

Abstract
Motivation
The computational search for promoters in prokaryotes remains an attractive problem in bioinformatics. Despite the attention it has received for many years, the problem has not been addressed satisfactorily. In any bacterial genome, the transcription start site is chosen mostly by the sigma (σ) factor proteins, which control the gene activation. The majority of published bacterial promoter prediction tools target σ70 promoters in Escherichia coli. Moreover, no σ-specific classification of promoters is available for prokaryotes other than for E. coli.
Results
Here, we introduce bTSSfinder, a novel tool that predicts putative promoters for five classes of σ factors in Cyanobacteria (σA, σC, σH, σG and σF) and for five classes of sigma factors in E. coli (σ70, σ38, σ32, σ28 and σ24). Comparing to currently available tools, bTSSfinder achieves higher accuracy (MCC = 0.86, F1-score = 0.93) compared to the next best tool with MCC = 0.59, F1-score = 0.79) and covers multiple classes of promoters.
Availability and Implementation
bTSSfinder is available standalone and online at http://www.cbrc.kaust.edu.sa/btssfinder.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The promoter is a chromosome region that determines where and affects how the transcription of a particular transcript is initiated. Promoter recognition is important in defining the transcription units responsible for specific pathways and gene regulation. Initiation of transcription is a dynamic partnership between RNA polymerase (RNAP) and promoter. In contrast to archaea and eukarya, bacteria have a single form of the RNAP core enzyme (E) (Schneider and Hasekorn, 1988). However, RNAP alone is not able to recognize and bind to promoters to initialize transcription. Different regulatory proteins called σ-factors are required that temporarily bind the RNAP core enzyme forming a holoenzyme (Eσ). The holoenzyme determines the RNAP-promoter binding specificity and transcription initiation site (TSS), depending on nutritional or environmental conditions or developmental stage (for reviews see: (Campagne et al., 2014; de Avila et al., 2011; Feklistov, 2013; Gruber and Gross, 2003; Imamura and Asayama, 2009; Ruff et al., 2015)).
Bacterial σ factors are classified into two families with distinct structure and function, termed as σ70 and σ54 in Escherichia coli. While most bacteria possess multiple members of the σ70 family, they contain a single representative of the σ54 family, which is involved in nitrogen metabolism. Surprisingly, cyanobacteria lack any σ54-like factors despite the majority of them having nitrogen fixation system. The number of σ70 family members can vary significantly between different species (from 1 to over 60). E. coli, for example, has seven σ factors (Gruber and Gross, 2003; Imamura and Asayama, 2009; Studholme and Buck, 2000; Wosten, 1998). In this study we focus on E. coli and three species of Cyanobacteria.
The most detailed promoter information for the E. coli genome is available in RegulonDB (Salgado et al., 2013) and EcoCyc database (Karp et al., 2002). RegulonDB holds 10 293 mapped TSSs (Release 8.8, 2015). For Cyanobacteria, the genome-wide promoter map for 3 species was identified: 12 797 for Nostoc 7120 (Mitschke et al., 2011a,b), 1471 for Synechococcus elongatus (Vijayan et al., 2011) and 351 for Synechocystis (Mitschke et al., 2011a,b). Interestingly, most of the TSSs in cyanobacteria are mapped to the non-coding RNA transcription units, rather than to protein-coding genes.
Lagging behind the explosion in genome/gene sequences and due to experimental costs, promoters and TSSs are mostly predicted computationally. In the last two decades, several computational tools were developed to identify bacterial promoters. The first attempt to predict bacterial promoters was by position weight matrices (PWM), which relied on the conservation of the -35 and the -10 elements for σ70, combined with the distribution of the distance between them (Hertz and Stormo, 1996; Huerta and Collado-Vides, 2003; Stormo, 2000). Tools developed using this approach have relatively low accuracy. Applying machine-learning approaches, e.g. support vector machines (SVM) and artificial neural networks (ANNs), increased accuracy. A method based on Sequence Alignment Kernel that achieved 17% average error rate on true and false promoter data was developed in (Gordon et al., 2003). In Gordon et al. (2006), a method was reported that employs an ensemble of SVMs with a variant of the mismatch string kernel in combination with a PWM and a model of the distribution of distances from TSS to gene start. The authors reported an average error rate of 11.6%, which they claim is ≈ 5% lower than the method reported in Gordon et al. (2003).
More complex algorithms were developed that incorporate series of ANNs (Knudsen, 1999) and interactive optimization of nodes (Jihoon et al., 1999). Ma et al. (2001) applied a procedure of preprocessing promoter sequences during training to extract features. Using a time-delay neural network, Reese (2001) developed NNPP2.2, a tool with high sensitivity, but poor specificity. Later, the distance between the TSS and the translation start site (TLS) was used as an additional feature for promoter recognition (TLS–NNPP) (Burden et al., 2005). This tool, although reduced the false positive rate, is only applicable to protein coding genes. Using some conserved hexamer motifs as promoter recognition features, Li and Lin (2006) developed a tool with the overall prediction sensitivity and specificity of 91% and 81%, respectively. Mann et al. (2007) reported that a combination of ANNs and hidden Markov models (HMMs) significantly increases the bacterial promoter prediction accuracy. Later, Rani and Bapi (2009) developed a tool where k-mers (k = 2,3,4,5) are used as discriminative features. This tool was reported to have much higher prediction accuracy. The PromPredict tool, based on the relative stability of DNA as a generic criterion for promoter prediction, was reported to achieve 58% precision in E. coli (Rangannan and Bansal, 2009). de Avila et al. (2011) published BacPP, which is designed to predict promoters of different σ classes from E. coli (more on this in the discussion section). Solovyev and Salamov (2011) developed BPROM for the recognition of E. coli σ70 promoters. This tool, based on the linear discriminant function (LDF) combines characteristics describing functional motifs and oligonucleotide composition and shows about 80% prediction accuracy. Song (2012) reported a variable-window Z-curve method based on the distribution of purine/pyrimidine, the distribution of amino/keto and the distribution of strong/weak hydrogen bonds. Depending on the false promoter sets for learning and testing, the accuracy of the method was reported to vary around 90–96% and 95–99% for E. coli and Bacillus subtilis, respectively.
Despite these efforts, all these tools tend to produce many false positives or show poor sensitivity, particularly when they are applied to long sequences or whole genomes. Therefore, most of the tools were only tested on short promoter segments of 50–70 bp surrounding known TSSs. Such tests are insufficient to adequately evaluate their prediction accuracy. Another significant restriction of these tools is that they are limited to the prediction of σ70 promoters in the model organism E. coli, and very rarely can extend to other bacterial species. Therefore, novel, more accurate and efficient tools are required for the computational recognition of different classes of promoters in a broader taxonomical scope.
In this study, we present a novel method for predicting TSSs in E. coli and three cyanobacterial species. We characterized promoters of E. coli for σ70, σ38, σ32, σ28 and σ24 factors, and Cyanobacteria for σA, σC, σH, σG and σF factors, and we developed promoter prediction models using this characterization. The prediction models are implemented in a tool, bTSSfinder, which is available as a standalone program as well as a web application.
Rationale
The main goal of this study was to develop a tool that predicts promoters for the different sigma classes in Cyanobacteria and E. coli. Success of any promoter prediction tool depends mainly on: (i) the features used to distinguish promoters from non-promoters, (ii) the size and diversity of the positive and negative datasets used for learning and (iii) the quality of both the positive and the negative datasets. Unfortunately, most studied characteristic features are not consistent within the same promoter class. Therefore, different studies have applied various combinations of features to improve the recognition of promoters. Furthermore, most of the reported tools were trained and tested on relatively small datasets, due to the lack of genome-wide TSSs maps with experimental validation. As for the quality of the datasets, here we face two problems: (i) the accuracy of experimental data on real TSSs varies significantly depending on the experimental method applied; (ii) the choice of the negative set (non-promoters) could have significant ramifications on the predictive power of the model, as it is a great challenge to define DNA regions that never serve as promoters.
To address these challenges: (i) we analyzed as many promoter sequences with experimentally validated TSS as available; (ii) from the whole pool of initially extracted negative samples (see below), we use different subsets of randomly chosen negative samples in both training and testing procedures and (iii) we checked different features that may allow a DNA region to serve as a potential promoter. We compare bTSSfinder with other available methods.
A note
Boundaries of promoter regions remain unclearly defined. The minimum promoter region that can initiate basal transcription spans −60 to +40 bp relative to the transcription start site (TSS, +1) is called the core promoter, whereas proximal promoters extend further upstream (Roy and Singer, 2015; Shahmuradov et al., 2003). In this study we consider a promoter region relative to the TSS location as a region spanning [-200,+51], where +1 position corresponds to the location of the TSS. When we predict such a promoter, we also predict the corresponding TSS at position +1, which makes prediction of TSSs and promoters in our case equivalent. We also considered wider promoters spanning regions [-1000,+101] relative to TSS located at position +1.
2 Methods
2.1 Materials
2.1.1 Collecting data
RegulonDB, version v8.0 contains 3597 experimentally validated TSSs for E. coli K12 MG1655 (accession NC_000913.2). Of them, 2979 TSSs were classified into seven σ classes: 1787 as σ70, 85 as σ54, 152 as σ38, 298 as σ32, 141 as σ28, 515 as σ24 and one as σ19. Due to their limited counts, σ54 and σ19 were excluded from further analysis.
For the non-marine cyanobacterium Nostoc sp. PCC 7120 (accession BA000019, referred to as Nostoc hereforth), Mitschke et al. (2011a,b) experimentally mapped 12 797 TSSs. The authors classified these promoters into four classes: 3955 gene TSSs (gTSS), 3854 antisense TSSs (aTSS), 3722 intergenic TSSs (iTSS) and 1266 non-coding DNA TSSs (nTSS). As for the freshwater cyanobacterium Synechocystis sp. PCC 6803 (accession NC_000911.1, referred to as Synechocystis hereforth), data for 351 TSSs were extracted from the Supplementary material of the respective article (Mitschke et al., 2011a,b). The data contain 172 gTSSs, 56 nTSSs and 123 aTSSs. For the freshwater cyanobacterium S. elongatus PCC 6301 (accession CP000100), we collected 1471 TSSs (Vijayan et al., 2011).
2.1.2 Preparing the positive set
Our preliminary assessment of the experimental sets revealed that some TSSs are located close to each other (a few nucleotides apart). To remove redundancy, intra-species pairwise comparison of all TSS positions was performed. For every TSS, starting from the 5′ end, we identified and discarded neighboring TSSs that were within 35 bp. The distance of 35 bp was chosen under the assumption that most signals involved in determining transcription start points are located in the short region between the -35 and the -10 boxes. After redundancy removal, the final TSS count was as follows: (i) E. coli: 1544 for σ70, 140 for σ38, 237 for σ32, 135 for σ28, 412 for σ24; (ii) Nostoc: 11 386, (iii) S. elongatus: 1471 and (iv) Synechocystis: 343.
Using the publicly available genomes (accessions above) and the above TSS annotation, we created two types of promoter sets: 251 bp promoters (200 bp upstream of TSS and 51 bp downstream) and 1101 bp promoters (1000 bp upstream and 101 bp downstream). Promoter sequences that do not satisfy the upstream length requirement for either set are excluded.
2.1.3 Preparing the negative set
To train and test the promoter prediction models, we generated two negative datasets with sequences of length 251 bp: one based on E. coli and another based on the three cyanobacterial species. The protocol for the generation of negative sets is described in the Supplementary material. The final counts for the negative sets were 8346 and 32 418 for E. coli and the three combined Cyanobacteria species, respectively.
2.1.4 Transcription factor binding sites (TFBSs)
Data on TFBSs were obtained from three sources: 2953 sites for E. coli from RegulonDB, 30 cyanobacterial sites from CollecTF (Kilic et al., 2014) and 63 sites from the literature. Due to the limited number of TFBSs for Cyanobacteria, we used both cyanobacterial and E. coli sets to compute the TFBS density in promoter regions.
2.2 Methodology
2.2.1 Computing PWMs
To build our PWMs for E. coli, we extracted initial PWMs from the literature for the -10, -35 elements for σ70, σ24, σ28, σ32 promoters and -15 and the AT-rich upstream elements (UP elements) for the σ70 promoters (Barnett et al., 2012; Dartigalongue et al., 2001; Djordjevic, 2011; Estrem et al., 1998; Huerta and Collado-Vides, 2003; Song et al., 2007). To create the PWMs for the -15 and the UP elements for σ24, σ28 and σ32 factors, we used the same initial PWMs of σ70. To the best of our knowledge, no σ38-promoter-specific PWM is available in published literature. However, it has been shown that most σ38 promoters are recognized by σ70 and vice versa (de Avila et al., 2011). Hence, we used the same initial PWMs as for σ70. Furthermore, by profiling the neighboring regions to known σ70 TSSs [TSS-6bp, TSS + 6bp], we discovered a new motif with the consensus AYYTNA (we named it TSS-motif). We propose that this new motif is another descriptive element for the recognition of promoters (see Supplementary material for PWMs and coverage).
As for the cyanobacterial species, there are no published PWMs to use as initial matrices. To overcome this limitation, we determined the orthology between the σ factors in E. coli and the σ factors in three cyanobacterial species using the BLAST software (Altschul et al., 1997). Where significant alignments are found, the initial PWMs from the respective σ factor in E. coli are used for the orthologous counterpart in the cyanobacterial species. The final PWMs, for all promoter elements for all σ factors in the four species, were computed using the Expectation Maximization (EM) algorithm (Cardon and Stormo, 1992) (see Supplementary material).
2.2.2 Classifying cyanobacterial promoters
EM was also used to classify the cyanobacterial promoters into different classes. Using the PWM for the σ70 -10 box in E. coli, we applied EM to the final set of cyanobacterial promoters (13 200 sequences, see the Materials section) with experimentally validated TSSs to obtain a subset that we denote as σA promoters. Following the same approach, we used PWMs for E. coli’s σ24, σ28, σ32 and σ38 to assign the unassigned cyanobacterial promoters to σG, σF, σH and σC classes, respectively. It should be noted that each round of classification is applied only to the sequences unassigned in the previous step.
2.2.3 Compiling and selecting features
Oligomer frequencies (triplets, tetramers, pentamers and hexamers) were used to calculate scores as described in Rani and Bapi (2009). We also used four physico-chemical properties of DNA: free energy, base stacking, melting temperature and entropy, as additional features to describe true and false promoter regions (see Supplementary material). We evaluated the predictive power of these features as well as the aforementioned promoter elements using Mahalanobis distance (D2), which we calculated based on the approach described by Afifi and Azen (2014). Based on these distances we selected the final set of features for use in the predictive model, as described in Section 3.2.
2.2.4 Building and testing the models
To obtain the model parameters in order to accomplish the best separation of promoter from non-promoter sequences, we applied Neural Network techniques using the VISAN tool (http://www.softberry.com/berry.phtml?topic=fdp.htm&no_menu=on). We estimated the performance of the predictive models using: sensitivity (Sn), specificity (Sp), Precision (the positive predictive value, P1), Accuracy (a measure of statistical bias, Ac), Negative predictive value (P2), the F1-score (the harmonic mean of Precision and Accuracy, F1) and the Mathew correlation coefficient (MCC). These statistical measures are briefly described in the Supplementary material.
2.2.5 bTSSfinder algorithm and tool
The algorithm of bTSSfinder is depicted in Figure 1. bTSSfinder scans the DNA sequence (251 bp at a time) and predicts position 201 as a potential TSS using the appropriate NN classifier. More details about the algorithm are given in the Results section (Section 3.3). The algorithm is implemented in the bTSSfinder tool.
Fig. 1.

Flow-chart of the algorithm implemented in the bTSSfinder program. T-10 is the threshold for the prediction of the -10 box (specific for every sigma class). Ttotal – the NN threshold for the selection of TSSs
3 Results
3.1 Classification of cyanobacterial promoters
The largest collection of experimentally validated promoters of E. coli in RegulonDB was classified into seven different sigma classes (σ70, σ54, σ38, σ32, σ28, σ24 and σ19). Unfortunately, no such classification exists for cyanobacterial promoters. We aim to classify these promoters into different sigma classes based on the -10 box, which is thought to be the most intrinsic characteristic in any given σ promoter class in bacteria. So far, the -10 and -35 boxes have been identified or predicted in a handful of promoters. Our preliminary comparison of E. coli and cyanobacterial promoters indicate that there is a level of conservation, based on which we used E. coli PWMs for the classification of cyanobacterial promoters.
Based on the inter-phyla orthology as revealed by the BLAST comparison (Supplementary Table S1), we propose the classification for cyanobacterial promoters into five classes: σA (analogous to σ70), σC (analogous to σ38), σF (analogous to σ28), σG (analogous to σ24) and σH (analogous to σ32).
Combining E. coli’s -10 box PWMs for the different σ factors with the EM algorithm (see Methods), we classified the 13 200 experimentally validated cyanobacterial promoters into 9895 σA promoters, 928 σG promoters, 686 σF promoters, 220 σH promoters, 355 σC promoters and 1116 unclassified (Supplementary Table S2). These significantly larger datasets than in previous studies (Imamura and Asayama, 2009) enabled us to update the models of the -35 and the -10 promoter elements of σF, σG, σH and σC in cyanobacteria. The training and the test sets for each σ factor promoter class were generated accordingly (Supplementary Table S2, see Supplementary material for consensuses and PWMs).
3.2 Features used for the prediction of promoters
We identified over 30 prospective features that may exert specificity for the different promoter classes. To cull the feature space to those with the highest predictive power, we calculated Mahalanobis distances for each feature and reduced the number to 19–21 features depending on the promoter class (Supplementary Table S3). To the best our knowledge, this is the first time a wide feature base was used for this type of problem. We group these selected features into the following:
Promoter elements: -10, -35, -15 and AT-rich UP elements, as well as the new TSSmotif proposed by us (see: Materials and Methods). UP-elements (length of 17 bp) are searched for upstream of the -35 box up to a distance of 130 bp.
Distances (d) between promoter elements: d(-10/-35) and d(-10/TSS) were used in other methods. In this study, we introduce a novel feature d(-15/-10). For all promoter classes, it is thought that the distance between the -10 box and the TSS varies from 3 to 12 bp; while the distance between the -15 and the -10 boxes varies between 0 and 10 bp. However, the variation in the distance between the -10 and the -35 boxes depends on promoter class (σ70: 15–22 bp, σ24: 12–19 bp, σ28: 10–12 bp, σ32: 13–15 bp).
Oligomer scores: Seven features were formulated based on the calculated scores for 3-mers, 4-mers, 5-mers and 6-mers in different segments of the promoter sequences (see Methods): (i) 3-mers in region [-20:+21], (ii) 4-mers/1 in [-100:+21], (iii) 4-mers/2 in [-100:+21], (iv) 5-mers/1 in [-100:+21], (v) 5-mers/2 in [-100:+21], (vi) 6-mers/1 in [-200:-1] and (vii) 6-mers/2 in [-100:+21].
Density of TFBSs: This group contains two features. TFBS density1, which is on the sense strand only, is calculated within the interval [-200:+51]; and TFBS density2 which is calculated within the interval [-200:-1] on both strands.
Physico-chemical properties of the promoter sequences: four features were chosen in the feature selection process: free energy, base stacking, entropy and melting temperature. All of which were computed in the region [-200:+20].
3.3 bTSSfinder: the bacterial promoter prediction tool
Using a combination of features for each promoter class (as outlined in Supplementary Table S3), we built 10 NN classifiers, one for each promoter class in E. coli and in cyanobacteria. Then, we implemented these models into the bTSSfinder program. bTSSfinder workflow is outlined in Figure 1. The program slides a window of 251 bp over the query sequence, one nucleotide at a time. For each window, position 201 is classified as TSS or non-TSS using the appropriate NN classifier based on a threshold that was predetermined during the training. Predictions that pass the qualifying threshold are labeled as putative TSSs. bTSSfinder performs additional filtering by discarding all but the highest-scoring TSS in intervals of a user-adjustable length (default 300 bp). Depending on user preference, bTSSfinder can report for a chosen phylum: (i) all predicted TSSs for all promoter classes, (ii) a user-selected promoter class and (iii) or the highest scoring TSS.
3.4 Evaluating bTSSfinder performance
We tested bTSSfinder on positive and negative sets for every promoter class in E. coli and cyanobacteria (Table 1). We observed good performance for all promoter classes in E. coli (251 bp, a single search window size). In the case of cyanobacteria, we observed the highest accuracy in σA promoters (F1-score: 0.94). The F1-score for the remaining cyanobacterial promoter classes ranged from 0.81 to 0.87. Although these results are considered high, they are somewhat less than what was achieved for σA promoters. This difference in performance may be due to the following: (i) σA (or orthologs in other species) and its promoter elements are highly conserved where for other classes they are not as conserved; (ii) the training set for σA promoter class is much larger if compared to other classes.
Table 1.
Testing results for five sigma classes of E. coli and cyanobacteria1
| σ class | TP | FN | TN | FP | Sn, % | Sp, % | P1, % | P2, % | F1, % |
|---|---|---|---|---|---|---|---|---|---|
| σ70 | 180 | 20 | 377 | 23 | 90.0 | 93.3 | 94.0 | 90.4 | 92 |
| σ38 | 37 | 3 | 75 | 5 | 92.5 | 93.8 | 93.7 | 92.6 | 93.1 |
| σ32 | 46 | 4 | 95 | 5 | 92.0 | 95.0 | 94.9 | 92.2 | 93.4 |
| σ28 | 31 | 4 | 69 | 1 | 88.6 | 98.6 | 98.4 | 89.6 | 93.2 |
| σ24 | 86 | 14 | 193 | 7 | 86.0 | 96.5 | 96.1 | 87.3 | 90.7 |
| σA | 921 | 79 | 1921 | 79 | 92.1 | 96.1 | 95.9 | 92.4 | 94 |
| σC | 36 | 14 | 96 | 4 | 72 | 96 | 94.7 | 75.0 | 81.8 |
| σF | 80 | 20 | 193 | 7 | 80.0 | 96.5 | 95.8 | 82.8 | 87.2 |
| σG | 72 | 28 | 188 | 12 | 72.0 | 94.0 | 92.3 | 77.1 | 80.9 |
| σH | 38 | 12 | 92 | 8 | 76 | 92 | 90.5 | 74.2 | 82.6 |
Test experiments for every sigma class were repeated 10 times for randomly selected negative sets and the means were taken.
3.5 Comparing bTSSfinder to other tools
We could only evaluate bTSSfinder against previously published promoter prediction tools for σ70 promoter class in E. coli. For fairness, we assessed all tools on a single testing dataset. The following promoter prediction tools were available for comparison: BPROM (Solovyev and Salamov, 2011), NNPP2 (Reese, 2001) and PromPredict (Rangannan and Bansal, 2009). All other promoter prediction tools that we checked were no longer available. A major drawback for these tools is that they were designed specifically for σ70 promoters. BPROM and NNPP2 were optimized for E. coli, while PromPredict was optimized for both E. coli and B. subtilis. We also tried to test BacPP, which is the first tool that attempted to predict the complete range of sigma promoters in E. coli (de Avila et al., 2011). The authors reported high prediction accuracy for BacPP, but these results were obtained from a small training and testing sets. Furthermore, this tool calculates the probabilities that a sigma factor might bind to every 80 bp window of the query and does not confer any predictions on the TSS. Given that we would have to make an educated guess as to where the TSS locations are as well as the shear number of promoters it predicts, we excluded this tool from our comparison. Results of the comparison for short (251 bp) sequences are presented in Table 2. Our comparison clearly indicates that bTSSfinder has significantly higher prediction accuracy.
Table 2.
Comparison of available promoter prediction programs tested on E. coli’s experimentally validated σ70 promoter sequences and a negative dataset of 251 bp sequences.
| Promoter prediction tool | Genes with ≥1 TSSpr | Total number of TSSpr | TP1 | FP | FN | Sn, % | P1,% | F1,% |
|---|---|---|---|---|---|---|---|---|
| bTTSSfinder | 197 | 355 | 143 | 212 | 57 | 71.5 | 40.3 | 51.5 |
| BPROM | 200 | 569 | 130 | 439 | 70 | 65.0 | 22.9 | 33.8 |
| NNPP2 | 175 | 460 | 109 | 351 | 91 | 54.5 | 23.7 | 33.0 |
| PromPredict | 74 | 149 | 0 | 149 | 200 | 0.0 | 0.0 | 0.0 |
Prediction is true, if distance between annotated and predicted TSSs is 50 bp or less.TSSpr: predicted TSS.
Using short sequences to predict TSSs is not sufficient in evaluating the accuracy and efficiency (especially the real false positive rate) of a prediction tool. It should also be tested on longer sequences. In fact, an ideal test should be genome-wide. Nonetheless, genome-wide TSS maps are scarce which renders the task of assessing such predictions unfeasible. First, we run the four programs on longer DNA sequences to search for putative σ70 TSSs in 200 test sequences of 1101 bp from E. coli (Table 3).
Table 3.
Comparison of available promoter prediction programs assessed on the 1100 bp upstream region of 200 E. coli σ70 promoters with experimentally validated TSSs.
| Promoter prediction tool | Tp | Fn | Tn | Fp | Sn % | Sp % | F1-score | MCC |
|---|---|---|---|---|---|---|---|---|
| bTTSSfinder1 | 183 | 17 | 189 | 11 | 91.5 | 94.5 | 0.93 | 0.86 |
| BPROM2 | 152 | 48 | 166 | 34 | 76.0 | 83.0 | 0.79 | 0.59 |
| NNPP22 | 109 | 91 | 176 | 24 | 54.5 | 88.0 | 0.66 | 0.45 |
| PromPredict2 | 0 | 200 | 200 | 0 | 0.0 | 100.0 | n.d. | n.d. |
Prediction is true, if the annotated TSS is exactly predicted.
Prediction is true, if distance between annotated and predicted TSSs is 50 bp or less.
n.d. not determined.
Our results highlight the scale of the problem that researchers encounter when they analyze long sequences. Specifically, the existence of multiple experimentally mapped TSSs (TSSmap), which are often in close proximity to each other, makes predicting the TSS (TSSpr) difficult. In previous studies, a TSSpr is considered a true positive if it was detected 500 bp or less away from a TSSmap. For example, the authors of PromPredict, a recent tool, considered a TSSpr as a true positive if it was within a distance of ±500 bp from a TSSmap. In this comparative analysis, a true positive is defined as a TSSpr that is within 50 bp away from a TSSmap (upstream or downstream). As presented in Table 3, bTSSfinder produced the best performance (Sn ≃ 72%, F1 ≃ 52%), followed by BPROM (Sn = 65%, F1 ≃ 34%) and NNPP2 (Sn ≃ 54%, F1 ≃ 33%). Surprisingly, PromPredict failed to produce a single true positive prediction (Se = 0, F1 = 0).
We also investigate if models optimized for E. coli can be used for Cyanobacteria and vice versa in bTSSfinder. We used a positive test set of 251 bp sequences from E. coli and searched for TSSs using the corresponding model for the cyanobacteria for a given σ class, and vice versa. Results of these cross-phylum experiment are presented in Table 4. For the cyanobacterial σA and σC test sets, applying E. coli’s σ70 and σ38 models reduced sensitivity but not significantly (92.3% using σA model versus 87.5% using σ70 model, 72% using σC model versus 68% using σ38 model). However, the opposite scenario had a significant impact on sensitivity (Table 4). Cross assessment of the models for the other sigma factors failed to reproduce the sensitivity achieved for their intended species. This perhaps can be explained by: (i) significant structural differences between promoters in E. coli (gammaproteobacteria) and the studied cyanobacterial species; (ii) biological differences in how transcription initiation points are detected in different bacterial phyla. In fact, we tested bTSSfinder and the other three tools on ten other bacterial species belonging to five different phyla: three Firmicutes, four Proteobacteria, one Spirochetes, Chlamydias and one CFB group. bTSSfinder consistently outperformed the other tools for each species albeit with sensitivity values averaging 59% (BPROM was next best with a sensitivity average of 49%). For details of this comparison consult the supplementary material (Supplementary Table S4).
Table 4.
Result of cross-phylum application of bTSSfinder on the positive dataset. Bold refers to sensitivity of the models applied to their intended species.
| Test sets | Sensitivity1 |
|
|---|---|---|
| bTSSfinder models for E. coli1 | bTSSfinder models for cyanobacteria1 | |
| σ70 | 89.5% | 66% |
| σA | 87.5% | 92.3% |
| σ38 | 90% | 35% |
| σC | 68% | 72% |
| σ32 | 92% | 54% |
| σH | 58% | 74% |
| σ28 | 88.6% | 42.6% |
| σF | 37% | 81% |
| σ24 | 86% | 31% |
| σG | 15.8% | 72.0% |
Sensitivity values obtained with the species-specific bTSSfinder parameters are given in bold.
We observed that some experimentally verified promoters did not pass the prediction thresholds. It has been reported that computational prediction tools have succeeded in predicting no more than 20% of known promoters (Hertz and Stormo, 1996). A later study that analyzed 599 σ70 promoters from E. coli showed that in over 50% of the cases, the true promoters do not produce the highest score, especially since true promoters are commonly found in TSS-dense regions (Huerta and Collado-Vides, 2003). To investigate this phenomenon, we analyzed the region ±300bp around experimental TSSs. For every base the NN score is calculated (using bTSSfinder) and those that satisfy the species and σ class-specific threshold were assessed against the experimentally mapped TSSs. In 90% of the test cases (425 in E. coli and 1300 in the cyanobacterial species), the TSSmap had higher score than threshold but only 10% passed filtering criteria to make it to the final predicted set due to the presence of a neighboring TSSpr that had higher NN score (Fig. 2; Fig S1). This may warrant an alternative approach to search for transcription start regions (TSRs) rather than points.
Fig. 2.
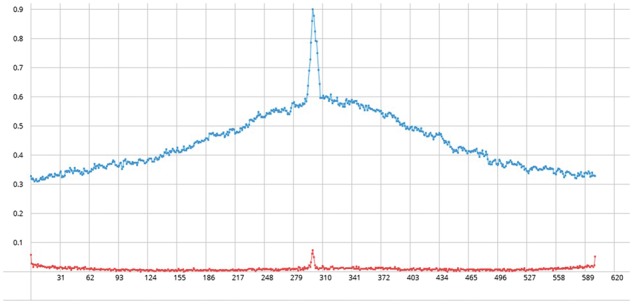
The scoring landscape of experimentally validated TSSs in E. coli. Top line: the distribution of NN scores that are higher than the threshold for every 300 bp upstream and downstream of a TSSmap. Bottom line: cases where the TSSpred is the TSSmap
3.6 Concluding remarks
The promoter prediction problem in prokaryotes is an old problem that has yet to achieve an adequate solution. Available tools tend to produce many false positives or have poor sensitivity, especially when applied to long sequences or whole genomes. These limitations are probably due to the following challenges:
Some in-vitro-strong promoters that are predicted computationally with high score are in fact not used in vivo at all, perhaps due to unknown repression mechanisms (Hertz and Stormo, 1996; Huerta and Collado-Vides, 2003).
Real TSSs tend to fall in promoter dense regions (Panyukov and Ozoline, 2013) with neighboring predicted TSSs that may produce higher prediction scores.
Some predicted TSSs may be evaluated as false positives due to the lack of experimentally-verified, comprehensive and precise TSS maps.
Scarcity of experimental data also means that training models using features extracted from the limited available data would naturally restrict their predictive power.
All methods, as far as we know, depend on promoter architecture and other physico-chemical properties in their model building. Yet, there are likely other ‘players’ that contribute to the transcription initiation process.
The choice of a negative dataset can be detrimental for the trained model since one cannot be certain about the total absence of TSSs in the negative dataset.
We believe that bTSSfinder is the first tool that can recognize promoters of different sigma classes from two bacterial phyla (Proteobacteria’s E. coli and Cyanobacteria). Nonetheless, promoter prediction, especially at the whole genome level, remains unresolved and this warrants further investigations in this field.
Supplementary Material
Acknowledgements
The authors thank Mohamad Jaber for some of the helpful discussions and feedback. We would also like to thank Ramzan Umarov for his help to use VISAN. All computational work in this study has been made using KAUST CBRC Dragon and Snapdragon compute cluster.
Funding
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under Awards No URF/1/1976-02 and FCS/1/2448-01.
Conflict of Interest: none declared.
References
- Afifi A.A., Azen S.P. (2014) Statistical Analysis: A Computer Oriented Approach. Academic press, Cambridge, Massachusetts, USA. [Google Scholar]
- Altschul S.F. et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett M.J. et al. (2012) Dual RpoH sigma factors and transcriptional plasticity in a symbiotic bacterium. J. Bacteriol., 194, 4983–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burden S. et al. (2005) Improving promoter prediction for the NNPP2.2 algorithm: a case study using Escherichia coli DNA sequences. Bioinformatics, 21, 601–607. [DOI] [PubMed] [Google Scholar]
- Campagne S. et al. (2014) Structural basis for -10 promoter element melting by environmentally induced sigma factors. Nat. Struct. Mol. Biol., 21, 269–276. [DOI] [PubMed] [Google Scholar]
- Cardon L.R., Stormo G.D. (1992) Expectation maximization algorithm for identifying protein-binding sites with variable lengths from unaligned DNA fragments. J. Mol. Biol., 223, 159–170. [DOI] [PubMed] [Google Scholar]
- Dartigalongue C. et al. (2001) Characterization of the Escherichia coli sigma E. regulon. J. Biol. Chem., 276, 20866–20875. [DOI] [PubMed] [Google Scholar]
- de Avila E.S.S. et al. (2011) BacPP: bacterial promoter prediction – a tool for accurate sigma-factor specific assignment in enterobacteria. J. Theor. Biol., 287, 92–99. [DOI] [PubMed] [Google Scholar]
- Djordjevic M. (2011) Redefining Escherichia coli sigma(70) promoter elements: -15 motif as a complement of the -10 motif. J. Bacteriol., 193, 6305–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrem S.T. et al. (1998) Identification of an UP element consensus sequence for bacterial promoters. Proc. Natl. Acad. Sci. U. S. A., 95, 9761–9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feklistov A. (2013) RNA polymerase: in search of promoters. Ann. N. Y. Acad. Sci., 1293, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon J.J. et al. (2006) Improved prediction of bacterial transcription start sites. Bioinformatics, 22, 142–148. [DOI] [PubMed] [Google Scholar]
- Gordon L. et al. (2003) Sequence alignment kernel for recognition of promoter regions. Bioinformatics, 19, 1964–1971. [DOI] [PubMed] [Google Scholar]
- Gruber T.M., Gross C.A. (2003) Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol., 57, 441–466. [DOI] [PubMed] [Google Scholar]
- Hertz G.Z., Stormo G.D. (1996) Escherichia coli promoter sequences: analysis and prediction. Methods Enzymol., 273, 30–42. [DOI] [PubMed] [Google Scholar]
- Huerta A.M., Collado-Vides J. (2003) Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J. Mol. Biol., 333, 261–278. [DOI] [PubMed] [Google Scholar]
- Imamura S., Asayama M. (2009) Sigma factors for cyanobacterial transcription. Gene Regul. Syst. Biol., 3, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jihoon Y. et al. (1999) Data-driven theory refinement algorithms for bioinformatics. In: International Joint Conference on Neural Networks, 1999. IJCNN '99. vol. 4066, p. 4064–4068.
- Karp P.D. et al. (2002) The ecocyc database. Nucleic acids research, 30, 56–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic S. et al. (2014) CollecTF: a database of experimentally validated transcription factor-binding sites in Bacteria. Nucleic Acids Res., 42, D156–D160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen S. (1999) Promoter2.0: for the recognition of PolII promoter sequences. Bioinformatics, 15, 356–361. [DOI] [PubMed] [Google Scholar]
- Li Q.Z., Lin H. (2006) The recognition and prediction of sigma70 promoters in Escherichia coli K-12. J. Theor. Biol., 242, 135–141. [DOI] [PubMed] [Google Scholar]
- Ma Q. et al. (2001) DNA sequence classification via an expectation maximization algorithm and neural networks: a case study. IEEE Trans. Syst. Man Cybern. C Appl. Rev., 31, 468–475. [Google Scholar]
- Mann S. et al. (2007) A pHMM-ANN based discriminative approach to promoter identification in prokaryote genomic contexts. Nucleic Acids Res., 35, e12–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschke J. et al. (2011a) An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U. S. A., 108, 2124–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschke J. et al. (2011b) Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc. Natl. Acad. Sci. U. S. A., 108, 20130–20135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyukov V.V., Ozoline O.N. (2013) Promoters of Escherichia coli versus promoter islands: function and structure comparison. PLoS One, 8, e62601.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangannan V., Bansal M. (2009) Relative stability of DNA as a generic criterion for promoter prediction: whole genome annotation of microbial genomes with varying nucleotide base composition. Mol. Biosyst., 5, 1758–1769. [DOI] [PubMed] [Google Scholar]
- Rani T.S., Bapi R.S. (2009) Analysis of n-gram based promoter recognition methods and application to whole genome promoter prediction. In Silico Biol., 9, S1–16. [PubMed] [Google Scholar]
- Reese M.G. (2001) Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput. Chem., 26, 51–56. [DOI] [PubMed] [Google Scholar]
- Roy A.L., Singer D.S. (2015) Core promoters in transcription: old problem, new insights. Trends Biochem. Sci., 40, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff E.F. et al. (2015) Initial events in bacterial transcription initiation. Biomolecules, 5, 1035–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado H. et al. (2013) RegulonDB v8.0: omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res., 41, D203–D213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G.J., Hasekorn R. (1988) RNA polymerase subunit homology among cyanobacteria, other eubacteria and archaebacteria. J. Bacteriol., 170, 4136–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahmuradov I.A. et al. (2003) PlantProm: a database of plant promoter sequences. Nucleic Acids Res., 31, 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev V., Salamov A. (2011) Automatic annotation of microbial genomes and metagenomic sequences. In: Li RW (ed), Metagenomics and its applications in agriculture, biomedicine and environmental studies. Nova Science Publishers, Hauppauge, NY, pp. 61–78. [Google Scholar]
- Song K. (2012) Recognition of prokaryotic promoters based on a novel variable-window Z-curve method. Nucleic Acids Res., 40, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W. et al. (2007) Sigma 28 promoter prediction in members of the Gammaproteobacteria. FEMS Microbiol. Lett., 271, 222–229. [DOI] [PubMed] [Google Scholar]
- Stormo G.D. (2000) DNA binding sites: representation and discovery. Bioinformatics, 16, 16–23. [DOI] [PubMed] [Google Scholar]
- Studholme D.J., Buck M. (2000) The biology of enhancer-dependent transcriptional regulation in bacteria: insights from genome sequences. FEMS Microbiol. Lett., 186, 1–9. [DOI] [PubMed] [Google Scholar]
- Vijayan V. et al. (2011) A high resolution map of a cyanobacterial transcriptome. Genome Biol., 12, R47.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosten M.M. (1998) Eubacterial sigma-factors. FEMS Microbiol. Rev., 22, 127–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.