

Abstract
The identification of an antigenic epitope by the immune system allows for the understanding of the protective mechanism of neutralizing antibodies that may facilitate the development of vaccines and peptide drugs. Peptide scanning is a simple and efficient method that straightforwardly maps the linear epitope recognized by a monoclonal antibody (mAb). Here, the authors present an epitope determination methodology involving serially truncated recombinant proteins, synthetic peptide design, and dot-blot hybridization for the antigenic recognition of nervous necrosis virus coat protein using a neutralizing mAb. This technique relies on the dot-blot hybridization of synthetic peptides and mAbs on a polyvinylidene fluoride (PVDF) membrane. The minimum antigenic region of a viral coat protein recognized by the RG-M56 mAb can be narrowed down by step-by-step trimmed peptide mapping onto a 6-mer peptide epitope. In addition, alanine scanning mutagenesis and residue substitution can be performed to characterize the binding significance of each amino acid residue making up the epitope. The residues flanking the epitope site were found to play critical roles in peptide conformation regulation. The identified epitope peptide may be used to form crystals of epitope peptide-antibody complexes for an x-ray diffraction study and functional competition, or for therapeutics.
Keywords: Immunology, Issue 121, peptide mapping, monoclonal antibody, linear epitope, dot-blot hybridization, alanine scanning, substitution, antigenic, neutralizing antibodies, polyvinylidene fluoride
Introduction
In the immune system, the recombination of V, D, and J segments allows for antibodies to create tremendous variations of complementarity determining regions (CDRs) for binding to various antigens to protect the host from pathogenic infection. The neutralizing defense of antibodies against antigens depends on the spatial complementarity between the CDRs of the antibodies and the epitopes of the antigens. Therefore, an understanding of this molecular interaction will assist prophylactic vaccine design and therapeutic peptide drug development. However, this neutralization interaction may be influenced both by multiple antigenic domains from one single antigen and by multiple CDRs of antibodies, which consequently make the epitope determination process more complex. Fortunately, the development of hybridoma technology, which fuses individual antibody-producing cells with myeloma cells, allows for a constantly dividing batch of cells to secrete one specific antibody, known as a monoclonal antibody (mAb)1. Hybridoma cells produce these pure, high-affinity mAbs to bind to a single antigenic domain of a specific antigen. With the relationship of the antigen-antibody established, several approaches, including peptide scanning, can be used to determine the epitope of an antigen using its corresponding mAb. Recent developments in synthetic peptide technology have made the peptide scanning technique more accessible and more convenient to perform. Briefly, a set of overlapping synthetic peptides are produced according to a target antigen sequence and are associated to a solid-supported membrane for mAb hybridization. Peptide scanning not only offers a simple way to map the antibody binding region, but also facilitates amino acid (aa) mutagenesis through residue scanning or substitution to evaluate the binding interaction between each aa residue of the epitope peptide and the CDRs of the antibody.
Here, the present study describes a protocol for the efficient identification of the linear epitope of the yellow grouper nervous necrosis virus (YGNNV) coat protein using a neutralizing mAb2,3,4. The protocol includes mAb preparation, construction and expression of serially truncated recombinant proteins, synthetic overlapping peptide design, dot-blot hybridization, alanine scanning, and substitution mutagenesis. Considering the high cost of peptide synthesis, the step of serially truncating the recombinant proteins of a desired target protein was modified, and the antigenic region was narrowed down to around 100 to 200 aa residues before the synthetic peptide array dot-blot analysis was performed.
Protocol
1. Preparation of Monoclonal Antibody
Culture the RG-M56 mouse monoclonal hybridoma cells2 in serum-free medium in 175T flasks at 37 ºC with 5% CO2 supplement. Collect the supernatant when the color of the medium turns yellow after five days of incubation. NOTE: Hybridoma cells were cultured in serum-free medium to avoid antibody contamination from fetal bovine serum.
Centrifuge the supernatant at 4,500 x g for 30 min at 4 ºC and discard the cell debris pellet.
Add 2 mL of protein G agarose (supplied as a 50% slurry) to a 5 mL column and equilibrate with 10 resin volumes (10 mL) of ice-cold PBS.
Load 200 mL of the antibody supernatant (step 1.2) onto the column and discard the pass-through.
Add 10 mL of ice-cold PBS to the column to wash it. Repeat twice.
Add 10 mL of 50 mM glycine, pH 2.7 to the column to elute the protein G-associated antibody. Collect 900 µL fractions in a microcentrifuge tube containing 100 µL of 10x neutralization buffer (1 M Tris, 1.5 M NaCl, and 1 mM EDTA, pH 8.0).
Store the purified antibody in 50% glycerol with 0.03% NaN3 at -20 ºC.
2. Construction and Expression of Serially Truncated Recombinant Proteins
- Prepare a PCR reaction mixture: 5 µL of 10x Pfu buffer, 0.2 mM of each dNTP, 0.2 µM forward primer3, 0.2 µM reverse primer3, 2 mM MgSO4, 1 ng of pET20b-1A593 plasmid DNA, and 2.5 U (unit) of Pfu DNA polymerase; add ddH2O to a final volume of 50 µL.
- Run samples in an automatic thermal cycler using the following parameters: Cycle 1 (94 ºC for 5 min); cycles 2-36 (94 ºC for 30 s, 63 ºC for 30 s, and 72 ºC for 60 s); and cycle 37 (72 ºC for 7 min).
Extract the polymerase chain reaction (PCR) products by using a PCR purification kit5 to facilitate the following restriction enzyme digestion.
- Digest the PCR-amplified DNA fragments with NdeI and XhoI restriction enzymes and ligate each of these DNA fragments into NdeI and XhoI enzyme-cleaved pET-20b(+) vector. Transform the constructs into Escherichia coli DH-5α-competent cells6.
- Prepare the digestion mixture in digestion buffer (20 mM Tris-acetate, 10 mM Mg(CH3COO)2, 50 mM KCH3COO, and 1 mM DTT, pH 7.9) with 1 µg of PCR-amplified DNA or pET-20b(+) vector DNA, and 2 U of NdeI and XhoI restriction enzymes in a final volume of 20 µL.
- Mix the digestion mixture gently and quickly spin down. Incubate at 37 ºC for 2 h in a dry bath to ensure the complete cutting of the restriction sites.
- Extract the restriction enzyme-digested DNA fragments using a PCR purification kit5 to facilitate the following plasmid construction.
- Prepare the ligation mixture in ligation buffer (66 mM Tris, 5 mM MgCl2, 1 mM ATP, and 5 mM DTT, pH 7.5) with 100 ng of predigested, PCR-amplified DNA, 10 ng of predigested pET-20b(+) vector DNA, and 5 U of T4 DNA ligase in a final volume of 10 µL.
- Mix the ligation mixture gently and quickly spin down. Incubate at 16 ºC for 18 h in a water bath.
- Put 10 µL of the ligation samples into 100 µL of the DH-5α-competent cells and mix gently before placing the microcentrifuge tube on ice for 30 min. Put the microcentrifuge tube in a dry bath at 42 ºC for 90 s to induce heat shock7. Immediately transfer the tube onto ice for 2 min.
- Add 900 µL of Luria-Bertani (LB) broth (1% Bacto tryptone, 0.5% Bacto yeast extract, and 0.5% NaCl, pH 7.0) to the tube. Incubate at 37 ºC with 150 rpm shaking for 45 min. Pellet the cells by centrifugation at 4,000 x g for 10 min and discard the supernatant.
- Resuspend the pellet with 50 µL of LB broth and spread each transformation onto pre-warmed LB plates containing 100 µg/mL ampicillin. Incubate the plates at 37 ºC for 16 h.
- Pick up a single colony using a 200 µL tip and place it into 3 mL of LB broth containing 100 µg/mL ampicillin in a loosely capped 15 mL tube. Incubate at 37 ºC with 150 rpm shaking for 12 h.
After sequence confirmation, transform 10 ng of DNA from these pET-20b(+) plasmids with varying lengths of YGNNV coat protein gene into the BL-21 (DE3) strain of E. coli by using the heat-shock method7. Follow steps 2.3.6-2.3.8 to perform the transformation.
Transfer a single colony from each transformed E. coli BL-21 cell into 3 mL of LB broth containing 100 µg/mL ampicillin in a loosely capped 15 mL tube. Incubate the culture at 37 ºC with 150 rpm shaking.
Cool the culture to 25 ºC when the OD600 of the culture is about 0.6 and add IPTG to a final concentration of 0.4 mM to induce the expression of the recombinant protein. Incubate the culture for an extra 4 h at 25 ºC with 200 rpm shaking.
Transfer 1 mL of the culture into a microcentrifuge tube. Pellet the cells by centrifugation at 12,000 x g for 1 min and discard the supernatant.
Resuspend the cell pellet in 100 µL of denaturation buffer (8 M urea, 20 mM sodium phosphate, and 0.5 M NaCl, pH 7.4) by pipetting up and down with a micropipette. Mix by vigorous vortexing. NOTE: The sample solutions should now be semi-transparent, a little sticky, and ready for the dot-blot hybridization assay.
3. Design and Synthesis of Overlapping Peptides
Design and synthesize3 serial 20-mer peptides that each overlap with its successor by 10 aa residues from the 195-338 aa region of the YGNNV coat protein to narrow down the epitope region of RG-M56 mAb by dot blotting.
Design and synthesize3 three 8-mer peptides (195VNVSVLCR202, 197VSVLCRWS204, and 199VLCRWSVR206) with an overlap of 6 aa residues onto the next synthetic peptide to narrow down the epitope region of 195-206 aa by dot blotting.
Design and synthesize3 7-mer (196NVSVLCR202 and 195VNVSVLC201), 6-mer (195VNVSVL200, 196NVSVLC201, and 197VSVLCR202), and 5-mer peptides (195VNVSV199, 196NVSVL200, 197VSVLC201, and 198SVLCR202) with an overlap of 6, 5, and 4 aa residues, respectively, onto their neighboring peptides to minimize the epitope region by dot blotting.
4. Dot-blot Hybridization
Dissolve each synthesized peptide in dimethyl sulfoxide (DMSO) to a final concentration of 10 mg/mL. NOTE: To overcome the varied solubility of synthetic peptides, all synthetic peptides should be dissolved in DMSO. DMSO is a good solvent to completely dissolve hydrophobic or hydrophilic peptides.
Soak the polyvinylidene fluoride (PVDF) membrane with methanol for 2 min. NOTE: PVDF membrane is up to 100% resistant to DMSO; others may not be so.
Equilibrate the PVDF membrane with modified Towbin buffer (25 mM Tris, 192 mM glycine, and 0.1% SDS, pH 8.3) for 2 min. NOTE: 10-20% (v/v) of methanol can be added to modified Towbin buffer to improve the transfer results.
Rinse a piece of chromatography paper with the modified Towbin buffer. Place the PVDF membrane onto the chromatography paper. Wait until the modified Towbin buffer has disappeared from the PVDF membrane surface before continuing to the next step.
Add 2 µL of each peptide sample to the membrane with a 10 µL tip. Air-dry the PVDF membrane on the chromatography paper for 10 min. Add each peptide sample slowly and gradually onto the membrane to avoid too much diffusion.
Block the membrane in TBST buffer (0.05% (v/v) Tween-20, 20 mM Tris, and 150 mM NaCl, pH 7.4) with 5% nonfat milk for 30 min at room temperature with gentle shaking.
Add RG-M56 mAb at a final dilution of 1:1,000 in TBST buffer with 5% nonfat milk to the membrane. Incubate the membrane at 37 ºC for 1 h with gentle shaking.
Remove the antibody solution. Wash the membrane in TBST buffer for 5 min with gentle shaking. Repeat twice.
Add the secondary antibody (goat anti-mouse IgG, Fc, conjugated alkaline phosphatase) at a final dilution of 1:5,000 in TBST buffer with 5% nonfat milk to the membrane. Incubate the membrane at 37 ºC for 1 h with gentle shaking.
Discard the antibody solution. Wash the membrane in TBST buffer for 5 min with gentle shaking. Repeat the wash step twice.
Develop the membrane with BCIP/NBT substrate solution at room temperature for 15 min in the dark. Stop the developing by washing the membrane with ddH2O when the signal appears.
Air-dry the membrane and capture the dot-blot image using an image system.
Measure the intensity of each dot blot using image analysis software3.
5. Alanine Scanning and Substitution
Design and synthesize3 alanine and methionine substitution peptides. Replace each aa residue with alanine for the 8-mer peptide 195VNVSVLCR202 and synthesize peptides 195ANVSVLCR202, 195VAVSVLCR202, 195VNASVLCR202, 195VNVAVLCR202, 195VNVSALCR202, 195VNVSVACR202, 195VNVSVLAR202, and 195VNVSVLCA202. Replace aa residue leucine 200 to methionine to create the SJNNV genotype epitope peptide 195VNVSVMCR202.
Follow Section 4 to perform alanine scanning and substitution mutagenesis dot blotting.
Representative Results
The goal of this experiment was to identify an epitope through dot blotting using mAb. To rapidly and efficiently narrow down the antigenic region recognized by mAb, the full-length and serially truncated YGNNV recombinant coat proteins with a 6xHis fusion tag at the C-terminus were expressed from an E. coli PET expression system10 (Figure 1A). The resulting recombinant proteins were spotted onto the PVDF membrane using RG-M56 mAb and anti-6xHis antibody for dot-blot hybridization. The dot blotting revealed positive signals against 1-338 aa (full-length), 51-338 aa, and 195-338 aa, but not 1-100 aa or 1-200 aa recombinant proteins (Figure 1B). The dot array hybridized against the anti-6xHis antibody and confirmed the expression of all recombinant proteins. These data indicate that the epitope of RG-M56 mAb recognition is located in a 144-aa recombinant protein near the C-terminus of YGNNV coat protein (195-338 aa).
Subsequently, serial 20-mer peptides with 10 aa-residues-long overlaps onto their neighbor were designed and synthesized from the sequence of 144-aa recombinant protein for peptide scanning to narrow down the epitope region. These synthetic peptides were spotted on a PVDF membrane and subjected to dot-blot hybridization using RG-M56 mAb. The result showed positive signals only on the peptide 195-214 aa and the positive control, 195-338 aa recombinant protein (Figure 2A). As the epitope is located within the peptide 195-214 aa region but not the peptide 205-224 aa region, three serial 8-mer peptides with 6-aa residues overlap from aa residue 195-206 (peptides 195-202 aa, 197-204 aa, and 199-206 aa) were designed and synthesized. Dot-blot hybridization results showed positive signals on the peptide 195-202 aa and the positive-control peptide 195-214 aa using RG-M56 mAb (Figure 2B).
Alanine scanning and substitution mutagenesis were performed to evaluate the specificity of each aa residue of the 8-mer epitope, 195VNVSVLCR202. Each aa residue of the 8-mer peptide was individually replaced with alanine. The alanine mutation peptide array was then placed onto a PVDF membrane using peptide 195-202 aa as a positive control. Dot-blot analysis indicated that the three replacing mutations, V197A, V199A, and C201A, abolished the binding affinity of RG-M56 mAb (Figure 3A). Although aa residue 200 in the SJNNV genotype epitope is methionine, as opposed to a leucine in the other four Betanodavirus genotype epitopes, the SJNNV genotype sequence, 195VNVSVMCR202, showed positive binding affinity against RG-M56 mAb, like that of the positive control (Figure 3A). This result indicates that the epitopes of all Betanodavirus genotypes can be recognized by RG-M56 mAb. The binding affinity of each aa residue participating to the epitope site was further quantified by measuring the signal intensity of the dot blot of each alanine substitution using image analysis software (Figure 3B). The intensities of the V197A, V199A, and C201A substitutions were reduced to 10.2%, 18.6%, and 8.5%, respectively, when compared with those of the positive control (100%), whereas the V195A, S198A, L200A, R202A, and L200M substitutions showed higher or similar intensities as that of the positive control. It is worth noting that the influencing strength of the N196A substitution is ambiguous, with a 37.4% reduction in the positive control. These results indicate that V197, V199, and C201 are essential residues for the binding of RG-M56 mAb.
The alanine scanning mutagenesis results reveal that the aa residues V195, N196, and R202 can be replaced with alanine, which implies that the epitope region could be further narrowed down at both termini. Therefore, the step-by-step trimmed peptide mapping through 7-mer, 6-mer, and 5-mer synthetic peptides step was designed to minimize the antigenic region. The positive signal was present on the 6-mer synthetic peptide 196NVSVLC201, but not on the 7-mer and 5-mer synthetic peptides (Figure 4). These data indicate that the minimal epitope of NNV coat protein recognized by RG-M56 mAb is the 6-mer peptide, 196NVSVLC201.
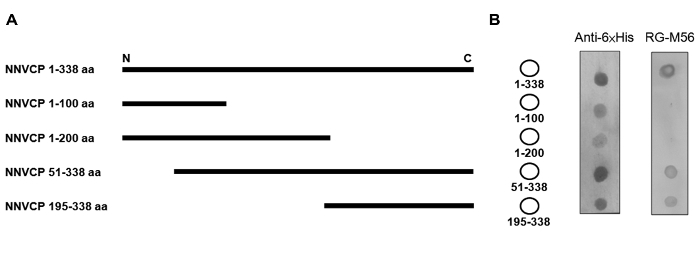 Figure 1: Reduction of the epitope region using serially truncated recombinant proteins and monoclonal antibody. (A) Map of serially truncated recombinant NNVCPs. NNVCP 1-338 aa is the full-length coat protein. (B) Dot-blot analysis of recombinant NNVCPs. Left: Map of the serially truncated YGNNV recombinant coat proteins on the PVDF membrane. Middle: The dot-blot analysis was performed using anti-6xHis antibody. Right: The dot-blot analysis was performed using RG-M56 mAb. NNVCP: nervous necrosis virus coat protein; aa: amino acid; PVDF: polyvinylidene fluoride; N: N-terminus; C: C-terminus; mAb: monoclonal antibody. Please click here to view a larger version of this figure.
Figure 1: Reduction of the epitope region using serially truncated recombinant proteins and monoclonal antibody. (A) Map of serially truncated recombinant NNVCPs. NNVCP 1-338 aa is the full-length coat protein. (B) Dot-blot analysis of recombinant NNVCPs. Left: Map of the serially truncated YGNNV recombinant coat proteins on the PVDF membrane. Middle: The dot-blot analysis was performed using anti-6xHis antibody. Right: The dot-blot analysis was performed using RG-M56 mAb. NNVCP: nervous necrosis virus coat protein; aa: amino acid; PVDF: polyvinylidene fluoride; N: N-terminus; C: C-terminus; mAb: monoclonal antibody. Please click here to view a larger version of this figure.
 Figure 2: Fine mapping of the epitope region using synthetic peptides. (A) Left: Amino acid sequences of the 20-mer synthetic peptides were aligned to NNVCP 195-338 aa; each preceding peptide had a 10 aa-residues-long overlap with the following peptide. Right: Map of the synthetic peptides on the PVDF membrane. Recombinant NNVCP 195-338 aa was used as a positive control. The dot-blot analysis was performed using RG-M56 mAb. (B) Left: Amino acid sequences of the 8-mer synthetic peptides, 195-202 aa, 197-204 aa, and 199-206 aa; each preceding peptide had a 6 aa-residues-long overlap with the following peptide. Synthetic peptide 195-214 aa was used as a positive control. Right: The dot-blot analysis was performed using RG-M56 mAb. Please click here to view a larger version of this figure.
Figure 2: Fine mapping of the epitope region using synthetic peptides. (A) Left: Amino acid sequences of the 20-mer synthetic peptides were aligned to NNVCP 195-338 aa; each preceding peptide had a 10 aa-residues-long overlap with the following peptide. Right: Map of the synthetic peptides on the PVDF membrane. Recombinant NNVCP 195-338 aa was used as a positive control. The dot-blot analysis was performed using RG-M56 mAb. (B) Left: Amino acid sequences of the 8-mer synthetic peptides, 195-202 aa, 197-204 aa, and 199-206 aa; each preceding peptide had a 6 aa-residues-long overlap with the following peptide. Synthetic peptide 195-214 aa was used as a positive control. Right: The dot-blot analysis was performed using RG-M56 mAb. Please click here to view a larger version of this figure.
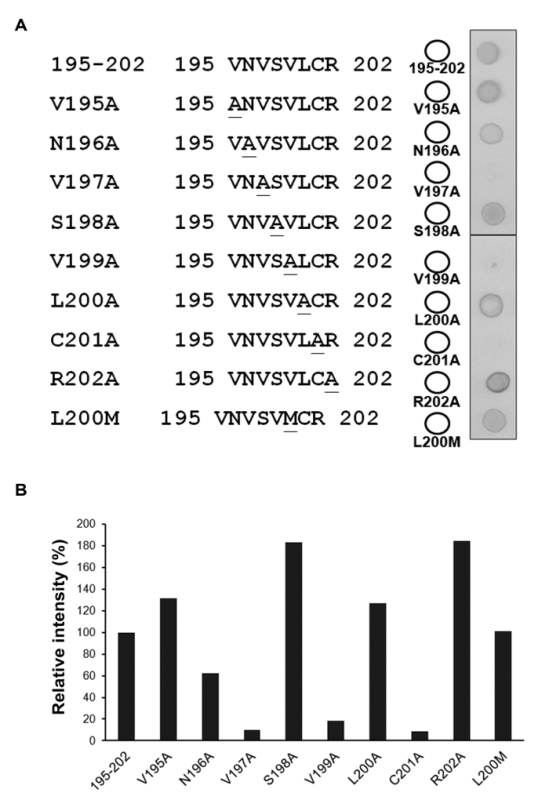 Figure 3: Alanine scanning and substitution mutagenesis of the 195VNVSVLCR202 epitope. (A) Amino acid sequences of substitution mutagenesis and dot-blot analysis. Each aa residue of the epitope region, 195-202 aa, was replaced individually with alanine. L200M substitution is the SJNNV genotype sequence at the Betanodavirus coat protein epitope region. Synthetic peptide 195-202 aa was used as a positive control. The replaced aa residues are underlined. (B) Quantification of the dot blot signal binding affinity. The signal intensity of the positive control (195-202 aa) was determined as 100%. Please click here to view a larger version of this figure.
Figure 3: Alanine scanning and substitution mutagenesis of the 195VNVSVLCR202 epitope. (A) Amino acid sequences of substitution mutagenesis and dot-blot analysis. Each aa residue of the epitope region, 195-202 aa, was replaced individually with alanine. L200M substitution is the SJNNV genotype sequence at the Betanodavirus coat protein epitope region. Synthetic peptide 195-202 aa was used as a positive control. The replaced aa residues are underlined. (B) Quantification of the dot blot signal binding affinity. The signal intensity of the positive control (195-202 aa) was determined as 100%. Please click here to view a larger version of this figure.
 Figure 4: Minimum epitope determination. The 7-mer (A), 6-mer (B), and 5-mer (C) synthetic peptides from 195-202 aa were used to identify the minimum epitope recognized by RG-M56 mAb. Synthetic peptide 195-202 aa was used as a positive control. Please click here to view a larger version of this figure.
Figure 4: Minimum epitope determination. The 7-mer (A), 6-mer (B), and 5-mer (C) synthetic peptides from 195-202 aa were used to identify the minimum epitope recognized by RG-M56 mAb. Synthetic peptide 195-202 aa was used as a positive control. Please click here to view a larger version of this figure.
Discussion
This protocol offers a rapid and straightforward technique to identify a mAb-recognized linear epitope. Taking into consideration the cost of peptide synthesis and the production efficiency of synthesizing peptides, the antigenic region of the virus coat protein was reduced by expressing serially truncated recombinant proteins before peptide scanning analysis. As such, the reliable and efficient E. coli pET expression system was used to produce these serially truncated recombinant proteins, as recombinant proteins with molecular weights between 10 to 50 kDa can be easily expressed through this system. In this way, the epitope can be easily narrowed down to a more manageable 100 to 200 aa region. The pET-20b(+) vector was specifically chosen, as it contains a sequence that codes for the expression of 6xHis-tags, allowing the produced 6xHis-tag fusion proteins to be immunodetected using an anti-6xHis antibody to confirm the expression of the recombinant proteins. The produced recombinant coat proteins were then analyzed using RG-M56 mAb via a dot-blot hybridization assay. An alternative method of recombinant protein epitope determination is to purify the expressed recombinant proteins via immobilized metal ion affinity chromatography10, separate the recombinant proteins with SDS-polyacrylamide gel electrophoresis, and perform Western blot analysis3.
To further and more finely map the epitope location determined by the results of dot-blot hybridization analysis using serially truncated recombinant proteins, overlapping synthetic peptides with different sizes were designed. Among the different possible lengths of synthetic peptide to synthesize, 20-mer with 10 aa-residues-long overlapping peptides were chosen first in the peptide scanning, both for their high synthesis purity (at around 90%) and for their peptide length, enough for the search for the continuous epitope recognized by B-cell antibody11. Note that the accuracy and purity of synthesized peptides deteriorate as the synthesized peptide is longer. In this way, the epitope region was rapidly reduced to around 10 aa residues in length. After serial 8-mer overlapping peptides were surveyed, the linear epitope region was defined to peptide 195-202 aa. Subsequently, alanine scanning of this 8-mer epitope peptide reveals the critical binding affinity strength of each aa residue, allowing the search for the minimal epitope. The essential roles of the V197, V199, and C201 aa residues implies that the linear epitope region covers at least 5 aa residues, from 197 to 201. Moreover, aa residues V195, N196, and R202 can be replaced with alanine without completely losing binding affinity, indicating that the epitope region may be reduced to 7, 6, or even 5 aa residues in length. Small peptide sequences can be readily and economically synthesized for the search of linear (continuous) epitopes. However, this synthetic peptide scanning technique is not suitable for the determination of a discontinuous epitope of an antibody, unless it is combined with epitope excision and mass spectrometric analyses12.
In this protocol, a dot-blot hybridization technique was used to search the linear epitope of a mAb. Dot-blot hybridization is a simple but effective method. In the beginning of the search, when the epitope region is narrowed down from a large-scale landscape, the main concern is to observe either a positive or negative signal after the hybridization of the antibody to a target membrane-bound protein, as most mAbs only bind to a specific epitope of an antigenic protein (Figures 1 and 2). However, when using dot-blot hybridization to explore the binding availability of each aa residue within the epitope region against the antibody, such as by alanine substitution mutagenesis, the signal intensity of each substituted aa residue determined by dot blotting should be factored into the overall binding significance. That signal intensity can be quantified easily using image analysis software (Figure 3B) or a densitometer. Alternatively, an enzyme-linked immuno-sorbent assay (ELISA) can be performed to quantify the degree of binding affinity and the resulting signal strength3.
In the previous study, the sole coat protein of non-envelope nervous necrosis virus was immuno-recognized by 10 mAbs with a high neutralization index value between 6.5 to 4.5 (log10 NI)2. The highly specific recognition ability of the mAbs were further used for the development of the one-step, rapid immunochromatographic diagnostic kit for the detection of NNV-infected fish13. The antigenic epitope of nervous necrosis virus coat protein was recognized by the RG-M18 mAb as an 8-mer peptide, 195VNVSVLCR2023, through which a novel NNV receptor was identified (unpublished data). In the present study, the epitope of nervous necrosis virus coat protein was further narrowed down to a 6-mer peptide, 196NVSVLC201,by the other mAb, RG-M56.
It is unexpected that two 7-mer peptides (196NVSVLCR202 and 195VNVSVLC201) containing 6-mer peptide 196NVSVLC201 are not recognized by RG-M56 mAb (Figure 4A). A reasonable interpretation is that, although the surrounding residues V195 and R202 may not directly contribute to the binding interaction between the epitope and antibody, the flanking residues influence the formation of the correct peptide conformation for antibody recognition. The appearance of V195 or R202 at their flanking terminus alone may contort the synthetic peptide conformation for antibody recognition and binding. The epitope conformation is driven by the strength of both termini, V195 and R202, which are balanced against each other in the 8-mer synthetic peptide, 195VNVSVLCR202,and counteracted in the 6-mer synthetic peptide, 196NVSVLC201. The aa residues, V195, N196, and R202, can be individually replaced with alanine without completely losing binding ability, and thus, the alanine scanning mutagenesis results indicate that these three aa residues may not play a significant role in the recognition and binding of RG-M56 mAb. However, after trimming one more residue from the 6-mer synthetic peptide, 196NVSVLC201, the 5-mer peptide, 197VSVLC201, without the N196 in the N-terminal flanking region, loses the ability to be recognized and bound by RG-M56 mAb (Figure 4C). This result suggests that the N196 residue may also play an important role in the flanking region of the epitope to stabilize the correct epitope conformation in order to facilitate the recognition and binding of RG-M56 mAb. The importance of flanking residues surrounding the epitope region had also been explored by other antigen-antibody binding studies. The significance of flanking aa residues surrounding the α-bungarotoxin epitope region for the binding of antibodies was investigated using different aa substitutions within the same cholinergic subsite. They were then evaluated as either essential, influential, or not influential14. It was also found that the specificity of the antibody-recognized epitope of carcinoma-associated epithelial mucins can be further influenced by the flanking aa residues. These effects may present conformational barriers that can impede the binding of an antibody to an epitope15.
The 6-mer epitope, 196NVSVLC201,has extremely hydrophobic features, with four hydrophobic residues, including two valines (197 and 199), one leucine (200), and one cysteine (201) (reduced form). Residues V197, V199, and C201 are critical for RG-M56 mAb recognition and binding, as determined by alanine scanning mutagenesis. The epitope region was situated at one of eight anti-parallel β-strands of the shell domain (S-domain) of the NNV coat protein16. Interestingly, the epitope does not appear on the outside protrusion domain, but hides in the jelly-roll structure of the S-domain under the other anti-parallel β-strands. The epitope, with its high hydrophobicity, may obtain a more stable microenvironment in this depression. Moreover, the 195VNVSVLCR2023 peptide of this epitope was found to hinder the propagation of the giant grouper nervous necrosis virus in grouper brain cells. Therefore, this epitope peptide was suggested to be a competitor involved in the receptor-binding domain required for viral entry3. It was hypothesized that peptide entry inhibitors comprising hydrophobic and/or amphipathic residues can alter the physical conformation and chemistry of cellular membrane interfaces and can impede the fusion of cellular and viral membranes17. Furthermore, many synthetic peptide entry inhibitors have demonstrated strong inhibitive properties against various virus infections17,18. Thus, the identified epitope peptide with hydrophobic residues and strong entry inhibition against NNV infection may facilitate the development of therapeutic peptide drugs.
Disclosures
The authors have no conflicts of interest related to this report.
Acknowledgments
The authors thank Miss Ching-Chun Lin and Miss Diana Lin of the Core Facility of the Institute of Cellular and Organismic Biology (ICOB) of Academia Sinica for offering their expertise on peptide synthesis and DNA sequencing, respectively. This study was supported by Academia Sinica.
References
- Milstein C, Kohler G. Clonal variations of myelomatous cells (proceedings) Minerva Med. 1977;68(50):3453. [PubMed] [Google Scholar]
- Lai YS, et al. In vitro neutralization by monoclonal antibodies against yellow grouper nervous necrosis virus (YGNNV) and immunolocalization of virus infection in yellow grouper Epinephelus awoara (Temminck & Schlegel) J Fish Dis. 2001;24(4):237–244. [Google Scholar]
- Chen CW, Wu MS, Huang YJ, Cheng CA, Chang CY. Recognition of Linear B-Cell Epitope of Betanodavirus Coat Protein by RG-M18 Neutralizing mAB Inhibits Giant Grouper Nervous Necrosis Virus (GGNNV) Infection. PLoS One. 2015;10(5):0126121. doi: 10.1371/journal.pone.0126121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y-S, et al. Propagation of yellow grouper nervous necrosis virus (YGNNV) in a new nodavirus-susceptible cell line from yellow grouper, Epinephelus awoara (Temminck & Schlegel), brain tissue. J Fish Dis. 2001;24(5):299–309. [Google Scholar]
- Lougee E, Morjaria S, Shaw O, Collins R, Vaughan R. A new approach to HLA typing designed for solid organ transplantation: epityping and its application to the HLA-A locus. Int J Immunogenet. 2013;40(6):445–452. doi: 10.1111/iji.12053. [DOI] [PubMed] [Google Scholar]
- Radulovich N, Leung L, Tsao MS. Modified gateway system for double shRNA expression and Cre/lox based gene expression. BMC Biotechnol. 2011;11:24. doi: 10.1186/1472-6750-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froger A, Hall JE. Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp. 2007. p. e253. [DOI] [PMC free article] [PubMed]
- Pronobis MI, Deuitch N, Peifer M. The Miraprep: A Protocol that Uses a Miniprep Kit and Provides Maxiprep Yields. PLoS One. 2016;11(8):e0160509. doi: 10.1371/journal.pone.0160509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzker ML. Emerging technologies in DNA sequencing. Genome Res. 2005;15(12):1767–1776. doi: 10.1101/gr.3770505. [DOI] [PubMed] [Google Scholar]
- Chiu CC, John JA, Hseu TH, Chang CY. Expression of ayu (Plecoglossus altivelis) Pit-1 in Escherichia coli: its purification and immunohistochemical detection using monoclonal antibody. Protein Expr Purif. 2002;24(2):292–301. doi: 10.1006/prep.2001.1558. [DOI] [PubMed] [Google Scholar]
- Atassi MZ. Antigenic structures of proteins. Their determination has revealed important aspects of immune recognition and generated strategies for synthetic mimicking of protein binding sites. Eur J Biochem. 1984;145(1):1–20. doi: 10.1111/j.1432-1033.1984.tb08516.x. [DOI] [PubMed] [Google Scholar]
- Opuni KF, et al. Mass spectrometric epitope mapping. Mass Spectrom Rev. 2016. [DOI] [PubMed]
- Chang CY, Chiu CC, Christopher John JA. In: The Aquaculture of Groupers. Liao IC, Leaño EM, editors. Taiwan Asian Fisheries Society, World Aquaculture Society, The Fisheries Society of Taiwan, National Taiwan Ocean University, Taiwan Press; 2008. pp. 207–224. [Google Scholar]
- Conti-Tronconi BM, et al. Alpha-bungarotoxin and the competing antibody WF6 interact with different amino acids within the same cholinergic subsite. Biochemistry. 1991;30(10):2575–2584. doi: 10.1021/bi00224a003. [DOI] [PubMed] [Google Scholar]
- Briggs S, Price MR, Tendler SJ. Fine specificity of antibody recognition of carcinoma-associated epithelial mucins: antibody binding to synthetic peptide epitopes. Eur J Cancer. 1993;29(2):230–237. doi: 10.1016/0959-8049(93)90181-e. [DOI] [PubMed] [Google Scholar]
- Chen NC, et al. Crystal Structures of a Piscine Betanodavirus: Mechanisms of Capsid Assembly and Viral Infection. PLoS Pathog. 2015;11(10):e1005203. doi: 10.1371/journal.ppat.1005203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badani H, Garry RF, Wimley WC. Peptide entry inhibitors of enveloped viruses: The importance of interfacial hydrophobicity. Biochim Biophys Acta. 2014. [DOI] [PMC free article] [PubMed]
- Qureshi NM, Coy DH, Garry RF, Henderson LA. Characterization of a putative cellular receptor for HIV-1 transmembrane glycoprotein using synthetic peptides. AIDS. 1990;4(6):553–558. doi: 10.1097/00002030-199006000-00009. [DOI] [PubMed] [Google Scholar]