NisP revealing a C-terminal autocleavage activity. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70 (6), 1499–1505. 10.1107/S1399004714004234. [DOI] [PubMed] [Google Scholar]
- Bryan P.; Alexander P.; Strausberg S.; Schwarz F.; Lan W.; Gilliland G.; Gallagher D. T. Energetics of folding subtilisin BPN′. Biochemistry 1992, 31 (21), 4937–4945. 10.1021/bi00136a003. [DOI] [PubMed] [Google Scholar]
- van der Donk W. A.; Nair S. K. Structure and mechanism of lanthipeptide biosynthetic enzymes. Curr. Opin. Struct. Biol. 2014, 29, 58–66. 10.1016/j.sbi.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Håvarstein L. S.; Diep D. B.; Nes I. F. A family of bacteriocin ABC transporters carry out proteolytic processing of their substrates concomitant with export. Mol. Microbiol. 1995, 16 (2), 229–240. 10.1111/j.1365-2958.1995.tb02295.x. [DOI] [PubMed] [Google Scholar]
- Franke C. M.; Tiemersma J.; Venema G.; Kok J. Membrane topology of the lactococcal bacteriocin ATP-binding cassette transporter protein LcnC. Involvement of LcnC in lactococcin A maturation. J. Biol. Chem. 1999, 274 (13), 8484–8490. 10.1074/jbc.274.13.8484. [DOI] [PubMed] [Google Scholar]
- Rincé A.; Dufour A.; Le Pogam S.; Thuault D.; Bourgeois C. M.; Le Pennec J. P. Cloning, expression, and nucleotide sequence of genes involved in production of lactococcin DR, a bacteriocin from Lactococcus lactis subsp. lactis. Appl. Environ. Microbiol. 1994, 60 (5), 1652–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings N. D.; Waller M.; Barrett A. J.; Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–509. 10.1093/nar/gkt953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes W. L.; Ferretti J. J.; Tagg J. R. Cloning of the gene encoding Streptococcin A-FF22, a novel lantibiotic produced by Streptococcus pyogenes, and determination of its nucleotide sequence. Appl. Environ. Microbiol. 1993, 59 (6), 1969–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross K. F.; Ronson C. W.; Tagg J. R. Isolation and characterization of the lantibiotic salivaricin A and its structural gene salA from Streptococcus salivarius 20P3. Appl. Environ. Microbiol. 1993, 59 (7), 2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore M. S.; Segarra R. A.; Booth M. C.; Bogie C. P.; Hall L. R.; Clewell D. B. Genetic structure of the Enterococcus faecalis plasmid pAD1-encoded cytolytic toxin system and its relationship to lantibiotic determinants. J. Bacteriol. 1994, 176 (23), 7335–7344. 10.1128/jb.176.23.7335-7344.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.; Qi F. X.; Novak J.; Krull R. E.; Caufield P. W. Effect of amino acid substitutions in conserved residues in the leader peptide on biosynthesis of the lantibiotic mutacin II. FEMS Microbiol. Lett. 2001, 195 (2), 139–144. 10.1111/j.1574-6968.2001.tb10511.x. [DOI] [PubMed] [Google Scholar]
- Ishii S.; Yano T.; Hayashi H. Expression and characterization of the peptidase domain of Streptococcus pneumoniae ComA, a bifunctional ATP-binding cassette transporter involved in quorum sensing pathway. J. Biol. Chem. 2006, 281 (8), 4726–4731. 10.1074/jbc.M512516200. [DOI] [PubMed] [Google Scholar]
- Wu K. H.; Tai P. C. Cys32 and His105 are the critical residues for the calcium-dependent cysteine proteolytic activity of CvaB, an ATP-binding cassette transporter. J. Biol. Chem. 2004, 279 (2), 901–909. 10.1074/jbc.M308296200. [DOI] [PubMed] [Google Scholar]
- Ishii S.; Yano T.; Ebihara A.; Okamoto A.; Manzoku M.; Hayashi H. Crystal structure of the peptidase domain of Streptococcus ComA, a bifunctional ATP-binding cassette transporter involved in the quorum-sensing pathway. J. Biol. Chem. 2010, 285 (14), 10777–10785. 10.1074/jbc.M109.093781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furgerson Ihnken L. A.; Chatterjee C.; van der Donk W. A. In vitro reconstitution and substrate specificity of a lantibiotic protease. Biochemistry 2008, 47, 7352–7363. 10.1021/bi800278n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Teng K.; Huan L.; Zhong J. Dissection of the bridging pattern of bovicin HJ50, a lantibiotic containing a characteristic disulfide bridge. Microbiol. Res. 2011, 166 (3), 146–154. 10.1016/j.micres.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Kuipers A.; Meijer-Wierenga J.; Rink R.; Kluskens L. D.; Moll G. N. Mechanistic dissection of the enzyme complexes involved in biosynthesis of lacticin 3147 and nisin. Appl. Environ. Microbiol. 2008, 74 (21), 6591–6597. 10.1128/AEM.01334-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishie M.; Sasaki M.; Nagao J.; Zendo T.; Nakayama J.; Sonomoto K. Lantibiotic transporter requires cooperative functioning of the peptidase domain and the ATP binding domain. J. Biol. Chem. 2011, 286 (13), 11163–11169. 10.1074/jbc.M110.212704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao J.; Aso Y.; Sashihara T.; Shioya K.; Adachi A.; Nakayama J.; Sonomoto K. Localization and interaction of the biosynthetic proteins for the lantibiotic, Nukacin ISK-1. Biosci., Biotechnol., Biochem. 2005, 69 (7), 1341–1347. 10.1271/bbb.69.1341. [DOI] [PubMed] [Google Scholar]
- Nishie M.; Shioya K.; Nagao J.; Jikuya H.; Sonomoto K. ATP-dependent leader peptide cleavage by NukT, a bifunctional ABC transporter, during lantibiotic biosynthesis. J. Biosci. Bioeng. 2009, 108 (6), 460–464. 10.1016/j.jbiosc.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Lin D. Y.; Huang S.; Chen J. Crystal structures of a polypeptide processing and secretion transporter. Nature 2015, 523 (7561), 425–430. 10.1038/nature14623. [DOI] [PubMed] [Google Scholar]
- Holland I. B.; Schmitt L.; Young J. Type 1 protein secretion in bacteria, the ABC-transporter dependent pathway. Mol. Membr. Biol. 2005, 22 (1–2), 29–39. 10.1080/09687860500042013. [DOI] [PubMed] [Google Scholar]
- Pao S. S.; Paulsen I. T.; Saier M. H. Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62 (1), 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saidijam M.; Bettaney K. E.; Leng D.; Ma P.; Xu Z.; Keen J. N.; Rutherford N. G.; Ward A.; Henderson P. J.; Szakonyi G.; et al. The MFS efflux proteins of Gram-positive and Gram-negative bacteria. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 77, 147–166. 10.1002/9780470920541.ch4. [DOI] [PubMed] [Google Scholar]
- Yan N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38 (3), 151–159. 10.1016/j.tibs.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Booth M. C.; Bogie C. P.; Sahl H. G.; Siezen R. J.; Hatter K. L.; Gilmore M. S. Structural analysis and proteolytic activation of Enterococcus faecalis cytolysin, a novel lantibiotic. Mol. Microbiol. 1996, 21 (6), 1175–1184. 10.1046/j.1365-2958.1996.831449.x. [DOI] [PubMed] [Google Scholar]
- Lohans C. T.; Li J. L.; Vederas J. C. Structure and biosynthesis of carnolysin, a homologue of enterococcal cytolysin with D-amino acids. J. Am. Chem. Soc. 2014, 136, 13150–13153. 10.1021/ja5070813. [DOI] [PubMed] [Google Scholar]
- Tang W.; Dong S.-H.; Repka L. M.; He C.; Nair S. K.; van der Donk W. A. Applications of the class II lanthipeptide protease LicP for sequence-specific, traceless peptide bond cleavage. Chem. Sci. 2015, 6, 6270–6279. 10.1039/C5SC02329G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkarev Z. O.; Finkina E. I.; Nurmukhamedova E. K.; Balandin S. V.; Mineev K. S.; Nadezhdin K. D.; Yakimenko Z. A.; Tagaev A. A.; Temirov Y. V.; Arseniev A. S.; et al. Isolation, structure elucidation, and synergistic antibacterial activity of a novel two-component lantibiotic lichenicidin from Bacillus licheniformis VK21. Biochemistry 2010, 49 (30), 6462–6472. 10.1021/bi100871b. [DOI] [PubMed] [Google Scholar]
- Lee M. V.; Ihnken L. A. F.; You Y. O.; McClerren A. L.; van der Donk W. A.; Kelleher N. L. Distributive and directional behavior of lantibiotic synthetases revealed by high-resolution tandem mass spectrometry. J. Am. Chem. Soc. 2009, 131, 12258–12264. 10.1021/ja9033507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeaux C. J.; Ha T.; van der Donk W. A. A price to pay for relaxed substrate specificity: a comparative kinetic analysis of the class II lanthipeptide synthetases ProcM and HalM2. J. Am. Chem. Soc. 2014, 136 (50), 17513–17529. 10.1021/ja5089452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Kelly W. L. Recent advances in thiopeptide antibiotic biosynthesis. Nat. Prod. Rep. 2010, 27 (2), 153–164. 10.1039/B922434C. [DOI] [PubMed] [Google Scholar]
- Freeman M. F.; Gurgui C.; Helf M. J.; Morinaka B. I.; Uria A. R.; Oldham N. J.; Sahl H. G.; Matsunaga S.; Piel J. Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides. Science 2012, 338, 387–390. 10.1126/science.1226121. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Yang X.; Wang H.; van der Donk W. A. High divergence of the precursor peptides in combinatorial lanthipeptide biosynthesis. ACS Chem. Biol. 2014, 9 (11), 2686–2694. 10.1021/cb500622c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Mukherjee S.; van der Donk W. A. Product formation by the promiscuous lanthipeptide synthetase ProcM is under kinetic control. J. Am. Chem. Soc. 2015, 137 (15), 5140–5148. 10.1021/jacs.5b01409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.; van der Donk W. A. Structural characterization of four prochlorosins: a novel class of lantipeptides produced by planktonic marine cyanobacteria. Biochemistry 2012, 51 (21), 4271–4279. 10.1021/bi300255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H.; Basu M. K.; Mitchell D. A. Expansion of ribosomally produced natural products: a nitrile hydratase- and Nif11-related precursor family. BMC Biol. 2010, 8, 70. 10.1186/1741-7007-8-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H.; Basu M. K. Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol. 2011, 193 (11), 2745–2755. 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour A.; Hindré T.; Haras D.; Le Pennec J. P. The biology of the lantibiotics of the lacticin 481 subgroup is coming of age. FEMS Microbiol. Rev. 2007, 31, 134–167. 10.1111/j.1574-6976.2006.00045.x. [DOI] [PubMed] [Google Scholar]
- Chatterjee C.; Miller L. M.; Leung Y. L.; Xie L.; Yi M.; Kelleher N. L.; van der Donk W. A. Lacticin 481 synthetase phosphorylates its substrate during lantibiotic production. J. Am. Chem. Soc. 2005, 127, 15332–15333. 10.1021/ja0543043. [DOI] [PubMed] [Google Scholar]
- Thibodeaux C. J.; Wagoner J.; Yu Y.; van der Donk W. A. Leader peptide establishes dehydration order, promotes efficiency, and ensures fidelity during lacticin 481 biosynthesis. J. Am. Chem. Soc. 2016, 138 (20), 6436–6444. 10.1021/jacs.6b00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uguen P.; Le Pennec J. P.; Dufour A. Lantibiotic biosynthesis: interactions between prelacticin 481 and its putative modification enzyme, LctM. J. Bacteriol. 2000, 182 (18), 5262–5266. 10.1128/JB.182.18.5262-5266.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y. O.; van der Donk W. A. Mechanistic investigations of the dehydration reaction of lacticin 481 synthetase using site-directed mutagenesis. Biochemistry 2007, 46, 5991–6000. 10.1021/bi602663x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y. O.; Levengood M. R.; Ihnken L. A.; Knowlton A. K.; van der Donk W. A. Lacticin 481 synthetase as a general serine/threonine kinase. ACS Chem. Biol. 2009, 4 (5), 379–385. 10.1021/cb800309v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H.; Gao Y.; Zhao F.; Wang J.; Teng K.; Zhang J.; Zhong J. Dissecting the catalytic and substrate binding activity of a class II lanthipeptide synthetase BovM. Biochem. Biophys. Res. Commun. 2014, 450 (2), 1126–1132. 10.1016/j.bbrc.2014.06.129. [DOI] [PubMed] [Google Scholar]
- Thibodeaux G. N.; van der Donk W. A. An engineered lantipeptide synthetase serves as a general leader peptide-dependent kinase. Chem. Commun. 2012, 48 (86), 10615–10617. 10.1039/c2cc34138g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton E. M.; Ross R. P.; Hill C.; Cotter P. D. Two-peptide lantibiotics: a medical perspective. Mini-Rev. Med. Chem. 2007, 7 (12), 1236–1247. 10.2174/138955707782795638. [DOI] [PubMed] [Google Scholar]
- Morgan S. M.; O’Connor P. M.; Cotter P. D.; Ross R. P.; Hill C. Sequential actions of the two component peptides of the lantibiotic lacticin 3147 explain its antimicrobial activity at nanomolar concentrations. Antimicrob. Agents Chemother. 2005, 49 (7), 2606–2611. 10.1128/AAC.49.7.2606-2611.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oman T. J.; van der Donk W. A. Insights into the mode of action of the two-peptide lantibiotic haloduracin. ACS Chem. Biol. 2009, 4, 865–874. 10.1021/cb900194x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuliffe O.; Hill C.; Ross R. P. Each peptide of the two-component lantibiotic lacticin 3147 requires a separate modification enzyme for activity. Microbiology 2000, 146 (9), 2147–2154. 10.1099/00221287-146-9-2147. [DOI] [PubMed] [Google Scholar]
- Thibodeaux G. N.; McClerren A. L.; Ma Y.; Gancayco M. R.; van der Donk W. A. Synergistic binding of the leader and core peptides by the lantibiotic synthetase HalM2. ACS Chem. Biol. 2015, 10 (4), 970–977. 10.1021/cb5009876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox C. R.; Coburn P. S.; Gilmore M. S. Enterococcal cytolysin: a novel two component peptide system that serves as a bacterial defense against eukaryotic and prokaryotic cells. Curr. Protein Pept. Sci. 2005, 6 (1), 77–84. 10.2174/1389203053027557. [DOI] [PubMed] [Google Scholar]
- Van Tyne D.; Martin M. J.; Gilmore M. S. Structure, function, and biology of the Enterococcus faecalis cytolysin. Toxins 2013, 5 (5), 895–911. 10.3390/toxins5050895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S. H.; Tang W.; Lukk T.; Yu Y.; Nair S. K.; van der Donk W. A. The enterococcal cytolysin synthetase has an unanticipated lipid kinase fold. eLife 2015, 4, e07607. 10.7554/eLife.07607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimafuji C.; Noguchi M.; Nishie M.; Nagao J. I.; Shioya K.; Zendo T.; Nakayama J.; Sonomoto K. In vitro catalytic activity of N-terminal and C-terminal domains in NukM, the post-translational modification enzyme of nukacin ISK-1. J. Biosci. Bioeng. 2015, 120 (6), 624–629. 10.1016/j.jbiosc.2015.03.020. [DOI] [PubMed] [Google Scholar]
- Ma H.; Gao Y.; Zhao F.; Zhong J. Individual catalytic activity of two functional domains of bovicin HJ50 synthase BovM. Wei Sheng Wu Xue Bao 2015, 55 (1), 50–58. [PubMed] [Google Scholar]
- Söding J.; Biegert A.; Lupas A. N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33 (Web Server), W244–W248. 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furgerson Ihnken L. A. Ph.D. Thesis; University of Illinois at Urbana-Champaign, 2009. Accessible through https://www.ideals.illinois.edu/. [Google Scholar]
- Tang W.; Jiménez-Osés G.; Houk K. N.; van der Donk W. A. Substrate control in stereoselective lanthionine biosynthesis. Nat. Chem. 2014, 7 (1), 57–64. 10.1038/nchem.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg N.; Goto Y.; Chen T.; van der Donk W. A. Characterization of the stereochemical configuration of lanthionines formed by the lanthipeptide synthetase GeoM. Biopolymers 2016, 106 (6), 834–842. 10.1002/bip.22876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul M.; Patton G. C.; van der Donk W. A. Mutants of the zinc ligands of lacticin 481 synthetase retain dehydration activity but have impaired cyclization activity. Biochemistry 2007, 46, 6268–6276. 10.1021/bi7000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.; Thibodeaux G. N.; van der Donk W. A. The enterococcal cytolysin synthetase coevolves with substrate for stereoselective lanthionine synthesis. ACS Chem. Biol. 2016, 11 (9), 2438–2446. 10.1021/acschembio.6b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Ni W.; van der Donk W. A. On the regioselectivity of thioether formation by lacticin 481 synthetase. Org. Lett. 2007, 9, 3343–3346. 10.1021/ol071301h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knerr P. J.; van der Donk W. A. Chemical synthesis of the lantibiotic lacticin 481 reveals the importance of lanthionine stereochemistry. J. Am. Chem. Soc. 2013, 135 (19), 7094–7097. 10.1021/ja4014024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Huo L.; Thibodeaux G. N.; van der Donk W. A. Synthesis and bioactivity of diastereomers of the virulence lanthipeptide cytolysin. Org. Lett. 2016, 18, 6188. 10.1021/acs.orglett.6b03246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L.; van der Donk W. A. Discovery and characterization of bicereucin, an unusual D-amino acid-containing mixed two-component lantibiotic. J. Am. Chem. Soc. 2016, 138 (16), 5254–5257. 10.1021/jacs.6b02513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk B.; Ensle P.; Müller W. M.; Süssmuth R. D. Deuterium labeled peptides give insights into the directionality of class III lantibiotic synthetase LabKC. J. Am. Chem. Soc. 2012, 134, 9922. 10.1021/ja3040224. [DOI] [PubMed] [Google Scholar]
- Rocap G.; Larimer F. W.; Lamerdin J.; Malfatti S.; Chain P.; Ahlgren N. A.; Arellano A.; Coleman M.; Hauser L.; Hess W. R.; et al. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 2003, 424 (6952), 1042–1047. 10.1038/nature01947. [DOI] [PubMed] [Google Scholar]
- Biller S. J.; Berube P. M.; Lindell D.; Chisholm S. W. Prochlorococcus: the structure and function of collective diversity. Nat. Rev. Microbiol. 2014, 13 (1), 13–27. 10.1038/nrmicro3378. [DOI] [PubMed] [Google Scholar]
- Yu Y.; Zhang Q.; van der Donk W. A. Insights into the evolution of lanthipeptide biosynthesis. Protein Sci. 2013, 22 (11), 1478–1489. 10.1002/pro.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. Ph.D. Thesis; University of Illinois at Urbana-Champaign, June 2015. Accessible through https://www.ideals.illinois.edu/. [Google Scholar]
- Patton G. C.; Paul M.; Cooper L. E.; Chatterjee C.; van der Donk W. A. The importance of the leader sequence for directing lanthionine formation in lacticin 481. Biochemistry 2008, 47, 7342–7351. 10.1021/bi800277d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao J.; Morinaga Y.; Islam M. R.; Asaduzzaman S. M.; Aso Y.; Nakayama J.; Sonomoto K. Mapping and identification of the region and secondary structure required for the maturation of the nukacin ISK-1 prepeptide. Peptides 2009, 30 (8), 1412–1420. 10.1016/j.peptides.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Oman T. J.; Knerr P. J.; Bindman N. A.; Velasquez J. E.; van der Donk W. A. An engineered lantibiotic synthetase that does not require a leader peptide on its substrate. J. Am. Chem. Soc. 2012, 134, 6952–6955. 10.1021/ja3017297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levengood M. R.; Patton G. C.; van der Donk W. A. The leader peptide is not required for post-translational modification by lacticin 481 synthetase. J. Am. Chem. Soc. 2007, 129, 10314–10315. 10.1021/ja072967+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N. I.; Sprules T.; Carpenter M. R.; Cotter P. D.; Hill C.; Ross R. P.; Vederas J. C. Structural characterization of lacticin 3147, a two-peptide lantibiotic with synergistic activity. Biochemistry 2004, 43 (11), 3049–3056. 10.1021/bi0362065. [DOI] [PubMed] [Google Scholar]
- Liu G.; Zhong J.; Ni J.; Chen M.; Xiao H.; Huan L. Characteristics of the bovicin HJ50 gene cluster in Streptococcus bovis HJ50. Microbiology 2009, 155 (2), 584–593. 10.1099/mic.0.022707-0. [DOI] [PubMed] [Google Scholar]
- Kreil G. D-amino acids in animal peptides. Annu. Rev. Biochem. 1997, 66, 337–345. 10.1146/annurev.biochem.66.1.337. [DOI] [PubMed] [Google Scholar]
- Kreil G. Peptides containing a D-amino acid from frogs and molluscs. J. Biol. Chem. 1994, 269 (15), 10967–10970. [PubMed] [Google Scholar]
- Heck S. D.; Siok C. J.; Krapcho K. J.; Kelbaugh P. R.; Thadeio P. F.; Welch M. J.; Williams R. D.; Ganong A. H.; Kelly M. E.; Lanzetti A. J.; et al. Functional consequences of posttranslational isomerization of Ser46 in a calcium channel toxin. Science 1994, 266 (5187), 1065–1068. 10.1126/science.7973665. [DOI] [PubMed] [Google Scholar]
- Jilek A.; Mollay C.; Tippelt C.; Grassi J.; Mignogna G.; Mullegger J.; Sander V.; Fehrer C.; Barra D.; Kreil G. Biosynthesis of a D-amino acid in peptide linkage by an enzyme from frog skin secretions. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (12), 4235–4239. 10.1073/pnas.0500789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikata Y.; Watanabe T.; Teramoto T.; Inoue A.; Kawakami Y.; Nishizawa Y.; Katayama K.; Kuwada M. Isolation and characterization of a peptide isomerase from funnel web spider venom. J. Biol. Chem. 1995, 270 (28), 16719–16723. 10.1074/jbc.270.28.16719. [DOI] [PubMed] [Google Scholar]
- Jungheim L. N.; Shepherd T. A.; Baxter A. J.; Burgess J.; Hatch S. D.; Lubbehusen P.; Wiskerchen M.; Muesing M. A. Potent human immunodeficiency virus type 1 protease inhibitors that utilize noncoded D-amino acids as p-2/p-3 ligands. J. Med. Chem. 1996, 39 (1), 96–108. 10.1021/jm950576c. [DOI] [PubMed] [Google Scholar]
- Tugyi R.; Uray K.; Ivan D.; Fellinger E.; Perkins A.; Hudecz F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (2), 413–418. 10.1073/pnas.0407677102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S.; Gu Z. Q.; Dunn D.; Tao M.; Josef K.; Tripathy R.; Bihovsky R.; Senadhi S. E.; O’Kane T. M.; McKenna B. A.; et al. D-amino acid containing, high-affinity inhibitors of recombinant human calpain I. J. Med. Chem. 1998, 41 (15), 2663–2666. 10.1021/jm980035y. [DOI] [PubMed] [Google Scholar]
- Imperiali B.; Fisher S. L.; Moats R. A.; Prins T. J. A conformational study of peptides with the general structure Ac-L-Xaa-Pro-D-Xaa-L-Xaa-NH2 - spectroscopic evidence for a peptide with significant beta-turn character in water and in dimethyl-sulfoxide. J. Am. Chem. Soc. 1992, 114 (9), 3182–3188. 10.1021/ja00035a002. [DOI] [Google Scholar]
- Cotter P. D.; O’Connor P. M.; Draper L. A.; Lawton E. M.; Deegan L. H.; Hill C.; Ross R. P. Posttranslational conversion of L-serines to D-alanines is vital for optimal production and activity of the lantibiotic lacticin 3147. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (51), 18584–18589. 10.1073/pnas.0509371102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck S. D.; Faraci W. S.; Kelbaugh P. R.; Saccomano N. A.; Thadeio P. F.; Volkmann R. A. Posttranslational amino acid epimerization: enzyme-catalyzed isomerization of amino acid residues in peptide chains. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (9), 4036–4039. 10.1073/pnas.93.9.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murkin A. S.; Tanner M. E. Dehydroalanine-based inhibition of a peptide epimerase from spider venom. J. Org. Chem. 2002, 67, 8389–8394. 10.1021/jo0204653. [DOI] [PubMed] [Google Scholar]
- Morinaka B. I.; Vagstad A. L.; Helf M. J.; Gugger M.; Kegler C.; Freeman M. F.; Bode H. B.; Piel J. Radical S-adenosyl methionine epimerases: regioselective introduction of diverse D-amino acid patterns into peptide natural products. Angew. Chem., Int. Ed. 2014, 53 (32), 8503–8507. 10.1002/anie.201400478. [DOI] [PubMed] [Google Scholar]
- Yang X.; van der Donk W. A. Post-translational introduction of D-alanine into ribosomally synthesized peptides by the dehydroalanine reductase NpnJ. J. Am. Chem. Soc. 2015, 137 (39), 12426–12429. 10.1021/jacs.5b05207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M. P.; Jack R. W.; Josten M.; Sahl H. G.; Jung G.; Ross R. P.; Hill C. Extensive post-translational modification, including serine to D-alanine conversion, in the two-component lantibiotic, lacticin 3147. J. Biol. Chem. 1999, 274 (53), 37544–37550. 10.1074/jbc.274.53.37544. [DOI] [PubMed] [Google Scholar]
- Navaratna M. A.; Sahl H. G.; Tagg J. R. Two-component anti-Staphylococcus aureus lantibiotic activity produced by Staphylococcus aureus C55. Appl. Environ. Microbiol. 1998, 64 (12), 4803–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini A.; Squartini A.; Nuti M. P. Nucleotide sequence and analysis of plasmid pMD136 from Pediococcus pentosaceus FBB61 (ATCC43200) involved in pediocin A production. Plasmid 2000, 43 (2), 111–122. 10.1006/plas.1999.1447. [DOI] [PubMed] [Google Scholar]
- Suda S.; Lawton E. M.; Wistuba D.; Cotter P. D.; Hill C.; Ross R. P. Homologues and bioengineered derivatives of LtnJ vary in ability to form D-alanine in the lantibiotic lacticin 3147. J. Bacteriol. 2012, 194 (3), 708–714. 10.1128/JB.06185-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel A. J.; Mu D.; Montalbán-López M.; Hendriks D.; Kuipers O. P. Designing and producing modified, new-to-nature peptides with antimicrobial activity by use of a combination of various lantibiotic modification enzymes. ACS Synth. Biol. 2013, 2 (7), 397–404. 10.1021/sb3001084. [DOI] [PubMed] [Google Scholar]
- Mu D.; Montalbán-López M.; Deng J.; Kuipers O. P. Substrate selectivity of the lantibiotic reductase LtnJ assessed by a collection of nisin derivatives as substrate. Appl. Environ. Microbiol. 2015, 81 (11), 3679–3687. 10.1128/AEM.00475-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict R. G.; Dvonch W.; Shotwell O. L.; Pridham T.; Lindenfelser L. A. Cinnamycin, an antibiotic from Streptomyces cinnamoneus. Antibiot. Chemother. 1952, 2, 591–594. [PubMed] [Google Scholar]
- Kessler H.; Steuernagel S.; Will M.; Jung G.; Kellner R.; Gillessen D.; Kamiyama T. The structure of the polycyclic nonadecapeptide Ro 09–0198. Helv. Chim. Acta 1988, 71 (8), 1924–1929. 10.1002/hlca.19880710811. [DOI] [Google Scholar]
- Wakamiya T.; Fukase K.; Naruse N.; Konishi M.; Shiba T. Lanthiopeptin, a new peptide effective against herpes-simplex virus - structural determination and comparison with Ro 09–0198, an immunopotentiating peptide. Tetrahedron Lett. 1988, 29 (37), 4771–4772. 10.1016/S0040-4039(00)80604-8. [DOI] [Google Scholar]
- Kaletta C.; Entian K. D.; Jung G. Prepeptide sequence of cinnamycin (Ro 09–0198): the first structural gene of a duramycin-type lantibiotic. Eur. J. Biochem. 1991, 199 (2), 411–415. 10.1111/j.1432-1033.1991.tb16138.x. [DOI] [PubMed] [Google Scholar]
- Kodani S.; Komaki H.; Ishimura S.; Hemmi H.; Ohnishi-Kameyama M. Isolation and structure determination of a new lantibiotic cinnamycin B from Actinomadura atramentaria based on genome mining. J. Ind. Microbiol. Biotechnol. 2016, 43 (8), 1159–1165. 10.1007/s10295-016-1788-9. [DOI] [PubMed] [Google Scholar]
- Kondo S.; Sezaki M.; Shimura M.; Sato K.; Hara T. Leucopeptin, a new peptide antibiotic. J. Antibiot. 1964, 17 (6), 262–263. [PubMed] [Google Scholar]
- Fredenhagen A.; Fendrich G.; Marki F.; Marki W.; Gruner J.; Raschdorf F.; Peter H. H. Duramycins B and C, two new lanthionine containing antibiotics as inhibitors of phospholipase A2. Structural revision of duramycin and cinnamycin. J. Antibiot. 1990, 43 (11), 1403–1412. 10.7164/antibiotics.43.1403. [DOI] [PubMed] [Google Scholar]
- Fredenhagen A.; Maerki F.; Fendrich G.; Maerki W.; Gruner J.; Van Oostrum J.; Raschdorf F.; Peter H. H. In Nisin and Novel Lantibiotics; Jung G., Sahl H.-G., Eds.; ESCOM: Leiden, The Netherlands, 1991; pp 131–140. [Google Scholar]
- Kessler H.; Steuernagel S.; Gillessen D.; Kamiyama T. Complete sequence determination and localization of one imino and three sulfide bridges of the nonadecapeptide Ro 09–0198 by homonuclear 2D-NMR spectroscopy. The DQF-RELAYED-NOESY-experiment. Helv. Chim. Acta 1987, 70 (3), 726–741. 10.1002/hlca.19870700322. [DOI] [Google Scholar]
- Zimmermann N.; Freund S.; Fredenhagen A.; Jung G. Solution structures of the lantibiotics duramycin B and C. Eur. J. Biochem. 1993, 216 (2), 419–428. 10.1111/j.1432-1033.1993.tb18159.x. [DOI] [PubMed] [Google Scholar]
- Hayashi F.; Nagashima K.; Terui Y.; Kawamura Y.; Matsumoto K.; Itazaki H. The structure of PA48009: the revised structure of duramycin. J. Antibiot. 1990, 43 (11), 1421–1430. 10.7164/antibiotics.43.1421. [DOI] [PubMed] [Google Scholar]
- Kessler H.; Seip S.; Wein T.; Steuernagel S.; Will M. In Nisin and Novel Lantibiotics; Jung G., Sahl H.-G., Eds.; ESCOM: Leiden, The Netherlands, 1991; pp 76–90. [Google Scholar]
- Wang Q. P.; VanDusen W. J.; Petroski C. J.; Garsky V. M.; Stern A. M.; Friedman P. A. Bovine liver aspartyl beta-hydroxylase. Purification and characterization. J. Biol. Chem. 1991, 266 (21), 14004–14010. [PubMed] [Google Scholar]
- Friedman M. Chemistry, biochemistry, nutrition, and microbiology of lysinoalanine, lanthionine, and histidinoalanine in food and other proteins. J. Agric. Food Chem. 1999, 47 (4), 1295–1319. 10.1021/jf981000+. [DOI] [PubMed] [Google Scholar]
- Hullin-Matsuda F.; Makino A.; Murate M.; Kobayashi T. Probing phosphoethanolamine-containing lipids in membranes with duramycin/cinnamycin and aegerolysin proteins. Biochimie 2016, 130, 81–90. 10.1016/j.biochi.2016.09.020. [DOI] [PubMed] [Google Scholar]
- Zhao M.; Li Z.; Bugenhagen S. 99mTc-labeled duramycin as a novel phosphatidylethanolamine-binding molecular probe. J. Nucl. Med. 2008, 49 (8), 1345–1352. 10.2967/jnumed.107.048603. [DOI] [PubMed] [Google Scholar]
- Richard A. S.; Zhang A.; Park S. J.; Farzan M.; Zong M.; Choe H. Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (47), 14682–14687. 10.1073/pnas.1508095112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton L. J.; Crow C.; Maraveyas A.; Madden L. A. Duramycin-induced calcium release in cancer cells. Anti-Cancer Drugs 2016, 27 (3), 173–182. 10.1097/CAD.0000000000000313. [DOI] [PubMed] [Google Scholar]
- Ökesli A.; Cooper L. E.; Fogle E. J.; van der Donk W. A. Nine post-translational modifications during the biosynthesis of cinnamycin. J. Am. Chem. Soc. 2011, 133 (34), 13753–13760. 10.1021/ja205783f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L.; Ökesli A.; Zhao M.; van der Donk W. A.. Insights into the biosynthesis of duramycin. Appl. Environ. Microbiol. 2017, DOI: 10.1128/AEM.02698-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somma S.; Merati W.; Parenti F. Gardimycin, a new antibiotic inhibiting peptidoglycan synthesis. Antimicrob. Agents Chemother. 1977, 11 (3), 396–401. 10.1128/AAC.11.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vértesy L.; Aretz W.; Bonnefoy A.; Ehlers E.; Kurz M.; Markus A.; Schiell M.; Vogel M.; Wink J.; Kogler H. Ala(0)-actagardine, a new lantibiotic from cultures of Actinoplanes liguriae ATCC 31048. J. Antibiot. 1999, 52 (8), 730–741. 10.7164/antibiotics.52.730. [DOI] [PubMed] [Google Scholar]
- Zimmermann N.; Metzger J. W.; Jung G. The tetracyclic lantibiotic actagardine. 1H-NMR and 13C-NMR assignments and revised primary structure. Eur. J. Biochem. 1995, 228 (3), 786–797. 10.1111/j.1432-1033.1995.tb20324.x. [DOI] [PubMed] [Google Scholar]
- Zimmermann N.; Jung G. The three-dimensional solution structure of the lantibiotic murein-biosynthesis-inhibitor actagardine determined by NMR. Eur. J. Biochem. 1997, 246 (3), 809–819. 10.1111/j.1432-1033.1997.00809.x. [DOI] [PubMed] [Google Scholar]
- Kettenring J. K.; Malabarba A.; Vekey K.; Cavalleri B. Sequence determination of actagardine, a novel lantibiotic, by homonuclear 2D NMR spectroscopy. J. Antibiot. 1990, 43 (9), 1082–1088. 10.7164/antibiotics.43.1082. [DOI] [PubMed] [Google Scholar]
- Holtsmark I.; Mantzilas D.; Eijsink V. G.; Brurberg M. B. Purification, characterization, and gene sequence of michiganin A, an actagardine-like lantibiotic produced by the tomato pathogen Clavibacter michiganensis subsp. michiganensis. Appl. Environ. Microbiol. 2006, 72 (9), 5814–5821. 10.1128/AEM.00639-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Bueno A.; van der Donk W. A. Heterologous production of the lantibiotic Ala(0)actagardine in Escherichia coli. Chem. Commun. 2012, 48 (89), 10966–10968. 10.1039/c2cc36336d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkel W. J.; Kamerbeek N. M.; Fraaije M. W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 124 (4), 670–689. 10.1016/j.jbiotec.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Boden R.; Borodina E.; Wood A. P.; Kelly D. P.; Murrell J. C.; Schafer H. Purification and characterization of dimethylsulfide monooxygenase from Hyphomicrobium sulfonivorans. J. Bacteriol. 2011, 193 (5), 1250–1258. 10.1128/JB.00977-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger S. K.; Williams D. E. Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol. Ther. 2005, 106 (3), 357–387. 10.1016/j.pharmthera.2005.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuliffe O.; Hill C.; Ross R. P. Identification and overexpression of ltnl, a novel gene which confers immunity to the two-component lantibiotic lacticin 3147. Microbiology 2000, 146 (1), 129–138. 10.1099/00221287-146-1-129. [DOI] [PubMed] [Google Scholar]
- Draper L. A.; Deegan L. H.; Hill C.; Cotter P. D.; Ross R. P. Insights into lantibiotic immunity provided by bioengineering of LtnI. Antimicrob. Agents Chemother. 2012, 56 (10), 5122–5133. 10.1128/AAC.00979-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso Y.; Okuda K.; Nagao J.; Kanemasa Y.; Thi Bich Phuong N.; Koga H.; Shioya K.; Sashihara T.; Nakayama J.; Sonomoto K. A novel type of immunity protein, NukH, for the lantibiotic nukacin ISK-1 produced by Staphylococcus warneri ISK-1. Biosci., Biotechnol., Biochem. 2005, 69 (7), 1403–1410. 10.1271/bbb.69.1403. [DOI] [PubMed] [Google Scholar]
- Okuda K.; Aso Y.; Nagao J.; Shioya K.; Kanemasa Y.; Nakayama J.; Sonomoto K. Characterization of functional domains of lantibiotic-binding immunity protein, NukH, from Staphylococcus warneri ISK-1. FEMS Microbiol. Lett. 2005, 250 (1), 19–25. 10.1016/j.femsle.2005.06.039. [DOI] [PubMed] [Google Scholar]
- Okuda K.; Yanagihara S.; Shioya K.; Harada Y.; Nagao J.; Aso Y.; Zendo T.; Nakayama J.; Sonomoto K. Binding specificity of the lantibiotic-binding immunity protein NukH. Appl. Environ. Microbiol. 2008, 74 (24), 7613–7619. 10.1128/AEM.00789-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke S.; Widdick D.; Bibb M.. A novel mechanism of immunity controls the onset of cinnamycin biosynthesis in Streptomyces cinnamoneus DSM 40646. J. Ind. Microbiol. Biotechnol. 2016, DOI: 10.1007/s10295-016-1869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda K.; Ohya M.; Kohno T.; Maeda T.; Endo S.; Wakamatsu K. Structure determination of an immunopotentiator peptide, cinnamycin, complexed with lysophosphatidylethanolamine by 1H-NMR. J. Biochem. 1996, 119 (2), 226–230. 10.1093/oxfordjournals.jbchem.a021226. [DOI] [PubMed] [Google Scholar]
- Wakamatsu K.; Choung S. Y.; Kobayashi T.; Inoue K.; Higashijima T.; Miyazawa T. Complex formation of peptide antibiotic Ro09–0198 with lysophosphatidylethanolamine: proton NMR analyses in dimethyl sulfoxide solution. Biochemistry 1990, 29 (1), 113–118. 10.1021/bi00453a013. [DOI] [PubMed] [Google Scholar]
- Shotwell O. L.; Stodola F. H.; Michael W. R.; Lindenfelser L. A.; Dworschack R. G.; Pridham T. G. Antibiotics against plant disease. III. Duramycin, a new antibiotic from Streptomyces cinnamomeus forma azacoluta. J. Am. Chem. Soc. 1958, 80, 3912–3915. 10.1021/ja01548a029. [DOI] [Google Scholar]
- Willey J. M.; Gaskell A. A. Morphogenetic signaling molecules of the streptomycetes. Chem. Rev. 2011, 111 (1), 174–187. 10.1021/cr1000404. [DOI] [PubMed] [Google Scholar]
- Ma H.; Kendall K. Cloning and analysis of a gene cluster from Streptomyces coelicolor that causes accelerated aerial mycelium formation in Streptomyces lividans. J. Bacteriol. 1994, 176 (12), 3800–3811. 10.1128/jb.176.12.3800-3811.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor T. J.; Kanellis P.; Nodwell J. R. The ramC gene is required for morphogenesis in Streptomyces coelicolor and expressed in a cell type-specific manner under the direct control of RamR. Mol. Microbiol. 2002, 45 (1), 45–57. 10.1046/j.1365-2958.2002.03004.x. [DOI] [PubMed] [Google Scholar]
- Hudson M. E.; Zhang D.; Nodwell J. R. Membrane association and kinase-like motifs of the RamC protein of Streptomyces coelicolor. J. Bacteriol. 2002, 184 (17), 4920–4924. 10.1128/JB.184.17.4920-4924.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K.; Miyake K.; Horinouchi S.; Beppu T. A gene cluster involved in aerial mycelium formation in Streptomyces griseus encodes proteins similar to the response regulators of two-component regulatory systems and membrane translocators. J. Bacteriol. 1993, 175 (7), 2006–2016. 10.1128/jb.175.7.2006-2016.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K.; Oinuma K.; Ikeda G.; Hosono K.; Ohnishi Y.; Horinouchi S.; Beppu T. AmfS, an extracellular peptidic morphogen in Streptomyces griseus. J. Bacteriol. 2002, 184 (5), 1488–1492. 10.1128/JB.184.5.1488-1492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller W. M.; Schmiederer T.; Ensle P.; Süssmuth R. D. In vitro biosynthesis of the prepeptide of type-III lantibiotic labyrinthopeptin A2 including formation of a C-C bond as a post-translational modification. Angew. Chem., Int. Ed. 2010, 49 (13), 2436–2440. 10.1002/anie.200905909. [DOI] [PubMed] [Google Scholar]
- Li H.; Xu H.; Zhou Y.; Zhang J.; Long C.; Li S.; Chen S.; Zhou J. M.; Shao F. The phosphothreonine lyase activity of a bacterial type III effector family. Science 2007, 315 (5814), 1000–1003. 10.1126/science.1138960. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Shao F.; Li Y.; Cui H.; Chen L.; Li H.; Zou Y.; Long C.; Lan L.; Chai J.; et al. A Pseudomonas syringae effector inactivates MAPKs to suppress PAMP-induced immunity in plants. Cell Host Microbe 2007, 1 (3), 175–185. 10.1016/j.chom.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Chen L.; Wang H.; Zhang J.; Gu L.; Huang N.; Zhou J. M.; Chai J. Structural basis for the catalytic mechanism of phosphothreonine lyase. Nat. Struct. Mol. Biol. 2008, 15 (1), 101–102. 10.1038/nsmb1329. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Li H.; Long C.; Hu L.; Xu H.; Liu L.; Chen S.; Wang D. C.; Shao F. Structural insights into the enzymatic mechanism of the pathogenic MAPK phosphothreonine lyase. Mol. Cell 2007, 28 (5), 899–913. 10.1016/j.molcel.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Young T. A.; Delagoutte B.; Endrizzi J. A.; Falick A. M.; Alber T. Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat. Struct. Biol. 2003, 10 (3), 168–174. 10.1038/nsb897. [DOI] [PubMed] [Google Scholar]
- Pereira S. F.; Goss L.; Dworkin J. Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 2011, 75 (1), 192–212. 10.1128/MMBR.00042-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann N. A.; van Herwerden E. F.; Hügelland M.; Süssmuth R. D. The supersized class III lanthipeptide stackepeptin displays motif multiplication in the core peptide. ACS Chem. Biol. 2016, 11 (1), 69–76. 10.1021/acschembio.5b00651. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Ökesli A.; van der Donk W. A. Mechanistic studies of Ser/Thr dehydration catalyzed by a member of the LanL lanthionine synthetase family. Biochemistry 2011, 50 (5), 891–898. 10.1021/bi101750r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg N.; Tang W.; Goto Y.; Nair S. K.; van der Donk W. A. Lantibiotics from Geobacillus thermodenitrificans. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (14), 5241–5246. 10.1073/pnas.1116815109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin A. M.; Beyrakhova K. A.; Cygler M. Structural insight into effector proteins of Gram-negative bacterial pathogens that modulate the phosphoproteome of their host. Protein Sci. 2015, 24 (5), 604–620. 10.1002/pro.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk J. M.; Völler G. H.; Krawczyk B.; Kretz J.; Brönstrup M.; Süssmuth R. D. Heterologous expression and engineering studies of labyrinthopeptins, class III lantibiotics from Actinomadura namibiensis. Chem. Biol. 2013, 20 (1), 111–122. 10.1016/j.chembiol.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Pesic A.; Henkel M.; Süssmuth R. D. Identification of the amino acid labionin and its desulfurised derivative in the type-III lantibiotic LabA2 by means of GC/MS. Chem. Commun. 2011, 47, 7401–7403. 10.1039/c1cc11573a. [DOI] [PubMed] [Google Scholar]
- Ross A. C.; Liu H.; Pattabiraman V. R.; Vederas J. C. Synthesis of the lantibiotic lactocin S using peptide cyclizations on solid phase. J. Am. Chem. Soc. 2010, 132 (2), 462–463. 10.1021/ja9095945. [DOI] [PubMed] [Google Scholar]
- Houssen W. E.; Wright S. H.; Kalverda A. P.; Thompson G. S.; Kelly S. M.; Jaspars M. Solution structure of the leader sequence of the patellamide precursor peptide, PatE1–34. ChemBioChem 2010, 11 (13), 1867–1873. 10.1002/cbic.201000305. [DOI] [PubMed] [Google Scholar]
- Roy R. S.; Kim S.; Baleja J. D.; Walsh C. T. Role of the microcin B17 propeptide in substrate recognition: solution structure and mutational analysis of McbA1–26. Chem. Biol. 1998, 5 (4), 217–228. 10.1016/S1074-5521(98)90635-4. [DOI] [PubMed] [Google Scholar]
- Völler G. H.; Krawczyk B.; Ensle P.; Süssmuth R. D. Involvement and unusual substrate specificity of a prolyl oligopeptidase in class III lanthipeptide maturation. J. Am. Chem. Soc. 2013, 135 (20), 7426–7429. 10.1021/ja402296m. [DOI] [PubMed] [Google Scholar]
- Luo H.; Hong S. Y.; Sgambelluri R. M.; Angelos E.; Li X.; Walton J. D. Peptide macrocyclization catalyzed by a prolyl oligopeptidase involved in alpha-amanitin biosynthesis. Chem. Biol. 2014, 21 (12), 1610–1617. 10.1016/j.chembiol.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluskens L. D.; Nelemans S. A.; Rink R.; de Vries L.; Meter-Arkema A.; Wang Y.; Walther T.; Kuipers A.; Moll G. N.; Haas M. Angiotensin-(1–7) with thioether bridge: an angiotensin-converting enzyme-resistant, potent angiotensin-(1–7) analog. J. Pharmacol. Exp. Ther. 2009, 328 (3), 849–854. 10.1124/jpet.108.146431. [DOI] [PubMed] [Google Scholar]
- Suda S.; Westerbeek A.; O’Connor P. M.; Ross R. P.; Hill C.; Cotter P. D. Effect of bioengineering lacticin 3147 lanthionine bridges on specific activity and resistance to heat and proteases. Chem. Biol. 2010, 17 (10), 1151–1160. 10.1016/j.chembiol.2010.08.011. [DOI] [PubMed] [Google Scholar]
- Hsu S. T.; Breukink E.; Tischenko E.; Lutters M. A.; De Kruijff B.; Kaptein R.; Bonvin A. M.; Van Nuland N. A. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat. Struct. Mol. Biol. 2004, 11 (10), 963–967. 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- Shioya K.; Harada Y.; Nagao J.; Nakayama J.; Sonomoto K. Characterization of modification enzyme NukM and engineering of a novel thioether bridge in lantibiotic nukacin ISK-1. Appl. Microbiol. Biotechnol. 2010, 86 (3), 891–899. 10.1007/s00253-009-2334-8. [DOI] [PubMed] [Google Scholar]